

Abstract
Drug resistance in Plasmodium falciparum remains a challenge for the malaria eradication programmes around the world. With the emergence of artemisinin resistance, the efficacy of the partner drugs in the artemisinin combination therapies (ACT) that include quinoline-based drugs is becoming critical. So far only few resistance markers have been identified from which only two transmembrane transporters namely PfMDR1 (an ATP-binding cassette transporter) and PfCRT (a drug-metabolite transporter) have been experimentally verified. Another P. falciparum transporter, the ATP-binding cassette containing multidrug resistance-associated protein (PfMRP2) represents an additional possible factor of drug resistance in P. falciparum. In this study, we identified a parasite clone that is derived from the 3D7 P. falciparum strain and shows increased resistance to chloroquine, mefloquine and quinine through the trophozoite and schizont stages. We demonstrate that the resistance phenotype is caused by a 4.1 kb deletion in the 5′ upstream region of the pfmrp2 gene that leads to an alteration in the pfmrp2 transcription and thus increased level of PfMRP2 protein. These results also suggest the importance of putative promoter elements in regulation of gene expression during the P. falciparum intra-erythrocytic developmental cycle and the potential of genetic polymorphisms within these regions to underlie drug resistance.
Introduction
Drug resistance of Plasmodium falciparum, the deadliest species of malaria parasites is a key threat to the disease elimination programmes worldwide (White, 2010). The first major defeat in the battle against malaria stemmed from the onset of chloroquine resistance in South East Asia and South America in the 1960s and a subsequent spread to Africa over the following 20 years (Wellems and Plowe, 2001; Wootton et al., 2002; Sa et al., 2009). In the 1970s through 1990s, the spread of P. falciparum-resistant strains compromised efficacy and use of other antimalarials such as sulphadoxine-pyrimethamine, mefloquine, quinine and other quinoline drugs (Wongsrichanalai et al., 2001; Rojanawatsirivet et al., 2004; Roper et al., 2004). The artemisinin combination therapy (ACT) that was implemented in late 1990s as the first line treatment halted the further spread of resistance to the ‘classical’ antimalarials in spite of their use as ACT partners (Nosten and White, 2007). However, the recent emergence of artemisinin resistance at the Thai-Cambodian and Thai-Myanmar borders presents a major threat to malaria control programmes and calls for more intense research into the mode of action and resistance mechanisms of not only artemisinin but also the partner drugs (Noedl et al., 2008; Dondorp et al., 2009; 2011). Identifying drug resistance markers in P. falciparum is essential for the general understanding of the molecular mechanisms but also for the purpose of surveillance and careful administration of suitable drug regiments (White and Olliaro, 1996; Price and Nosten, 2001; Wellems and Plowe, 2001; Dondorp et al., 2011).
Over the last two decades, much attention has been paid to P. falciparum genes encoding drug/metabolite transporter proteins and ATP-binding cassette (ABC) transporters for their potential as drug resistance factors (Gardner et al., 2002; Sauvage et al., 2009). These include P. falciparum chloroquine resistance transporter (PfCRT) and multi-drug resistance protein (PfMDR1) that were originally identified as main factors of chloroquine resistance and recently linked with tolerance to other quinoline-based antimalarial drugs (Fidock et al., 2000; Djimde et al., 2001; Price et al., 2004; Martin et al., 2009; Setthaudom et al., 2011). A single-nucleotide polymorphism (SNP) that changes the 76th codon from lysine to threonine in PfCRT is one of the most frequent mutation directly associated with chloroquine resistance in Asian, African and South American isolates (Djimde et al., 2001; Bray et al., 2005; Lakshmanan et al., 2005). Amplification of the pfmdr1 gene in Thai isolates associates with P. falciparum resistance to mefloquine, artesunate, halofantrine, quinine and dihydroartemisinin while the presence of the N86Y mutation enhances parasite sensitivity to mefloquine but predisposes it to chloroquine resistance (Price et al., 1999; 2004; Djimde et al., 2001; Sidhu et al., 2005). Although the mutations and copy number polymorphisms (CNP) in the two drug-resistance genes appear to be among the main contributors, their prevalence alone is often not sufficient to explain drug resistance. This suggests an involvement of other genetic factors that contribute to the resistance phenotype as a part of the widely recognized complex genetic traits (Mu et al., 2003; Anderson et al., 2005; Lehane et al., 2011).
Additional members of the ABC-transporter gene family, namely the ABCC subfamily, termed multidrug resistance-associated protein (MRP) were suggested to play a role in malaria drug resistance. The P. falciparum genome encodes two MRPs homologues (pfmrp1 and pfmrp2) and analogously to other eukaryotes, both of these proteins are suggested to function in the redox metabolism pathway transporting glutathione (GSSG/GSH) and glutathione conjugates (GS-X) across biological membranes (Jones and George, 2005). It was suggested that PfMRP1 (PFA0590w) mediates drug resistance since knockout P. falciparum cell lines exhibit increased accumulation of glutathione and increased sensitivity to chloroquine, quinine, artemisinin, piperaquine and primaquine (Raj et al., 2009). In these experiments, the pfmrp1 deletion parasite lines are unable to survive beyond 5% parasitaemia in the culture, suggesting that this protein is crucial for normal physiological growth (Raj et al., 2009). In field isolates from Tanzania, the mutant pfmrp1 gene carrying the 1466 K allele was found to be associated with recrudescent infections following sulphadoxine-pyrimethamine treatment in children (Dahlstrom et al., 2009b). Analogously, treatment with the artemether–lumefantrine combination resulted in selection of the I876V polymorphism of pfmrp1 gene in African parasites in recurrent infections (Dahlstrom et al., 2009a). Recently, a non-synonymous SNP causing F1390I substitution in pfmrp1 was found among Thai-Myanmar field isolates and was shown to be associated with increased in vitro IC50s of various antimalarials such as artemisinin, mefloquine and lumefantrine (Veiga et al., 2011). The role of the second orthologue, pfmrp2, in P. falciparum drug resistance is unknown. Although SNPs in the pfmrp2 gene were detected in culture-adapted strains HB3, Dd2, D10 and 7G8 as well as in 4 African field isolates, none of these could be associated with differential drug sensitivities (Mu et al., 2003; Robinson et al., 2011).
In this study, we provide evidence for a potential role of PfMRP2 in drug resistance of P. falciparum by investigating two 3D7 isogenic clones that exhibit differences in drug sensitivities. We identified a structural polymorphism (∼4.1 kb deletion) in the 5′ upstream region of pfmrp2 that results in the shift of its peak expression to the late stages of the intra-erythrocytic developmental cycle (IDC) (trophozoite and schizont stages). This transcriptional shift results in increased levels of PfMRP2 in the trophozoite and schizont stages that coincide with a decreased sensitivity of the mutant strain to mefloquine, chloroquine and quinine. Here we provide first evidence that PfMRP2 is potentially involved in drug resistance via polymorphism in its transcriptional regulatory elements.
Results
Characterization of drug sensitivity of 3D7 clones
At the onset of our study, we wished to characterize drug sensitivities of isogenic clones of the 3D7 strain of P. falciparum with the goal of uncovering genetic variation(s) responsible for drug resistance in malaria parasites. To generate these clones, we carried out a limiting dilution assay as previously described (Rovira-Graells et al., 2012). A chi-square test determined clone independence as the probability that the wells positive for parasite growth were non-randomly distributed across the 96-well plate (P-value = 3.3 × 10−5; data not shown). The 3D7 origin of all derived clones was verified by genotyping (Fig. S1). Using a standard microscopy-based drug survival assay, we identified two clones (6A and 11C) that show significant differences in sensitivities to antimalarial drugs, chloroquine (CQ), mefloquine (MEF) (Fig. 1). The observed drug resistance phenotypes were specific to later developmental stages of the P. falciparum IDC, namely trophozoite and schizont. The 50% inhibitory concentration (IC50) of the 6A clone for MEF was found to be higher by 2.4- and 1.8-fold compared to 11C at the trophozoite (P-value = 0.060) and schizont (P-value = 0.007) stages respectively (Fig. 1A). Decreased susceptibility to CQ was also observed in 6A compared to 11C at the trophozoite stage (1.9-fold, P-value = 0.021) (Fig. 1B). The difference in IC90 values for both clones shared a similar pattern in divergence of IC50 values, with the 6A clone exhibiting resistance to MEF at an even higher statistical significance. In contrast, no significant difference in drug sensitivities was observed between the clones when treated for 48 h starting at the ring stage for both drugs. In the next step we wished to confirm these initial findings using an additional parasite survival detection method and with that expand this analysis to a wider panel of antimalarial drugs. For this we utilized flow cytometry detection of parasite survival in the drug sensitivity assays, as previously described (Malleret et al., 2011). Hence we further confirmed that clone 6A exhibits a decreased sensitivity to MEF and CQ by 1.4-fold (P-value = 0.007) and 1.5-fold (P-value = 0.015) compared to clone 11C at the schizont stage respectively (Fig. S2). Five additional antimalarial drugs comprising of quinine (QN) and amodiaquine (AMO) which belong to the class of quinolines; pyrimethamine (PYR), an antifolate drug; lumefantrine (LUM); and artemisinin (ART) were tested. In addition to MEF and CQ, 6A displayed increased tolerance to QN (1.3-fold, P-value = 0.023) and a borderline tolerance to LUM (1.2-fold, P-value = 0.070) compared to 11C at the schizont stage. There were no statistically significant differences in drug sensitivities to AMO, PYR and ART between the two clones at both ring and schizont stages (Fig. S2). This indicates that the elevated resistance observed in 6A at the mature stages is specific to amino-methanol quinoline drugs. In addition, we carried out the chloroquine sensitivity assay in the presence of verapamil which is known to reverse CQ-resistance that is conferred by PfCRT mutations (Fidock et al., 2000; Sidhu et al., 2002; Bray et al., 2005; Lakshmanan et al., 2005; Martin et al., 2009). We found that the presence of 0.8 μM verapamil did not alter the sensitivity of 6A to CQ (Fig. S2). Hence the drug resistance phenotype of 6A is likely independent of PfCRT.
Figure 1.

Drug inhibitory concentrations of clones 11C and 6A. IC50 and IC90 values (nM) of clones 11C (red) and 6A (blue) when exposed to mefloquine (A) or chloroquine (B) drugs at ring [10 h post invasion (hpi)], trophozoite (24 hpi) and schizont (34 hpi) stages. Values are determined from drug assays done in triplicate. Error bars represent the standard deviations of the IC50/IC90 measurements. *P-value < 0.05; **P-value < 0.01; ***P-value < 0.001.
The IC50 values of the clones 6A and 11C treated at the ring stage were comparable to previously reported values for the CQ and MEF-sensitive 3D7 strain of P. falciparum (Rathod et al., 1997; Pickard et al., 2003; Chong et al., 2006; Vivas et al., 2007, Wisedpanichkij et al., 2009, WHO, 2010). This observation is consistent with the drug susceptible status of the original 3D7 population used for the limiting dilution. On the other hand, the IC50 values of the resistant trophozoites and/or schizonts of clone 6A approached the levels of clinically relevant resistance for CQ (defined as IC50 > 80 nM) (Valderramos and Fidock, 2006; Kreidenweiss et al., 2008) and MEF (defined as IC50 > 30 nM) (Pickard et al., 2003; WHO, 2010; Wang et al., 2012). This indicates that the molecular mechanism(s) that mediates this stage-specific resistance phenotype is potentially relevant to clinical drug resistance.
Genome-wide studies of clones 6A and 11C
To characterize genetic differences between the two clones, we performed high-throughput whole-genome sequencing on the Illumina Mi-seq platform using the genomic DNA extracted from the clones. We obtained high-quality sequence data with 12-fold genomic coverage encompassing 98.4% and 97.8% of the P. falciparum genome for clone 6A and 11C respectively. A total of 67 SNPs were identified between the clones (Table S2). Our results agree with the previous study of genetic stability of the core P. falciparum genome in the in vitro culture that identified less than 50 SNPs among the 3D7 clones under short-term culture (Bopp et al., 2013). In light of the established mutation rate of 1.0 to 9.7 × 10−9 mutations per base per generation (Bopp et al., 2013), we determined that these two clones are separated by 297 to 2851 generations in the continuous in vitro culture of the original 3D7 clone. Seventeen non-synonymous SNPs were found in 10 genes including a lipase, a mitochondrial ribosomal protein L29/L47, a fikk protein kinase, a protein phosphatase, an exported hyp8 protein and five hypothetical proteins (Table S2). In 11C, we observed a G-> A mutation at the 4179th position in the ORF of PFL1410c that encodes an ABC transporter protein, PfMRP2. This mutation leads to the establishment of a potential ‘opal’ TGA stop codon that, however, may not terminate translation (Mourier et al., 2005; Lobanov et al., 2006; Roseler et al., 2012) (see Discussion). None of the known drug resistance gene markers – pfcrt, pfmdr1, pfgch1, pfdhps, pfdhfr, pfatp6, pfnhe-1 and pfmrp1 harboured any SNPs in their ORFs, thus excluding their involvement in the differential drug sensitivities between the two clones. To detect larger structural polymorphisms including copy number polymorphisms (CNP) we carried out comparative genomic hybridization (CGH) using the long oligonucleotide P. falciparum DNA microarray that comprises of 5402 intergenic and 11 372 intragenic probes for the 5343 P. falciparum genes as described (Hu et al., 2007). A total of 8 segments including 28 genes exhibited copy number variations between 6A and 11C (Fig. 2A and Table S1). Of these, 10 genes belong to the variable surface antigen (VSA) families, var and rifin. Two major regions that comprised the majority of the detected CNPs were located at the subtelomeric regions of chromosomes 2 and 12 spanning 38.7 kb and 75.3 kb respectively. These CNPs occur frequently in culture-adapted laboratory strains and contain mainly genes involved in host–parasite interactions (Kemp et al., 1985; Biggs et al., 1989) and thus are likely not responsible for the drug resistance. Additional CNPs involving four genes (Rab11a GTPase, sporozoite asparagine rich protein, tubulin-tyrosine ligase and 28SrRNA) were detected by CGH. Although none of these four genes were previously linked with drug resistance, their role in the observed differential drug sensitivities cannot be completely ruled out. Finally we observed an allele loss at the 5′ upstream region of PFL1410c that was represented by a low CGH signal in 6A. As mentioned above, this gene encodes PfMRP2, an ABC transporter, whose role in drug resistance has been established in many eukaryotic systems.
Figure 2.

Genomic and transcriptomic differences between clone 6A and 11C.A. Distribution of the CGH microarray signal plotted as log2 ratios between clones 6A and 11C over the 14 P. falciparum chromosomes with red peaks representing the statistically significant CNPs. Red lines with log2 ratios > 0 indicates higher copy (above x-axis) number and log2 ratios < 0 (below x-axis) indicate lower copy number in clone 6A relative to 11C. Genes associated with the CNPs are shown in the grey boxes. The double asterisk (**) indicates that the CNP is detected at the non-coding 5′ region of the gene instead of within the coding sequence of the gene.B. Heat maps showing the mean-centred microarray log2 expression ratios of the 4478 P. falciparum genes ordered by fast fourier transformation over the 48 h IDC for clones 6A and 11C.C. Heat map showing the log2 expression ratios of the 23 differentially expressed (DE) genes detected by Statistical Analysis of Microarrays (SAM) with FDR < 5% over the IDC [depicted by hours post invasion (hpi)]. The DE genes with corresponding detected CNPs are indicated with blue or yellow symbols. R: ring; T: trophozoite; S: schizont.
Next, we carried out genome-wide gene expression analyses with the goal of identifying key gene expression differences that can be linked with the differential drug sensitivities. For that we measured mRNA abundance for all 5343 P. falciparum genes of the two clones by sampling highly synchronized cultures at 8 h regular intervals across the 48 h IDC. This allowed us to reconstruct the IDC transcriptional cascades for both clones and thus evaluate transcriptional differences in all developmental stages (Fig. 2B). Using a spearman rank correlation-based method to estimate the ‘parasite age’ in each cell sample (for details see Mok et al., 2011), we determined that both parasite clone cultures progressed identically through the IDC. This allowed us to compare the total mRNA abundance levels in each experimental time point directly. For this we carried out the Statistical Analysis for Microarray (SAM) (see Experimental procedures). With the exclusion of the VSA gene families (var, rif and stevor), only 23 genes (0.4% of genome) were found to be significantly differentially expressed (DE) between the 2 clones at < 5% false discovery rate (Fig. 2C). Of these, 12 DE genes were associated with the deletions on chromosome 2 and 12 (Fig. 2A and C). The other DE genes mainly involve members of small multi-gene families such as PHIST proteins, FIKK protein kinases and acyl-coA synthetase-like proteins, many of which have been implicated in host–parasite interactions and previously found to exhibit transcriptional variability between P. falciparum clones in general (Rovira-Graells et al., 2012). Hence their involvement in the observed drug resistance is unlikely.
Importantly, the pfmrp2 gene (PFL1410c) was also found among the DE genes (Fig. 2C). The most remarkable feature of the differential expression of pfmrp2 is the temporal shift of the peak mRNA expression that is reflected by their anti-correlated IDC expression profiles (Pearson correlation coefficient = −0.8). Essentially, in 11C, the mRNA abundance of pfmrp2 peaks in the ring stage and subsequently declines through the later trophozoite and schizont stages (Figs 2C and 4B). This expression profile presumably represents the wild-type expression as it is found in the parental-3D7 population (data not shown) and other P. falciparum strains (Bozdech et al., 2003; Llinas et al., 2006). In 6A, pfmpr2 transcription is suppressed in the ring stage, activated through trophozoites and finally reaches its peak in schizonts. This temporal transcriptional shift is highly unusual between P. falciparum populations (Llinas et al., 2006). Accordingly, the timing of gene expression between 6A and 11C was highly conserved with 4050 (98.7%) genes exhibiting Pearson Correlation coefficient (PCC) of at least 0.3 while only 12 genes (0.3%) display anti-correlation with PCC < −0.3 (data not shown). Most importantly, this temporal shift leads to marked increase of pfmrp2 mRNA levels in the trophozoite and schizont stages that coincide with the stage-specific MEF, CQ, QN and LUM-resistance profile of 6A (Fig. 1).
Figure 4.
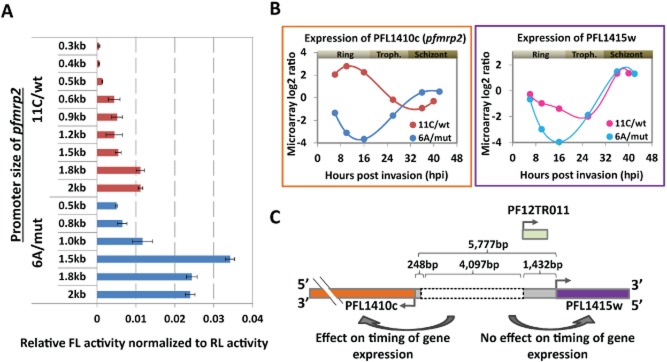
Differential promoter activity of pfmrp2 gene in clone 6A/mut and 11C/wt by luciferase reporter assay and expression profiling of pfmrp2.A. Transcriptional activity of PFL1410c (pfmrp2) promoter constructs of clones 6A/mut (blue) and 11C/wt (red) in the ring stage measured by the firefly luciferase reporter assay. Bars represent the firefly luciferase (FL) activity normalized to the level of renilla luciferase (RL) activity for each cell line transfected with each promoter construct of ∼0.3–2.1 kb from the start codon of the pfmrp2 gene. Error bars indicate the standard deviation of the FL activity over three independent transfection experiments.B. Transcriptional profiles of PFL1410c (pfmrp2) and PFL1415w genes in clones 6A/mut and 11C/wt over the 48 h asexual life cycle measured by microarray log2 ratios. Time points in terms of hours post invasion (hpi) for each clone reflect the age of the parasite as calculated from the best fit spearman rank correlations to the reference transcriptome which was previously generated (Foth et al., 2011).C. Schematic diagram depicts the location of the breakpoint sites and the deletion segment (grey dotted box) within the 5′ upstream sequence of the PFL1410c (pfmrp2) gene in clone 6A/mut compared to clone 11C/wt and relative to the neighbouring genesPFL1415w. The numbers represent the distances between the deletion break points and the start codons of both PFL1410c (pfmrp2) and PFL1415w genes. A non-coding RNA, PF12TR011 (light green box), is potentially transcribed within the 1432 bp upstream of the PFL1415w (PlasmoDB version 9).
4.1 kb deletion in the promoter region of pfmrp2
To characterize the detected pfmrp2 CNP, we used long-range PCR with a panel of primer pairs (Table S3) spanning different sections of the 14 829 bp upstream of the pfmrp2 start codon (Fig. 3A). While the PCR reactions for all primer pairs yielded DNA products of expected sizes for genomic DNA from 11C, no expected size products were obtained from 6A. The only two PCR products from 6A were generated by primers 1.9 and 1.12 and both of these were ∼4.1 kb smaller than expected (Fig. 3A). These results indicate that the 5′ upstream region of the pfmrp2 gene in the clone 6A contains a ∼ 4.1 kb deletion located between the 20th and 6212th position upstream of the pfmrp2 start codon. Due to that, in the subsequent text, we refer to the clones as 6A/mut (6A mutant) and 11C/wt (11C wild-type). The subsequent sequence analysis confirmed these observations showing that the promoter region of 6A/mut was interrupted after a 45-nucleotide-long poly (dA:dT) homopolymer immediately following the 248 nucleotides upstream of the pfmrp2 start codon (Fig. 3B). Interestingly, the 5′ end of this 6A/mut deletion mapped to 4345 nt upstream of the start codon which falls into another poly (dA:dT) region comprised of 24 nucleotides. Interestingly this deletion shortens the distance between pfmrp2 and its neighbouring gene (PFL1415w) to 1432 bp between the two start codons of both genes (see below).
Figure 3.
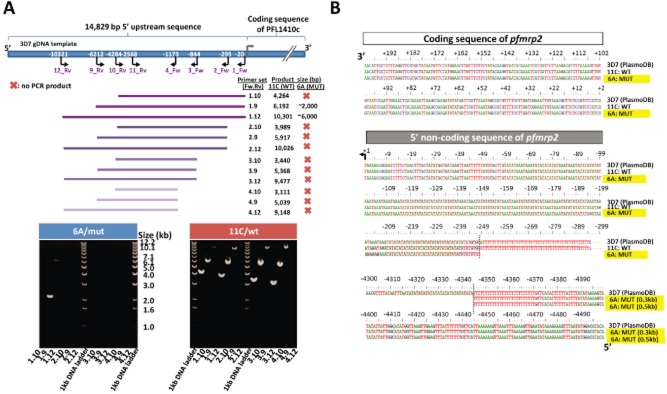
Deletion of the pfmrp2 gene 5′ upstream sequence identified by PCR and sequencing.A. A map of the 5′ upstream region of the 3D7 pfmrp2 gene showing the panel of used primer pairs (purple) and their positions relative to the gene start codon. Listed are the sizes of the PCR products obtained for clones 11C/wt and 6A/mut for the various primer pairs. Bottom panel: Agarose gel images of PCR products obtained for the corresponding primer pairs. The images show full-length PCR products obtained for clone 11C/wt for all primer pairs and the truncated products for the primer pairs 1.9 and 1.12 from 6A/mut.B. Sequence alignment of clone 11C/wt and clone 6A/mut compared to 3D7 sequence (PlasmoDB version 8.1). The sequence alignment depicts the 200 bp coding sequence of the pfmrp2 gene at the 5′ end and its upstream, non-coding region. The grey dotted lines indicate the break points of the 6A/mut deletion. The numbers indicate the base position relative to the start codon. Two DNA constructs of 6A/mut of 0.3 and 0.5 kb in size were sequenced to identify the deletion break point as −4345th bp from the start codon.
Transcriptional activity of the mutant and wild-type promoter of pfmrp2
To determine the effect of this ∼4.1 kb deletion on the transcriptional activity of the putative promoter of pfmrp2, we carried out transient transfections involving a dual-luciferase reporter gene assay (Experimental procedures). DNA fragments ranging from 320 bp to 2110 bp of the 5′ promoter sequence of pfmrp2 from both 6A/mut and 11C/wt were cloned upstream of the firefly luciferase gene in the pPf86 plasmid (Fig. S3) (Militello and Wirth, 2003) and transiently transfected to P. falciparum cells. Monitoring the firefly luciferase activity we observed significant differences in the expression levels between the cell lines transfected with constructs from 6A/mut and 11C/wt, comparatively. For the majority of the fragments, the promoter constructs from 6A/mut yielded significantly higher luciferase activity (2.1- to 6.2-fold) compared to 11C/wt (Fig. 4A) (P-value < 0.05; Student's T-test). The only exception was the 655 bp/640 bp from 6Amut/11Cwt construct whose activity is comparable between the two clones.
Both promoter regions showed increasing transcriptional activity with the size of their corresponding fragment up to ∼1.8 kb. In both cases, the size-dependent increase in transcriptional activity followed a stepwise profile. For the 11C/wt promoter, there were two main significant increases in transcriptional activity between the 507 bp and 640 bp (3.3-fold, P-value = 0.025) and between 1511 bp and 1775 bp (2.0-fold, P-value = 0.001) constructs respectively (Fig. 4A). Similarly for the 6A/mut, we observed significantly elevated luciferase levels between the 655 bp and 984 bp (1.8-fold, P-value = 0.033) and 984 bp and 1377 bp (2.9-fold, P-value = 1 × 10−4) constructs. Surprisingly, addition of another 409 bases to the 1377 bp fragment (1786 bp) was marked by a decline of the transcriptional activity (1.4-fold, P-value = 6 × 10−4). Taken together, these data suggest that the 4.1 kb deletion created a new transcriptionally activating DNA element 5′ upstream of the pfmrp2 gene that has a different (possibly higher) transcriptional initiation activity compared to the endogenous promoter. The stepwise profile of transcriptional activity within the panels of deletion constructs for 11C/wt indicates the presence of DNA regulatory elements within the 1775 bp 5′ upstream sequence of the pfmrp2 gene. Next, we searched for DNA promoter motifs within the 5777 bp upstream sequence of pfrmrp2 gene based on the list of putative PfApiAP2 target genes motifs (Campbell et al., 2010). We identified a total of 41 motifs assigned to several ApiAP2 proteins (Campbell et al., 2010) that were present within the segments coinciding with significant changes in the promoter activity (Fig. S4). Moreover many of the motifs occur in these regions in multiple copies. Due to that no clear association between any of the motifs and transcription could be made. More in-depth functional studies are needed to determine which of these predicted elements play a role in transcriptional regulation of the pfmrp2 gene.
As described above, the pfmrp2 mRNA profile in 6A/mut is essentially a mirror image of the wild-type profile, shifting its stage-specific expression from rings to schizonts (Fig. 4B). Interestingly, PFL1415w, located 5777 bp upstream of pfmrp2 (on the opposite strand) has also a schizont-specific transcriptional profile which is unaffected by the 4.1 kb deletion in 6A/mut. This suggests that in 6A/mut, the ∼1.4 kb promoter region of PFL1415w gene is sufficient for transcriptional regulation of PFL1415c. In 6A/mut, pfmrp2 (PFL1410c) acquired a highly similar mRNA expression profile to PFL1415w, which may indicate that the 1432 bp sequence upstream of PFL1415w that was ‘spliced’ to 248 bp upstream from pfmrp2 may harbour a bidirectional promoter element which drives the expression of the pfmrp2 gene in a similar fashion (Fig. 4C). It must be also noted that a recent study identified a non-coding RNA, PF12TR011 (PF3D7_1229200) that is expressed from a region between 250 and 1420 bp upstream of the PFL1415w gene and exhibits a maximum transcript expression at the trophozoite stage (Mourier et al., 2008). It is possible that this ncRNA may function in the regulation of a schizont stage-specific gene expression of both PFL1415w and PFL1410c (pfmrp2) in 6A/mut and thus may contribute to the drug resistance phenotypes.
Characterizations of PfMRP2 protein expression and intracellular localizations
Next, we generated anti-PfMRP2 polyclonal antibodies by mice immunization with a specific peptide that corresponds to the hydrophilic 13-amino-acid sequence epitope at the N-terminus of PfMRP2 (Fig. 5A). Western blot analyses were subsequently carried out with total protein lysates from equal numbers of highly synchronized parasites collected every 6 h over the 48 h IDC from both 6A/mut and 11C/wt (Fig. 5B). The immunoblotting revealed a doublet protein band at molecular weight ∼250 kDa, corresponding to the expected size (248 kDa) of PfMRP2 protein. In addition, we observed ∼130 kDa and < 50 kDa bands that may represent processed forms of PfMRP2 (the full-length and processed form of PfMRP2 were detected by four independent antibodies) (Fig. S5A). To quantify the levels of protein expression, we calculated the sum of the PfMRP2 band intensities for each lane measured by densitometry (Bio-Rad Densitometer, USA) and normalized these optical density (OD) values to the loading control of the 42 kDa Pfactin I protein, PFL2215w (Fig. 5C). In 11C/wt, the PfMRP2 protein signal was most intense from the mid-rings to mid-trophozoite stages. On the other hand, in 6A/mut, the highest intensity of PfMRP2 was detected in late trophozoites and schizonts. These protein profiles reflect the temporal shifts in pfmrp2 mRNA levels between the two clones. Importantly, there were higher levels of PfMRP2 protein in 6A/mut through mid-trophozoites and late schizonts compared to 11C/wt (Fig. 5B). This coincides well with the decreased sensitivity of 6A/mut to MEF, CQ, QN, and possibly LUM in trophozoite and/or schizont stages.
Figure 5.

Characterization of protein expression levels of PfMRP2 in clone 6A/mut and 11C/wt and localization in the parasite by immunofluorescence assay.A. Immunoblot image of the PfMRP2 protein bands detected for each clone sampled at 6 h intervals over the 48 h asexual cycle. Total protein amounts corresponding to equal number of parasites per time point sample were resolved on the SDS-PAGE, blotted and subsequently reacted with the anti-PfMRP2 polyclonal peptide antibody. The same samples were probed with actin as a loading control.B. Relative protein expression levels of PfMRP2 measured by densitometer (Bio-Rad, USA). The protein profiles represent the summed abundance of all bands determined by their optical density (OD) over the IDC for clone 6A/mut (blue) and 11C/wt (red). For this plot the PfMRP2 OD values were normalized by the normalized by OD values for actin obtained identically.C. Immunofluorescence images of PfMRP2 localization labelled with Alexa488 during the development from ring to trophozoite and schizont stages in the wild-type clone 11C parasite. Nucleus was stained with DAPI (4′,6′-diamidino-2-phenylindole). The bottom panel shows dual labelling of the parasite at early schizont stage with anti-PfMRP2 (Alexa488) antibody and digestive vacuole-specific PfCRT antibody (Alexa594).
PfMRP2 protein localization in the P. falciparum parasites over the 48 h IDC was characterized by immunofluorescence microscopy (IFA) using the derived polyclonal anti-PfMRP2 antibodies. In all asexual blood stages of 11C/wt, the PfMRP2 IFA signal exhibited a punctate pattern suggesting its association with membranous vesicles within the parasite cytoplasm (Fig. 5D). In the ring stage, PfMRP2-associated compartments concentrated to the periphery of the parasite, while in trophozoite and schizont stages these were more dispersed throughout the cytoplasm. The signal also appeared discrete and punctuated around individual merozoites in the mature schizonts just prior to rupture. No differences in the MRP2 localization pattern between the 11C/wt and 6A/mut parasites were observed (Fig. S5B). Colocalization studies have shown no overlaps between the IFA patterns of PfCRT and PfMRP2 which indicates that PfMRP2 does not associate with the parasite digestive vacuole. The punctuate IFA pattern was reproduced by all four independent polyclonal anti-PfMRP2 antibodies (Fig. S5C) and no labelling was observed using the pre-immune sera (Fig. S5D). This further confirms the specificity of the derived antibody and hence the localization of the PfMRP2 protein to putative membranous structures. The localization of PfMRP2 in membrane-bound vesicles is consistent with the expected intracellular pattern observed for PfMRP1 (Raj et al., 2009) and possibly plasma membrane association observed for both PfMRP1 and PfMRP2 (Kavishe et al., 2009). Similarly, mammalian MRPs have been shown to localize to both the plasma membrane (Zaman et al., 1994) and internal vesicular structures (Rajagopal and Simon, 2003).
Discussion
Genetic polymorphism of promoter regions and differential expression
Here we show that a large deletion (∼4.1 kb) in the upstream promoter region can alter expression of the pfmrp2 gene in such a way that the timing of its transcription shifts to the later stages of the IDC. This results in increased levels of the transcript and the protein product in these stages. Extensive research over the last decade has firmly established the transcriptional flexibility of Plasmodium parasites that contributes to phenotypic plasticity of this pathogen. There are significant transcriptional variations between P. falciparum strains even after long-term culture-adaptation (Llinas et al., 2006), isogenic subclones (Mok et al., 2007; Rovira-Graells et al., 2012) as well as freshly culture-adapted P. falciparum isolates (Mackinnon et al., 2009). Several recent studies also demonstrated the existence of distinct transcriptional profiles in in vivo P. falciparum isolates that likely reflect diverse physiological states (Daily et al., 2007) which may be associated with malaria infection characteristics such as a severe disease (Milner et al., 2012) or artemisinin resistance (Mok et al., 2011). Transcriptional changes that occur among P. falciparum strains or isolates comprise not only of genes involved in the parasite pathogenesis such as factors of host parasite interactions, invasion and sexual differentiation, but also include genes associated with the parasite metabolism, growth and regulation. Hence, a comprehensive understanding of the gene expression variability between malaria parasite populations is one of the key issues for the understanding of clinical aspects of malaria disease and variation in drug sensitivities.
Presently, very little is known about the molecular mechanisms of transcriptional regulation that drives variability in gene expression in the P. falciparum parasite. In a recent work, Rovira-Graells et al. (2012) investigated transcriptional differences between a set of isogenic clones of P. falciparum, grown under homogenous conditions (Rovira-Graells et al., 2012). It was shown that the differentially expressed genes are not restricted to factors of host–parasite interaction but include genes associated with the lipid and protein metabolism, erythrocyte remodelling and gene regulation (including the PfApiAP2 family of transcription factors). Comparative analyses with several previously completed epigenetic studies led to the conclusion that a large proportion of these transcriptional changes are driven by stochastic switches between hetero and euchromatin status of the chromosomal loci containing these genes (Rovira-Graells et al., 2012). Similar to other eukaryotes, the P. falciparum heterochromatin domains are marked by histone 3 lysine 9 trimethylation (H3K9me3) (Lopez-Rubio et al., 2009) and binding to heterochromatin-binding protein (HP1) (Flueck et al., 2009). However, several recent studies demonstrated that the H3K9me3 domains are restricted mainly to the subtelomeres and few intrachromosomal islands, while the vast majority of the P. falciparum genome is in a euchromatic state (Salcedo-Amaya et al., 2009; Bartfai et al., 2010). Hence, hetero-euchromatin epigenetic switches cannot explain all transcriptional differences between the parasite strains and isolates, in particular for those genes within the euchromatin. Here, we demonstrate that structural polymorphisms (such as deletions) within the cis-regulatory elements of genes could contribute to transcriptional and thus phenotypic differences between P. falciparum populations.
DNA regulatory elements have been previously suggested to play a potential role in P. falciparum drug resistance. Waller et al. (2003) showed that truncation in the 3′ untranslated region (UTR) of pfcrt leads to decreases in the gene expression and with that that a lower IC50 value to chloroquine (Waller et al., 2003). Also a segment of the UTR of pfmdr1 was shown to be able to drive transcription that can be induced by chloroquine and other quinoline derivatives (Myrick et al., 2004). Together with our findings these results substantiate further studies of genetic polymorphisms within intragenic regions as potential markers of drug resistance as well as other important malaria parasite phenotypes.
Mutagenic effect of low complexity sequences within the P. falciparum genome
The break point sites of the 4.1 kb deletion in the pfmrp2 promoter were found to be associated with two homologous low complexity sequences consisting of TA repeats (n > 13) followed by T:A homopolymers (> 25 nt). These types of repeats are ubiquitous in the P. falciparum intergenic regions, particularly within 2 kb upstream of the translational start sites (e.g. putative promoters) that are also characterized by an extreme A::T content (∼80–90%) (Gardner et al., 2002). It is well established that low complexity sequence regions (LCRs) are fast evolving between and within individual species and thus play an important role in evolution (Huntley and Golding, 2006). In P. falciparum, LCRs contribute to adaptive processes by varying amino acid sequences of proteins (Zilversmit et al., 2010) and regulation of gene expression (Frugier et al., 2010). With the increasing volume of available genomic information, LCRs have been found in genes associated with a broader spectrum of functionalities such as adaptive host–parasite interactions (cell adhesion, rosseting, host cytoplasm remodelling) as well as stress and DNA damage responses (Tan et al., 2010). The LCRs are known to promote high levels of genetic polymorphisms in their vicinity (Haerty and Golding, 2011). The increased level of genetic variability is presumably a reflection of the LCR genetic instability, which is driven by DNA polymerase slippages as well as additional ‘mutagenic’ properties. Besides the enrichment of SNPs, the presence of LCR is frequently associated with larger structural polymorphisms caused by uneven cross-over events or by chromosome breakage and healing mechanisms such as microhomology-mediated end joining (MMEJ), single-strand annealing (SSA) or non-homologous end joining (NHEJ) repair mechanisms (Shuman and Glickman, 2007; McVey and Lee, 2008). While all previous LCR studies in P. falciparum were mainly focused on coding regions, here we demonstrate that such polymorphisms could be also found in the upstream non-coding regions where they could affect regulatory elements of transcription. With the rising applications of high-throughput sequencing technologies it will be worth exploring the frequency and the biological significance of such polymorphisms in malaria parasite populations.
PfMRP2 protein expression in 3D7 clones
Within the ORF of the pfmrp2 gene, we observed a single base mutation at the 4,179th nucleotide from the gene's start site in clone 11C/wt (but surprisingly not in 6A/mut). This is the first report of the particular SNP within pfmrp2 with respect to sequence data of the 71 strains in PlasmoDB version 9.3. Although this mutation would establish a non-sense OPAL TGA stop codon at the 1393rd amino acid in 11C/wt, we detected a 250 kDa size band corresponding to the expected size of PfMRP2, showing that the full-length protein is expressed. Previous reports have identified at least four such mutations in other P. falciparum protein-coding genes that despite containing the OPAL TGA stop codon, their corresponding protein products were not truncated (Mourier et al., 2005; Lobanov et al., 2006; Roseler et al., 2012). Instead, in the presence of a selenocysteine insertion sequence (SECIS) element in the 3′ UTR of the genes, the TGA codon utilizes the selenocysteine amino acid via the Pf tRNAsec (Mourier et al., 2005; Roseler et al., 2012). Using the SECISaln software we have also identified a putative non-canonical SECIS element located between the canonical stop codon and 150 bp downstream of the gene (see Fig. S6). Hence, our results suggest that the pfmrp2 gene in the clone 11C/wt encodes a full-length selenoprotein. Although the role of utilization of Pf tRNAsec in P. falciparum remains uncharacterized, it is tempting to speculate that this mechanism may play a role in post-translational regulation of PfMRP2 and thus drug resistance mechanisms.
PfMRP2 plays a role in drug resistance of P. falciparum
The MRPs belong to the ABC transporter superfamily that facilitates active transport of solutes across biological membranes (Paumi et al., 2009). The MRPs represent a specialized group of pumps transporting glutathione conjugates as a final step of xenobiotic detoxification. Hence, MRPs have been linked with drug resistance in many cellular systems ranging from tumours (Zaman et al., 1994) to microbes (Paumi et al., 2009). The resistance mechanisms are typically linked with direct efflux of the GSH-conjugated drugs from the cytoplasm or their sequestration into membranous compartments such as lysosomes mediated by the MRP transporters directly. PfMRP1 was previously shown to mediate drug resistance by direct efflux of GSH-drug adducts out of the P. falciparum parasite (Raj et al., 2009). Here we provide evidence that the second P. falciparum homologue, PfMRP2, can also play a role in malaria parasite resistance, in particular to quinolines. As an alternative mechanism to direct drug transport, PfMRP2 could also mediate drug resistance via a transport of reduced glutathione (GSH) that is crucial for haem detoxification (Meierjohann et al., 2002). The mode of action of both CQ and MEF involves binding to free haem and thereby inhibiting its polymerization to haemozoin. This would lead to accumulation of the toxic free haem and subsequently to oxidative stress and membrane damage within the parasite (Sullivan et al., 1998).
In conclusion, it is becoming progressively clear that drug resistance of malaria parasites is often underlined by complex genetic traits that involve larger numbers of genes than originally thought. Here we provide evidence of an additional molecular factor that could contribute to these phenotypes; PfMRP2, a novel drug transporter. Moreover, these results show that drug resistance could be caused by genetic polymorphisms at the gene promoter regions by which they affect the transcriptional patterns/levels of the affected genes. In the future, it will be interesting to investigate whether genetic polymorphisms at the pfmrp2 promoter, or promoters of other resistance markers could be associated with drug efficacy in natural infections.
Experimental procedures
Limiting dilution and in vitro tissue culture of Plasmodium falciparum
Single clones was derived from the 3D7 P. falciparum strain by the limiting dilution method as described (Rosario, 1981). Parasites were maintained in human red blood cells as described (Trager and Jensen, 1976) and synchronized with 5% sorbitol. Tightly synchronized cultures of the clones 6A and 11C were harvested subsequently in 8 hourly time-courses to obtain six time point samples for RNA and protein to measure their transcriptional profiles.
In vitro drug sensitivity assays (modified schizont maturation assay)
Chloroquine (CQ) and mefloquine (MEF) drug sensitivity assays were carried out on highly synchronized cultures of the clones at the ring [14 h post invasion (hpi)], trophozoite (24 hpi) and schizont (34 hpi) stages while quinine (QN), lumefantrine (LUM), amodiaquine (AMO), pyrimethamine (PYR), artemisinin (ART), verapamil (VER) + CQ assays were done on ring (10 hpi) and schizont (30 hpi) stages. To minimize any discrepancies in parasite age between the clones at the start of each assay, both clones were grown concurrently and sorbitol synchronizations were always carried out in parallel. Stock solutions were prepared from mefloquine hydrochloride (SIGMA, USA) and chloroquine diphosphate (SIGMA, UK), quinine (SIGMA, Germany), lumefantrine (SIGMA, China), amodiaquin dihydrochloride dihydrate (SIGMA, USA), verapamil hydrochloride (SIGMA, China), artemisinin (SIGMA, USA) and pyrimethamine (SIGMA, Germany). Treatment was carried out on parasites at 1% haematocrit and 3% parasitaemia with 8–10 serially diluted drug concentrations. Upon invasion of new rings in the control, the % parasitaemia (rings) of the giemsa-stained smears were counted for at least 300 RBCs by microscopy. The 50% and 90% inhibitory concentration were determined by non-linear regression analysis using HN-NonLin V1.1 (http://malaria.farch.net). The IC values for each drug were also determined by flow cytometry. After labelling the parasites with Hoechst 33324 (SIGMA, Singapore) and dihydroethidium (SIGMA, Singapore), the dye signal incorporation was detected by the LSR II flow cytometer using the UV (305 nm) and the blue lasers (488 nm) as described (Malleret et al., 2011), At least three biological replicate assays were carried out for each IDC stage, clone and drug, except for the assay involving chloroquine and verapamil where the assays were done in duplicates.
Genotyping of clones with msp1, msp2 and glurp genes
To validate the identities of parental 3D7 and the clones 6A and 11C as being 3D7 in origin, the standard method of genotyping using the highly polymorphic surface antigens genes msp1 (merozoite surface protein 1), msp2 (merozoite surface protein 2) and glurp (glutamate rich protein) by nested PCR was carried out as previously described (Snounou et al., 1999; Snounou, 2002).
Aminoallyl-dUTP coupled DNA synthesis for comparative genomic hybridization (CGH)
One millilitre of infected pRBCs at 10% parasitaemia was harvested and genomic DNA extraction by phenol-chloroform method was carried out as described (MR4/ATCC, 2004). Synthesis of genomic DNA coupled with aminoallyl-dUTP by exo-klenow enzyme was done as previously described (Bozdech et al., 2003).
cDNA synthesis by reverse transcription for transcriptional profiling
RNA from the P. falciparum samples that were harvested from in vitro cultures (0.4 ml to 2 ml) were isolated as described (Bozdech et al., 2013). Reverse transcription was then performed on 10 μg of starting total RNA to generate single-strand aa-dUTP labelled cDNA (Bozdech et al., 2013).
Fluorophore labelling of DNA and dual-channel microarray hybridization
Two micrograms of each sample (cDNA and klenow-DNA products) was labelled with Cy 5 fluorophore. For transcriptional profiling, equal amounts of the cy5-labelled cDNA time point samples was hybridized together with the cy3-labelled cDNA generated from a 3D7 reference pool consisting of all parasite asexual stages. Replicate microarrays were carried out for each time point sample. For CGH, equal amount of the cy5-labelled klenow-DNA product of each clone was hybridized against the cy3-labelled DNA of parental 3D7 on the microarray chip having a total of 15 667 probes. Information of these probes have been deposited in NCBI's Gene Expression Omnibus (Edgar et al., 2002) through GEO Platform Accession Nos GPL11248 and GPL11250.
Statistical analysis of microarray data
The signal intensities for all probes across each array were Lowess Normalized and subsequently filtered for quality control in Acuity 4.0 software (Axon Instruments, USA). For all array features, only ‘spots’ with at least 95% pixels median intensity above the local background intensity plus 2 standard deviations for either channel were included for downstream analysis. Microarray data from this study has been deposited into GEO series number GSE44129.
CNP analysis using Genomic Alteration Detection Analysis (GADA)
CNP analysis of the 3D7 clones was carried out with the criteria that the T-coefficient greater than 7.0 in at least one probe between the two clones were observed using the Genomic Alteration Detection Analysis (GADA) v0.8 program in R package (Pique-Regi et al., 2009).
Detection of differentially expressed genes between the clones
The log2 ratios were averaged between the technical hybridization replicates and then averaged to obtain log2 ratios for each gene. Only genes with less than 40% missing ratios were analysed and missing data were imputed by KNN (10th nearest neighbour). The parasite age was estimated by finding the best fit Spearman Rank Correlation value to a reference transcriptome. VSAs were removed and analysed separately prior to identification of differentially expressed genes. The SAM (statistical analysis of microarrays) approach for time-course data with all 924 possible permutation and FDR cut-off of less than 5% was used.
High-throughput sequencing of the clones
Two micrograms of genomic DNA from each of the clones was sequenced using Illumina Mi-seq sequencing technology with 250 bp paired-end reads. The sequence data were aligned to the P. falciparum 3D7 genome (PlasmoDB version 9.1) and reads that did not map to the reference genome and/or PCR duplicates were removed using Samtool and Picard. The reads were realigned around indels using GATK RealignerTargetCreator and base quality scores were recalibrated using GATK Table-Recalibration. Identification of all possible SNPs in clones was carried out using GATK UnifiedGenotyper (Min Base quality score ≥ 20) by calling variants in the sequence data. This was followed by using GATK VariantFiltration for hard-filtering based on quality scores, read depth etc. to obtain good quality SNPs. SNPs that failed any of these cut-off filters were removed – variant quality as function of depth QD < 2.0, strand bias (P < 10e-6), mapping quality < 40, depth of read < 5, MappingQualityRankSum < −12.5, ReadPosRankSum < −8.0. Genetic variants were then annotated using snpEFF.
Generation of anti-PFMRP2 polyclonal antibody
Four polyclonal antibodies were raised in parallel in mice against the hydrophilic amino acid sequence KNHTNKFHKRKKE, between positions 101th to 113th near the N-terminal of the protein (Biosciences Peptide Synthesis Facility, School of Biological Sciences, Nanyang Technological University). The epitope was selected using the Bcell Parker Hydrophilicity Prediction software.
SDS-PAGE of protein samples and Western blotting
One millilitre of 10% infected pRBC was harvested every 6 h over the 48 h IDC from tightly synchronized in vitro cultures of the clones and lysed with saponin and loading buffer. Ten microlitres of supernatant from the lysed samples were resolved by a 6% polyacrylamide gel and transferred onto the membrane at 4°C overnight at 40 V. After blocking with 5% non-fat milk (Bio-Rad, USA), the membrane was probed with PfMRP2 polyclonal mouse serum antibody in a 1:1000 dilution and subsequently with anti-mouse HRP-conjugated secondary IgG antibody in a 1:3000 dilution. Proteins bands were visualized with chemiluminescent Western blotting substrate (Santa Cruz, USA) and analysed with GS-800™ Densitometer and the Quantity One® Analysis Software (Bio-Rad, USA). The membrane was re-probed with actin-specific antibodies as the loading control.
Immunofluorescence Assay (IFA) of MRP2 protein
IFA was carried out with parasites at different IDC stages by incubation of the paraformaldehyde-fixed cells with 1:200 dilution of either anti-PfMRP2 serum or the negative control (mouse pre-immune serum) at 4°C for 16–18 h. Incubation with secondary antibody of 1:800 dilution of Alexa Fluor® 488 or 594 goat anti-mouse monoclonal antibody was performed in the dark at room temperature for 1 h. 1:1000 dilution of the rabbit anti-PfCRT antibody from MR4 was used in the colocalization experiment. Fluorescence images were taken using Zeiss LSM 510 confocal microscope (Carl Zeiss, Switzerland) and processed using ImageJ software v1.43.
Identification of SECIS element downstream of the pfmrp2 gene
SECIS element was identified using the SECISaln software 1.0 (Chapple et al., 2009) which uses the SECISearch tool to find putative loose canonical and non-canonical SECIS elements based on an overall structure energy cut-off of −7.
Polymerase chain reaction (PCR)
Twelve primer pairs were designed for various sites within the ∼15 kb upstream region from the ATG translation start site of the pfmrp2 (PFL1410c) gene. Three additional primer pairs were designed spanning the ORF sequence of the gene. The list of primer sequences used is stated in Table S3. PCR was carried out using DNA isolated from clone 6A and 11C. Long-range PCR using the Expand Long Template PCR system (Roche, Switzerland) was used to generate DNA products up to 15 kb in size. The PCR products were analysed by agarose gel electrophoresis. High fidelity DNA polymerase (Roche, Switzerland) was used to amplify DNA products which were purified by gel extraction (Qiagen, USA) for DNA sequencing using BigDye® Terminator v3.1 cycle sequencing method (1st BASE, Singapore). The sequencing results were analysed by blast search to the 3D7 genome (PlasmoDB database version 8.2) and multiple sequence alignment carried out using BioEdit Sequence Alignment Editor software V7.9.0 (Hall, 1999).
Generation of luciferase constructs
The plasmid backbone used in the luciferase experiments came from a construct, pPf86, which contains the pfhsp86 1.8 kb promoter upstream of the firefly luciferase gene (Militello and Wirth, 2003). Restriction digestion with enzymes NcoI and XhoI (New England Biolabs, USA) was used to replace the pfhsp86 promoter with the promoter of interest. A panel of constructs of various sizes of up to 2 kb upstream of pfmrp2 were amplified from the genomic DNA of clone 11C/wt and clone 6A/mut with primers (see Table S3) containing the restriction sites. PCR products were then digested with NcoI and XhoI enzymes, and ligated with the pPf86 plasmid backbone. The ligated products were transformed into DH5α Escherichia coli competent cells. The constructs were then sequenced.
Transient transfection of P. falciparum parasites with firefly and renilla luciferase constructs containing the pfmrp2 promoter
The pPf86 vector backbone containing various lengths of the pfmrp2 promoter were transfected into the highly synchronized early ring stage (< 6 hpi) 3D7 parasites at 15% parasitaemia by electroporation as described (Militello and Wirth, 2003). All parasite samples were also transfected with the plasmid expressing renilla luciferase (RL) which was used as reference for normalizing firefly luciferase (FL) activity. One hundred micrograms of total plasmid DNA (50 μg of FL plasmid and 50 μg RL plasmid) was used for each transfection. The flasks were gassed and maintained in culture until sample collection in the subsequent generation.
Screening of luciferase activity
The cells were harvested at each of the three stages: mid-rings, mid-trophozoites and mid-schizonts and screened for level of firefly and renilla luciferase activity for the following transfectants: (i) insert/pPf86 plasmid (target of interest), (ii) the original pPf86 plasmid (positive control) and (iii) without plasmid (negative control). At the time of harvest, the cells were lysed with 0.15% saponin and the parasite pellet washed twice with PBS to remove any traces of ghost RBCs. The Dual Luciferase Reporter Assay system (Promega, USA) was used to determine both firefly (target expression) and renilla (reference expression) luciferase activity. The transfection experiment was carried out in triplicates across different cell generations and the averaged luciferase activities were determined. The LUC readings for the negative control (without plasmid) were taken as the background LUC levels. Firefly LUC activity was measured in relative light units (RLUs) and the normalized LUC was calculated by the following formula:
where FLuc is firefly luciferase and RLuc is renilla luciferase.
Acknowledgments
This work was supported by grant of the Singapore National Medical Council (NMRC) # IRG10may058.
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher's web-site.
Supplementary
Supplementary
References
- Anderson TJ, Nair S, Qin H, Singlam S, Brockman A, Paiphun L. Nosten F. Are transporter genes other than the chloroquine resistance locus (pfcrt) and multidrug resistance gene (pfmdr) associated with antimalarial drug resistance? Antimicrob Agents Chemother. 2005;49:2180–2188. doi: 10.1128/AAC.49.6.2180-2188.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartfai R, Hoeijmakers WA, Salcedo-Amaya AM, Smits AH, Janssen-Megens E, Kaan A, et al. H2A.Z demarcates intergenic regions of the Plasmodium falciparum epigenome that are dynamically marked by H3K9ac and H3K4me3. PLoS Pathog. 2010;6:e1001223. doi: 10.1371/journal.ppat.1001223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggs BA, Kemp DJ. Brown GV. Subtelomeric chromosome deletions in field isolates of Plasmodium falciparum and their relationship to loss of cytoadherence in vitro. Proc Natl Acad Sci USA. 1989;86:2428–2432. doi: 10.1073/pnas.86.7.2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bopp SE, Manary MJ, Bright AT, Johnston GL, Dharia NV, Luna FL, et al. Mitotic evolution of Plasmodium falciparum shows a stable core genome but recombination in antigen families. PLoS Genet. 2013;9:e1003293. doi: 10.1371/journal.pgen.1003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdech Z, Llinas M, Pulliam BL, Wong ED, Zhu J. DeRisi JL. The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 2003;1:E5. doi: 10.1371/journal.pbio.0000005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdech Z, Mok S. Gupta AP. DNA microarray-based genome-wide analyses of Plasmodium parasites. Methods Mol Biol. 2013;923:189–211. doi: 10.1007/978-1-62703-026-7_13. [DOI] [PubMed] [Google Scholar]
- Bray PG, Martin RE, Tilley L, Ward SA, Kirk K. Fidock DA. Defining the role of PfCRT in Plasmodium falciparum chloroquine resistance. Mol Microbiol. 2005;56:323–333. doi: 10.1111/j.1365-2958.2005.04556.x. [DOI] [PubMed] [Google Scholar]
- Campbell TL, De Silva EK, Olszewski KL, Elemento O. Llinas M. Identification and genome-wide prediction of DNA binding specificities for the ApiAP2 family of regulators from the malaria parasite. PLoS Pathog. 2010;6:e1001165. doi: 10.1371/journal.ppat.1001165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapple CE, Guigo R. Krol A. SECISaln, a web-based tool for the creation of structure-based alignments of eukaryotic SECIS elements. Bioinformatics. 2009;25:674–675. doi: 10.1093/bioinformatics/btp020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong CR, Chen X, Shi L, Liu JO. Sullivan DJ., Jr A clinical drug library screen identifies astemizole as an antimalarial agent. Nat Chem Biol. 2006;2:415–416. doi: 10.1038/nchembio806. [DOI] [PubMed] [Google Scholar]
- Dahlstrom S, Ferreira PE, Veiga MI, Sedighi N, Wiklund L, Martensson A, et al. Plasmodium falciparum multidrug resistance protein 1 and artemisinin-based combination therapy in Africa. J Infect Dis. 2009a;200:1456–1464. doi: 10.1086/606009. [DOI] [PubMed] [Google Scholar]
- Dahlstrom S, Veiga MI, Martensson A, Bjorkman A. Gil JP. Polymorphism in PfMRP1 (Plasmodium falciparum multidrug resistance protein 1) amino acid 1466 associated with resistance to sulfadoxine-pyrimethamine treatment. Antimicrob Agents Chemother. 2009b;53:2553–2556. doi: 10.1128/AAC.00091-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daily JP, Scanfeld D, Pochet N, Le Roch K, Plouffe D, Kamal M, et al. Distinct physiological states of Plasmodium falciparum in malaria-infected patients. Nature. 2007;450:1091–1095. doi: 10.1038/nature06311. [DOI] [PubMed] [Google Scholar]
- Djimde A, Doumbo OK, Cortese JF, Kayentao K, Doumbo S, Diourte Y, et al. A molecular marker for chloroquine-resistant falciparum malaria. N Engl J Med. 2001;344:257–263. doi: 10.1056/NEJM200101253440403. [DOI] [PubMed] [Google Scholar]
- Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondorp AM, Fairhurst RM, Slutsker L, Macarthur JR, Breman JG, Guerin PJ, et al. The threat of artemisinin-resistant malaria. N Engl J Med. 2011;365:1073–1075. doi: 10.1056/NEJMp1108322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M. Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, et al. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell. 2000;6:861–871. doi: 10.1016/s1097-2765(05)00077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flueck C, Bartfai R, Volz J, Niederwieser I, Salcedo-Amaya AM, Alako BT, et al. Plasmodium falciparum heterochromatin protein 1 marks genomic loci linked to phenotypic variation of exported virulence factors. PLoS Pathog. 2009;5:e1000569. doi: 10.1371/journal.ppat.1000569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foth BJ, Zhang N, Chaal BK, Sze SK, Preiser PR. Bozdech Z. Quantitative time-course profiling of parasite and host cell proteins in the human malaria parasite Plasmodium falciparum. Mol Cell Proteomics. 2011;10:M110.006411. doi: 10.1074/mcp.M110.006411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frugier M, Bour T, Ayach M, Santos MA, Rudinger-Thirion J, Theobald-Dietrich A. Pizzi E. Low complexity regions behave as tRNA sponges to help co-translational folding of plasmodial proteins. FEBS Lett. 2010;584:448–454. doi: 10.1016/j.febslet.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haerty W. Golding GB. Increased polymorphism near low-complexity sequences across the genomes of Plasmodium falciparum isolates. Genome Biol Evol. 2011;3:539–550. doi: 10.1093/gbe/evr045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- Hu G, Llinas M, Li J, Preiser PR. Bozdech Z. Selection of long oligonucleotides for gene expression microarrays using weighted rank-sum strategy. BMC Bioinformatics. 2007;8:350. doi: 10.1186/1471-2105-8-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntley MA. Golding GB. Selection and slippage creating serine homopolymers. Mol Biol Evol. 2006;23:2017–2025. doi: 10.1093/molbev/msl073. [DOI] [PubMed] [Google Scholar]
- Jones PM. George AM. Multidrug resistance in parasites: ABC transporters, P-glycoproteins and molecular modelling. Int J Parasitol. 2005;35:555–566. doi: 10.1016/j.ijpara.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Kavishe RA, van den Heuvel JM, van de Vegte-Bolmer M, Luty AJ, Russel FG. Koenderink JB. Localization of the ATP-binding cassette (ABC) transport proteins PfMRP1, PfMRP2, and PfMDR5 at the Plasmodium falciparum plasma membrane. Malar J. 2009;8:205. doi: 10.1186/1475-2875-8-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp DJ, Corcoran LM, Coppel RL, Stahl HD, Bianco AE, Brown GV. Anders RF. Size variation in chromosomes from independent cultured isolates of Plasmodium falciparum. Nature. 1985;315:347–350. doi: 10.1038/315347a0. [DOI] [PubMed] [Google Scholar]
- Kreidenweiss A, Kremsner PG. Mordmuller B. Comprehensive study of proteasome inhibitors against Plasmodium falciparum laboratory strains and field isolates from Gabon. Malar J. 2008;7:187. doi: 10.1186/1475-2875-7-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmanan V, Bray PG, Verdier-Pinard D, Johnson DJ, Horrocks P, Muhle RA, et al. A critical role for PfCRT K76T in Plasmodium falciparum verapamil-reversible chloroquine resistance. EMBO J. 2005;24:2294–2305. doi: 10.1038/sj.emboj.7600681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehane AM, van Schalkwyk DA, Valderramos SG, Fidock DA. Kirk K. Differential drug efflux or accumulation does not explain variation in the chloroquine response of Plasmodium falciparum strains expressing the same isoform of mutant PfCRT. Antimicrob Agents Chemother. 2011;55:2310–2318. doi: 10.1128/AAC.01167-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas M, Bozdech Z, Wong ED, Adai AT. DeRisi JL. Comparative whole genome transcriptome analysis of three Plasmodium falciparum strains. Nucleic Acids Res. 2006;34:1166–1173. doi: 10.1093/nar/gkj517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobanov AV, Delgado C, Rahlfs S, Novoselov SV, Kryukov GV, Gromer S, et al. The Plasmodium selenoproteome. Nucleic Acids Res. 2006;34:496–505. doi: 10.1093/nar/gkj450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Rubio JJ, Mancio-Silva L. Scherf A. Genome-wide analysis of heterochromatin associates clonally variant gene regulation with perinuclear repressive centers in malaria parasites. Cell Host Microbe. 2009;5:179–190. doi: 10.1016/j.chom.2008.12.012. [DOI] [PubMed] [Google Scholar]
- Mackinnon MJ, Li J, Mok S, Kortok MM, Marsh K, Preiser PR. Bozdech Z. Comparative transcriptional and genomic analysis of Plasmodium falciparum field isolates. PLoS Pathog. 2009;5:e1000644. doi: 10.1371/journal.ppat.1000644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVey M. Lee SE. MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–538. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malleret B, Claser C, Ong AS, Suwanarusk R, Sriprawat K, Howland SW, et al. A rapid and robust tri-color flow cytometry assay for monitoring malaria parasite development. Sci Rep. 2011;1:118. doi: 10.1038/srep00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RE, Marchetti RV, Cowan AI, Howitt SM, Broer S. Kirk K. Chloroquine transport via the malaria parasite's chloroquine resistance transporter. Science. 2009;325:1680–1682. doi: 10.1126/science.1175667. [DOI] [PubMed] [Google Scholar]
- Meierjohann S, Walter RD. Muller S. Regulation of intracellular glutathione levels in erythrocytes infected with chloroquine-sensitive and chloroquine-resistant Plasmodium falciparum. Biochem J. 2002;368:761–768. doi: 10.1042/BJ20020962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Militello KT. Wirth DF. A new reporter gene for transient transfection of Plasmodium falciparum. Parasitol Res. 2003;89:154–157. doi: 10.1007/s00436-002-0721-5. [DOI] [PubMed] [Google Scholar]
- Milner DA, Jr, Pochet N, Krupka M, Williams C, Seydel K, Taylor TE, et al. Transcriptional profiling of Plasmodium falciparum parasites from patients with severe malaria identifies distinct low versus high parasitemic clusters. PLoS ONE. 2012;7:e40739. doi: 10.1371/journal.pone.0040739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok BW, Ribacke U, Winter G, Yip BH, Tan CS, Fernandez V, et al. Comparative transcriptomal analysis of isogenic Plasmodium falciparum clones of distinct antigenic and adhesive phenotypes. Mol Biochem Parasitol. 2007;151:184–192. doi: 10.1016/j.molbiopara.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Mok S, Imwong M, Mackinnon MJ, Sim J, Ramadoss R, Yi P, et al. Artemisinin resistance in Plasmodium falciparum is associated with an altered temporal pattern of transcription. BMC Genomics. 2011;12:391. doi: 10.1186/1471-2164-12-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourier T, Pain A, Barrell B. Griffiths-Jones S. A selenocysteine tRNA and SECIS element in Plasmodium falciparum. RNA. 2005;11:119–122. doi: 10.1261/rna.7185605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourier T, Carret C, Kyes S, Christodoulou Z, Gardner PP, Jeffares DC, et al. Genome-wide discovery and verification of novel structured RNAs in Plasmodium falciparum. Genome Res. 2008;18:281–292. doi: 10.1101/gr.6836108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MR4/ATCC. Methods in malaria research. In: Ljungström I, Perlmann H, Schlichtherle M, Scherf A, editors; Wahlgren M, editor. Methods in Malaria Research. Manassas, VA: Malaria Research and Reference Reagent Resource Center (MR4); 2004. pp. 167–168. [Google Scholar]
- Mu J, Ferdig MT, Feng X, Joy DA, Duan J, Furuya T, et al. Multiple transporters associated with malaria parasite responses to chloroquine and quinine. Mol Microbiol. 2003;49:977–989. doi: 10.1046/j.1365-2958.2003.03627.x. [DOI] [PubMed] [Google Scholar]
- Myrick A, Munasinghe A, Patankar S. Wirth DF. Mapping of the Plasmodium falciparum multidrug resistance gene 5′-upstream region, and evidence of induction of transcript levels by antimalarial drugs in chloroquine sensitive parasites. Mol Microbiol. 2004;49:671–683. doi: 10.1046/j.1365-2958.2003.03597.x. [DOI] [PubMed] [Google Scholar]
- Noedl H, Se Y, Schaecher K, Smith BL, Socheat D. Fukuda MM. Evidence of artemisinin-resistant malaria in western Cambodia. N Engl J Med. 2008;359:2619–2620. doi: 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- Nosten F. White NJ. Artemisinin-based combination treatment of falciparum malaria. Am J Trop Med Hyg. 2007;77:181–192. [PubMed] [Google Scholar]
- Paumi CM, Chuk M, Snider J, Stagljar I. Michaelis S. ABC transporters in Saccharomyces cerevisiae and their interactors: new technology advances the biology of the ABCC (MRP) subfamily. Microbiol Mol Biol Rev. 2009;73:577–593. doi: 10.1128/MMBR.00020-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickard AL, Wongsrichanalai C, Purfield A, Kamwendo D, Emery K, Zalewski C, et al. Resistance to antimalarials in Southeast Asia and genetic polymorphisms in pfmdr1. Antimicrob Agents Chemother. 2003;47:2418–2423. doi: 10.1128/AAC.47.8.2418-2423.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pique-Regi R, Ortega A. Asgharzadeh S. Joint estimation of copy number variation and reference intensities on multiple DNA arrays using GADA. Bioinformatics. 2009;25:1223–1230. doi: 10.1093/bioinformatics/btp119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price RN. Nosten F. Drug resistant falciparum malaria: clinical consequences and strategies for prevention. Drug Resist Updat. 2001;4:187–196. doi: 10.1054/drup.2001.0195. [DOI] [PubMed] [Google Scholar]
- Price RN, Cassar C, Brockman A, Duraisingh M, van Vugt M, White NJ, et al. The pfmdr1 gene is associated with a multidrug-resistant phenotype in Plasmodium falciparum from the western border of Thailand. Antimicrob Agents Chemother. 1999;43:2943–2949. doi: 10.1128/aac.43.12.2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price RN, Uhlemann AC, Brockman A, McGready R, Ashley E, Phaipun L, et al. Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet. 2004;364:438–447. doi: 10.1016/S0140-6736(04)16767-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj DK, Mu J, Jiang H, Kabat J, Singh S, Sullivan M, et al. Disruption of a Plasmodium falciparum multidrug resistance-associated protein (PfMRP) alters its fitness and transport of antimalarial drugs and glutathione. J Biol Chem. 2009;284:7687–7696. doi: 10.1074/jbc.M806944200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal A. Simon SM. Subcellular localization and activity of multidrug resistance proteins. Mol Biol Cell. 2003;14:3389–3399. doi: 10.1091/mbc.E02-11-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathod PK, McErlean T. Lee PC. Variations in frequencies of drug resistance in Plasmodium falciparum. Proc Natl Acad Sci USA. 1997;94:9389–9393. doi: 10.1073/pnas.94.17.9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson T, Campino SG, Auburn S, Assefa SA, Polley SD, Manske M, et al. Drug-resistant genotypes and multi-clonality in Plasmodium falciparum analysed by direct genome sequencing from peripheral blood of malaria patients. PLoS ONE. 2011;6:e23204. doi: 10.1371/journal.pone.0023204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojanawatsirivet C, Congpuong K, Vijaykadga S, Thongphua S, Thongsri K, Bangchang KN, et al. Declining mefloquine sensitivity of Plasmodium falciparum along the Thai-Myanmar border. Southeast Asian J Trop Med Public Health. 2004;35:560–565. [PubMed] [Google Scholar]
- Roper C, Pearce R, Nair S, Sharp B, Nosten F. Anderson T. Intercontinental spread of pyrimethamine-resistant malaria. Science. 2004;305:1124. doi: 10.1126/science.1098876. [DOI] [PubMed] [Google Scholar]
- Rosario V. Cloning of naturally occurring mixed infections of malaria parasites. Science. 1981;212:1037–1038. doi: 10.1126/science.7015505. [DOI] [PubMed] [Google Scholar]
- Roseler A, Prieto JH, Iozef R, Hecker B, Schirmer RH, Kulzer S, et al. Insight into the selenoproteome of the malaria parasite Plasmodium falciparum. Antioxid Redox Signal. 2012;17:534–543. doi: 10.1089/ars.2011.4276. [DOI] [PubMed] [Google Scholar]
- Rovira-Graells N, Gupta AP, Planet E, Crowley VM, Mok S, Ribas de Pouplana L, et al. Transcriptional variation in the malaria parasite Plasmodium falciparum. Genome Res. 2012;22:925–938. doi: 10.1101/gr.129692.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sa JM, Twu O, Hayton K, Reyes S, Fay MP, Ringwald P. Wellems TE. Geographic patterns of Plasmodium falciparum drug resistance distinguished by differential responses to amodiaquine and chloroquine. Proc Natl Acad Sci USA. 2009;106:18883–18889. doi: 10.1073/pnas.0911317106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salcedo-Amaya AM, van Driel MA, Alako BT, Trelle MB, van den Elzen AM, Cohen AM, et al. Dynamic histone H3 epigenome marking during the intraerythrocytic cycle of Plasmodium falciparum. Proc Natl Acad Sci USA. 2009;106:9655–9660. doi: 10.1073/pnas.0902515106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvage V, Aubert D, Escotte-Binet S. Villena I. The role of ATP-binding cassette (ABC) proteins in protozoan parasites. Mol Biochem Parasitol. 2009;167:81–94. doi: 10.1016/j.molbiopara.2009.05.005. [DOI] [PubMed] [Google Scholar]
- Setthaudom C, Tan-ariya P, Sitthichot N, Khositnithikul R, Suwandittakul N, Leelayoova S. Mungthin M. Role of Plasmodium falciparum chloroquine resistance transporter and multidrug resistance 1 genes on in vitro chloroquine resistance in isolates of Plasmodium falciparum from Thailand. Am J Trop Med Hyg. 2011;85:606–611. doi: 10.4269/ajtmh.2011.11-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuman S. Glickman MS. Bacterial DNA repair by non-homologous end joining. Nat Rev Microbiol. 2007;5:852–861. doi: 10.1038/nrmicro1768. [DOI] [PubMed] [Google Scholar]
- Sidhu AB, Verdier-Pinard D. Fidock DA. Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science. 2002;298:210–213. doi: 10.1126/science.1074045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhu AB, Valderramos SG. Fidock DA. pfmdr1 mutations contribute to quinine resistance and enhance mefloquine and artemisinin sensitivity in Plasmodium falciparum. Mol Microbiol. 2005;57:913–926. doi: 10.1111/j.1365-2958.2005.04729.x. [DOI] [PubMed] [Google Scholar]
- Snounou G. Genotyping of Plasmodium spp. Nested PCR. Methods Mol Med. 2002;72:103–116. doi: 10.1385/1-59259-271-6:103. [DOI] [PubMed] [Google Scholar]
- Snounou G, Zhu X, Siripoon N, Jarra W, Thaithong S, Brown KN. Viriyakosol S. Biased distribution of msp1 and msp2 allelic variants in Plasmodium falciparum populations in Thailand. Trans R Soc Trop Med Hyg. 1999;93:369–374. doi: 10.1016/s0035-9203(99)90120-7. [DOI] [PubMed] [Google Scholar]
- Sullivan DJ, Jr, Matile H, Ridley RG. Goldberg DE. A common mechanism for blockade of heme polymerization by antimalarial quinolines. J Biol Chem. 1998;273:31103–31107. doi: 10.1074/jbc.273.47.31103. [DOI] [PubMed] [Google Scholar]
- Tan JC, Tan A, Checkley L, Honsa CM. Ferdig MT. Variable numbers of tandem repeats in Plasmodium falciparum genes. J Mol Evol. 2010;71:268–278. doi: 10.1007/s00239-010-9381-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trager W. Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- Valderramos SG. Fidock DA. Transporters involved in resistance to antimalarial drugs. Trends Pharmacol Sci. 2006;27:594–601. doi: 10.1016/j.tips.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga MI, Ferreira PE, Jornhagen L, Malmberg M, Kone A, Schmidt BA, et al. Novel polymorphisms in Plasmodium falciparum ABC transporter genes are associated with major ACT antimalarial drug resistance. PLoS ONE. 2011;6:e20212. doi: 10.1371/journal.pone.0020212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivas L, Rattray L, Stewart LB, Robinson BL, Fugmann B, Haynes RK, et al. Antimalarial efficacy and drug interactions of the novel semi-synthetic endoperoxide artemisone in vitro and in vivo. J Antimicrob Chemother. 2007;59:658–665. doi: 10.1093/jac/dkl563. [DOI] [PubMed] [Google Scholar]
- Waller KL, Muhle RA, Ursos LM, Horrocks P, Verdier-Pinard D, Sidhu AB, et al. Chloroquine resistance modulated in vitro by expression levels of the Plasmodium falciparum chloroquine resistance transporter. J Biol Chem. 2003;278:33593–33601. doi: 10.1074/jbc.M302215200. [DOI] [PubMed] [Google Scholar]
- Wang Z, Parker D, Meng H, Wu L, Li J, Zhao Z, et al. In vitro sensitivity of Plasmodium falciparum from China-Myanmar border area to major ACT drugs and polymorphisms in potential target genes. PLoS ONE. 2012;7:e30927. doi: 10.1371/journal.pone.0030927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellems TE. Plowe CV. Chloroquine-resistant malaria. J Infect Dis. 2001;184:770–776. doi: 10.1086/322858. [DOI] [PubMed] [Google Scholar]
- White NJ. Artemisinin resistance – the clock is ticking. Lancet. 2010;376:2051–2052. doi: 10.1016/S0140-6736(10)61963-0. [DOI] [PubMed] [Google Scholar]
- White NJ. Olliaro PL. Strategies for the prevention of antimalarial drug resistance: rationale for combination chemotherapy for malaria. Parasitol Today. 1996;12:399–401. doi: 10.1016/0169-4758(96)10055-7. [DOI] [PubMed] [Google Scholar]
- WHO. Global Report on Antimalarial Drug Efficacy and Drug Resistance: 2000–2010. Geneva: World Health Organization; 2010. [Google Scholar]
- Wisedpanichkij R, Chaijaroenkul W, Sangsuwan P, Tantisawat J, Boonprasert K. Na-Bangchang K. In vitro antimalarial interactions between mefloquine and cytochrome P450 inhibitors. Acta Trop. 2009;112:12–15. doi: 10.1016/j.actatropica.2009.05.018. [DOI] [PubMed] [Google Scholar]
- Wongsrichanalai C, Sirichaisinthop J, Karwacki JJ, Congpuong K, Miller RS, Pang L. Thimasarn K. Drug resistant malaria on the Thai-Myanmar and Thai-Cambodian borders. Southeast Asian J Trop Med Public Health. 2001;32:41–49. [PubMed] [Google Scholar]
- Wootton JC, Feng X, Ferdig MT, Cooper RA, Mu J, Baruch DI, et al. Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum. Nature. 2002;418:320–323. doi: 10.1038/nature00813. [DOI] [PubMed] [Google Scholar]
- Zaman GJ, Flens MJ, van Leusden MR, de Haas M, Mulder HS, Lankelma J, et al. The human multidrug resistance-associated protein MRP is a plasma membrane drug-efflux pump. Proc Natl Acad Sci USA. 1994;91:8822–8826. doi: 10.1073/pnas.91.19.8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilversmit MM, Volkman SK, DePristo MA, Wirth DF, Awadalla P. Hartl DL. Low-complexity regions in Plasmodium falciparum: missing links in the evolution of an extreme genome. Mol Biol Evol. 2010;27:2198–2209. doi: 10.1093/molbev/msq108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary
Supplementary