

Abstract
Protein export to the bacterial periplasm is achieved by SecYEG, an inner membrane heterotrimer. SecY and SecE are encoded by essential genes, while SecG is not essential for growth under standard laboratory conditions. Using a quantitative and sensitive export assay, we show that SecG plays a critical role for the residual export mediated by mutant signal sequences; the magnitude of this effect is not proportional to the strength of the export defect. In contrast, export mediated by wild-type signal sequences is only barely retarded in the absence of SecG. When probed with mutant signal sequences, secG loss of function mutations display a phenotype opposite to that of prlA mutations in secY. The analysis of secG and prlA single and double mutant strains shows that the increased export conferred by several prlA alleles is enhanced in the absence of SecG. Several combinations of prlA alleles with a secG deletion cannot be easily constructed. This synthetic phenotype is conditional, indicating that cells can adapt to the presence of both alleles. The biochemical basis of this phenomenon is linked to the stability of the SecYE dimer in solubilized membranes. With prlA alleles that can be normally introduced in a secG deletion strain, SecG has only a limited effect on the stability of the SecYE dimer. With the other prlA alleles, the SecYE dimer can often be detected only in the presence of SecG. A possible role for the maintenance of SecG during evolution is proposed.
INTRODUCTION
A complex protein machine, the preprotein translocase, promotes protein export across the Escherichia coli inner membrane (1, 2). The core of the translocase consists of three inner membrane proteins, SecY, SecE, and SecG. The largest protein, SecY, has 10 transmembrane helices that surround a central pore through which the preprotein is transported (3, 4). The SecYEG complex has been largely conserved across evolution (5, 6), although differences in the length and number of transmembrane segments are observed with SecE and SecG (3). SecY and SecE form a tight complex, whereas SecG appears to be less tightly bound (7).
A large number of signal sequence mutations have been shown to interfere with export (8, 9). Mutations exerting the strongest effects usually introduce charged residues in the central hydrophobic core of signal sequences. Suppressor mutations, called prl mutations, restore the export of mutant signal sequences to various extents; they have been isolated in most genes encoding components of the translocase, including prlA mutations in secY, prlG mutations in secE, and prlH mutations in secG (10–15). The strongest suppressors are prlA alleles that can even promote the export of periplasmic proteins whose signal sequences are completely deleted (16). These prl mutations are believed to exert their effect by facilitating the opening of the central pore of the translocase (17, 18). This effect is normally triggered by wild-type signal sequences. In addition, several prl mutations have been shown to destabilize the interactions between SecY and SecE (19).
In E. coli, the SecY and SecE proteins are encoded by essential genes (20, 21). In contrast, a deletion of secG had little if any effect on growth under standard laboratory conditions (22–24). Indeed, the export kinetics of proteins encoded by wild-type genes was only slightly slower in the absence of SecG, as documented by pulse-chase experiments (22–24). The contribution of SecG was much stronger with in vitro translocation assays, in which SecG had a pronounced stimulatory effect on export (25, 26). A significant effect of SecG on protein export was also observed in vivo with mutant signal sequences. For instance, mutant MalE signal sequences decrease maltose fermentation, as monitored on MacConkey-maltose indicator plates, and the absence of secG resulted in a strongly enhanced Mal− phenotype (23). Furthermore, several secG mutations, including null alleles, were isolated as suppressors of toxic chimeric proteins containing a mammalian signal sequence fused to the mature portion of alkaline phosphatase. In these cases, suppression was associated with a marked reduction in the kinetics of export of the chimeric protein (23, 27).
The in vivo assays described above were performed in vastly different time scales, from seconds in the case of pulse-chase experiments to hours in the case of colony growth and fermentation on indicator plates. We compare here the kinetics of protein export in the presence and in the absence of SecG using an experimental system that allowed us to examine under the same conditions wild-type efficient signal sequences as well as mutant inefficient ones (28). Our results confirm that export mediated by wild-type signal sequences shows a very limited dependence on SecG. In contrast, the residual export mediated by mutant signal sequences can be dramatically decreased in the absence of SecG. There was, however, no correlation between the strength of the export defects and their enhancement in the absence of SecG. Several pseudorevertants of mutant signal sequences were identified in the course of this study. Two of these were almost as efficient as their wild-type counterparts, but one was essentially SecG independent while export mediated by the other one remained strongly SecG dependent.
Since several secG alleles confer a phenotype opposite to that conferred by prl mutations (29), we attempted to determine whether epistatic effects could be detected with secG and prlA alleles. Surprisingly, several prlA alleles showed a synthetic phenotype with a secG deletion. The binding of these mutant SecY proteins to SecE was much reduced, particularly in the absence of SecG. In conclusion, SecG contributes both to signal sequence interaction with the translocase and to the intrinsic stability of the translocase in the E. coli inner membrane.
MATERIALS AND METHODS
Reagents.
Liquid and solid media were prepared as described previously (23). Antibiotics were used at the following concentrations: ampicillin (Ap), 200 μg/ml; chloramphenicol (Cm), 30 μg/ml; kanamycin (Kn), 40 μg/ml; and tetracycline (Tc), 7.5 μg/ml. The anti-OmpA rabbit antiserum (Covalab) was raised against SDS-PAGE-purified OmpA. Anti-PhoA antibodies were from Millipore (AB1204) or from Biodesign/Meridian Life Science (K59134R). Anti-MalE antiserum was from New England BioLabs (E8030S). Mouse monoclonal antibody 10C1, raised against the last cytoplasmic loop of SecY, was a gift from I. Collinson. E. coli polar lipids were purchased from Avanti polar lipids and used as described previously (27).
Bacterial strains and plasmids.
The E. coli strains used in this study are described in Table 1 and were constructed by transduction with bacteriophage P1 or by transformation. The pACYC184-derived plasmids carrying the different secG alleles have been described (27). Two sets of prlA alleles have been used, those isolated as suppressors of several different signal sequence mutations (12, 14) and those isolated as suppressors of malE18 in a secG1 strain (23). These mutants were characterized by sequencing the secY gene of the suppressor strains (Table 1).
TABLE 1.
E. coli strains
| Strain | Relevant genotype/description | Reference(s) or source |
|---|---|---|
| MC1000 | F− araD139 Δ(ara-leu)7697 Δ(lac)X74 rpsL150 galE galK relA1 thi | 50 |
| MC4100 | F− araD139 Δ(argF-lac)U169 flhD5301 fruA25 relA1 rpsL150 rbsR22 Δ(fimB-fimE)632 deoC1 thi | 51 |
| CAG12072 | MG1655 zgj-203::Tn10 | 52 |
| CAG12131 | MG1655 leuO::Tn10kan | 52 |
| DB504 | MC4100 malE18-1 Δara714 | Lab collection |
| KN370 | C600 recD1009 ΔsecG::Kn | 22 |
| DHB3 | MC1000 malFΔ3 phoAΔ(PvuII) phoR | 36 |
| DB617 | DHB3 zgj-203::Tn10 | This work |
| DB618 | DHB3 zgj-203::Tn10 ΔsecG::Kn | This work |
| DB619 | DHB3 zgj-203::Tn10 secG1 | 23 |
| DB620 | DHB3 zgj-203::Tn10 secG15 | 27 |
| MM1 | MC4100 malE10-1 | 31 |
| MM2 | MC4100 malE14-1 | 31 |
| MM3 | MC4100 malE16-1 | 31 |
| MM4 | MC4100 malE18-1 | 31 |
| MM5 | MC4100 malE19-1 | 31 |
| Mph42 | MC1000 phoR | 53 |
| Mph53 | Mph42 phoA82 | 54 |
| Mph55 | Mph42 phoA93 | 54 |
| Mph56 | Mph42 phoA73 | 54 |
| OF734 | MG1655 recD::Tn10 rbsB16R | 32 |
| OF735 | MG1655 recD::Tn10 rbsB17D | 32 |
| DB686 | Mph42 phoA68 Δara714 ΔompA zgj-203::Tn10 | A. Rietsch |
| DB685 | DB686 ΔsecG::Kn | This work |
| DB638 | DB504 ΔsecG::Kn | This work |
| DB687 | DB504 ΔsecG::Kn pDB613 (malE14::TnphoA) | This work |
| KJ195 | MC4100 gspA::Tn10 | Lab collection |
| prlA3 | MC4100 secY F67C | 12 |
| prlA6 | MC4100 secY I408N | 12 |
| prlA7 | MC4100 secY L407R | 12 |
| prlA207 | MC4100 secY I278S | 12 |
| prlA304 | MC410 0 secY I90N | 12 |
| prlA502 | DB504 secY G69D, identical to prlA9 | Reference 23 and this work |
| prlA517 | DB504 secY G81E | Reference 23 and this work |
| prlA535 | DB504 secY V411D | Reference 23 and this work |
| prlA538 | DB504 secY A75P | Reference 23 and this work |
| prlA543 | DB504 secY A88E | Reference 23 and this work |
| prlA545 | DB504 secY A71V | Reference 23 and this work |
| DB559 | B ompT gal hsdS ΔsecG::Kn | 27 |
The coding regions of the PhoA signal sequences from strains Mph42 and Mph53 to Mph56 were cloned as described for pBADPHOA (30). The coding regions of the MalE signal sequences (amino acids 1 to 29; cleavage after 26th residue) were amplified from strains MC4100 and MM1 to MM5 (31) with primers MbpUP, 5′CCTAGCTAGCAGGAGGAATTCATTATGAAAATAAAAACAG, and MbpDON, 5′GGGGTACCTCGATTTTGGCG. The coding regions of the RbsB signal sequences (amino acids 1 to 31; cleavage after 25th residue) were amplified from strains MC4100, OF734, and OF735 (32) with primers RbpUP, 5′CCTAGCTAGCAGGAGGAATTCACCATGAACATGAAGAAACTGGC, and RbpDON, 5′AAAGGTACCAGCGCGATGGTGTC. The PCR fragments were digested with NheI and KpnI and cloned in the cognate sites of pBADslAP (30). The coding region of OmpA was amplified with primers pOAUP, 5′GAGGTCATGAAGAAGACAGCTATCGCG, and OADON, 5′CTAGTCTAGACCTGCGGCTGAGTTACAACG. The PCR fragment was digested with BspHI and XbaI and cloned between the NcoI and XbaI sites of pBAD24. The mature portion of OmpA was amplified with primer slOA, 5′GAGGTACCATGGTACACTGGTGC, and OADON. The PCR fragment was digested with KpnI and XbaI and cloned in pBAD24 (33). NheI-KpnI fragments were used to fuse the signal sequences in frame with the mature portion of OmpA.
The appropriate portions of secY from the chromosomal prlA alleles were PCR amplified and used to replace the wild-type sequence in pBADHisEYG, a pBAD24 vector with a hexahistidine tag at the N terminus of SecE (34); all inserts were sequenced. Vectors that express only SecE and SecY were prepared by deletion of the HindIII or XbaI-KpnI fragments that encode SecG.
PhoA assay.
PhoA activity was determined by determining the rate of p-nitro-phenyl-phosphate hydrolysis (28).
Pulse-labeling and immunoprecipitation.
Labeling and immunoprecipitation were performed as described previously (28). Cells from saturated cultures in LB medium were diluted 1:50 and grown overnight in M63 medium containing 0.2% (vol/vol) glycerol as a carbon source and all amino acids except methionine and cysteine at a final concentration of 50 mg/liter, diluted 1:40, and grown at 37°C to an A600 of 0.2. Cells were induced with 0.2% arabinose, and 1-ml samples were labeled with 50 μCi/ml of [35S]methionine plus cysteine (IS-103; Hartmann, Braunschweig, Germany) for 1 min. Labeling was stopped by transferring the samples to chilled tubes containing 0.1 ml of 0.2% (wt/vol) methionine. Cells were centrifuged for 2 min at 13,000 × g at 4°C, resuspended in 50 μl of SDS buffer (1% [wt/vol] SDS, 10 mM Tris [pH 8], 1 mM EDTA), and heated for 2 min at 90°C. After 5 min at room temperature, 800 μl of KI buffer (50 mM Tris [pH 8], 150 mM NaCl, 2% [vol/vol] Triton X-100) was added. After 10 min on ice, the lysates were centrifuged for 10 min at 13,000 × g. Antiserum was added to 300 μl of lysates. After 12 h on ice, immune complexes were bound to an excess of fixed Staphylococcus aureus cells (IgGsorb) for 1 h on ice. The cells were washed twice with 1 ml of HB (50 mM Tris [pH 8], 1 M NaCl, 1% Triton X-100, 1 mM EDTA) and once with 1 ml of 10 mM Tris, pH 8. Pellets were resuspended in 50 μl of SB+ (50 mM Tris [pH 6.8], 5% [vol/vol] β-mercaptoethanol, 1% SDS, 0.0025% [wt/vol] bromophenol blue, and 8.5% [vol/vol] glycerol) and boiled for 2 min, and eluates were loaded onto 10% SDS-polyacrylamide gels. Gels were fixed, dried, and autoradiographed. Quantification was performed with a Cyclone MS storage phosphor screen and image analysis with the Optiquant v. 4.00 software (Packard, Meriden, CT).
Pulse-labeling and detection of hexahistidine-tagged membrane protein complexes.
Cells were grown, induced for 1 min with 0.2% arabinose, labeled for 5 min, and lysed as described previously (27). Membranes were solubilized in the presence of E. coli polar lipids, 2 mM MgCl2 and 1% dodecylmaltoside. After incubation at 4°C for 30 min with 20 μl of nickel-nitrilotriacetic acid (Ni-NTA)–agarose matrix (Qiagen), the samples were washed three times with the solubilization buffer. The first and third washes were performed at 4°C. The second wash was performed either at 4°C or at 37°C for 5 min. The proteins were eluted twice for 20 min at 37°C in 20 μl of SBX (80 mM Tris [pH 6.8], 1% [vol/vol] β-mercaptoethanol, 2% SDS, 0.01% [wt/vol] bromophenol blue, 10% [vol/vol] glycerol, and 10 mM EDTA). Eluates were loaded onto 0.5 M NaCl “high-Tris” SDS–19.6% polyacrylamide gels (35) and electrophoresed overnight at 30 mA. Gels were analyzed as described above or with Fujifilm MultiGauge v3.0 software.
RESULTS
Enzymatic assay of protein export in vivo.
The E. coli alkaline phosphatase encoded by the phoA gene is a powerful tool to study protein export as well as the topology of inner membrane integral proteins. Indeed, folding of the enzyme into an active conformation requires the formation of two intramolecular disulfide bonds (36, 37). This oxidation normally occurs only in the bacterial periplasm, although cytoplasmic oxidation can be achieved, for instance, in trxB mutant strains (38). Assay of PhoA activity offers an extremely large dynamic range, which is critical to measure the residual activity of signal sequence mutations causing a strong secretion defect. In addition, the kinetics of enzyme accumulation upon induction of chimeric proteins expression provided a useful tool to explain the selective effects of secG suppressor mutations on the export mediated by toxic mammalian signal sequences (23, 27). We have fused several wild-type and mutant signal sequences to the mature portion of PhoA and expressed the chimeric proteins from the arabinose PBAD promoter (33). Transcriptional induction upon addition of arabinose occurs within minutes, and the accumulation of enzyme activity reaches its steady-state level within 60 to 80 min.
The signal sequences of MalE, PhoA, and RbsB all promote a rapid and efficient export of PhoA in secG+ cells, as judged by the accumulation of enzymatic activity upon induction (Fig. 1A, D, and G). We have analyzed three different secG alleles, a null allele (22), and the secG1 (F43S) and secG15 (L59R) mutants, which can both confer a detectable Sec phenotype (23, 27). There was essentially no effect of the secG mutations on export mediated by the three wild-type signal sequences. In striking contrast, the residual export mediated by the malE10 (L10P) mutant signal sequence was drastically reduced in the secG mutant strains; a slightly higher export was observed in strains expressing secG1 or secG15 compared to the null allele, confirming that these mutations do not entirely abolish secG function. Similar results were obtained with malE14 (A14E), phoA73 (L14Q), and rbsB16 (S16R) (Fig. 1 B, C, E, and H), as well as with several other mutant signal sequences (data not shown).
FIG 1.

Effects of secG mutations on PhoA export. The indicated wild-type and mutant signal sequences were fused to the mature portion of PhoA and expressed from the arabinose PBAD promoter. The kinetics of PhoA export after induction with 0.2% arabinose was measured in isogenic strains carrying the indicated secG alleles (DB617 to DB620). Each curve represents the average results from three independent cultures; error bars indicate the standard deviations (SD).
During the cloning of the chimeric genes, we isolated several revertants that formed dark blue colonies on 5-bromo-4-chloro-3-indolylphosphate (XP)-arabinose indicator plates. The phoA82 mutation deletes two residues in the hydrophobic core of the PhoA signal sequence and was not expected to revert. This pseudoreversion generated a Lys-to-Glu substitution near the end of the signal sequence and probably exerts its effect by improving the charge distribution around the residual hydrophobic core. In a secG+ strain, this revertant was almost as effective in promoting export as the phoA+ signal sequence, although the kinetics of export was slower. In contrast, export was severely decreased in strains carrying either one of the secG mutant alleles (Fig. 1F). Another pseudorevertant was isolated during the cloning of rbsB16 (S16R), in which the mutant Arg residue was replaced by a His one, a residue that would not have been predicted to be active in the hydrophobic core of a signal sequence. Surprisingly, this substitution generated a signal sequence that was nearly as effective as the wild-type one, and its export activity was the same in secG+ and secG mutant strains (Fig. 1I). Similar results were obtained with a pseudorevertant of malE16 (T16K) that contains both the original mutation and an S13F substitution (data not shown).
We have compared the residual export activity of several mutant signal sequences derived from the phoA, malE, and rbsB ones in secG+ and secG null strains. In all cases, export was reduced in the absence of SecG. The magnitude of this effect varied, ranging from a 2-fold to a >20-fold decrease (Fig. 2). There was no correlation between the strength of a given signal sequence defect and its dependence on SecG for residual export. For instance, phoA73 (286 U) has a weaker effect on export than phoA82 (83 U), but its residual export is much more strongly reduced in the absence of SecG (36-fold versus 2-fold).
FIG 2.

The additional export defect in ΔsecG strains does not correlate with the intrinsic defect of individual signal sequence mutations. PhoA export was measured in cultures induced for 60 min. For each signal sequence, the activities in the secG+ and ΔsecG strains (DB617 and DB618) are connected by a line. With the three wild-type signal sequences (*), as well as with the RbsB16 pseudorevertant (S16H, Ψ), the horizontal lines reflect an equivalent export in the presence and in the absence of SecG.
Pulse-chase labeling assay of protein export in vivo.
Signal sequence activity is commonly assessed by short pulse-labeling followed by the determination of the relative amounts of uncleaved precursor and mature protein. To verify that our kinetic assay faithfully measures signal sequence activity, we have also measured signal sequence processing in secG+ and secG null strains by pulse-chase labeling and immunoprecipitation (Fig. 3). With wild-type MalE and PhoA, the amount of precursor was approximately 3 times higher at the end of the 20-s pulse in the secG null strain than in the secG+ strain. After a 3-min chase, there was no longer any detectable difference between the two strains and essentially all the precursors were processed (Fig. 3A, top panels). Considering the difference in time scale of the two assays, the slight delay in export kinetics observed in the secG null strain by pulse-labeling would not be detected in the PhoA enzymatic assay. Pulse-chase experiments were also performed with two signal sequence mutations that confer weak export defects, allowing sufficient signal sequence cleavage to measure both the mature and precursor forms of the mutant proteins (Fig. 3A, bottom panels). With malE16 (T16K), the levels of the mature MalE were barely detectable at the end of the pulse-labeling in secG+ and secG null strains. Export readily occurred during the chase in secG+ cells but not in secG null cells. Similar results were obtained with phoA73 (L14Q). With most of the other signal sequence mutations, export was too low to accurately determine the amount of cleaved mature proteins, even in secG+ cells.
FIG 3.

Kinetics of export measured by pulse-chase and immunoprecipitation. (A) Kinetics of MalE and PhoA export in secG+ and ΔsecG strains. The proteins, expressed from the chromosomal genes, contained either the wild-type alleles or the indicated signal sequence mutations. Cells were pulse-labeled at 37°C for 30 s and chased for 3 and 10 min. Lysates were immunoprecipitated with anti-MalE or anti-PhoA antibodies. The percentages of mature proteins were calculated from the intensities of the mature and precursor bands. The values represent the averages from two independent cultures; the error bars indicate ranges. (B) Hybrid proteins containing the indicated signal sequences fused to the mature portion of OmpA were expressed from the PBAD promoter in strain DB686 (secG+) or DB685 (ΔsecG), in which the chromosomal ompA gene is deleted. After 5 min of induction with 0.2% arabinose, the cultures were pulse-labeled for 20 s and chased for 10 min. Lysates were immunoprecipitated with anti-OmpA antibodies. The values represent the percentages of mature OmpA and are the averages from three independent cultures; error bars indicate the SD.
Our observations are not limited to export substrates containing the mature portion of PhoA. Several signal sequences were fused to the mature portion of OmpA. Their export was also analyzed by pulse-chase and immunoprecipitation (Fig. 3B). OmpA export during a 20-s pulse was slightly reduced in secG null cells, but there was no significant difference after a 10-min chase. Similar results were obtained with the PhoA-OmpA chimera, although export efficiency was reduced, indicating that the mature portion can influence signal sequence activity (data not shown). The export of the PhoA82rev-OmpA chimera was efficient in secG+ cells, particularly after a 10-min chase. In contrast, this protein was very poorly exported in secG null cells. The RbsB+ and RbsB16rev (S16H) signal sequences had similar high export activity when fused to OmpA, and their export efficiency was only slightly decreased in secG null cells.
The PhoA enzyme accumulation assay is less efficient than a pulse-chase to measure slight secretion defects since kinetic differences of fewer than a few minutes are not easily detected. In contrast, export of chimera with low signal sequence residual activity is readily determined enzymatically because of the large dynamic range of the PhoA assay (0.5 to 2,000 units). In conclusion, our results indicate that the presence of SecG has only a slight effect on export mediated by wild-type signal sequences but plays a major role in the residual activity of mutant signal sequences.
In vivo interactions between prlA and secG mutant alleles.
prlA mutations increase the export efficiency of mutant signal sequences. Since secG mutations exert an opposite effect, it seemed interesting to determine whether epistasis could be detected in cells containing both prlA and secG mutations. Members of two sets of partially overlapping prlA mutations have been analyzed (12, 23). The amino acid substitutions introduced in the second set were determined by sequencing the secY gene. Mutations like prlA3 and prlA545 can be introduced by P1-mediated transduction in strains carrying a secG null or secG point mutation alleles at the expected frequency, indicating that both mutant alleles can be present in the same cells. When export mediated by the MalE14 signal sequence was measured in these double mutant strains, the values obtained were intermediate between those observed in prlA secG+ and in prlA+ secG single mutant strains (Fig. 4, top panels). This suggests that SecY and SecG act together during the signal sequence interaction(s) with the translocase. However, the magnitude of the export increase mediated by these prlA mutations was significantly higher in secG mutant strains (Fig. 4, bottom panels).
FIG 4.

Combined effects of secG and prlA mutations on PhoA export. The MalE14 mutant signal sequence was fused to the mature portion of PhoA and expressed from the arabinose PBAD promoter (pDB613). The level of PhoA export 60 min after induction with 0.2% arabinose was measured in isogenic strains derived from DB687 (ΔsecG). (A) prlA3 mutant strain; (B) prlA545 mutant strain. The indicated secG alleles were expressed from pACYC184-derived plasmids (27). The prlA alleles were introduced by cotransduction with the gspA::Tn10 marker. Each value represents the average of results from two to four independent cultures assayed twice; error bars indicate the SD. In the bottom panels, the ratios of mean PhoA export in the prlA mutant and prlA+ strains are indicated.
Several prlA alleles could not be introduced normally by cotransduction with a linked gspA::Tn10 marker in secG null strains. With a P1 lysate grown on a prlA502, gspA::Tn10 strain, for instance, 24/24 Tcr transductants retained the wild-type secY+ allele, even though secY alleles are normally cotransduced with this marker at a frequency of 90 to 95%. This suggested that the simultaneous presence of some prlA and secG null or nearly null alleles confers a synthetic phenotype. To test this hypothesis, we attempted to introduce the secG::Kn null allele in the prlA strains by selecting for Knr transductants. Although secG null transductants can be recovered under these conditions, their frequency was drastically reduced (Fig. 5). This reduction depended on the individual prlA alleles. A 200-fold decrease was observed with prlA517, prlA543, prlA207, and prlA304, whereas a small but reproducible 2- to 4-fold decrease was observed with prlA6. Although they are unexpectedly low, the frequencies observed are significantly higher than those of reversion or of spontaneous mutations, unless the target size is more than 100 genes. These frequencies suggest that cells expressing these prlA alleles can somehow adapt to the absence of SecG.
FIG 5.

Transduction frequencies of the ΔsecG allele in prlA strains. Representative members of two sets of prlA alleles (12, 23) were analyzed in each panel. All strains are derivatives of DB504 that contain the gspA::Tn10 marker and the indicated prlA alleles. Transductions were carried out with P1 lysates grown on strain DB638, to introduce the secG null allele, and on strain CAG12131, to determine the transduction efficiency of each culture. The numbers of Knr secG null transductants were normalized to those of Knr leuO transductants. Different lysates grown on CAG12131 were used in each panel. Each value represents the average of results from three independent cultures; error bars indicate the range.
We have isolated strains carrying both the secG::Kn null allele and each of the four prlA alleles that cannot be easily transduced in secG::Kn strains. When export mediated by the MalE14 signal sequence was measured in these double mutant strains, the values obtained were high and intermediate between those observed in the prlA+ secG+ parent strain and in prlA secG+ single mutant strains (Fig. 6, top panels). Strikingly, the magnitude of the export increase mediated by these prlA mutations was more than 40-fold higher in secG mutant strains (Fig. 6, bottom panels). Thus, the wild-type SecG protein somehow counteracts the permissive effect of the prlA mutations. This effect is modest with prlA alleles than can be freely introduced in secG mutant strains but very high with those that cannot be easily transduced.
FIG 6.

Enhanced effects of some prlA mutations on PhoA export in the absence of secG. The prlA alleles were introduced in DB504 by cotransduction with the gspA::Tn10 marker, and pDB613 was introduced by transformation. KnR secG null and control Knr leuO transductants were isolated, and PhoA export was assayed 60 min after induction with 0.2% arabinose. Each value represents the average result for three independent transductants assayed twice; error bars indicate the SD. In the bottom panels, the ratios of mean PhoA export in the prlA mutant and prlA+ strains are indicated.
In vitro interactions between SecY and SecE in the presence or in the absence of SecG.
prl mutations in prlA or prlG have been shown to destabilize the interaction of SecY with SecE in solubilized membranes (19). It seemed therefore possible that secG mutations also affect the stability of SecYE heterodimers, thus accounting for the difficulty of introducing a secG null allele in several of the prlA mutant strains. To test this hypothesis, inducible plasmids that contain selected prlA alleles with or without secG were constructed. All plasmids expressed a His-tagged SecE, to allow the affinity purification of SecE from solubilized membranes. The amount of SecY bound to SecE, in the presence or in the absence of SecG, could then be quantified. This method seemed particularly suitable to assess the affinity of different SecY proteins for SecE, since different temperatures and/or different detergents can be used to solubilize the membranes.
Membranes of induced cells expressing either SecY+ or PrlA545 were solubilized on ice and kept at 4°C during the purification procedure. Under these conditions, stoichiometric amounts of both SecY proteins remained bound to SecE, both in the presence and in the absence of SecG (Fig. 7A, left panel). When the complexes were incubated at 37°C, the amount of SecY that remained bound to SecE was reduced approximately 2-fold in the presence of SecG. The absence of SecG had only a modest effect on SecY+ binding, whereas binding of the PrlA545 protein was reduced approximately 3-fold. In contrast, binding of the PrlA517 protein to SecE was strongly dependent on the presence of SecG, even at 4°C. We have determined by pulse-labeling and immunoprecipitation that equivalent amounts of SecY accumulated with the prlA+ and prlA517 plasmids either in the presence or in the absence of SecG (Fig. 7A, right panel). The labeling period was the same as that used to prepare the membranes. The binding of PrlA7 to SecE was not affected by the absence of SecG at 4°C and was reduced approximately 3-fold at 37°C. Binding of PrlA207 to SecE was also not affected by the absence of SecG at 4°C but was barely detectable at 37°C (Fig. 7B and C). Since the cells express the prlA+ allele from the chromosomal gene, it is likely that these very low levels reflect mainly the presence of the SecY+ protein bound to SecE. These results confirm the observation that prlA alleles affect the stability of the SecYE dimer (19). Furthermore, they indicate that SecG can contribute to the stability of the SecYE dimer and that this contribution is dependent on the nature of individual prlA alleles. Finally, the correlation between the efficiency of introduction of the secG null allele in a given prlA strain (Fig. 5) and the stability of the different SecYE dimers (Fig. 7) provides a biochemical explanation for this genetic phenomenon.
FIG 7.

Stability of the different SecYE dimers in solubilized membranes. DB559 cells carrying derivatives of pBAD-HisEYG were induced and labeled with [35S]methionine as described previously (27). Isolated membranes were solubilized by dodecylmaltoside in the presence of E. coli polar lipids. His-tagged SecE was purified on Ni2+-agarose. The complexes were either kept all the time at 4°C or washed once at 37°C for 5 min. (A) Left panel, comparison of SecY+, PrlA517 (G81E), and PrlA545 (A71V). Right panel, cultures labeled as described for the preparation of membranes or chased for an additional 5 min were lysed and immunoprecipitated with an anti-SecY antibody; the amounts of accumulated SecY were normalized to that detected at the end of the labeling period with the SecY+-SecG+ encoding plasmid. (B) Comparison of SecY+, PrlA7 (L407R), and PrlA207 (I278S). While experiments performed at 4°C were highly reproducible, binding of SecY+ at 37°C in the absence of SecG was more variable, as can be seen by comparison with results shown in panel A. (C) Autoradiogram of part of the experiments quantified in panel B.
DISCUSSION
This work was prompted by the discrepancy between the effects observed in vivo and in vitro upon removal of SecG (23, 24, 39). SecG function in vitro is particularly evident in the absence of a proton motive force (40). In addition, the effect of SecG can be produced by substitution of the SecDF complex (26). Our results confirm that SecG has only a minimal effect in vivo on the export of all wild-type E. coli proteins tested so far; this effect can be detected only with short pulse-labeling. The conservation of this nonessential gene during evolution is therefore puzzling. In the Gram-positive bacterium S. aureus, SecG is also nonessential for growth and for mouse infection, but it is critical for the normal accumulation of several exoproteins (41). Distant SecG homologues are also present in Archaea and eukaryotes. In yeast, deletion of the two SecG homologues (Sbh1 and Sbh2) is viable but confers a temperature-sensitive phenotype (42). In Drosophila melanogaster, a mutation in the Sec61β/secG promoter that essentially abolishes its expression is compatible with basal cell functions, including protein translocation. However, a developmental arrest was proposed to result from the defective secretion of Gurken and cuticle proteins (43); a recent study showed that Gurken is normally translocated in the endoplasmic reticulum (ER) lumen of the mutant flies (44). This suggests that a later step of the secretion pathway, possibly common to Gurken and cuticle proteins, is defective in Sec61β mutant embryos. Sec61β may therefore be required for the translocation or for the membrane integration of a secretory cofactor(s). In any event, the sequence conservation among these SecG homologues is much lower than that observed with the SecY and SecE homologues. This agrees with the notion that the SecG homologues play an accessory role in protein export.
Nevertheless, even a very slight growth advantage, hardly detectable under standard laboratory conditions, would ensure the maintenance of a “nonessential gene” over millions of years of evolution (45). We showed here that the residual export mediated by mutant signal sequences, as well as the efficient export of one pseudorevertant, can strongly depend on SecG. This suggests an additional possibility for the conservation of SecG function during evolution. About one-third of the E. coli genes encode envelope proteins that are targeted to the inner membrane, the periplasm, or the outer membrane. Thus, many different proteins are translocase substrates. Since horizontal gene transfer plays a major role in the evolution of bacterial genomes, newly imported foreign envelope genes harbor signal sequences that evolved with the translocase of their original host and may not be efficient substrates for the E. coli translocase. Upon transfer to the new host, the integration or export of the new envelope proteins could be much more dependent on SecG than those of the endogenous envelope genes.
Prl mutations have been isolated in all major components of the translocase, in the ATPase motor SecA as well as in SecYEG, the core inner membrane constituents of the translocation channel (15, 46). The current biochemical model of the Prl phenotype suggests that prl mutations, in a direct or indirect way, contribute to the opening of the translocation channel by displacement of the pore formed by the TM2a helix of SecY or by stabilization of the open complex (17, 18). Several prl mutations in secY and secE have been shown to have an additional effect on the SecYEG complex, because they weaken the interaction of SecY with SecE (19). Our results show that the stability of this complex can also be influenced by SecG, as suggested previously (26). This result appeared surprising since SecG is bound to a side of SecY opposite to that involved in SecE binding (Fig. 8). The prlA alleles that tolerate normally the absence of SecG can be grouped on a side of the SecYEG complex (Fig. 8, top panel). A top view of the complex shows that they are distributed more or less evenly around the plug domain (bottom panel). The prlA alleles that prevent the introduction of a secG deletion appear to be more widely distributed, even though most of them also surround the plug domain; the two alleles that map to the TM2 domain are located toward the membrane, on the outside of the complex and far away from both SecE and SecG. The Methanocaldococcus jannaschii SecG and SecE have only a single transmembrane domain. SecG makes only limited contacts with SecY but is close to the C-terminal end of SecE (3). It seems difficult to envision how single amino acid substitutions in SecY could make its binding to SecE so dependent on the presence of SecG. It is also possible that additional interactions of SecY involve the second transmembrane domain of the E. coli SecG and/or the N-terminal portion of SecE. In addition, the crystal structures show SecYEG only in a closed form. Taken together, our results are compatible with the notion that the SecYEG complex is very dynamic and that all three Sec proteins contribute to its stability. This was first suggested by the observation that SecG undergoes a topological inversion during translocation, an event that requires the MPIase glycolipid (47, 48).
FIG 8.
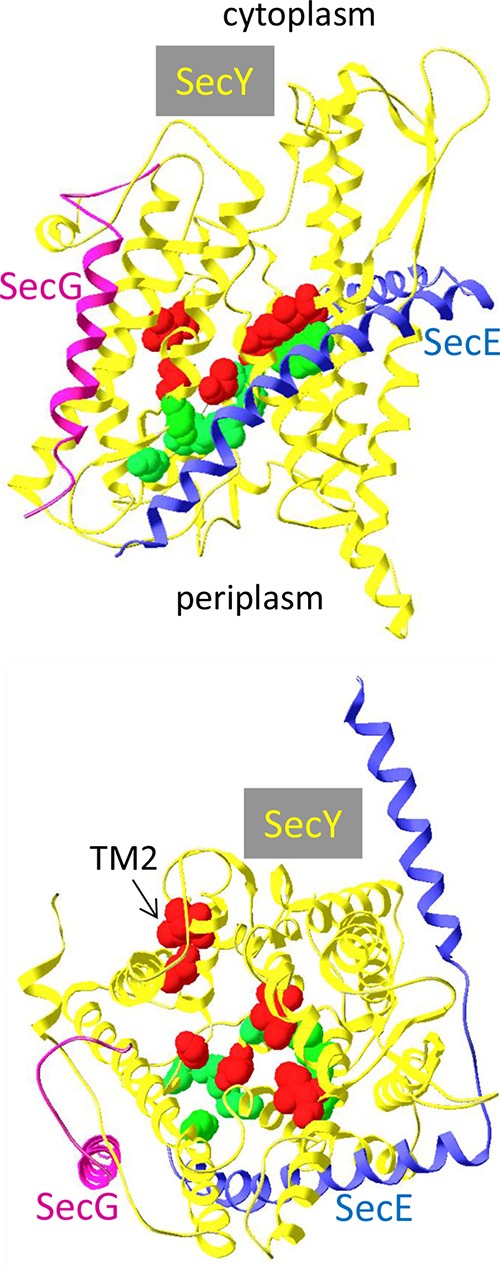
Localization of the prlA alleles on the crystal structure of the M. jannaschii SecYEG complex. Top, lateral view from the membrane plane; bottom, from the cytoplasm. Each subunit is color coded and shown as a ribbon. The side chains of residues modified by prlA mutations are indicated by their volume. Residues that interfere with the introduction of a ΔsecG allele by P1 transduction are shown in red. Residues that do not affect the introduction of a ΔsecG allele are shown in green. The structure (PDB ID 1RHZ [3]) was drawn with DeepViewSwiss-Pdbviewer v4.0.4.
We had isolated a set of prlA mutations that increase MalE18 export sufficiently to allow growth on minimal maltose plates in a secG1 (F43S) strain (23). While there was a definite overlap with previously isolated prlA mutations (for instance, the G69D substitution is present in both sets [prlA502 and prlA9]), our set was significantly different in two aspects. First, only 1 of the 11 mutations mapped to TM7 or TM10, whereas 10/24 previously known mutations mapped to these domains. Second, our set appeared to be slightly enriched for mutations in the hinge and TM2 domains (3/11), a region of SecY where only one prl mutation had been previously isolated. With approximately one-half the prlA alleles, the introduction of a secG deletion was more difficult than anticipated. These alleles are not restricted to a domain of SecY. For instance, the prlA3, prlA502, and prlA545 mutations all affect residues located in or near the TM2a plug domain (residues 67, 69, and 71). While the G69D substitution severely interferes with introduction of a secG deletion, the F67C and A71V substitutions behave like SecY+ in this assay. There is also no strict correlation with a classification of prlA alleles (10). The biochemical basis for this synthetic phenotype is not known. It may be related to the interaction of phospholipids metabolism and SecG function (49). Although cells containing certain pairs of secY and secG alleles appear at a frequency that can be as low as 10−3, they grow normally once isolated.
Our current model is that SecG helps the opening step of translocation (plug displacement). The transition from a closed ground state to an activated open state is catalyzed by a signal sequence and SecG. Wild-type signal sequences are probably sufficiently efficient in this initial step to allow normal export in the absence of SecG. With weak or mutant signal sequences, the contribution of SecG becomes critical. When toxic signal sequences are fused to an export reporter, the specific reduction in export kinetics provided by the SecG suppressor mutations is then sufficient to allow growth (24, 27). The properties of pseudorevertants of mutant signal sequences are compatible with this model. It is possible that SecG also contributes to later stages of protein translocation, such as elongation and/or lateral opening, to allow the integration of transmembrane domains in the lipid bilayer.
ACKNOWLEDGMENTS
This work was supported by the State of Geneva and, initially, by the Swiss National Science Foundation.
We thank C. Georgopoulos, P. Linder, M. Strubin, and Y. Mattenberger for helpful discussions and I. Collinson for the anti-SecY antibody.
REFERENCES
- 1.Veenendaal AK, van der Does C, Driessen AJ. 2004. The protein-conducting channel SecYEG. Biochim Biophys Acta 1694:81–95. doi: 10.1016/j.bbamcr.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 2.Rusch SL, Kendall DA. 2007. Oligomeric states of the SecA and SecYEG core components of the bacterial Sec translocon. Biochim Biophys Acta 1768:5–12. doi: 10.1016/j.bbamem.2006.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van den Berg B, Clemons WM Jr, Collinson I, Modis Y, Hartmann E, Harrison SC, Rapoport TA. 2004. X-ray structure of a protein-conducting channel. Nature 427:36–44. doi: 10.1038/nature02218. [DOI] [PubMed] [Google Scholar]
- 4.Clemons WM Jr, Menetret JF, Akey CW, Rapoport TA. 2004. Structural insight into the protein translocation channel. Curr Opin Struct Biol 14:390–396. doi: 10.1016/j.sbi.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 5.Pohlschroder M, Prinz WA, Hartmann E, Beckwith J. 1997. Protein translocation in the three domains of life: variations on a theme. Cell 91:563–566. doi: 10.1016/S0092-8674(00)80443-2. [DOI] [PubMed] [Google Scholar]
- 6.Osborne AR, Rapoport TA, van den Berg B. 2005. Protein translocation by the Sec61/SecY channel. Annu Rev Cell Dev Biol 21:529–550. doi: 10.1146/annurev.cellbio.21.012704.133214. [DOI] [PubMed] [Google Scholar]
- 7.Joly JC, Leonard MR, Wickner WT. 1994. Subunit dynamics in Escherichia coli preprotein translocase. Proc Natl Acad Sci U S A 91:4703–4707. doi: 10.1073/pnas.91.11.4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schatz PJ, Beckwith J. 1990. Genetic analysis of protein export in Escherichia coli. Annu Rev Genet 24:215–248. doi: 10.1146/annurev.ge.24.120190.001243. [DOI] [PubMed] [Google Scholar]
- 9.Danese PN, Silhavy TJ. 1998. Targeting and assembly of periplasmic and outer-membrane proteins in Escherichia coli. Annu Rev Genet 32:59–94. doi: 10.1146/annurev.genet.32.1.59. [DOI] [PubMed] [Google Scholar]
- 10.Emr SD, Hanley-Way S, Silhavy TJ. 1981. Suppressor mutations that restore export of a protein with a defective signal sequence. Cell 23:79–88. doi: 10.1016/0092-8674(81)90272-5. [DOI] [PubMed] [Google Scholar]
- 11.Stader J, Gansheroff LJ, Silhavy TJ. 1989. New suppressors of signal-sequence mutations, prlG, are linked tightly to the secE gene of Escherichia coli. Genes Dev 3:1045–1052. doi: 10.1101/gad.3.7.1045. [DOI] [PubMed] [Google Scholar]
- 12.Osborne RS, Silhavy TJ. 1993. PrlA suppressor mutations cluster in regions corresponding to three distinct topological domains. EMBO J 12:3391–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bost S, Belin D. 1997. prl mutations in the Escherichia coli secG gene. J Biol Chem 272:4087–4093. doi: 10.1074/jbc.272.7.4087. [DOI] [PubMed] [Google Scholar]
- 14.Smith MA, Clemons WM Jr, DeMars CJ, Flower AM. 2005. Modeling the effects of prl mutations on the Escherichia coli SecY complex. J Bacteriol 187:6454–6465. doi: 10.1128/JB.187.18.6454-6465.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flower AM. 2007. The SecY translocation complex: convergence of genetics and structure. Trends Microbiol 15:203–210. doi: 10.1016/j.tim.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 16.Derman AI, Puziss JW, Bassford PJ, Beckwith J. 1993. A signal sequence is not required for protein export in prlA mutants of Escherichia coli. EMBO J 12:879–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li W, Schulman S, Boyd D, Erlandson K, Beckwith J, Rapoport TA. 2007. The plug domain of the SecY protein stabilizes the closed state of the translocation channel and maintains a membrane seal. Mol Cell 26:511–521. doi: 10.1016/j.molcel.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 18.Maillard AP, Lalani S, Silva F, Belin D, Duong F. 2007. Deregulation of the SecYEG translocation channel upon removal of the plug domain. J Biol Chem 282:1281–1287. doi: 10.1074/jbc.M610060200. [DOI] [PubMed] [Google Scholar]
- 19.Duong F, Wickner W. 1999. The PrlA and PrlG phenotypes are caused by a loosened association among the translocase SecYEG subunits. EMBO J 18:3263–3270. doi: 10.1093/emboj/18.12.3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishiyama K, Kabuyama Y, Akimaru J, Matsuyama S, Tokuda H, Mizushima S. 1991. SecY is an indispensable component of the protein secretory machinery of Escherichia coli. Biochim Biophys Acta 1065:89–97. doi: 10.1016/0005-2736(91)90015-Z. [DOI] [PubMed] [Google Scholar]
- 21.Schatz PJ, Bieker KL, Ottemann KM, Silhavy TJ, Beckwith J. 1991. One of three transmembrane stretches is sufficient for the functioning of the SecE protein, a membrane component of the E. coli secretion machinery. EMBO J 10:1749–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishiyama K, Hanada M, Tokuda H. 1994. Disruption of the gene encoding p12 (SecG) reveals the direct involvement and important function of SecG in the protein translocation of Escherichia coli at low temperature. EMBO J 13:3272–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bost S, Belin D. 1995. A new genetic selection identifies essential residues in SecG, a component of the Escherichia coli protein export machinery. EMBO J 14:4412–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flower AM, Hines LL, Pfennig PL. 2000. SecG is an auxiliary component of the protein export apparatus of Escherichia coli. Mol Gen Genet 263:131–136. doi: 10.1007/s004380050039. [DOI] [PubMed] [Google Scholar]
- 25.Nishiyama K, Mizushima S, Tokuda H. 1993. A novel membrane protein involved in protein translocation across the cytoplasmic membrane of Escherichia coli. EMBO J 12:3409–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duong F, Wickner W. 1997. Distinct catalytic roles of the SecYE, SecG and SecDFyajC subunits of preprotein translocase holoenzyme. EMBO J 16:2756–2768. doi: 10.1093/emboj/16.10.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bost S, Silva F, Rudaz C, Belin D. 2000. Both transmembrane domains of SecG contribute to signal sequence recognition by the Escherichia coli protein export machinery. Mol Microbiol 38:575–587. doi: 10.1046/j.1365-2958.2000.02153.x. [DOI] [PubMed] [Google Scholar]
- 28.Belin D. 2010. In vivo analysis of protein translocation to the Escherichia coli periplasm. Methods Mol Biol 619:103–116. doi: 10.1007/978-1-60327-412-8_6. [DOI] [PubMed] [Google Scholar]
- 29.Khatib K, Belin D. 2002. A novel class of secA alleles that exert a signal-sequence-dependent effect on protein export in Escherichia coli. Genetics 162:1031–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belin D, Guzman LM, Bost S, Konakova M, Silva F, Beckwith J. 2004. Functional activity of eukaryotic signal sequences in Escherichia coli: the ovalbumin family of serine protease inhibitors. J Mol Biol 335:437–453. doi: 10.1016/j.jmb.2003.10.076. [DOI] [PubMed] [Google Scholar]
- 31.Bedouelle H, Bassford PJJ, Fowler AV, Zabin I, Beckwith J, Hofnung M. 1980. Mutations which alter the function of the signal sequence of the maltose binding protein of Escherichia coli. Nature 285:78–81. doi: 10.1038/285078a0. [DOI] [PubMed] [Google Scholar]
- 32.Francetic O, Kumamoto CA. 1996. Escherichia coli SecB stimulates export without maintaining export competence of ribose-binding protein signal sequence mutants. J Bacteriol 178:5954–5959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Collinson I, Breyton C, Duong F, Tziatzios C, Schubert D, Or E, Rapoport T, Kuhlbrandt W. 2001. Projection structure and oligomeric properties of a bacterial core protein translocase. EMBO J 20:2462–2471. doi: 10.1093/emboj/20.10.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brundage L, Hendrick JP, Schiebel E, Driessen AJ, Wickner W. 1990. The purified E. coli integral membrane protein SecY/E is sufficient for reconstitution of SecA-dependent precursor protein translocation. Cell 62:649–657. doi: 10.1016/0092-8674(90)90111-Q. [DOI] [PubMed] [Google Scholar]
- 36.Boyd D, Manoil C, Beckwith J. 1987. Determinants of membrane protein topology. Proc Natl Acad Sci U S A 84:8525–8529. doi: 10.1073/pnas.84.23.8525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Manoil C, Mekalanos JJ, Beckwith J. 1990. Alkaline phosphatase fusions: sensors of subcellular location. J Bacteriol 172:515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Derman AI, Prinz WA, Belin D, Beckwith J. 1993. Mutations that allow disulfide bond formation in the cytoplasm of Escherichia coli. Science 262:1744–1747. doi: 10.1126/science.8259521. [DOI] [PubMed] [Google Scholar]
- 39.Hanada M, Nishiyama KI, Mizushima S, Tokuda H. 1994. Reconstitution of an efficient protein translocation machinery comprising SecA and the three membrane proteins, SecY, SecE, and SecG (p12). J Biol Chem 269:23625–23631. [PubMed] [Google Scholar]
- 40.Hanada M, Nishiyama K, Tokuda H. 1996. SecG plays a critical role in protein translocation in the absence of the proton motive force as well as at low temperature. FEBS Lett 381:25–28. doi: 10.1016/0014-5793(96)00066-X. [DOI] [PubMed] [Google Scholar]
- 41.Sibbald MJ, Winter T, van der Kooi-Pol MM, Buist G, Tsompanidou E, Bosma T, Schafer T, Ohlsen K, Hecker M, Antelmann H, Engelmann S, van Dijl JM. 2010. Synthetic effects of secG and secY2 mutations on exoproteome biogenesis in Staphylococcus aureus. J Bacteriol 192:3788–3800. doi: 10.1128/JB.01452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Finke K, Plath K, Panzner S, Prehn S, Rapoport TA, Hartmann E, Sommer T. 1996. A second trimeric complex containing homologs of the Sec6lp complex functions in protein transport across the ER membrane of S. cerevisiae. EMBO J 15:1482–1494. [PMC free article] [PubMed] [Google Scholar]
- 43.Valcarcel R, Weber U, Jackson DB, Benes V, Ansorge W, Bohmann D, Mlodzik M. 1999. Sec61beta, a subunit of the protein translocation channel, is required during Drosophila development. J Cell Sci 112:4389–4396. [DOI] [PubMed] [Google Scholar]
- 44.Kelkar A, Dobberstein B. 2009. Sec61beta, a subunit of the Sec61 protein translocation channel at the endoplasmic reticulum, is involved in the transport of Gurken to the plasma membrane. BMC Cell Biol 10:11. doi: 10.1186/1471-2121-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cao TB, Saier MH Jr. 2003. The general protein secretory pathway: phylogenetic analyses leading to evolutionary conclusions. Biochim Biophys Acta 1609:115–125. doi: 10.1016/S0005-2736(02)00662-4. [DOI] [PubMed] [Google Scholar]
- 46.Huie JL, Silhavy TJ. 1995. Suppression of signal sequence defects and azide resistance in Escherichia coli commonly result from the same mutations in secA. J Bacteriol 177:3518–3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nishiyama KI, Suzuki T, Tokuda H. 1996. Inversion of the membrane topology of SecG coupled with SecA-dependent preprotein translocation. Cell 85:71–81. doi: 10.1016/S0092-8674(00)81083-1. [DOI] [PubMed] [Google Scholar]
- 48.Moser M, Nagamori S, Huber M, Tokuda H, Nishiyama K. 2013. Glycolipozyme MPIase is essential for topology inversion of SecG during preprotein translocation. Proc Natl Acad Sci U S A 110:9734–9739. doi: 10.1073/pnas.1303160110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Flower AM. 2001. SecG function and phospholipid metabolism in Escherichia coli. J Bacteriol 183:2006–2012. doi: 10.1128/JB.183.6.2006-2012.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Casadaban MJ, Cohen SN. 1980. Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J Mol Biol 138:179–207. doi: 10.1016/0022-2836(80)90283-1. [DOI] [PubMed] [Google Scholar]
- 51.Casadaban MJ. 1976. Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J Mol Biol 104:541–555. doi: 10.1016/0022-2836(76)90119-4. [DOI] [PubMed] [Google Scholar]
- 52.Singer M, Baker TA, Schnitzler G, Deischel SM, Goel M, Dove W, Jaacks KJ, Grossman AD, Erickson JW, Gross CA. 1989. A collection of strains containing genetically linked alternating antibiotic resistance elements for genetic mapping of Escherichia coli. Microbiol Rev 53:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Michaelis S, Inouye H, Oliver D, Beckwith J. 1983. Mutations that alter the signal sequence of alkaline phosphatase in Escherichia coli. J Bacteriol 154:366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Michaelis S, Hunt JF, Beckwith J. 1986. Effects of signal sequence mutations on the kinetics of alkaline phosphatase export to the periplasm in Escherichia coli. J Bacteriol 167:160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]