

Abstract
Purpose
Torsades de pointes (TdP) tachycardias are triggered, polymorphic ventricular arrhythmias arising from early afterdepolarizations (EADs) and increased dispersion of repolarization. Ranolazine is a new agent which reduces pathologically elevated late INa but also IKr. Aim of this study was to evaluate the effects of ranolazine in a validated isolated Langendorff-perfused rabbit heart model.
Methods
TdP was reproducibly induced with d-sotalol (10−4 mol/L) and low potassium (K) (1.0 mmol/L for 5 min, pacing at CL 1000 ms). In 10 hearts, ECG and 8 epi- and endocardial monophasic action potentials were recorded. Action potential duration (APD) was measured at 90% repolarization and dispersion defined as APD max–min.
Results
D-sotalol prolonged APD90 and increased dispersion of APD90, simultaneously causing EADs and induction of TdP. The combination of d-sotalol and two concentrations of ranolazine did not increase dispersion of ventricular APD90 as compared to vehicle. Ranolazine at 5 μmol/L did not cause additional induction of EADs and/or TdP but also did not significantly suppress arrhythmogenic triggers. The higher concentration of ranolazine (10 μmol/L) in combination with d-sotalol caused further prolongation of APD90, at the same time reduction in APD90 dispersion. In parallel, the incidence of EADs was reduced and an antitorsadogenic effect was seen.
Conclusions
In the healthy isolated rabbit heart (where late INa is not elevated), ranolazine does not cause proarrhythmia but exerts antiarrhythmic effects in a dose-dependent manner against d-sotalol/low K-induced TdP. This finding—despite additional APD prolongation—supports the safety of a combined use of both drugs and merits clinical investigation.
Introduction
Drug-induced Torsades de Pointes (TdP) can be caused by multiple compounds and has become an important issue in current pharmacological development. Manifestation of TdP is a classic proarrhythmic effect of class III antiarrhythmic drugs such as sotalol. Although occurring rarely, TdP represents a potentially lethal polymorphic ventricular tachycardia.
TdP usually manifest in conjunction with QT-interval prolongation or if patients present with particular risk factors. Many antiarrhythmic drugs have the ability to prolong action potential (AP) duration (APD) and refractoriness by inhibition of K-currents when most needed, at fast heart rates, thereby compromising their antiarrhythmic effects. Reverse use-dependence of APD is a physiological capacity of the healthy heart wherein the APD usually shortens at increasing heart rate. Some antiarrhythmic drugs also exhibit a reverse use-dependent effect meaning the ability of the drug to preferentially prolong cardiac APD rather at slow than at fast heart rates [1,2].
Under normal conditions, most Na channels open only transiently and become quickly inactivated, thereby producing the peak Na current which produces the upstroke of the AP. There is increasing recognition of the importance of this small late component of INa (late INa) due to Na channels that remain active, inactivate with much slower kinetics, or reopen. The amplitude of this current is small in healthy myocardium but may persist for hundreds of milliseconds (i.e., during the whole AP) [3]. Therefore, the long persistence and its slow inactivation kinetics make late INa relevant [4,5] when increased as it has been shown in pathological conditions such as ischemia, hypoxia, elevated reactive oxygen, heart failure, and nitrogen species [6–10].
Late INa has been shown to act as a potent contributor to severe arrhythmias and its pharmacological inhibition seems to be of antiarrhythmic potential [11] (for review see [3]). An elevated late INa causes prolongation of the cardiac AP making early afterdepolarizations (EADs) more likely to occur [12,13]. Additionally, cellular Na cycling is tightly integrated with Ca homeostasis, as Na modulates the transport direction of the Na/Ca exchanger (NCX) in addition to the membrane potential. Cellular Na overload and AP prolongation stimulate the reverse mode NCX facilitating the efflux of Na in exchange for influx of Ca [14]. The net result is an elevated diastolic Ca concentration making late afterdepolarizations (DADs) more likely to occur via “leaky” cardiac ryanodine receptors (RyR2) of the sarcoplasmic reticulum and NCX which possibly generates a depolarizing current (transient inward current, ITI) that give rise to delayed afterdepolarizations (DADs) [6,13–16].
Ranolazine is currently the most potent clinical inhibitor of late INa [3]. In multicellular tissue and in cardiac myocytes, ranolazine was shown to cause direct and indirect concentration-, voltage-, as well as frequency-dependent inhibition of late INa [17–20]. In cardiomyocytes isolated from failing canine hearts, ranolazine was found to be 38-fold more potent in inhibiting late than peak INa; 50% inhibition of peak and late INa occur at concentrations of 244 and 6.5 μmol/L, respectively [17]. However, it remains unclear how this drug produces its selective effect on late INa.
It is important to note that ranolazine also has inhibitory effects on IKr which would rather prolong cardiac APD [20,21]. This is very likely the basis for a moderate QT-interval prolongation in patients treated with ranolazine [22]. Because of concerns regarding QT-interval prolongations, ranolazine is currently contraindicated in combination with class III antiarrhythmic drugs apart of amiodarone. On the basis of the particular pharmacological action that this drug has, the present study was designed to investigate the net effect of ranolazine on APD and ventricular APD dispersion in healthy isolated rabbit hearts where late INa is not relevantly elevated [18]. Furthermore, another objective of this study was to investigate the dose-dependent safety of ranolazine in combination with sotalol, both of which are known to inhibit K-channels, under physiological conditions and in the presence of low K in order to test for TdP formation.
Methods
Animals and Materials
New Zealand white rabbits (3.5–5.0 kg) were used. All procedures regarding care and use of animals were in accordance with institutional guidelines. Rabbits were heparinized and anaesthetized with sodium thiopental (50 mg/kg/i.v). Hearts were rapidly removed and immersed in ice-cold Tyrode’s solution. The aorta was cannulated and perfused with oxygenized Tyrode’s solution at 37°. Finally, the heart was transferred and mounted on a modified Langendorff apparatus. The intrinsic heart rate was slowed by mechanical ablation of the AV node leading to complete AV block and a ventricular rhythm of 40–50 pbm. The heart was then paced by means of a monophasic action potential (MAP) combination catheter positioned into the right ventricle as described previously [23,24].
MAP Recordings
A ring with 6 evenly spaced cantilever arms holding silver–silver chloride contact MAP electrodes was mounted around the central stub of the Langendorff setup. Two electrodes were placed on the right ventricle, and the remaining four were evenly spread over the left ventricular epicardium. Two additional MAP’s were recorded by means of the right ventricular MAP combination catheter used for pacing and another catheter introduced to the left ventricular endocardium. Data were acquired in digital form (1000 data points/s) and stored on a computer hard disk for later analyses [23,24].
Measurement of Action Potential Duration and Dispersion of Ventricular Repolarization
Action potential duration was measured at 90% repolarization (APD90) by means of a validated automatic and interactive software program (LabView 2.2 National Instruments, Austin TX, USA) [1,2]. An average APD90 of all analyzable MAP locations was calculated. Dispersion of APD90 was defined as the range of action potential durations measured at 90% from all electrodes (APD90 max.–APD90 min). Activation time was defined as the average time between the pacing artifact and the fastest MAP upstroke. Only recordings with at least 6 MAP signals of high quality including both endocardial signals were considered for the final analysis. The stability of MAP recordings was excellent over the time course of experiments.
Protocols
Experiments were performed in a paired manner meaning that vehicle, d-sotalol and finally d-sotalol and ranolazine were exposed to the same heart after washout periods. After achieving steady state at CL of 1000 ms, the pacing CL was decreased stepwise to 800, 500, and 400 ms. Steady state at a certain CL was considered after 100 beats, which had to be undisturbed by premature ventricular contractions for at least 25 beats. After completion of the baseline recordings in 20 experiments, d-sotalol was added to the buffer solution: d-sotalol was applied at a concentration of 10−4 mol/L, and the stimulation protocol was repeated again. Then, the solution was switched to a low K solution containing 1.2 mmol/L and stimulation to a CL of 1000 ms. When triggered arrhythmias ensued, pacing was discontinued so as not to disturb these. After a washout period, the same protocol was repeated as before but now with a solution containing either ranolazine at 5 or 10 μmol/L ion addition to d-sotalol.
Statistics
All data are shown as mean ± SEM. Student’s t-test or two-way repeated-measure ANOVA with post hoc tests was used to test for significance. A two-sided P value of <0.05 was considered significant.
Results
Effects on Whole Heart Action Potential Duration
Isolated rabbit hearts were superfused and paced with decreasing cycle lengths. Action potential duration measured at 90% repolarization (APD90) was prolonged from 205 ± 24 ms to 238 ±22 ms (CL 800 ms) in the presence of 10−4 mol/L d-sotalol (Figure 1B–C, P < 0.05 vs. vehicle, n = 5 each). Similarly, the combination of d-sotalol and 5 μmol/L ranolazine prolonged APD90 to 251 ± 25 ms (CL 800 ms, P < 0.01 vs. vehicle, n = 5). However, APD90 was not different between hearts that were either treated with d-sotalol alone or in combination with 5 μmol/l ranolazine (P = 0.52 RM-ANOVA).
Figure 1.
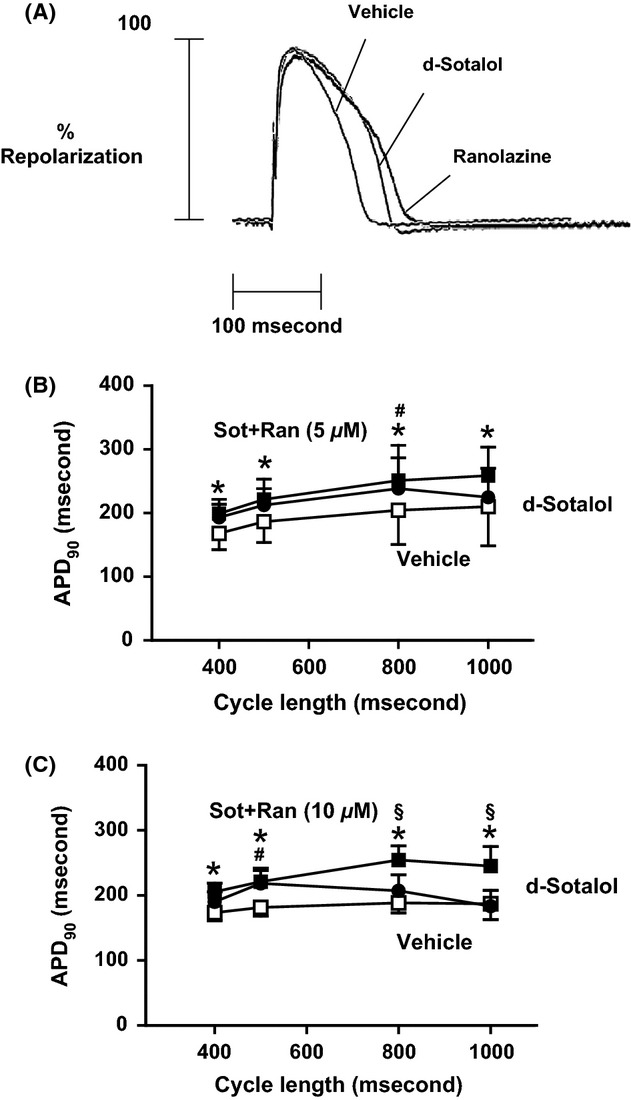
Effects of sotalol and ranolazine on APD90. (A) Original tracings of representative action potentials at a CL of 800 ms. (B) Sotalol exhibits significant APD prolongation but not in combination with 5 μmol/L ranolazine compared with vehicle (n = 5 each). (C) Sotalol exhibits significant APD prolongation which was further prolonged by addition of 10 μmol/L ranolazine (n = 5 each). P < 0.05: *vehicle versus sot+ran, #vehicle versus sot, and §sot+ran versus sot.
Conversely, a higher concentration of ranolazine at 10 μmol/L led to a further prolongation of APD90 when added to d-sotalol compared with d-sotalol alone (Figure 1C, n = 5 each). When paced with a cycle length of 800 ms, the APD90 was further prolonged from 207 ± 11 ms in the presence of d-sotalol to 255 ± 10 ms after addition of 10 μmol/L ranolazine (P < 0.05, n = 5 each).
The same observation holds true when changing to a low potassium Tyrode’s solution. There was no further prolongation of APD90 when adding 5 μmol/L ranolazine to d-sotalol compared with d-sotalol alone (Figure 2A, n = 5 each, P = 0.1 RM-ANOVA). However, addition of 10 μmol/L ranolazine to d-sotalol prolonged APD90 compared with d-sotalol alone (Figure 2B, n = 5 each, P < 0.01 RM-ANOVA).
Figure 2.
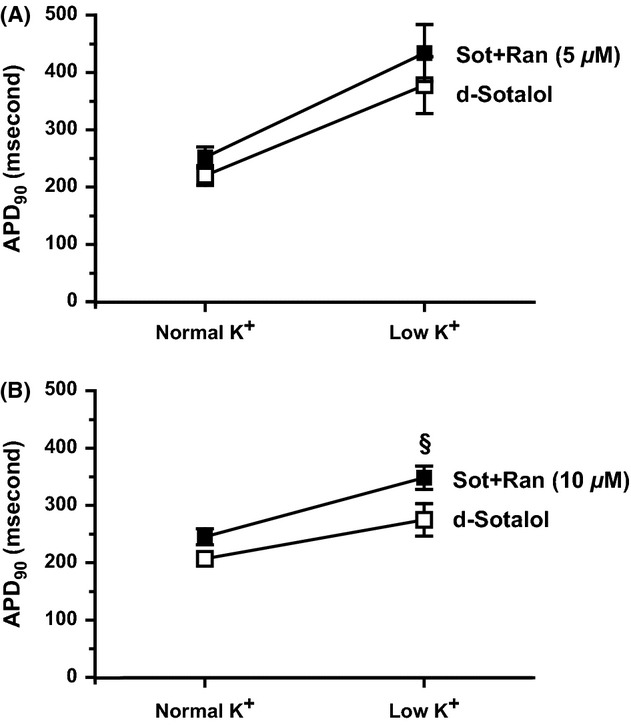
Effects of transient low K on APD90 of sotalol- and ranolazine-treated hearts. K was lowered to 25% for 5 min. (A) No significant differences of APD90 were detected between d-sotalol-treated and d-sotalol- and 5 μmol/L ranolazine-treated hearts (n = 5 each). (B) APD90 was significantly increased when 10 μmol/L ranolazine were added to d-sotalol compared with d-sotalol alone (n = 5 each, P < 0.05, sot+ran vs. sot). $ d-sot: normal K versus low K P < 0.05. + sot+ran: normal K versus low K P < 0.05.
Effects on Whole Heart Action Potential Dispersion
Dispersion of APD90 was increased after temporarily lowering K concentration (1.0 mmol/L for 5 min) during bradycardic pacing at CL 1000 ms. D-sotalol alone and in combination with either 5 or 10 μmol/L ranolazine largely increased APD90 dispersion under low K concentrations (Figures3A,C, P < 0.05 vs. baseline, n = 5 each). Before TdP was induced, steady-state pacing was carried out at CLs of 400, 500, 800, and 1000 ms. Figure 3B depicts that d-sotalol markedly increased dispersion of APD90 at a CL of 800 ms from 37 ± 4 ms to 72 ± 10 ms (P < 0.01). Addition of 5 μmol/L ranolazine to d-sotalol reversed this effect to 48 ± 8 ms which was not statistical different to vehicle (Figure 3B, P = 0.26 RM-ANOVA). Conversely, addition of 10 μmol/L ranolazine to d-sotalol reduced APD90 dispersion compared with d-sotalol alone (Figure 3D, P < 0.05 RM-ANOVA).
Figure 3.
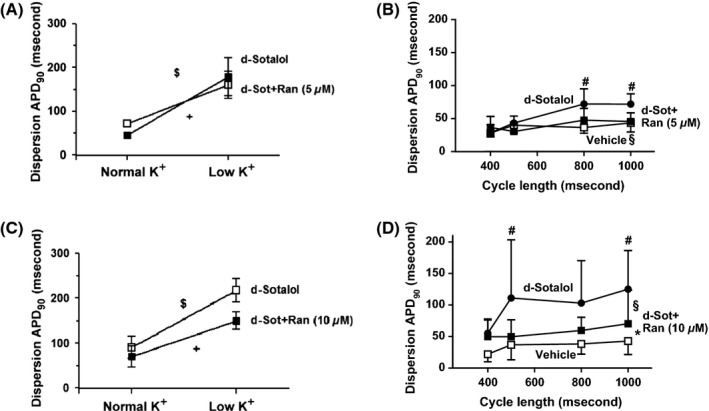
Effects of sotalol and ranolazine on APD90 dispersion upon transient low K. (A) Sotalol alone or in combination with 5 μmol/L ranolazine significantly increased APD90 dispersion upon transient low K (P < 0.05 vs. normal K). (B) Sotalol increased APD90 dispersion compared with vehicle, whereas 5 μmol/L ranolazine did not (n = 5 each, P < 0.05). (C) Sotalol alone or in combination with 10 μmol/L ranolazine significantly increased APD90 dispersion upon transient low K (P < 0.05 vs. normal K). (D) Sotalol increased APD90 dispersion compared with vehicle which could be significantly reduced by addition of 10 μmol/L ranolazine (n = 5 each, P < 0.05). P < 0.05: *vehicle versus sot+ran, #vehicle versus sot, and §sot+ran versus sot.
Effects on EAD and TdP Inducibility
No EADs were seen under steady-state conditions with normal potassium concentration. Upon lowering K to 25% for 5 min, EADs and TdP could be observed in all experiments. EADs were counted for 120s in each experiment. Interestingly, there was no difference in the amount of EADs in the presence of d-sotalol alone or in combination with 5 μmol/L ranolazine. An average of 108 ± 5 EADs/120s occurred in the presence of d-sotalol compared with 104 ± 14/120s when 5 μmol/L ranolazine was added to the solution (Figure 4E, P = 0.79). However, the higher concentration of ranolazine was capable to largely reduce the occurrence of EADs triggered by low K solution and d-sotalol. In these experiments, addition of 10 μmol/L ranolazine largely reduced the occurrence of EADs from 99 ± 13/120 s to 45 ± 12/120 s (Figure 4G, P < 0.05).
Figure 4.

Effects of sotalol and ranolazine on formation of EADs and TdPs. (A) Representative original tracing showing EADs in a d-sotalol-treated heart. (B–D) Original recordings showing that TdP occurred in d-sotalol-treated hearts, to a lower extend when 5 μmol/L ranolazine was added and could be suppressed in the presence of 10 μmol/L ranolazine. (E) Mean values of EADs in hearts treated with sotalol alone or in combination with 5 μmol/L ranolazine. (F) Mean values of TdP incidence in hearts treated with sotalol alone or in combination with 5 μmol/L ranolazine. (G) Mean values of EADs in hearts treated with sotalol alone or in combination with 10 μmol/L ranolazine (§P < 0.05). (H) Mean values of TdP incidence in hearts treated with sotalol alone or in combination with 10 μmol/L ranolazine (§P < 0.05).
TdP was defined as runs of polymorphic VT that consist of at least three beats. Under normal conditions without low K solution, no TdP was observed. Upon induction, TdP coincided with increased EADs, APD, and dispersion. As Figure 4F presents, hearts that were treated with d-sotalol alone developed 9 ± 3 TdP/experiment. Addition of 5 μmol/L ranolazine caused a trend toward a reduction in the incidence of TdP to 5 ± 3/experiment that was, however, not statistically significant (P = 0.37). In another series of experiments, we doubled the concentration of ranolazine to 10 μmol/L. Interestingly, now ranolazine effectively suppressed the incidence of TdP from 4 ± 1/experiment to 0.6 ± 0.6/experiment (Figure 4H, P < 0.05).
Discussion
The present study was designed to investigate the electrophysiological consequences and safety of a combination of the class III antiarrhythmic drug d-sotalol and ranolazine in a validated isolated Langendorff-perfused rabbit heart model with the following major findings: (1) d-sotalol prolonged APD and significantly increased the APD dispersion thereby causing induction of EADs and TdP. (2) The combination of d-sotalol with 5 μmol/L ranolazine did not further prolong APD and lacked the induction of EADs and/or TdP but, however, did not significantly suppress arrhythmogenic triggers. (3) The higher concentration of ranolazine (10 μmol/L) in combination with d-sotalol caused further prolongation of the APD, but significantly reduced APD dispersion and most importantly the incidence of EADs and triggered arrhythmias and consequently exhibited antitorsadogenic effects.
The Isolated Langendorff-Perfused Rabbit Heart Model
Our isolated Langendorff-perfused rabbit heart model provides a simple crystalloid-perfused isolated heart that allows for simultaneous recordings of multiple MAP signals from the myocardium (both endocardium and epicardium of both ventricles) [25]. As a whole heart model, it is therefore well suited to observe the generation of arrhythmias from local early afterdepolarizations (EADs) and in the circumstances of an increased dispersion of repolarization such as torsade de pointes (TdP) arrhythmias usually are [26,27]. Verification of the occurrence of TdP arrhythmias can then be performed using the simultaneous multilead volume-conducted ECG of the model. This has been used to correlate even simple ECG indexes, such as an increased dispersion of the QT interval and an increased T wave area, to the occurrence of triggered arrhythmias [23,24]. The same ECG indexes may also be measured in patients and may serve as measures of proarrhythmic risk in patients treated with drugs that prolong action potential duration.
Differential Modulation of the APD
D-sotalol is a class III antiarrhythmic potassium channel blocker affecting the rapidly activating delayed rectifier K current (IKr) but has also beta-blocking capacities. D-sotalol is commonly used for rhythm-control treatment of atrial fibrillation (AF). The combination of class III antiarrhythmic drugs apart of amiodarone with ranolazine is currently contraindicated making our findings of clinical relevance in particular due to the fact that recent studies suggest that ranolazine may also be of therapeutically interest for the treatment of AF [3,28–32].
D-sotalol caused a significant prolongation of the APD in our model. Addition of 5 μmol/L ranolazine did not pronounce this effect while 10 μmol/L caused a further prolongation. This finding of a dose-dependent effect of ranolazine on the cardiac APD is remarkable. Although ranolazine also has at least weak peak INa inhibiting effects, this compound has an up to 38-fold higher potency for late INa than peak INa with an IC50 of 6.5 vs. 244 μmol/L [17] in ventricular myocytes with a therapeutic range of ranolazine of 2–8 μmol/L. Inhibition effects of ranolazine on late INa was demonstrated both indirectly and directly in multicellular myocardium and in isolated myocytes in several diseased conditions [17–19]. This capacity would lead to an abbreviation of the APD due to inhibition of a net inward current which has not been observed in our study. Similarly to d-sotalol, ranolazine also inhibits IKr in cardiomyocytes with a potency of ∼12 μmol/L [20,21]. Blocking IKr causes prolongation of the ventricular APD, whereas inhibition of late INa has the opposite effect and abbreviates the AP. Therefore, the net effect of ranolazine on APD is mainly driven by the relative magnitude of the reductions in late INa (inward) and IKr (outward) currents during the repolarization period [3]. Of note, it has been also shown that ranolazine exerts differential effects on APD throughout the ventricular wall due to differences in late INa amplitude [20]. Because we used healthy rabbit hearts where late INa amplitude is small [18], ranolazine caused no APD abbreviation in contrast to models with elevated late INa such as heart failure, ischemia and when late INa is pharmacologically elevated [10,17–19,33]. It is, however, interesting to note that potent blockade of IKr by ranolazine outweighed its effect on late INa leading to a further net APD prolongation when d-sotalol was combined with 10 μmol/L ranolazine. Our results are in line with the findings of a recent study investigating the effects of ranolazine in addition to different class III antiarrhythmic drugs. Frommeyer and coworkers also report that 10 μmol/L ranolazine further increased the APD compared with d-sotalol alone without investigating lower concentrations of ranolazine [34]. The dose dependency revealed in our study might be attributed to the different IC50 values of ranolazine for IKr and late INa. On the one hand, 5 μmol/L ranolazine is closed to the reported IC50 of 6.5 μmol/L to inhibit late INa making a potent inhibition very likely, while the IC50 to inhibit IKr is about 12 μmol/L [17,20,21]. On the other hand, 10 μmol/L ranolazine is closer to the IC50 to inhibit IKr making a more potent inhibition of this current and consequent APD prolongation very likely.
Antitorsadogenic Effects of Ranolazine
Drug-induced TdP has emerged as one of the most significant concerns in the safety of compounds and a major obstacle to new drug development. Occurrence of TdP is the classic proarrhythmic effect of class III drugs as d-sotalol that has been causally linked to an increased dispersion of ventricular repolarization [35]. D-sotalol was shown to cause a rate-dependent and concentration-dependent increase in dispersion of ventricular repolarization in our isolated rabbit heart model [23,24]. Both drugs, d-sotalol and ranolazine, are capable to inhibit IKr, thereby prolonging the QT interval of the ECG in patients (modestly with ranolazine) [22]. This is the reason why it is important to determine whether the combination causes a more pronounced and thus probably dangerous dispersion of ventricular APD than d-sotalol alone. Here, we show that the increased dispersion of ventricular APD well investigated for d-sotalol does not become potentiated in combination with ranolazine. The contrary is the case; ranolazine was capable to reduce the increased dispersion of ventricular APD as it was also reported by Frommeyer and coworkers earlier [34]. Another study shows that endogenous (physiological) late INa, although small in amplitude, indeed modulates ventricular repolarization, APD, and arrhythmogenesis when repolarization reserve was decreased by treatments of rabbit hearts with E-4031, 4-aminopyridine, and BaCl2 to decrease Ito, IKr, and IK1, respectively [36]. In their experiments, tetrodotoxin or ranolazine significantly decreased APD, transmural dispersion of repolarization, beat-to-beat variability, and the QT interval. Taken together with our findings, it can be stated that endogenous late INa seems to be arrhythmogenic in hearts with decreased repolarization reserve and that inhibition of this small current using ranolazine reduces arrhythmogenic triggers. Accordingly, we found a decreased occurrence of EADs when 10 μmol/L ranolazine was added to d-sotalol.
Episodes of TdP was induced by means of bradycardia, a high concentration of d-sotalol, and transient reduction in K concentrations in the perfusate. The induction was reproducible when the protocol was repeated. All of the inducing factors as well as the intermediate CL and particularly the polymorphic undulating pattern of the simultaneous ECG suggest that the arrhythmias induced are similar to TdP observed in the clinical setting [23,24]. Transmural differences of late INa and hence APD might increase transmural dispersion of repolarization and QT interval, which underlies the development of TdP tachyarrhythmias [37]. The potential of ranolazine to suppress EADs induced by IKr blockers which are considered as triggers for the initiation of proarrhythmia has been reported in a previous study [19]. It may be attributed to the inhibition of the physiologically late INa, which becomes a more pronounced contributor of ventricular repolarization when IKr and/or IKs are reduced [36]. Probably different amplitudes of late INa throughout the LV wall may also account for our observations. Late INa has been supposed to be largest in midmyocardial myocytes that can be characterized by having APs that prolong disproportionately relative compared with the APs of other LV myocytes in response to drugs that prolong QT interval [38]. Accordingly, ranolazine produced a preferential abbreviation of midmyocardial APD, thereby causing a reduction in transmural dispersion of repolarization [20]. Finally, our findings confirm previous reports showing a potent suppression of TdP in vitro in isolated rabbit hearts pretreated with d-sotalol, in vivo in anesthetized rabbits treated with the IKr blocker clofilium and in a chronic atrioventricular block dog model where TdP was induced using dofetilide [34,39,40]. The latter one also found a dose dependency of ranolazine to suppress arrhythmogenic triggers.
Clinical Implications
Ranolazine has been reported to cause a slight prolongation of the ECG QT interval in patients [22]. In general, extended QT intervals increase the risk of sudden cardiac death. Because of this, ranolazine is contraindicated in combination with class III antiarrhythmic drugs apart of amiodarone. Our findings challenge this contraindication due to the fact that addition of ranolazine in both concentrations did neither potentiate arrhythmogenic triggers nor increase the likelihood of TdP. The contrary is the case, 10 μmol/L ranolazine in addition to d-sotalol sufficiently suppressed the occurrence of TdP in our model although further APD prolongation was present. This finding further substantiates that ranolazine is a safe drug despite of its IKr-blocking properties at least in our rabbit model. Combination of ranolazine with amiodarone has been reported in studies to be safe [31] and is approved. To our knowledge, there is no study in men which investigates the safety and efficacy of a combination with d-sotalol and ranolazine for the treatment of arrhythmias. Only one clinical trials was recently performed (HARMONY, EudraCT-Nr: 2011-001134-42), investigating ranolazine alone or in combination with dronedarone with respect to atrial fibrillation burden in subjects with paroxysmal atrial fibrillation and implanted pacemakers. Another trial RAFFAELLO (EudraCT-Nr: 2011-002789-18) is a dose-ranging study testing the efficacy and safety of three doses of ranolazine versus placebo in maintaining sinus rhythm after successful electrical cardioversion in subjects with persistent atrial fibrillation.
Application of ranolazine does not cause an increase in cardiovascular death or all-cause mortality in the Metabolic Efficiency with Ranolazine for Less Ischemia in Non-ST-Elevation Acute Coronary Syndromes–Thrombolysis In Myocardial Infarction 36 (MERLIN–TIMI 36) trial. Antiarrhythmic effects of ranolazine were observed in this study by using continuous Holter monitoring. There was a ∼36% reduction in ventricular tachycardia lasting ≥8 beats in ranolazine-treated patients [11]. Most importantly for our study, the incidence of sudden cardiac death was significantly elevated in the placebo group with a QTc of more than 450 ms, but not in patients taking ranolazine. Moreover, in patients with impaired ejection fraction (EF < 40%), sudden cardiac death was markedly but not significantly (P = 0.07) reduced by ∼45% in patients treated with ranolazine.
Taken together, ranolazine appears to have antiarrhythmic ventricular effects in patients after admission for ACS. Nevertheless, studies specifically designed to evaluate the potential role of ranolazine as an antiarrhythmic agent on ventricular arrhythmias and in combination with other antiarrhythmic drugs are warranted.
Conclusion
Our results in the healthy rabbit in vitro model clearly show that ranolazine in addition to d-sotalol does not potentiate the proarrhythmic risk of d-sotalol related to IKr blockade although APD is further prolonged by the combination with 10 μmol/L ranolazine. This is likely explained by its inhibitory effects on late INa of ranolazine. Clinical studies are clearly needed to investigate the role and safety of this drug on top of class III antiarrhythmic agents in order to prevent proarrhythmia but also as a new treatment option of ventricular and atrial arrhythmias.
Conflict of Interest
Dres. Sossalla and Maier receive speaker’s honoraria from Berlin-Chemie.
Acknowledgments
S.S. is supported by the German Heart Foundation/German Foundation of Heart Research through a research grant. S.S. and L.S.M. are funded by the Deutsche Forschungsgemeinschaft (DFG) through the SFB 1002 (TP A03).
We thank Christian Bauer and Christiane Schulz for their excellent support.
References
- 1.Dorian P, Newman D. Rate dependence of the effect of antiarrhythmic drugs delaying cardiac repolarization: An overview. Europace. 2000;2:277–285. doi: 10.1053/eupc.2000.0114. [DOI] [PubMed] [Google Scholar]
- 2.Wu L, Ma J, Li H, et al. Late sodium current contributes to the reverse rate-dependent effect of IKr inhibition on ventricular repolarization. Circulation. 2011;123:1713–1720. doi: 10.1161/CIRCULATIONAHA.110.000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sossalla S, Maier LS. Role of ranolazine in angina, heart failure, arrhythmias, and diabetes. Pharmacol Ther. 2012;133:311–322. doi: 10.1016/j.pharmthera.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Belardinelli L, Shryock JC, Fraser H. Inhibition of the late sodium current as a potential cardioprotective principle: Effects of the late sodium current inhibitor ranolazine. Heart. 2006;92(Suppl 4):iv6–iv14. doi: 10.1136/hrt.2005.078790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Makielski JC, Farley AL. Na+ current in human ventricle: Implications for sodium loading and homeostasis. J Cardiovasc Electrophysiol. 2006;17(Suppl 1):S15–S20. doi: 10.1111/j.1540-8167.2006.00380.x. [DOI] [PubMed] [Google Scholar]
- 6.Maltsev VA, Sabbah HN, Higgins RS, Silverman N, Lesch M, Undrovinas AI. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998;98:2545–2552. doi: 10.1161/01.cir.98.23.2545. [DOI] [PubMed] [Google Scholar]
- 7.Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM. Intracellular Na+ concentration is elevated in heart failure but Na+/K+ pump function is unchanged. Circulation. 2002;105:2543–2548. doi: 10.1161/01.cir.0000016701.85760.97. [DOI] [PubMed] [Google Scholar]
- 8.Valdivia CR, Chu WW, Pu J, et al. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005;38:475–483. doi: 10.1016/j.yjmcc.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 9.Ju YK, Saint DA, Gage PW. Hypoxia increases persistent sodium current in rat ventricular myocytes. J Physiol. 1996;497:337–347. doi: 10.1113/jphysiol.1996.sp021772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song Y, Shryock JC, Wagner S, Maier LS, Belardinelli L. Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J Pharmacol Exp Ther. 2006;318:214–222. doi: 10.1124/jpet.106.101832. [DOI] [PubMed] [Google Scholar]
- 11.Scirica BM, Morrow DA, Hod H, et al. Effect of ranolazine, an antianginal agent with novel electrophysiological properties, on the incidence of arrhythmias in patients with non ST-segment elevation acute coronary syndrome: Results from the Metabolic Efficiency With Ranolazine for Less Ischemia in Non ST-Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN-TIMI 36) randomized controlled trial. Circulation. 2007;116:1647–1652. doi: 10.1161/CIRCULATIONAHA.107.724880. [DOI] [PubMed] [Google Scholar]
- 12.Maltsev VA, Silverman N, Sabbah HN, Undrovinas AI. Chronic heart failure slows late sodium current in human and canine ventricular myocytes: Implications for repolarization variability. Eur J Heart Fail. 2007;9:219–227. doi: 10.1016/j.ejheart.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song Y, Shryock JC, Belardinelli L. An increase of late sodium current induces delayed afterdepolarizations and sustained triggered activity in atrial myocytes. Am J Physiol Heart Circ Physiol. 2008;294:H2031–H2039. doi: 10.1152/ajpheart.01357.2007. [DOI] [PubMed] [Google Scholar]
- 14.Bers DM. Excitation-contraction coupling and cardiac contractile force. 2nd edn. Dordrecht, Netherlands: Kluwer Academic Publishers; 2001. [Google Scholar]
- 15.Lederer WJ, Tsien RW. Transient inward current underlying arrhythmogenic effects of cardiotonic steroids in Purkinje fibres. J Physiol. 1976;263:73–100. doi: 10.1113/jphysiol.1976.sp011622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sossalla S, Fluschnik N, Schotola H, et al. Inhibition of elevated Ca2+/calmodulin-dependent protein kinase II improves contractility in human failing myocardium. Circ Res. 2010;107:1150–1161. doi: 10.1161/CIRCRESAHA.110.220418. [DOI] [PubMed] [Google Scholar]
- 17.Undrovinas AI, Belardinelli L, Undrovinas NA, Sabbah HN. Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J Cardiovasc Electrophysiol. 2006;17(Suppl 1):S169–S177. doi: 10.1111/j.1540-8167.2006.00401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sossalla S, Wagner S, Rasenack EC, et al. Ranolazine improves diastolic dysfunction in isolated myocardium from failing human hearts–role of late sodium current and intracellular ion accumulation. J Mol Cell Cardiol. 2008;45:32–43. doi: 10.1016/j.yjmcc.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 19.Song Y, Shryock JC, Wu L, Belardinelli L. Antagonism by ranolazine of the pro-arrhythmic effects of increasing late INa in guinea pig ventricular myocytes. J Cardiovasc Pharmacol. 2004;44:192–199. doi: 10.1097/00005344-200408000-00008. [DOI] [PubMed] [Google Scholar]
- 20.Antzelevitch C, Belardinelli L, Zygmunt AC, et al. Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation. 2004;110:904–910. doi: 10.1161/01.CIR.0000139333.83620.5D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rajamani S, Shryock JC, Belardinelli L. Rapid kinetic interactions of ranolazine with HERG K+ current. J Cardiovasc Pharmacol. 2008;51:581–589. doi: 10.1097/FJC.0b013e3181799690. [DOI] [PubMed] [Google Scholar]
- 22.Chaitman BR. Ranolazine for the treatment of chronic angina and potential use in other cardiovascular conditions. Circulation. 2006;113:2462–2472. doi: 10.1161/CIRCULATIONAHA.105.597500. [DOI] [PubMed] [Google Scholar]
- 23.Zabel M, Hohnloser SH, Behrens S, Woosley RL, Franz MR. Differential effects of D-sotalol, quinidine, and amiodarone on dispersion of ventricular repolarization in the isolated rabbit heart. J Cardiovasc Electrophysiol. 1997;8:1239–1245. doi: 10.1111/j.1540-8167.1997.tb01014.x. [DOI] [PubMed] [Google Scholar]
- 24.Zabel M, Hohnloser SH, Behrens S, Li YG, Woosley RL, Franz MR. Electrophysiologic features of torsades de pointes: Insights from a new isolated rabbit heart model. J Cardiovasc Electrophysiol. 1997;8:1148–1158. doi: 10.1111/j.1540-8167.1997.tb01001.x. [DOI] [PubMed] [Google Scholar]
- 25.Zabel M, Portnoy S, Franz MR. Electrocardiographic indexes of dispersion of ventricular repolarization: An isolated heart validation study. J Am Coll Cardiol. 1995;25:746–752. doi: 10.1016/0735-1097(94)00446-W. [DOI] [PubMed] [Google Scholar]
- 26.Milberg P, Frommeyer G, Kleideiter A, et al. Antiarrhythmic effects of free polyunsaturated fatty acids in an experimental model of LQT2 and LQT3 due to suppression of early afterdepolarizations and reduction of spatial and temporal dispersion of repolarization. Heart Rhythm. 2011;8:1492–1500. doi: 10.1016/j.hrthm.2011.03.058. [DOI] [PubMed] [Google Scholar]
- 27.Milberg P, Pott C, Fink M, et al. Inhibition of the Na+/Ca2+ exchanger suppresses torsades de pointes in an intact heart model of long QT syndrome-2 and long QT syndrome-3. Heart Rhythm. 2008;5:1444–1452. doi: 10.1016/j.hrthm.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 28.Sossalla S, Sohns C, Seegers J, Luthje L, Vollmann D, Zabel M. Ranolazine maintained sinus rhythm in a patient with refractory symptomatic atrial fibrillation. Cardiovasc Ther. 2013;31:303–306. doi: 10.1111/1755-5922.12017. [DOI] [PubMed] [Google Scholar]
- 29.Frommeyer G, Milberg P, Uphaus T, Kaiser D, Kaese S, Breithardt G, Eckardt L. Antiarrhythmic effect of ranolazine in combination with class-III drugs in an experimental whole heart model of atrial fibrillation. Cardiovasc Ther. 2013;31:e63–e71. doi: 10.1111/1755-5922.12035. [DOI] [PubMed] [Google Scholar]
- 30.Burashnikov A, Di Diego JM, Zygmunt AC, Belardinelli L, Antzelevitch C. Atrium-selective sodium channel block as a strategy for suppression of atrial fibrillation: Differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation. 2007;116:1449–1457. doi: 10.1161/CIRCULATIONAHA.107.704890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miles RH, Murdock DK. Ranolazine verses amiodarone for prophylaxis against atrial fibrillation following coronary artery bypass surgery. Heart Rhythm. 2010;7:258. [Google Scholar]
- 32.Sossalla S, Kallmeyer B, Wagner S, et al. Altered Na+ currents in atrial fibrillation effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J Am Coll Cardiol. 2010;55:2330–2342. doi: 10.1016/j.jacc.2009.12.055. [DOI] [PubMed] [Google Scholar]
- 33.Toischer K, Hartmann N, Wagner S, et al. Role of late sodium current as a potential arrhythmogenic mechanism in the progression of pressure-induced heart disease. J Mol Cell Cardiol. 2013;61:111–122. doi: 10.1016/j.yjmcc.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frommeyer G, Kaiser D, Uphaus T, et al. Effect of ranolazine on ventricular repolarization in class III antiarrhythmic drug-treated rabbits. Heart Rhythm. 2012;9:2051–2058. doi: 10.1016/j.hrthm.2012.08.029. [DOI] [PubMed] [Google Scholar]
- 35.Surawicz B. Electrophysiologic substrate of torsade de pointes: Dispersion of repolarization or early afterdepolarizations? J Am Coll Cardiol. 1989;14:172–184. doi: 10.1016/0735-1097(89)90069-7. [DOI] [PubMed] [Google Scholar]
- 36.Wu L, Rajamani S, Li H, January CT, Shryock JC, Belardinelli L. Reduction of repolarization reserve unmasks the proarrhythmic role of endogenous late Na(+) current in the heart. Am J Physiol Heart Circ Physiol. 2009;297:H1048–H1057. doi: 10.1152/ajpheart.00467.2009. [DOI] [PubMed] [Google Scholar]
- 37.Antzelevitch C, Belardinelli L. The role of sodium channel current in modulating transmural dispersion of repolarization and arrhythmogenesis. J Cardiovasc Electrophysiol. 2006;17(Suppl 1):S79–S85. doi: 10.1111/j.1540-8167.2006.00388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Antzelevitch C, Sicouri S, Litovsky SH, et al. Heterogeneity within the ventricular wall. Electrophysiology and pharmacology of epicardial, endocardial, and M cells. Circ Res. 1991;69:1427–1449. doi: 10.1161/01.res.69.6.1427. [DOI] [PubMed] [Google Scholar]
- 39.Wang WQ, Robertson C, Dhalla AK, Belardinelli L. Antitorsadogenic effects of ({+/-})-N-(2,6-dimethyl-phenyl)-(4[2-hydroxy-3-(2-methoxyphenoxy)propyl]-1-piperazine (ranolazine) in anesthetized rabbits. J Pharmacol Exp Ther. 2008;325:875–881. doi: 10.1124/jpet.108.137729. [DOI] [PubMed] [Google Scholar]
- 40.Antoons G, Oros A, Beekman JD, et al. Late Na+ current inhibition by ranolazine reduces torsades de pointes in the chronic atrioventricular block dog model. J Am Coll Cardiol. 2010;55:801–809. doi: 10.1016/j.jacc.2009.10.033. [DOI] [PubMed] [Google Scholar]