

Abstract
The human pathogen Campylobacter jejuni is naturally competent for transformation with its own DNA. Genes required for efficient transformation in C. jejuni include those similar to components of type II secretion systems found in many Gram-negative bacteria (R. S. Wiesner, D. R. Hendrixson, and V. J. DiRita, J Bacteriol 185:5408–5418, 2003, http://dx.doi.org/10.1128/JB.185.18.5408-5418.2003). Two of these, ctsE and ctsP, encode proteins annotated as putative nucleotide binding nucleoside triphosphatases (NTPases) or nucleoside triphosphate (NTP) binding proteins. Here we demonstrate that the nucleotide binding motifs of both proteins are essential for their function in transformation of C. jejuni. Localization experiments demonstrated that CtsE is a soluble protein while CtsP is membrane associated in C. jejuni. A bacterial two-hybrid screen identified an interaction between CtsP and CtsX, an integral membrane protein also required for transformation. Topological analysis of CtsX by the use of LacZ and PhoA fusions demonstrated it to be a bitopic, integral membrane protein with a cytoplasmic amino terminus and a periplasmic carboxyl terminus. Notwithstanding its interaction with membrane-localized CtsX, CtsP inherently associates with the membrane, requiring neither CtsX nor several other Cts proteins for this association.
INTRODUCTION
The Gram-negative bacterium Campylobacter jejuni is a leading cause of bacterial gastroenteritis worldwide (1). C. jejuni often colonizes the avian intestinal tract; consequently, a common route of infection is through consumption of contaminated poultry (2).
A number of Campylobacter species are naturally competent for transformation, meaning that they can take up macromolecular DNA from the environment and incorporate it heritably into their genomes (3, 4). The ability to acquire DNA from the environment may contribute to the extensive genetic diversity observed among strains of C. jejuni (5, 6). Horizontal gene transfer in vivo has been demonstrated during experimental infection of chicks, a natural host for this pathogen (7).
Multiple genes whose products are involved in natural transformation of C. jejuni have been identified (8–12). Using transposon mutagenesis and a genetic screen for loss of competence, we isolated mutations that mapped to 11 genes encoded in C. jejuni strain 81-176 (8). Mutations in these result in a reduction in transformability to levels 4 orders of magnitude below the levels seen with the wild type (8). Among these are six genes arranged in a likely operon, some of which encode proteins similar to components of type II secretion and type IV pilus biogenesis systems and homologous to proteins important for natural transformation in other organisms (13). Two of these, ctsE and ctsP, encode putative nucleoside triphosphatases (NTPases) or nucleoside triphosphate (NTP) binding proteins, according to the annotated genome of C. jejuni strain 11168 (8, 14).
CtsE is a member of the type II secretion/type IV secretion system superfamily of NTPases collectively referred to as the PulE-VirB11 family (15, 16). Members of this family are involved in diverse processes, including secretion, pilus biogenesis, competence for natural transformation, and conjugation (15). PulE-VirB11 family members share several elements, including the nucleotide-binding motifs—Walker boxes A and B—an Asp box, and a His box (17–19). CtsE also has a tetracysteine motif conserved among the GspE and PilB/HofB subfamilies (20, 21).
ATPase activity has been demonstrated in vitro for several members of the PulE-VirB11 family (22–26). It is hypothesized that this activity powers the transformation process, though this has yet to be conclusively shown. Phylogenetic analyses placed CtsE in the type II secretion subfamily of NTPases; in one such analysis, CtsE fell between the ComG1 subfamily of Gram-positive NTPases involved in competence and the PilT and PilU subfamily involved in retraction of type IV pili (15, 16). Another analysis placed CtsE in the Gram-positive competence subfamily, closely related to the GspE subfamily of type II secretion machinery (16). Although ATPase activity has not been demonstrated for most of the type II secretion machinery, it has been hypothesized.
CtsE is one of two putative identified NTP binding proteins important for natural transformation of C. jejuni. The other is CtsP (8), which has Walker box A and B nucleotide binding motifs but lacks other characteristics of the PulE-VirB11 superfamily, including the His box, the Asp box, and the tetracysteine motif. According to BLAST analyses, CtsP exhibits weak homology to several ATPases, including PilT homologues, ClpX ATP binding subunits, and members of the AAA family of ATPases. The Walker boxes of CtsP resemble those of the AAA+ superfamily, but CtsP does not appear to have the minimal AAA consensus sequence (18, 27).
In this study, we characterized CtsE and CtsP, the two putative NTPases/NTP binding proteins required for natural transformation of C. jejuni. We also carried out analysis of a third gene product encoded in the cts gene cluster, CtsX, unique to the C. jejuni transformation system. CtsX lacks significant sequence homology to other proteins and shares no clearly conserved domains beyond a transmembrane domain. We determined the subcellular localization of each of these proteins, and we investigated the roles of the nucleotide binding motifs in CtsE and CtsP. Further analysis investigated protein-protein interactions among constituents of the type II-like Cts system in C. jejuni (8), revealing association between CtsP and CtsX.
MATERIALS AND METHODS
Bacterial strains and media.
Bacterial strains used in this work are listed in Table 1. C. jejuni was routinely cultured on Mueller-Hinton (MH) agar with 10% sheep's blood under microaerophilic conditions (5% CO2, 10% O2, balanced with N2) in a trigas incubator at 37°C. When necessary, media were supplemented with the following antibiotics at the indicated concentrations: trimethoprim (10 μg ml−1), kanamycin (as noted; either 30 or 150 μg ml−1), streptomycin (100 μg ml−1), and chloramphenicol (as noted; either 20 or 30 μg ml−1). All C. jejuni strains were stored at −80°C in MH broth with 20% glycerol.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s)a | Source or reference |
|---|---|---|
| Strains | ||
| E. coli DH5α | supE44 ΔlacU169 (ϕ80lacZΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | Laboratory strain |
| E. coli JM101 | F′ traD36 proA+B+lacIqZΔM15 Δ(lax-proAB) glnV thi | New England Biolabs |
| E. coli DH5α pRK212.1 | Contains conjugative plasmid for transferring DNA into Campylobacter | 28 |
| E. coli Sp850 | λ− e14− relA1 spoT1 cyaA1400(::kan) thi-1 | E. coli Genetic Stock Center, Yale |
| E. coli TG1 | ΔhsdS Δ(lac-proAB) supE thi [F0traD36 proAB+ lacIqZΔM15] | Laboratory strain |
| C. jejuni 81-176 | Clinical isolate | 29 |
| C. jejuni DRH153 | 81-176 astA::aphA3; Knr | 30 |
| C. jejuni DRH212 | 81-176 rpsLSm | 30 |
| C. jejuni RSW46 | DRH212 ctsX | 8 |
| C. jejuni RSW53 | 81-176 ceuB::solo | 8 |
| C. jejuni RSW57 | 81-176 ctsD::solo | 8 |
| C. jejuni RSW58 | 81-176 ctsF::solo | 8 |
| C. jejuni RSW115 | DRH212 ctsP | 8 |
| C. jejuni RSW136 | DRH212 ctsE | 8 |
| Plasmids | ||
| pT18 | Encodes T18 fragment of CyaA | 31 |
| pT25 | Encodes T25 fragment of CyaA | 31 |
| pT18-Zip | Encodes T18 fragment in frame with 35-aa leucine zipper | 31 |
| pT25-Zip | Encodes T25 fragment in frame with 35-aa leucine zipper | 31 |
| pTrcLacZ | pTrc99A with the lacZ gene (lacks codons Met 1 to Ser 7) | 32 |
| pTrcPhoA | pTrc99A with the phoA gene (lacks codons Met 1 to Met 26) | 32 |
| pECO102 | pRY112 derivative with cat promoter in XhoI-BamHI sites | 8 |
| pRSW176 | pT18 with ctsP in XhoI-ClaI sites | This study |
| pRSW184 | pT25 with ctsX in PstI-BamHI sites | This study |
| pRSW186 | pT25 with ctsE in PstI-BamHI sites | This study |
| pRSW191 | pT18 with ctsE in XhoI-ClaI sites | This study |
| pRSW206 | pECO102 with N-terminal His tag in frame with BamHI site | This study |
| pRSW208 | pECO102 with ctsP and a C-terminal FLAG tag | |
| pRSW211 | pECO102 with C-terminal FLAG tag in frame with XhoI site | This study |
| pRSW223 | pRSW211 with ctsE in BamHI-XhoI site | This study |
| pRSW228 | pRSW208 with E81A point mutation | This study |
| pRSW246 | pRSW223 with K296Q point mutation | This study |
| pRSW248 | pTrcLacZ with coding sequence for 1st 50 aa of ctsX in NcoI-XmaI site | This study |
| pRSW249 | pTrcLacZ with coding sequence for 1st 100 aa of ctsX in NcoI-XmaI site | This study |
| pRSW250 | pTrcPhoA with coding sequence for 1st 14 aa of ctsX in NcoI-XmaI site | This study |
| pRSW251 | pTrcLacZ with coding sequence for 1st 14 aa of ctsX in NcoI-XmaI site | This study |
| pRSW252 | pTrcPhoA with coding sequence for 1st 50 aa of ctsX in NcoI-XmaI site | This study |
| pRSW253 | pTrcPhoA with coding sequence for 1st 100 aa of ctsX in NcoI-XmaI site | This study |
| pRSW254 | pTrcPhoA with coding sequence of ctsX in NcoI-XmaI site | This study |
| pRSW255 | pTrcLacZ with coding sequence of ctsX in NcoI-XmaI site | This study |
| pRSW256 | pRSW211 with ctsX in BamHI-XhoI sites | This study |
| pJMB1 | pRSW206 with ctsX in BamHI-XhoI | This study |
aa, amino acids; Kn, kanamycin.
Escherichia coli strains were routinely cultured at 37°C in Luria-Bertani (LB) broth or agar. When necessary, the following antibiotics were used at the indicated concentrations: ampicillin (100 μg ml−1), chloramphenicol (30 μg ml−1), kanamycin (50 μg ml−1), and tetracycline (12.5 μg ml−1). All E. coli strains were stored at −80°C in LB broth–20% glycerol.
Construction of FLAG fusion proteins.
To express FLAG fusion proteins in C. jejuni, we modified pECO102 by mutagenesis with PFU turbo (8). Two primers were made that annealed to pECO102 and would insert the coding sequence for the FLAG tag and a stop codon downstream of the XhoI site. (All primers used in this study are indicated in Table 2). pECO102 was amplified by PCR with these primers, and the template DNA was digested with DpnI. E. coli DH5α was transformed with the reaction mixture, and clones were screened by PCR. Candidate plasmids were sequenced to confirm insertion of the FLAG sequence, and one, pRSW211, was used for subsequent cloning. To construct in-frame fusion proteins with a C-terminal FLAG tag, PCR primers were designed to amplify the coding sequence of the protein of interest from the second amino acid through the last amino acid (excluding the stop codon). The 5′ primer had BamHI sites and the 3′ primer had XhoI sites for in-frame insertion into pRSW211. All subsequent clones were verified by sequence determination.
TABLE 2.
Oligonucleotide primers used in this study
| Plasmid | Sequencea | Site | Primer nameb |
|---|---|---|---|
| pRSW176 | 5′ CCCATCGATACCCTTAATAAGCCATTCTCTAAAGCACTAAGCTTT 3′ | ClaI | Cj1473c pT18 3′ |
| pRSW176 | 5′ CCGCTCGAGGAGTAAAATTATTCCATTTAGAGAAGAAATTTTTC 3′ | XhoI | Cj1473c pT18 5′ |
| pRSW184 | 5′ CGCGGATCCCTACTTTCCATCTAATTCCATTAAACC 3′ | BamHI | Cj1472c pT25 3′ |
| pRSW184 | 5′ AAAACTGCAGGGCAAGAAAGAATTAAAGAGCTTGAGCTTAGG 3′ | PstI | Cj1472c pT25 5′ |
| pRSW186 | 5′ CGCGGATCCTCATCTTACAACCCTTAAAAGCTCATC 3′ | BamHI | Cj1471c pT25 3′ |
| pRSW186 | 5′ AAAACTGCAGGGGAAAGTAGAATGGATAAAATTTTTCAAGC 3′ | PstI | Cj1471c pT25 5′ |
| pRSW191 | 5′ CCCATCGATACTCTTACAACCCTTAAAAGCTCATCTATAC 3′ | ClaI | Cj1471c pT18 3′ |
| pRSW191 | 5′ CCGCTCGAGGGAAAGTAGAATGGATAAAATTTTTCAAGC 3′ | XhoI | Cj1471c pT18 5′ |
| pRSW208 | 5′ CGCGGATCCAGTAAAATTATTCCATTTAGAGAAGAA 3′ | BamHI | Cj1473c comp strt |
| pRSW208 | 5′ CCGCTCGAGTTATTTGTCATCATCGTCCTTATAATCCCTTAATAAGCCATTCTCTAAAGCACT 3′ | XhoI | FLAG |
| pRSW211 | 5′ ATCAAGCTTATCGATACCGTCGACCTCGAGGATTATAAGGACGATGATGACAAATAAGGGGGGCCCGGTACCCAGCTTTT3′ | pECO102 FLAG tag 1 | |
| pRSW211 | 5′ AAAAGCTGGGTACCGGGCCCCCCTTATTTGTCATCATCGTCCTTATAATCCTCGAGGTCGACGGTATCGATAAGCTTGAT3′ | pECO102 FLAG tag 2 | |
| pRSW223 | 5′ CGCGGATCCCAAAGTAGAATGGATAAAATTTTTCAAGCT 3′ | BamHI | Cj1471c comp strt |
| pRSW223 | 5′ CCGCTCGAGTCTTACAACCCTTAAAAGCTCATCTAT 3′ | XhoI | Cj1471c no stp cod |
| pRSW228 | 5′ CAAGGTCAGAGTTAAATCATTCTTGATGCAGTGGGAATGTATGATTATGCAA3′ | CtsP E81A 1 | |
| pRSW228 | 5′ TTGCATAATCATACATTCCCACTGCATCAAGAATGATTAAACTCTGACCTTG3′ | CtsP E81A 2 | |
| pRSW246 | 5′ AAACACGCGTATAAAGTTGTGCTTTGACCACTACCTGTAGGACCGGTTAAT3′ | Cj1471c K296Q 1 | |
| pRSW246 | 5′ATTAACCGGTCCTACAGGTAGTGGTCAAAGCACAACTTTA TACGCGTGTTT3′ | Cj1471c K296Q 2 | |
| pRSW248 | 5′ CTCATCCATGGCGCAAGAAAGAATTAAAGAGCTTGAGCTT 3′ | NcoI | CtsX fusion 5′ |
| pRSW248 | 5′ TCCCCCCGGGAATATTTTTTTGCTTATTATATTTTTGCAT 3′ | XmaI | CtsX 50 aa fusion 3′ |
| pRSW249 | 5′ CTCATCCATGGCGCAAGAAAGAATTAAAGAGCTTGAGCTT 3′ | NcoI | CtsX fusion 5′ |
| pRSW249 | 5′ TCCCCCCGGGTTTAACTTCTTCTAGTTCTTTATATAACTT 3′ | XmaI | CtsX 100 aa fusion 3′ |
| pRSW250 | 5′ ACAATTTCACACAGGAAACAGACCATGCAAGAAAGAATTAAAGAGCTTGAGCTTAGGTATAAATACTTTCCTGTTCTGGAAAAC CGGGCTGCTCAG 3′ | Nterm 14aa PhoA1 | |
| pRSW250 | 5′ CTGAGCAGCCCGGTTTTCCAGAACAGGAAAGTATTTATACCTAAGCTCAAGCTCTTTAATTCTTTCTTGCATGGTCTGTTTCCTGTG TGAAATTGT 3′ | Nterm 14aa PhoA2 | |
| pRSW251 | 5′ ACAATTTCACACAGGAAACAGACCATGCAAGAAAGAATTAAAGAGCTTGAGCTTAGGTATAAATACTTTCTGGCCGTCGTTTTACA ACGGGGATCC 3′ | Nterm 14aa LacZ1 | |
| pRSW251 | 5′ GGATCCCCGTTGTAAAACGACGGCCAGAAAGTATTTATACCTAAGCTCAAGCTCTTTAATTCTTTCTTGCATGGTCTGTTTCCT GTGTGAAATTGT 3′ | Nterm 14aa LacZ2 | |
| pRSW252 | 5′ CTCATCCATGGCGCAAGAAAGAATTAAAGAGCTTGAGCTT 3′ | NcoI | CtsX fusion 5′ |
| pRSW252 | 5′ TCCCCCCGGGAATATTTTTTTGCTTATTATATTTTTGCAT 3′ | XmaI | CtsX 50 aa fusion 3′ |
| pRSW253 | 5′ CTCATCCATGGCGCAAGAAAGAATTAAAGAGCTTGAGCTT 3′ | NcoI | CtsX fusion 5′ |
| pRSW253 | 5′ TCCCCCCGGGTTTAACTTCTTCTAGTTCTTTATATAACTT 3′ | XmaI | CtsX 100 aa fusion 3′ |
| pRSW254 | 5′ CTCATCCATGGCGCAAGAAAGAATTAAAGAGCTTGAGCTT 3′ | NcoI | CtsX fusion 5′ |
| pRSW254 | 5′ TCCCCCCGGGCTTTCCATCTAATTCCATTAAACCATA 3′ | XmaI | CtsX full fusion 3′ |
| pRSW255 | 5′ CTCATCCATGGCGCAAGAAAGAATTAAAGAGCTTGAGCTT 3′ | NcoI | CtsX fusion 5′ |
| pRSW255 | 5′ TCCCCCCGGGCTTTCCATCTAATTCCATTAAACCATA 3′ | XmaI | CtsX full fusion 3′ |
| pRSW256 | 5′ CGCGGATCCCAAGAAAGAATTAAAGAGCTTGAGCTT 3′ | BamHI | Cj1472c comp strt |
| pRSW256 | 5′ CCGCTCGAGCTTTCCATCTAATTCCATTAAACCATA 3′ | XhoI | Cj1472c no stp cod |
| pJMB1 | 5′ CGCGGATCCAGTAAAATTATTCCCATTTAGAGAAGAA 3′ | BamHI | CtsX comp strt + BamHI |
| pJMB1 | 5′ CCGCTCGAGTTACCTTAATAAGCCATTCTCTAAAGA 3′ | XhoI | CtsX comp no stp cod +XhoI |
Italic sequence characters represent restriction enzyme cut sites.
Abbreviations: aa, amino acids; comp strt, complement start codon; no stp cod, no stop codon.
Construction of Walker box point mutants.
Point mutants were constructed in the Walker boxes of ctsE and ctsP by PFU mutagenesis (Stratagene). Both ctsE and ctsP were cloned into pRSW211 to create C-terminal FLAG fusions pRSW223 (CtsE-FLAG) and pRSW208 (CtsP-FLAG). The conserved lysine residue (K296) of Walker box A of CtsE-FLAG was mutated by changing the coding sequence from AAA to CAA, resulting in an amino acid change to a glutamine (K296Q); this plasmid was designated pRSW246. The conserved glutamic acid residue (E81A) of Walker box B of CtsP-FLAG was mutated by changing the coding sequence from GAA to GCA, resulting in an amino acid change to an alanine (E81A); this plasmid was designated pRSW228. DH5α pRK212.1 was transformed with these constructs, and conjugations were performed as described previously (28, 33). pRSW208 and pRSW228 were conjugated into strain DRH212 ΔctsP (RSW115), while pRSW223 and pRSW246 were conjugated into strain DRH212 ΔctsE (RSW136) (8). Transconjugants were verified by PCR. Expression of the fusion proteins was detected in whole-cell lysates by SDS-PAGE and Western blot analysis as discussed below.
Construction of His fusion proteins.
To express a 6×His-tagged version of CtsX, genomic DNA from C. jejuni strain 81-176 was used as a template to amplify ctsX from the second codon to the stop codon using a forward primer with a BamHI site and a reverse primer with an XhoI site flanking the coding sequence. This fragment was cloned into the BamHI and XhoI sites of pBW206, a derivative of pECO102 that contains an N-terminal 6×His tag immediately upstream of the BamHI site, creating JMB1. This plasmid was transformed into DH5α pRK212.1 and transferred by conjugation into the ΔctsX strain as described previously (28, 33). Transconjugants were verified by PCR, and the stability of the fusion proteins was assessed using SDS-PAGE and Western blot analysis of whole-cell lysates.
Transformation assays.
Transformation assays were performed as described previously (3, 8). Briefly, C. jejuni was grown 16 to 18 h on MH agar plates supplemented with appropriate antibiotics. Cells were resuspended in MH broth to an optical density at 600 nm of 0.5, and 500-μl aliquots were added to 13-mm-long test tubes containing 1 ml of solidified agar. After incubation for 3 h at 37°C in 5% CO2, 1 μg of DRH153 (81-176 astA::aphA3) DNA was added and the cultures were incubated for an additional 4 h (30). The number of transformants and the total number of bacteria were determined by dilution plating on MH agar with appropriate antibiotics. Transformation efficiency values represent the number of transformants per total number of bacteria per microgram of DNA. Transformations were conducted in triplicate, and the transformation efficiency data represent averages of the results from three samples from one experiment.
Cell fractionations.
Cell fractionation was carried out as previously described for C. jejuni with a few modifications (34). C. jejuni was cultured on MH agar plates under microaerophilic conditions at 37°C for 16 to 18 h. Cells were resuspended from plates in MH broth and centrifuged (10,000 × g, 10 min at 4°C), and the resulting pellet was resuspended in 10 mM HEPES (pH 7.4). After one freeze-thaw cycle in a dry ice-ethanol bath, cells were sonicated with a microtip in a Branson digital sonifier at 30% amplitude six times for 10 s each time. Cell debris was pelleted by centrifugation (10,000 × g for 10 min at 4°C in a Sorvall tabletop centrifuge), and the supernatants were subjected to ultracentrifugation to form membrane pellets (100,000 × g for 70 min at 4°C with an SW41 rotor). The supernatant was removed and saved as the soluble fraction; the pellet (membrane fraction) was washed once in 10 mM HEPES and subsequently resuspended in an appropriate volume of 10 mM Tris-HCl (pH 8.0). The soluble fraction contains cytoplasmic and periplasmic contents, while the membrane fraction contains both inner and outer membranes. The same procedure was followed for fractionation of E. coli. Protein concentrations were determined with the Bio-Rad protein assay or with the ThermoScientific BCA assay per the manufacturer's instructions. Equal concentrations of proteins were then analyzed by SDS-PAGE and Western blot analysis.
Membrane flotation.
C. jejuni was cultured on MH agar plates under microaerophilic conditions at 37°C for 16 to 18 h. Cells were resuspended from plates in MH broth and centrifuged (Sorvall tabletop centrifuge; 10,000 × g for 10 min at 4°C), and the resulting pellet was resuspended in P buffer (100 mM sodium phosphate [pH 7.6], 50 mM MgCl2, 10 mM EDTA). Cells were lysed by passage through a French press. Lysates were incubated with DNase for 30 min and centrifuged (5,000 rpm for 5 min at 4°C) to remove cell debris. The supernatant was subjected to ultracentrifugation (100,000 × g for 1 h at 4°C; SW41 rotor), and a sample of the supernatant, containing the cytosolic and periplasmic proteins, was collected. The pellet was resuspended in P buffer and centrifuged (100,000 × g for 1 h at 4°C; SW41 rotor). The supernatant was discarded, and the pellet was resuspended in P buffer and mixed with 81% sucrose (dissolved in P buffer) to reach a final concentration of 71%. The sample was overlaid with 52% and 42% sucrose and subjected to ultracentrifugation (18 h at 100,000 × g and 4°C; SW41 rotor). After ultracentrifugation, the presence of the membrane bands was noted. Fractions (1.8 ml) were taken, starting from the top of the sample, and were labeled 1 to 6 sequentially. The sucrose was diluted in P buffer, and proteins were precipitated using 2% deoxycholate and trichloroacetic acid (TCA) overnight at 4°C. After the proteins were precipitated, fractions were subjected to ultracentrifugation (1.5 h at 100,000 × g and 4°C; SW41 rotor). Pellets from each fraction were washed with acetone to remove residual TCA and subsequently with 100% ether to remove lipids. Pellets were vacuum dried and resuspended in water for testing in the isocitrate dehydrogenase assay or resuspended in 6× sample buffer for SDS-PAGE and immunoblot analysis.
SDS-PAGE and immunoblotting.
To detect FLAG fusion proteins, samples were diluted 1:1 with 2× SDS sample buffer and boiled for 5 min before being loaded onto 12% polyacrylamide gels for separation by SDS-PAGE. For His-CtsX-containing samples, samples were diluted 1:6 with 6× SDS sample buffer and were separated on a 15% polyacrylamide gel. Samples were normalized by protein concentration. Proteins were transferred to nitrocellulose membranes (Hoefer semidry; 200 mA for 2 h), and, after blocking with 5% milk–Tris-buffered saline (TBS), membranes were probed with primary antibody. For FLAG-tagged fusion proteins, an anti-FLAG M2 monoclonal antibody–peroxidase conjugate (Sigma) was used at 1:1,000. This was developed using Western Lighting Chemiluminescence Reagent Plus (Perkin-Elmer Life Sciences). For the His-tagged fusion protein, THE His tag antibody (Genscript) was used at 1:1,500. A rat anti-mouse IgG1 horseradish peroxidase (HRP)-conjugated antibody (Southern Biotech) was used at 1:3,500 for the secondary antibody, and blots were developed using SuperSignal West Pico chemiluminescent substrate (ThermoScientific).
Isocitrate dehydrogenase assays.
To demonstrate efficient fractionation, fractions were tested for the presence of isocitrate dehydrogenase, a cytoplasmic protein. Isocitrate dehydrogenase activity was assayed as described previously (34). The reaction mixture contained 100 μl 10 mM MgCl2, 100 μl 50 mM Tris-HCl (pH 8.0), 100 μl 10 mM NADP, 550 to 600 μl distilled H2O, and 0 to 50 μl of the cell fraction. The reaction was started by the addition of 100 μl of 50 mM sodium isocitrate, and the optical density at 340 nm was read at 15-s intervals for 3 min. The specific activity of isocitrate dehydrogenase was calculated using an absorption coefficient of NADPH of 6.22 mM−1 cm−1 at 340 nm. The percentage (of the total unfractionated sample) of isocitrate dehydrogenase activity present in each fraction was calculated. For all fractionations, a minimum of 90% of the isocitrate dehydrogenase activity was present in the cytoplasmic fraction.
Identification of interacting proteins with the bacterial-two hybrid system.
A bacterial two-hybrid system was used to identify potential interactions among Cts proteins. This system is based on functional complementation between two fragments of the catalytic domain of adenylate cyclase from Bordetella pertussis (31). Genes ctsE, ctsP, ctsX, ctsF, ctsD, ctsR, ctsG, ctsW, and dprA were PCR amplified from the coding sequence of the second amino acid through the stop codon and cloned into the PstI and BamHI sites of pT25 to generate in-frame T25 fusion proteins. The same genes were amplified with primers containing XhoI and ClaI sites from the second amino acid to the last amino acid, excluding the stop codon, and inserted into the XhoI and ClaI sites of pT18 to create in-frame fusion proteins corresponding to the adenylate cyclase T18 domain. The insertions in these plasmids were verified by PCR and confirmed by sequencing.
E. coli strain Sp850 (cya) was transformed with all combinations of Cts-pT18 and Cts-pT25 plasmids, and transformants were screened for protein-protein interactions by growth on LB agar plates at 30°C with 100 μg ml−1 ampicillin, 30 μg ml−1 chloramphenicol, 40 μg ml−1 X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside), and 0.5 mM ITPG (isopropyl-β-d-thiogalactopyranoside). The negative control contained Sp850 with pT18 and pT25, and the positive control was Sp850 with pT18-Zip and pT25-Zip (31). Assays were done in triplicate, and the data represent averages of the results of three biological replicate experiments. Standard deviations are indicated.
Topology analysis of CtsX.
LacZ and PhoA fusions of ctsX were made using pTrcLacZ and pTrcPhoA (32). Translational fusions were generated at amino acid residues 14, 50, and 89 and at amino acid residue 195 (the final amino acid in the full-length CtsX protein). These sites were chosen after analysis of CtsX hydrophobicity by the use of several computer programs (Kyte Doolittle, EMBL) which predicted a transmembrane segment at roughly amino acids 22 to 42. PCR products were cloned into the NcoI and XmaI sites in pTrcLacZ and pTrcPhoA. Plasmids were screened by PCR and verified by sequencing. E. coli strain TG1 was transformed with these plasmids to carry out alkaline phosphatase assays and β-galactosidase assays using standard methods (35). Strains were assayed in triplicate, and data represent averages of the results of three biological replicate experiments. Standard deviations are indicated.
Membrane extractability of CtsP and CtsX fusion proteins.
For membrane solubility studies, cells were fractionated as described previously. Purified membranes were diluted 1:1 into (i) 10 mM Tris-HCl (pH 8.0), (ii) 1 M NaCl, (iii) 2 M NaCl, (iv) 3 M urea, (v) 5 M urea, (vi) 0.5 M Na2CO3 (pH 3.0), (vii) 0.5 M Na2CO3 (pH 11.0), or (viii) 1 M KCl and rocked on a Nutator mixer for 30 min at 4°C. Membranes were subsequently collected by ultracentrifugation (1 h at 100,000 × g and 4°C; SW41 rotor) and were resuspended in 50 μl of 10 mM Tris (pH 8.0). The supernatant was removed and precipitated by addition of 400 μl of acetone, incubation at −20°C for 1 h, and centrifugation at 16,000 × g at 4°C. Precipitated proteins were air dried for 30 min and resuspended in 50 μl of 10 mM Tris-HCl (pH 8.0). Equal volumes of soluble and membrane fractions were resuspended in SDS sample buffer and loaded onto polyacrylamide gels for separation by SDS-PAGE and Western blot analysis to detect CtsX or CtsP as described above.
RESULTS
The Walker boxes of CtsE and CtsP are required for function in vivo.
Walker boxes A and B are conserved nucleotide binding motifs important for NTPase function (19), and both motifs are present in CtsP and CtsE. To determine the contribution of CtsP and CtsE for natural transformation of C. jejuni, we tested whether Walker box motifs are important in vivo. Carboxyl-terminal FLAG fusions to each protein were expressed in C. jejuni ΔctsP and ΔctsE mutants; the fusion proteins complemented the respective mutant alleles to near-wild-type levels of transformation, similar to the complementation observed with untagged alleles of ctsP or ctsE (Fig. 1) (8).
FIG 1.
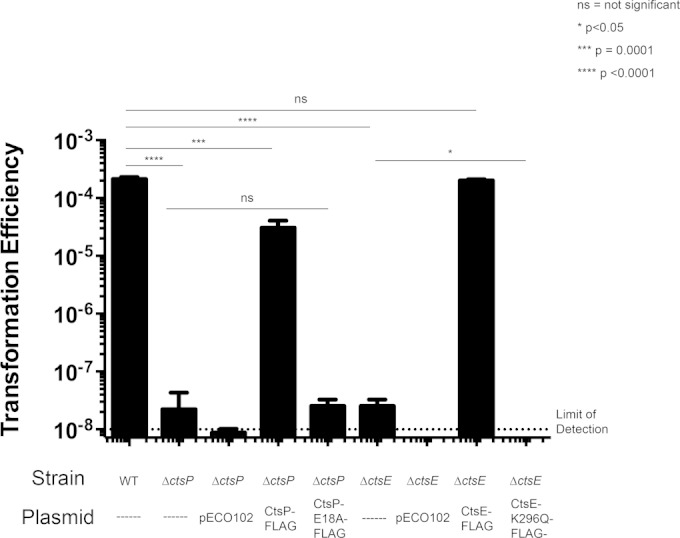
Transformation efficiency of C. jejuni CtsE and CtsP mutants. Strains are complemented with pECO102, pECO102 expressing the wild-type coding sequence with a C-terminal FLAG tag, or pECO102 expressing the coding sequence with a Walker box mutation and a C-terminal FLAG tag. The data represent averages of the results from three samples per strain from one experiment. Experiments were repeated at least three times with similar results. Error bars indicate standard deviations. The limit of detection is indicated with a dashed line. Stars indicate P values calculated using one-way analysis of variance (ANOVA) with Dunnett's multiple-comparison test.
Walker box A (GXXXXGK[S/T], where X is any amino acid) forms a loop structure (the P-loop) in which the lysine can directly contact the phosphoryl group of the bound nucleotide (36, 37). This Walker box motif is important for the function of many PulE-VirB11 superfamily members, and mutation of invariant residues abolishes ATP binding in a number of proteins (17, 38–40). The conserved lysine codon (K296) of Walker box A in ctsE was changed by site-directed mutagenesis to encode a glutamine in CtsE-FLAG (CtsE-FLAG K296Q), which was expressed in a ΔctsE strain to determine whether it could restore transformation. While the FLAG-tagged version of CtsE could complement a CtsE deletion mutant, the Walker box A point mutant in CtsE (CtsE-FLAG K296Q) could not (Fig. 1). Immunoblotting confirmed that CtsE-FLAG K296Q was expressed at levels similar to CtsE-FLAG levels (data not shown).
Several alleles of the gene encoding CtsP-FLAG with alterations to the same Walker box A lysine proved unstable (data not shown), preventing functional assessment of Walker box A in CtsP; we thus targeted Walker box B in CtsP for mutagenesis. The conserved sequence of Walker box B is hhhhDEXX, where h is a hydrophobic amino acid and X is any amino acid. The coding sequence for the conserved glutamic acid residue (E81) was changed by site-directed mutagenesis to encode an alanine residue in CtsP-FLAG, which was stable (by immunoblotting; data not shown). CtsP-FLAG E81A was unable to complement a ΔctsP mutant (Fig. 1). Through these site-directed mutagenesis studies, we have determined that intact nucleotide binding motifs are required for the function of both CtsE and CtsP in transformation.
Localization of CtsE and CtsP.
Members of the PulE-VirB11 superfamily of NTPases are hypothesized to act at the inner membrane of Gram-negative bacteria through association with membrane-localized proteins within their respective transport systems. To assess localization of CtsP-FLAG and CtsE-FLAG, C. jejuni strains expressing either fusion protein were fractionated into soluble and insoluble fractions as described previously (34). The soluble fraction contains the cytoplasmic and periplasmic contents, and the insoluble fraction contains the inner and outer membranes. Activity of isocitrate dehydrogenase, a cytoplasmic enzyme, was monitored to determine the purity of the fractions (34). Immunoblot analysis of these fractions using anti-FLAG monoclonal antibody revealed different subcellular locations for these two putative NTPases. CtsP-FLAG was detected primarily in the membrane fraction, while CtsE-FLAG was located primarily in the soluble fraction (Fig. 2A and B). To determine whether the cytoplasmic localization of CtsE-FLAG resulted from competition for membrane binding partners with the chromosomally encoded version of CtsE, we assessed localization of CtsE-FLAG in a strain ΔctsE background. The tagged protein was still observed primarily in the cytoplasmic fraction in the absence of native CtsE (Fig. 2B).
FIG 2.
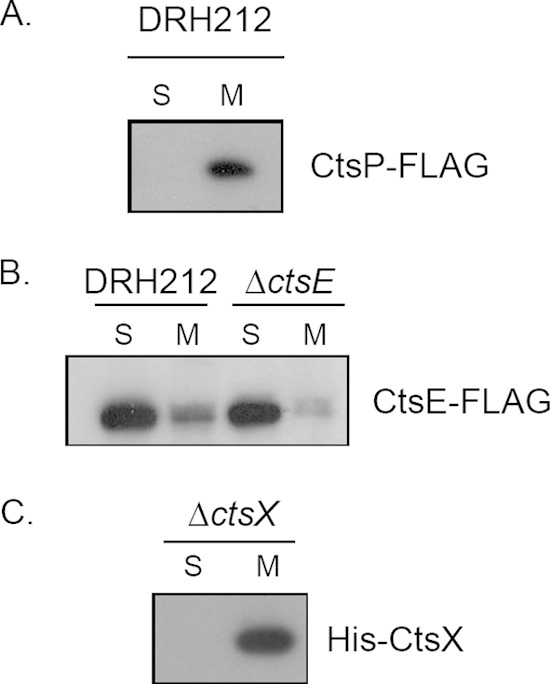
Localization of CtsE, CtsP, and CtsX fusion proteins. Cellular fractions of DRH212 expressing CtsP-FLAG (A), CtsE-FLAG (B), or His-CtsX (C) from pECO102 or the ΔctsE mutant expressing CtsE-FLAG (B) were separated by 12% or 15% SDS-PAGE. The presence of the CtsP-FLAG and CtsE-FLAG fusion proteins was detected by immunoblotting using a monoclonal antibody against FLAG. His-CtsX was detected using a monoclonal antibody against His. S, soluble fraction; M, membrane fraction.
Bacterial two-hybrid screen for identification of Cts interaction partners.
CtsP localizes to the membrane fraction of C. jejuni but does not have a predicted hydrophobic region that could serve as a transmembrane domain. We hypothesized that interaction with another protein might be responsible for the membrane localization of CtsP. To explore this, we used a bacterial two-hybrid system based on functional reconstitution of adenylate cyclase activity (31). For two-hybrid analysis, we tested competence genes ctsE, ctsX, ctsF, ctsD, and ctsR, all of which are encoded contiguously with ctsP, and ctsG, ctsW, and dprA, which are not linked to ctsP. Genes were cloned into both pT18 and pT25, two plasmids that provide different domains of adenylate cyclase. We tested for interactions between all Cts proteins. Two different protein-protein interactions among Cts proteins were identified (Fig. 3). One was between CtsE-T18 and T25-CtsE, which is consistent with observations that GspE proteins in other type II transport systems form multimers (26, 41, 42), suggesting that CtsE may form a multimonomer complex.
FIG 3.
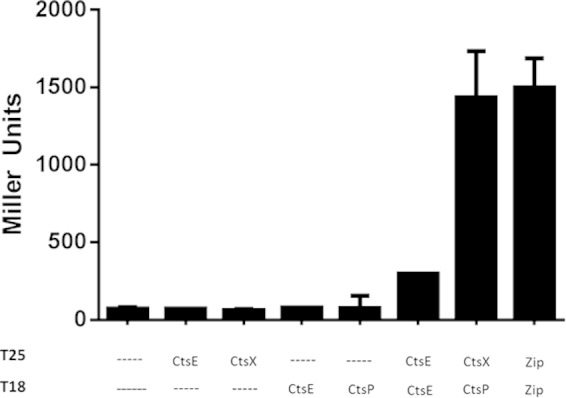
β-Galactosidase assays of bacterial two-hybrid-system interactions. When fused to proteins that interact, the T25 and T18 fragments of adenylate cyclase are able to associate, leading to cyclic AMP (cAMP) synthesis and resulting in transcription of lacZ. For a positive control, T25 and T18 were fused to a leucine zipper (Zip) (31). Activities represent averages of the results from three samples from one experiment which was repeated at least three times with similar results. Error bars indicate standard deviations.
The other interaction we identified was between CtsP-T18 and T25-CtsX (Fig. 3); ctsX is encoded immediately downstream of ctsP (8). Based on Kyte-Doolittle hydropathy analysis, CtsX is predicted to be a membrane protein by virtue of a single putative transmembrane helix from amino acid 21 to amino acid 40. To test this experimentally, we expressed a His-CtsX fusion protein in C. jejuni DRH212. Cells were fractionated into soluble and membrane fractions as discussed above for CtsE and CtsP, and fraction purity was assessed by isocitrate dehydrogenase assays (data not shown). His-CtsX was detected by immunoblotting with an anti-His monoclonal antibody. As predicted, the majority of the His-CtsX fusion protein was located in the membrane fraction (Fig. 2C).
The membrane topology of CtsX was experimentally determined using β-galactosidase and alkaline phosphatase fusions to the CtsX coding sequence. Alkaline phosphatase is active only when transported outside the cytoplasm (35), whereas β-galactosidase is active in the cytoplasm. Portions of the CtsX coding sequence at different positions on both sides of the predicted transmembrane region of CtsX were fused in frame with lacZ and phoA reporter genes in pTrcLacZ and pTrcPhoA (32) (Fig. 4).
FIG 4.

Topology studies of CtsX. Reporter protein activity levels of E. coli TG1 expressing four fragments of ctsX in a translational fusion with lacZ (left) or with phoA (right). Data represent averages of the results from three samples from one experiment that was repeated at least three times with similar results. Error bars indicate standard deviations. Stars represent P values calculated using Student's unpaired t test.
The first 14 amino acids of CtsX directed high-level β-galactosidase activity but low-level alkaline phosphatase activity, indicating that the N terminus of the protein resides in the cytoplasm (Fig. 4). In contrast, fragments of CtsX, including the putative transmembrane domain, directed low β-galactosidase levels but high alkaline phosphatase activity levels (Fig. 4), consistent with there being a transmembrane domain in the protein as predicted by the Kyte-Doolittle plot. Based on these data, we predict that small portion of the N terminus of CtsX is localized to the cytosol, with the remainder of the protein within the periplasmic space.
Localization of CtsP in the absence of other Cts proteins.
Because CtsP lacks an obvious membrane localization signal and interacts with CtsX, which is membrane localized, we hypothesized that membrane localization of CtsP depends on CtsX. However, CtsP-FLAG localized to the membrane fraction in mutant cells lacking ctsX (Fig. 5A), as well as in mutants lacking other transformation genes, including ctsF, ctsE, ctsD, ctsR, and ceuB (8) (Fig. 5B and data not shown). To test whether membrane localization of CtsP requires any C. jejuni proteins, we expressed CtsP-FLAG in E. coli JM101. Membrane localization was still observed (Fig. 5C), suggesting either that membrane localization is an intrinsic feature of CtsP or that a protein with which it interacts and colocalizes is also present in E. coli.
FIG 5.
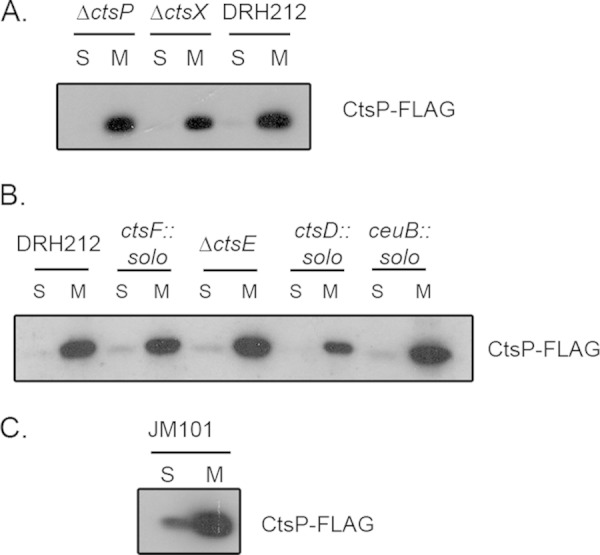
Localization of CtsP in the absence of other Cts proteins. Localization of CtsP-FLAG is indicated. Cellular fractions of DRH212 or cts mutants (A or B) or cellular fractions from E. coli JM101 expressing CtsP-FLAG from pECO102 (C) were separated by 12% SDS-PAGE. The presence of the CtsP-FLAG fusion protein was detected by immunoblotting using a monoclonal antibody against the FLAG epitope. S, soluble fraction; M, membrane fraction.
To rule out the possibility that the overexpressed protein is simply insoluble and thus pellets with membranes in the fractionation experiments described above, we tested membrane association of CtsP using a membrane flotation assay. Membrane flotation assays utilize a sucrose gradient to separate the membranes from any insoluble proteins that would pellet during ultracentrifugation. Using this assay, we observed that, while some CtsP did become insoluble (in fraction 6), the majority was located in membrane-containing fractions (fractions 2 to 5). This was true when CtsP-FLAG was expressed in wild-type cells as well as in the ΔctsP, ΔctsX, or ΔctsE strains. Even when CtsP-FLAG was expressed in E. coli, it was primarily localized to the membrane-containing fraction and not the insoluble fraction (Fig. 6).
FIG 6.

Membrane flotation of CtsP-FLAG. Soluble and membrane fractions of DRH212, cts mutants (ΔctsP, ΔctsE, and ΔctsX), and E. coli containing the fusion protein CtsP-FLAG expressed on pECO102 were obtained. The membrane fractions were resuspended in a sucrose gradient to separate the membranes from the insoluble material and subjected to ultracentrifugation, and fractions were taken moving sequentially down the gradient (numbered 1 to 6, where 1 is the top of the gradient and 6 contains the insoluble proteins). These fractions were separated via 12% SDS-PAGE. The presence of the CtsP-FLAG fusion protein was detected by immunoblotting using a monoclonal antibody against the FLAG epitope. S, soluble fraction; M, membrane fraction.
Membrane extractability of CtsP and CtsX.
Given its fractionation with the membrane of both C. jejuni and E. coli, we hypothesized that CtsP is a peripheral membrane protein, perhaps associating with the membrane by interacting with the polar head of the phospholipid bilayer. In contrast, CtsX, with its more obvious transmembrane domain and the fusion protein's ability to localize alkaline phosphatase to the periplasmic space (Fig. 4), appears to be an integral membrane protein. To further characterize CtsP and CtsX as integral or peripheral membrane proteins, we analyzed the avidity of their association with C. jejuni membranes by employing different extraction procedures commonly used to release peripherally associated membrane proteins (43).
Membranes of C. jejuni expressing CtsP-FLAG or His-CtsX were treated with high concentrations of salt (1 and 2 M NaCl, 1 M KCl) or urea (3 M, 5 M) or extremes of pH (Na2CO3 at pH 3.0 or pH 11.0) for 30 min at 4°C as described in Materials and Methods. Control membranes were treated with 10 mM Tris (pH 8.0). CtsP-FLAG was partially extracted from the membranes in the presence of high salt concentrations (1 and 2 M NaCl, 1 M KCl), while CtsX remained predominantly membrane associated (Fig. 7). Treatment of the membranes with urea (3 M, 5 M) nearly completely solubilized CtsP-FLAG, while His-CtsX was extracted only partially by this treatment (Fig. 7). Membrane treatment with 0.5 M Na2CO3 at pH 11 also extracted CtsP-FLAG into the soluble fraction (Fig. 7). In contrast, Na2CO3 treatment was not sufficient to affect His-CtsX localization, and it remained in the membrane fraction after the treatment (Fig. 7). These results suggest that while His-CtsX is an integral membrane protein, the more ready extraction of CtsP-FLAG suggests that it is not integral but is peripheral to the membrane, interacting with it perhaps through an as-yet-undefined mechanism.
FIG 7.

Membrane extractability of CtsP-FLAG and His-CtsX. Membranes were incubated with the indicated reagents for 30 min at 4°C and sedimented by ultracentrifugation. Separated fractions were examined by 12% or 15% SDS-PAGE. Fusion proteins were detected by immunoblotting with anti-FLAG M2 antibody for CtsP-FLAG and with an anti-6×His antibody for His-CtsX. S, soluble fraction after washes; M, membrane fraction.
DISCUSSION
The goal of this study was to characterize two putative NTPases/NTP binding proteins involved in natural transformation of C. jejuni. One of these, CtsE, belongs to a family of well-studied NTPases involved in transport of macromolecules in a number of systems, including type II secretion, type IV secretion, and type IV pilus biogenesis (15, 16). Members of this family have ATPase activity in vitro (22–26), although the exact ATP-dependent step in transport is unclear. Given the homology of CtsE to other members of this family and the importance of an intact Walker box A for CtsE to function in natural transformation, it is likely that CtsE has ATPase activity critical for its role in transformation.
The other putative NTPase/NTP binding protein, CtsP, has little homology to other NTPases and cannot be assigned to a defined family. CtsP Walker box A mutants were all unstable in C. jejuni, but a stable mutant protein lacking Walker box B was unable to correct the transformation defect of a ΔctsP mutant. We suggest that CtsP may bind and hydrolyze nucleotides and that this property is necessary for natural transformation of C. jejuni, although further work is needed to determine whether CtsP has ATPase activity.
Transformation in C. jejuni involves a number of proteins with similarity to those required for pilus biogenesis and natural transformation in Neisseria gonorrhoeae and Vibrio cholerae (8, 44–48). In these species, transformation is facilitated by the production of a pilus that is produced and retracted through the power of two ATPases. Transformation in N. gonorrhoeae requires the activity of two ATPases, PilF and PilT (44, 49), and in V. cholerae it requires PilB and PilT (47). Both N. gonorrhoeae ATPases are members of the PulE-VirB11 superfamily (15, 16). PilF is required for elaboration of the pilus, while PilT is required for retraction (44, 50). As noted, while CtsE appears to fall into this family, CtsP does not. However, unlike these two systems, C. jejuni has never been shown to produce a pilus (8) and lacks a homolog to the major structural subunits found in these systems, PilE in N. gonorrhoeae and PilA in V. cholerae. Instead, C. jejuni contains a number of genes that may encode pseudopilins, ctsG, ctsT, and Cjj81176_1096; two of these have been shown to function in transformation (8). Whether a pilus or pseudopilus is responsible for DNA binding or uptake across the outer membrane, requiring the actions of CtsE and CtsP, has yet to be determined. In the V. cholerae transformation system, pseudopilins are thought to initiate pilus formation whereas the major pilin subunit, PilA, is then responsible for the pilus structure (47). Loss of any of these proteins leads to an approximately 3-log decrease in transformation efficiency (47). Similarly to the V. cholerae system, the pseudopilins in C. jejuni may be sufficient to induce transformation. A second possibility is that, unlike the process in the V. cholerae system, the pseudopilins also form enough of a pilus-like structure to facilitate transformation even without the presence of a major pilin subunit.
CtsE appears to be predominantly cytosolic in C. jejuni, whereas CtsP is predominantly membrane associated. A CtsE homologue, ComGA, involved in transformation of Bacillus subtilis localizes to the membrane in that species, where it behaves as a peripheral membrane protein (51). In some other bacteria, CtsE homologues (generally termed GspE proteins) become membrane localized by interacting with an integral membrane protein generally termed GspL (52, 53) but no GspL homologue was identified in the C. jejuni genome sequence. It is difficult to imagine how CtsE could play a role in the early stages of transformation unless it interacts with proteins at the membrane. In the Vibrio cholerae system, a green fluorescent protein (GFP)-tagged version of the CtsE homolog, PilB, forms dynamic foci at the membrane that transiently overlap other components of the Vibrio transformation apparatus (47). Perhaps similarly to the Vibrio system, a transient, or very weak, association of CtsE occurs with the membrane and this could have been disrupted during fractionation; this might account for the small amount of CtsE-FLAG observed in the membrane fraction.
Unlike CtsE, CtsP localizes to the membrane in C. jejuni, where it may interact with other components of the transformation machinery. By bacterial two-hybrid analysis, we detected an interaction between CtsP and CtsX, another protein necessary for efficient C. jejuni transformation. CtsX is encoded immediately downstream of CtsP in a putative operon (8). CtsX is an integral membrane protein with its amino terminus exposed to the cytoplasm and to residues 50 to 195 in the periplasm. Interaction with CtsX is not necessary for CtsP localization to the membrane; neither is interaction with several other components of the transformation machinery. CtsP behaves as a peripheral membrane protein, and it may associate directly with the membrane. This possibility is strengthened by the membrane localization of CtsP-FLAG observed during expression in E. coli. If membrane localization required interaction with another protein, it would have to be present in E. coli as well and would likely not be involved in natural transformation, a process that has not been described in E. coli. As noted above, type II secretion system GspE family members, with which CtsE shares homology, associate with the membrane through interaction with another protein, GspL. When these GspE family members are expressed in E. coli, cytoplasmic localization is observed (54), which differs from our observed localization of CtsP to membrane fractions in E. coli. Further study is needed to confirm that CtsP directly associates with the membrane. Furthermore, we were able to replicate the membrane association phenotype using membrane flotation. If this association were an artifact of the initial fractionation procedure, we would expect to see CtsP-FLAG associate predominately with the insoluble aggregate and not float with the membranes. One possibility that has yet to be discounted definitively is that the CtsP-FLAG localization (as well as that of CtsE-FLAG) is due to studying plasmid-encoded proteins rather than the chromosomally encoded native protein. But the behavior of the plasmid-encoded, FLAG-tagged proteins mitigates this concern, because both CtsE-FLAG and CtsP-FLAG restore the transformation efficiency of their respective mutant strains to near-wild-type levels.
CtsX and CtsP represent novel components of the C. jejuni transformation machinery. Homologues of CtsX have not been identified in other transformation systems, and BLAST analysis does not provide obvious clues about its function. As it resides largely in the periplasm, perhaps its C terminus interacts with other components of the transformation machinery in that compartment. CtsX and CtsP are encoded in a putative operon between the type II secretion/type IV pilus biogenesis system homologues ctsD, ctsE, and ctsF. Given the CtsX/CtsP interaction, we hypothesize that these two proteins comprise a component of type II secretion/type IV pilus biogenesis systems specifically involved in C. jejuni competence. Perhaps the interaction is required for assembly or translocation of other transformation components or allows assembly of a structure needed specifically for DNA uptake and not pilus biogenesis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jeremy Ellermeir for helpful contributions and discussions.
This work was supported in part by NIAID R01AI069383 (to V.J.D.). J.M.B. and R.S.E. were trainees of the University of Michigan Predoctoral Genetics Training Program supported by T32GM07544.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02434-14.
REFERENCES
- 1.Bereswill S, Kist M. 2003. Recent developments in Campylobacter pathogenesis. Curr Opin Infect Dis 16:487–491. doi: 10.1097/00001432-200310000-00017. [DOI] [PubMed] [Google Scholar]
- 2.Young KT, Davis LM, DiRita VJ. 2007. Campylobacter jejuni: molecular biology and pathogenesis. Nat Rev Microbiol 5:665–679. doi: 10.1038/nrmicro1718. [DOI] [PubMed] [Google Scholar]
- 3.Wang Y, Taylor DE. 1990. Natural transformation in Campylobacter species. J Bacteriol 172:949–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson DL, Bell JA, Young VB, Wilder SR, Mansfield LS, Linz JE. 2003. Variation of the natural transformation frequency of Campylobacter jejuni in liquid shake culture. Microbiology 149(Pt 12):3603–3615. doi: 10.1099/mic.0.26531-0. [DOI] [PubMed] [Google Scholar]
- 5.Dingle KE, Colles FM, Wareing DR, Ure R, Fox AJ, Bolton FE, Bootsma HJ, Willems RJ, Urwin R, Maiden MC. 2001. Multilocus sequence typing system for Campylobacter jejuni. J Clin Microbiol 39:14–23. doi: 10.1128/JCM.39.1.14-23.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suerbaum S, Lohrengel M, Sonnevend A, Ruberg F, Kist M. 2001. Allelic diversity and recombination in Campylobacter jejuni. J Bacteriol 183:2553–2559. doi: 10.1128/JB.183.8.2553-2559.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Boer P, Wagenaar JA, Achterberg RP, van Putten JP, Schouls LM, Duim B. 2002. Generation of Campylobacter jejuni genetic diversity in vivo. Mol Microbiol 44:351–359. doi: 10.1046/j.1365-2958.2002.02930.x. [DOI] [PubMed] [Google Scholar]
- 8.Wiesner RS, Hendrixson DR, DiRita VJ. 2003. Natural transformation of Campylobacter jejuni requires components of a type II secretion system. J Bacteriol 185:5408–5418. doi: 10.1128/JB.185.18.5408-5418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bacon DJ, Alm RA, Hu L, Hickey TE, Ewing CP, Batchelor RA, Trust TJ, Guerry P. 2002. DNA sequence and mutational analyses of the pVir plasmid of Campylobacter jejuni 81-176. Infect Immun 70:6242–6250. doi: 10.1128/IAI.70.11.6242-6250.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larsen JC, Szymanski C, Guerry P. 2004. N-linked protein glycosylation is required for full competence in Campylobacter jejuni 81-176. J Bacteriol 186:6508–6514. doi: 10.1128/JB.186.19.6508-6514.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takata T, Ando T, Israel DA, Wassenaar TM, Blaser MJ. 2005. Role of dprA in transformation of Campylobacter jejuni. FEMS Microbiol Lett 252:161–168. doi: 10.1016/j.femsle.2005.08.052. [DOI] [PubMed] [Google Scholar]
- 12.Jeon B, Zhang Q. 10 August 2007. Cj0011c, a periplasmic single- and double-stranded DNA binding protein, contributes to natural transformation in Campylobacter jejuni. J Bacteriol doi: 10.1128/JB.01012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen I, Christie PJ, Dubnau D. 2005. The ins and outs of DNA transfer in bacteria. Science 310:1456–1460. doi: 10.1126/science.1114021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parkhill J, Wren BW, Mungall K, Ketley JM, Churcher C, Basham D, Chillingworth T, Davies RM, Feltwell T, Holroyd S, Jagels K, Karlyshev AV, Moule S, Pallen MJ, Penn CW, Quail MA, Rajandream MA, Rutherford KM, van Vliet AH, Whitehead S, Barrell BG. 2000. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 403:665–668. doi: 10.1038/35001088. [DOI] [PubMed] [Google Scholar]
- 15.Planet PJ, Kachlany SC, DeSalle R, Figurski DH. 2001. Phylogeny of genes for secretion NTPases: identification of the widespread tadA subfamily and development of a diagnostic key for gene classification. Proc Natl Acad Sci U S A 98:2503–2508. doi: 10.1073/pnas.051436598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peabody CR, Chung YJ, Yen MR, Vidal-Ingigliardi D, Pugsley AP, Saier MH Jr. 2003. Type II protein secretion and its relationship to bacterial type IV pili and archaeal flagella. Microbiology 149(Pt 11):3051–3072. doi: 10.1099/mic.0.26364-0. [DOI] [PubMed] [Google Scholar]
- 17.Possot O, Pugsley AP. 1994. Molecular characterization of PulE, a protein required for pullulanase secretion. Mol Microbiol 12:287–299. doi: 10.1111/j.1365-2958.1994.tb01017.x. [DOI] [PubMed] [Google Scholar]
- 18.Patel S, Latterich M. 1998. The AAA team: related ATPases with diverse functions. Trends Cell Biol 8:65–71. [PubMed] [Google Scholar]
- 19.Walker JE, Saraste M, Runswick MJ, Gay NJ. 1982. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J 1:945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Possot OM, Letellier L, Pugsley AP. 1997. Energy requirement for pullulanase secretion by the main terminal branch of the general secretory pathway. Mol Microbiol 24:457–464. doi: 10.1046/j.1365-2958.1997.3451726.x. [DOI] [PubMed] [Google Scholar]
- 21.Robien MA, Krumm BE, Sandkvist M, Hol WG. 2003. Crystal structure of the extracellular protein secretion NTPase EpsE of Vibrio cholerae. J Mol Biol 333:657–674. doi: 10.1016/j.jmb.2003.07.015. [DOI] [PubMed] [Google Scholar]
- 22.Rivas S, Bolland S, Cabezon E, Goni FM, de la Cruz F. 1997. TrwD, a protein encoded by the IncW plasmid R388, displays an ATP hydrolase activity essential for bacterial conjugation. J Biol Chem 272:25583–25590. doi: 10.1074/jbc.272.41.25583. [DOI] [PubMed] [Google Scholar]
- 23.Bhattacharjee MK, Kachlany SC, Fine DH, Figurski DH. 2001. Nonspecific adherence and fibril biogenesis by Actinobacillus actinomycetemcomitans: TadA protein is an ATPase. J Bacteriol 183:5927–5936. doi: 10.1128/JB.183.20.5927-5936.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herdendorf TJ, McCaslin DR, Forest KT. 2002. Aquifex aeolicus PilT, homologue of a surface motility protein, is a thermostable oligomeric NTPase. J Bacteriol 184:6465–6471. doi: 10.1128/JB.184.23.6465-6471.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sexton JA, Pinkner JS, Roth R, Heuser JE, Hultgren SJ, Vogel JP. 2004. The Legionella pneumophila PilT homologue DotB exhibits ATPase activity that is critical for intracellular growth. J Bacteriol 186:1658–1666. doi: 10.1128/JB.186.6.1658-1666.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Camberg JL, Sandkvist M. 2005. Molecular analysis of the Vibrio cholerae type II secretion ATPase EpsE. J Bacteriol 187:249–256. doi: 10.1128/JB.187.1.249-256.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogura T, Wilkinson AJ. 2001. AAA+ superfamily ATPases: common structure–diverse function. Genes Cells 6:575–597. doi: 10.1046/j.1365-2443.2001.00447.x. [DOI] [PubMed] [Google Scholar]
- 28.Figurski DH, Helinski DR. 1979. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc Natl Acad Sci U S A 76:1648–1652. doi: 10.1073/pnas.76.4.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korlath JA, Osterholm MT, Judy LA, Forfang JC, Robinson RA. 1985. A point-source outbreak of campylobacteriosis associated with consumption of raw milk. J Infect Dis 152:592–596. doi: 10.1093/infdis/152.3.592. [DOI] [PubMed] [Google Scholar]
- 30.Hendrixson DR, Akerley BJ, DiRita VJ. 2001. Transposon mutagenesis of Campylobacter jejuni identifies a bipartite energy taxis system required for motility. Mol Microbiol 40:214–224. doi: 10.1046/j.1365-2958.2001.02376.x. [DOI] [PubMed] [Google Scholar]
- 31.Karimova G, Pidoux J, Ullmann A, Ladant D. 1998. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci U S A 95:5752–5756. doi: 10.1073/pnas.95.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blank TE, Donnenberg MS. 2001. Novel topology of BfpE, a cytoplasmic membrane protein required for type IV fimbrial biogenesis in enteropathogenic Escherichia coli. J Bacteriol 183:4435–4450. doi: 10.1128/JB.183.15.4435-4450.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guerry P, Yao R, Alm RA, Burr DH, Trust TJ. 1994. Systems of experimental genetics for Campylobacter species. Methods Enzymol 235:474–481. doi: 10.1016/0076-6879(94)35163-5. [DOI] [PubMed] [Google Scholar]
- 34.Myers JD, Kelly DJ. 2005. A sulphite respiration system in the chemoheterotrophic human pathogen Campylobacter jejuni. Microbiology 151(Pt 1):233–242. doi: 10.1099/mic.0.27573-0. [DOI] [PubMed] [Google Scholar]
- 35.Manoil C. 1991. Analysis of membrane protein topology using alkaline phosphatase and beta-galactosidase gene fusions. Methods Cell Biol 34:61–75. doi: 10.1016/S0091-679X(08)61676-3. [DOI] [PubMed] [Google Scholar]
- 36.Fry DC, Kuby SA, Mildvan AS. 1986. ATP-binding site of adenylate kinase: mechanistic implications of its homology with ras-encoded p21, F1-ATPase, and other nucleotide-binding proteins. Proc Natl Acad Sci U S A 83:907–911. doi: 10.1073/pnas.83.4.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hyde SC, Emsley P, Hartshorn MJ, Mimmack MM, Gileadi U, Pearce SR, Gallagher MP, Gill DR, Hubbard RE, Higgins CF. 1990. Structural model of ATP-binding proteins associated with cystic fibrosis, multidrug resistance and bacterial transport. Nature 346:362–365. doi: 10.1038/346362a0. [DOI] [PubMed] [Google Scholar]
- 38.Korangy F, Julin DA. 1992. Alteration by site-directed mutagenesis of the conserved lysine residue in the ATP-binding consensus sequence of the RecD subunit of the Escherichia coli RecBCD enzyme. J Biol Chem 267:1727–1732. [PubMed] [Google Scholar]
- 39.Turner LR, Lara JC, Nunn DN, Lory S. 1993. Mutations in the consensus ATP-binding sites of XcpR and PilB eliminate extracellular protein secretion and pilus biogenesis in Pseudomonas aeruginosa. J Bacteriol 175:4962–4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sandkvist M, Bagdasarian M, Howard SP, DiRita VJ. 1995. Interaction between the autokinase EpsE and EpsL in the cytoplasmic membrane is required for extracellular secretion in Vibrio cholerae. EMBO J 14:1664–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turner LR, Olson JW, Lory S. 1997. The XcpR protein of Pseudomonas aeruginosa dimerizes via its N-terminus. Mol Microbiol 26:877–887. doi: 10.1046/j.1365-2958.1997.6201986.x. [DOI] [PubMed] [Google Scholar]
- 42.Py B, Loiseau L, Barras F. 1999. Assembly of the type II secretion machinery of Erwinia chrysanthemi: direct interaction and associated conformational change between OutE, the putative ATP-binding component and the membrane protein OutL. J Mol Biol 289:659–670. doi: 10.1006/jmbi.1999.2803. [DOI] [PubMed] [Google Scholar]
- 43.Pozidis C, Chalkiadaki A, Gomez-Serrano A, Stahlberg H, Brown I, Tampakaki AP, Lustig A, Sianidis G, Politou AS, Engel A, Panopoulos NJ, Mansfield J, Pugsley AP, Karamanou S, Economou A. 2003. Type III protein translocase: HrcN is a peripheral ATPase that is activated by oligomerization. J Biol Chem 278:25816–25824. doi: 10.1074/jbc.M301903200. [DOI] [PubMed] [Google Scholar]
- 44.Freitag NE, Seifert HS, Koomey M. 1995. Characterization of the pilF-pilD pilus-assembly locus of Neisseria gonorrhoeae. Mol Microbiol 16:575–586. doi: 10.1111/j.1365-2958.1995.tb02420.x. [DOI] [PubMed] [Google Scholar]
- 45.Tønjum T, Freitag NE, Namork E, Koomey M. 1995. Identification and characterization of pilG, a highly conserved pilus-assembly gene in pathogenic Neisseria. Mol Microbiol 16:451–464. doi: 10.1111/j.1365-2958.1995.tb02410.x. [DOI] [PubMed] [Google Scholar]
- 46.Drake SL, Koomey M. 1995. The product of the pilQ gene is essential for the biogenesis of type IV pili in Neisseria gonorrhoeae. Mol Microbiol 18:975–986. doi: 10.1111/j.1365-2958.1995.18050975.x. [DOI] [PubMed] [Google Scholar]
- 47.Seitz P, Blokesch M. 2013. DNA-uptake machinery of naturally competent Vibrio cholerae. Proc Natl Acad Sci U S A 110:17987–17992. doi: 10.1073/pnas.1315647110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seitz P, Pezeshgi Modarres H, Borgeaud S, Bulushev RD, Steinbock LJ, Radenovic A, Dal Peraro M, Blokesch M. 2014. ComEA is essential for the transfer of external DNA into the periplasm in naturally transformable Vibrio cholerae cells. PLoS Genet 10:e1004066. doi: 10.1371/journal.pgen.1004066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brossay L, Paradis G, Fox R, Koomey M, Hebert J. 1994. Identification, localization, and distribution of the PilT protein in Neisseria gonorrhoeae. Infect Immun 62:2302–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wolfgang M, Lauer P, Park HS, Brossay L, Hebert J, Koomey M. 1998. PilT mutations lead to simultaneous defects in competence for natural transformation and twitching motility in piliated Neisseria gonorrhoeae. Mol Microbiol 29:321–330. doi: 10.1046/j.1365-2958.1998.00935.x. [DOI] [PubMed] [Google Scholar]
- 51.Chung YS, Dubnau D. 1998. All seven comG open reading frames are required for DNA binding during transformation of competent Bacillus subtilis. J Bacteriol 180:41–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sandkvist M. 2001. Type II secretion and pathogenesis. Infect Immun 69:3523–3535. doi: 10.1128/IAI.69.6.3523-3535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sandkvist M. 2001. Biology of type II secretion. Mol Microbiol 40:271–283. doi: 10.1046/j.1365-2958.2001.02403.x. [DOI] [PubMed] [Google Scholar]
- 54.Filloux A. 2004. The underlying mechanisms of type II protein secretion. Biochim Biophys Acta 1694:163–179. doi: 10.1016/j.bbamcr.2004.05.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.