

Abstract
Rett syndrome (RS) is an X-linked neurodevelopmental disorder mostly involving mutations in the gene for methyl-CpG-binding protein 2 (MECP2). Ganglioside abnormalities were previously found in cerebrum and cerebellum in RS patients. We evaluated total lipid distribution in cerebrum/brainstem, hippocampus, and cerebellum in male mice carrying either the Mecp2tm1.1Bird knockout mutation or the Mecp2308/y deletion mutation. The concentration of the neuronal enriched ganglioside GD1a was significantly lower in the cerebrum/brainstem of Mecp2tm1.1Bird mice than in that of age matched controls, but was not reduced in the Mecp2308/y mice. No other differences in brain lipid content, including myelin-enriched cerebrosides, were detected in mice with either type of Mecp2 mutation. These findings indicate that the poor motor performance previously reported in the RS mutant mice is not associated with major brain lipid abnormalities and that most previous brain lipid abnormalities observed in RS patients were not observed in the Mecp2tm1.1Bird or the Mecp2308/y RS mice.
Keywords: Mecp2; Rett; mouse models; gangliosides, GD1a; myelin
Introduction
Rett syndrome (RS) is a progressive X-linked neurodevelopmental disorder afflicting mostly females and involves microcephaly, reduced muscle tone, repetitive hand movements, anxiety, autistic-like behavior, and epileptic seizures (1-3). About 80% of females with RS have mutations in the gene for methyl-CpG-binding protein 2 (MECP2) (3, 4). The MeCP2 protein is a transcription regulator, which can both activate and repress transcription (1, 5). Mouse mutants were produced that express either partial deletion (Mecp2308/y) or extensive deletion (Mecp2tm1.1Bird) of the MECP2 gene (1, 6). The behavioral motor abnormalities occur earlier and are more severe in male mice carrying the Mecp2tm1.1Bird mutation than in male mice carrying the Mecp2308/y mutation. It is not clear how alterations in the MECP2 gene contribute to the behavioral, neurodevelopmental, and neurochemical abnormalities in either patients or in mice with RS, but multiple mechanisms involving gene-environmental interactions are likely involved (1, 6-8).
Previous studies showed lipid and ganglioside abnormalities in frontal and temporal cortex, cerebellum, and in cerebrospinal fluid of patients with RS (9-11). Some of these brain lipid changes were associated with neuropathology involving Purkinje cell loss and gliosis in cerebellum, and gliosis, synaptic and myelin abnormalities in frontal cortex (9, 10, 12, 13). The content of galactocerebroside, an excellent lipid marker for myelin content (14, 15), was reduced in temporal white matter of some Rett patients, but was not reduced in white matter from frontal cortex suggesting local rather than global myelin abnormalities (10). The status of myelin content in RS cerebellum is ambiguous. Cerebellar cerebrosides were reported elevated in one study of RS patients (9), whereas histological evidence of cerebellar myelin and white matter loss was reported in other studies of RS patients (12, 13). The neuronal-enriched ganglioside GD1a was also reduced in frontal gray matter and white matter of some RS patients (10). However, most of the lipid changes found in RS patients and mice were subtle at best and were not clearly associated with documented morphological changes. Morphological changes were recently found, however, in cortex, hippocampus, and cerebellum of Mecp2 mutant mice (16). Furthermore, subtle changes in brain phospholipids were also recently reported in the Mecp2tm1.1Bird mutant mice (17). Viewed together, these findings suggest that lipid abnormalities might contribute to RS pathogenesis.
Lipids can provide important information on the structure and function of the nervous system. Gangliosides are sialic acid-containing glycosphingolipids that are enriched in neural membranes and are sensitive indicators of neural membrane integrity. Gangliosides GD1a and GT1b are enriched in neuronal and synaptic membranes, whereas ganglioside GD3 is enriched in reactive glial cells and is a good marker for neurodegeneration (18-21). In the mouse cerebellum, GD1a is enriched in granule cells while LD1/GT1a is enriched in Purkinje cells (22, 23). Changes in these gangliosides are indicative of changes in granule cells and Purkinje cells, respectively (19, 20, 22, 24). GM1 and cerebrosides are enriched in myelin and can be indicative of CNS myelin content (15, 25, 26). Although cholesterol and phospholipids are present in most neural membranes, cardiolipin is enriched in the inner mitochondrial membrane and plays an important role in energy metabolism (25, 27). Hence, changes in brain lipid content and distribution can be indicative of neuropathology.
In light of the lipid and the morphological changes in the brain of RS patients and Mecp2 mutant mice, we examined total lipid composition in cerebrum/brainstem, hippocampus, and cerebellum in two strains of mice with RS. The concentration of ganglioside GD1a was significantly lower in the cerebrum/brainstem of Mecp2tm1.1Bird male mice than in the control male mice suggesting a disturbance in synaptic integrity in this brain region of this strain. Although motor performance is worse in the mutant male RS mice than in control male mice, no significant abnormalities were detected for the content or distribution of most major cerebellar lipid classes in the RS mutant mice to include myelin-enriched lipids. These findings indicate that with the exception of reduced GD1a in the Mecp2tm1.1Bird mice, the lipid abnormalities reported in humans with RS are not observed in mice with RS.
Experimental Procedure
Mice
The inbred B6.129S-Mecp2tm1Hzo/J (Mecp2308/y) Rett mice were originally obtained from the Jackson Laboratory (Bar Harbor, Maine) and were generated as previously described(1). Mixed background Mecp2tm1.1Bird/J (C57BL/6J.129S/CD1) mice were obtained as a gift from Michael Greenberg (Harvard University) and were generated as previously described (6). The mice were maintained through brother-sister inbreeding and kept in the Animal Care Facility of Boston College with all procedures in strict adherence with the NIH Guide for the care and use of laboratory animals and approved by the Institutional Animal Care and Use Committee. The mice were housed in plastic cages with Sani-chip bedding (P.J. Murphy Forest Products Corp., Montville, N.J.) and kept on a 12-hr light/dark cycle at approximately 22°C . All cages and water bottles were changed once per week. Only males were used for these studies since female Rett mice have a less severe disease phenotype than male Rett mice (1, 6).
Mouse Genotyping
DNA from 30 day-old Mecp2tm1.1Bird and Mecp2308/y mice was isolated from ∼3 mm of tail using the Qiagen DNeasy tail tissue protocol. PCR amplification for both strains was performed using 1 μL of DNA (∼50−100 ng). For the Mecp2tm1.1Bird mice, two separate PCR reactions (A & B) were run that also included the amplification of a control gene. The PCR amplification was set up similar to that of the JAX genotype protocol for the Mecp2tm1.1Bird mice with the following modifications: 2.4 μL of 5X GoTaq Buffer, 0.3 μL dNTPs (10 mM mix), 0.2 μL control forward primer (5'-CTAggCCACAgAATTgAAAgATCT-3'), 0.2 μL control reverse primer (5'-gTAggTggAAATTCTagCATCATCC-3), 1 μL Mecp2 forward primer (5'-ggTAAAgACCCATgTgACCC-3'), 1 μL Mecp2 reverse Ra primer (5'-TCCACCTAgCCTgCCTgTAC-3') (reaction A), 1 μL Mecp2 reverse Rb primer (5'-ggCTTgCCACATgACAA-3') (reaction B), 0.06 μL GoTaq DNA Polymerase (Promega) and 5.84 μL water for a 12 μL reaction volume. For the Mecp2tm1.1Bird mice PCR amplification, the concentration of each primer was 20 mM. The control primer set amplified an internal control 324 bp fragment band (IL-2 gene) from the wild-type allele. The Mecp2 forward primer along with the reverse Ra primer (reaction A) amplified a 400bp fragment from the disrupted Rett allele, whereas the Mecp2 forward primer along with the reverse Rb primer (reaction B) amplified a 416 bp fragment from the wild-type allele. The DNA was amplified using the following protocol for reactions A and B: Initial denaturation 94°C for 3 min, followed by 35 cycles of denaturation at 94°C for 30 s; annealing 63°C for 1 min (for reaction A) and 57.5°C for 1 min (for reaction B); extension at 72°C for 1 min; and a final extension at 72°C for 5 min following the last cycle.
The PCR reaction for the Mecp2308/y mice, was set up similar to that of the JAX genotype protocol for the Mecp2308/y mice with the following modifications: 5μL of 5X GoTaq Buffer, 0.5 μL dNTPs (10 mM mix), 5 μL forward primer (5’-AACGGGGTAGAAAGCCTG-3’), 2.5 μL AR Primer (5’-TGATGGGGTCCTCAGAGC-3’), 2.5 μL BR primer (5’-ATGCTCCAGACTGCCTTG-3’), 0.25μL GoTaq DNA Polymerase (Promega) and 8.25 μL water for a 25μL reaction volume. For the Mecp2308/y mice PCR amplification, the concentration of each primer was 10 mM. DNA was amplified using the following protocol: Initial denaturation at 94°C for 2 min, followed by 31 cycles of denaturation at 94°C for 45 s; annealing 62°C for 45 s; extension at 72°C for 45 s, and a final extension at 72°C for 5 min following the last cycle. The forward primer along with the AR primer amplified a 396 bp fragment from the wild type allele, whereas the forward primer along with the BR primer amplified a 318 bp fragment from the knockout allele. PCR products (12−15 μL) were separated on 1% agarose gels containing ethidium bromide, visualized with UV light, and analyzed using the FluroChem8900 Software. PCR reactions A and B for the Mecp2tm1.1Bird were run and analyzed separately.
Lipid Isolation, Purification, and Quantitation
Total lipids were extracted with chloroform (CHCl3) and methanol (CH3OH) 1:1 by volume and were purified from the lyophilized brain tissue using modifications of previously described procedures (28-30). Neutral and acidic lipids were separated using DEAE-Sephadex (A-25, Pharmacia Biotech, Upsala, Sweden) column chromatography as previously described (31). The total lipid extract, suspended in CHCl3:CH3OH:dH20, 30:60:8 by volume (solvent A), was applied to a DEAE-Sephadex column (1.2 mL bed volume) that had been equilibrated prior with solvent A. The column was washed twice with 20 mL solvent A and the entire neutral lipid fraction, consisting of the initial eluent plus washes, was collected. This fraction contained cholesterol, phosphatidylcholine, phosphatidylethanolamine and plasmologens, sphingomyelin, and cerebrosides. Next, acidic lipids were eluted from the column with 35 mL of solvent B (CHCl3:CH3OH:0.8 M Na acetate, 30:60:8 by volume).
The acidic lipid fraction, containing gangliosides, was dried under rotary evaporation and was then “Folch” partitioned to separate the acidic phospholipids lipids and sulfatides into the lower organic phase and the gangliosides into the upper aqueous phase as previously described (28, 32, 33). The amount of sialic acid in the ganglioside fraction was determined by the resorcinol assay (34). The ganglioside fraction was further purified with base treatment and desalting, as previously described (29, 35). After the ganglioside fraction (Folch upper phase) was transferred, the acidic phospholipid fraction (Folch lower phase) was evaporated under a stream of nitrogen and resuspended in 10 mL of CHCl3:CH3OH (1:1 by volume). This fraction contained cardiolipin, phosphatidylserine, phosphatidylinositol, and sulfatides.
High-performance thin-layer chromatography
All lipids were analyzed qualitatively by high-performance thin-layer chromatograph (HPTLC) according to previously described methods (29, 31, 36, 37). To enhance precision, an internal standard (oleoyl alcohol) was added to the neutral and the acidic lipid standards and samples as previously described (31). Purified lipid standards were either purchased from Matreya Inc. (Pleasant Gap, PA, USA), or Sigma (St. Louis, MO, USA). For gangliosides, the HPTLC plates (E. Merck, Darmstadt, Germany) were developed and visualized as in Figure 1 and as previously described (29). For neutral and acidic phospholipids, the plates were developed to a height of either 4.5 cm or 6 cm, respectively with chloroform: methanol: acetic acid: formic acid: water (35:15:6:2:1 by volume), and then both were developed to the top with hexanes: diisopropyl ether: acetic acid (65:35:2 by volume) as shown in Figures 2 and 3, and as previously described (25, 31). Neutral and acidic phospholipids were visualized by charring with 3% cupric acetate in 8% phosphoric acid solution, followed by heating in an oven at 165°C for 7 minutes. The percentage distribution and density of individual bands was determined as previously described (29). Briefly, the HPTLC plates were scanned on a Personal Densitometer SI with ImageQuant software (Molecular Dynamics). The total brain ganglioside distribution was normalized to 100% and the percentage distribution values were used to calculate sialic acid concentration of individual gangliosides (μg/100 mg dry wt) as we previously described (24). For the neutral and the acidic phospholipids, each lipid was normalized to the internal standard (oleyl alcohol) and its concentration was quantified using a standard curve of each respective lipid over the range of 1.0 to 4.0 μg (31).
Figure 1.

High-performance thin-layer chromatogram of brain gangliosides in Mecp2tm1.1Bird −/y and +/y mice. Approximately 1.5 μg ganglioside sialic acid were spotted per lane. The plate was developed by a single ascending run with chloroform: methanol: water (55:45:10 by volume) containing 0.02% calcium chloride. The bands were visualized with the resorcinol-HCl spray.
Figure 2.
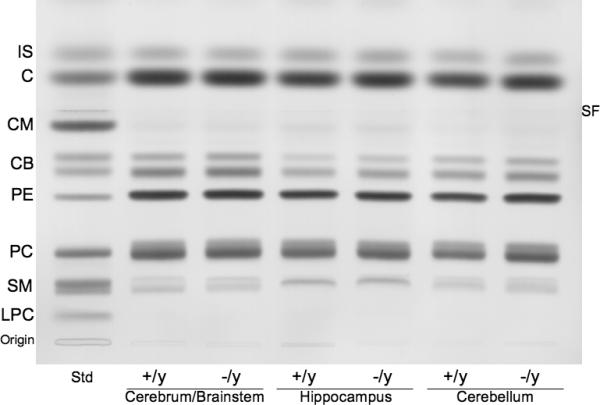
High-performance thin-layer chromatogram of brain neutral lipids in Mecp2tm1.1Bird −/y and +/y mice. The amount of neutral lipids spotted per lane was equivalent to approximately 70 μg brain dry weight. The plate was developed and the lipid bands visualized as described in methods. Std indicates neutral lipid standards. IS, internal standard (oleoyl alcohol); C, cholesterol; CM, ceramide; CB, cerebroside (doublet); PE, phosphatidylethanolamine; PC, phosphatidylcholine; SM, sphingomyelin. SF, indicates the solvent front.
Figure 3.
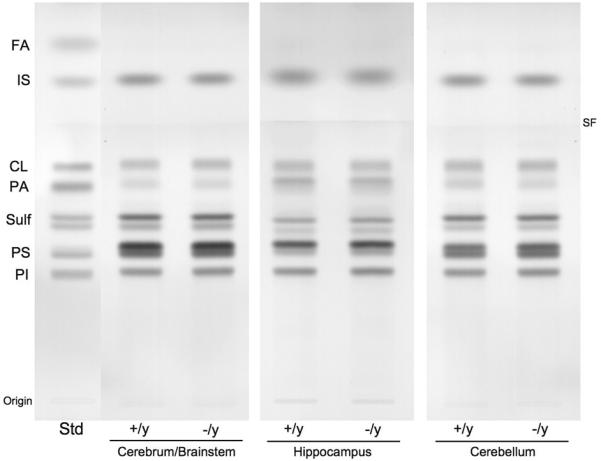
High-performance thin-layer chromatogram of brain acidic lipids in Mecp2tm1.1Bird −/y and +/y mice. The amount of acidic lipids spotted per lane was equivalent to approximately 200 μg brain dry weight. The plate was developed and the lipid bands visualized as described in methods. Std indicates acidic lipid standards. FA, fatty acids; IS, internal standard; CL, cardiolipin; PA, phosphatidic acid; Sulf, sulfatides (doublet); PS, phosphatidylserine; PI, phosphatidylinositol. SF, indicates the solvent front.
Representative standard lipid mixtures for neutral and acidic lipids are shown on the HPTLC in Figures 2 and 3, respectively. The lipid methodology we employed in these studies is capable of detecting relatively small differences in brain lipid levels (less than 10%) as we previously showed (19, 25, 29, 32).
Statistical Analysis
All data were analyzed by the two-tailed Student's t-Test to calculate statistical significance between the control mice and the RS mice.
Results
Our objective was to determine if the brain lipid changes reported previously in patients with RS also occurred in mice with RS. The neurological phenotype appears earlier and is more severe in the male Mecp2tm1.1Bird mice than in the male Mecp2308/y mice (1, 6). Since the neurological phenotype is also more severe in male mice than female mice, our analysis was restricted to males. Due to differences in age of phenotype onset, we evaluated the Mecp2tm1.1Bird mice at 68 days of age and the Mecp2308/y mice at 321 days of age (Table 1). The cerebrum/brainstem weight was significantly lower by about 13% in the Mecp2tm1.1Bird −/y mice than in the +/y mice, but was not lower than normal in the Mecp2308/y mice. No differences for hippocampus or cerebellum weight were detected between the +/y and −/y mice for either Mecp2 strain. No significant differences for total ganglioside content were found between the +/y and −/y mice across the brain regions in either Mecp2 strain (Table 1). The qualitative and quantitative distribution of the individual ganglioside species in the various brain regions is shown in Figure 1 and Table 2. The thin-layer chromatographic ganglioside distribution is shown only for Mecp2tm1.1Bird mice in Figure 1, as this distribution was similar to that found in the Mecp2308/y mice. GM2 and GM3 were present in only trace amounts in both the +/y and −/y mice of either strain and were not included in the analysis. Ganglioside GD1a was significantly lower, by about 15%, in the cerebrum/brainstem of the Mecp2tm1.1Bird −/y mice than in the +/y mice. No other statistically significant changes in ganglioside concentration were found among the brain regions between the +/y and −/y mice in either Mecp2 strain. We also examined whole brain ganglioside content and distribution in young (30−35 day-old) Mecp2308/y −/y and +/y mice, but no differences were found (data not shown). The differences in ganglioside content and distribution between younger and older mice and among the different brain regions were reported previously in other mouse strains (26, 35, 37). Hence, with the exception of reduced GD1a in cerebrum/brainstem of the Mecp2tm1.1Bird −/y mice, no significant abnormalities were found for the content or distribution of gangliosides in the Mecp2tm1.1Bird or the Mecp2308/y mutant male mice.
Table I.
Brain ganglioside content in Mecp2Bird and Mecp2308/y micea
| Strain (Age) |
Mecp2Bird (68 ± 2 days) |
Mecp2308/y (321 ± 13 days) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Region | Cerebrum/Brainstem |
Hippocampus |
Cerebellum |
Cerebrum/Brainstem |
Cerebellum |
|||||
| Genotype | +/y |
−/y |
+/y |
−/y |
+/y |
−/y |
+/y |
−/y |
+/y |
−/y |
| Brain Dry weight (mg) | 76.8 ± 3.5 | 66.7 ± 1.3 * | 6.4 ± 0.3 | 6.4 ± 0.4 | 10.8 ± 0.9 | 11.2 ± 0.1 | 73.4 ± 2.6 | 79.0 ± 3.3 | 10.6 ± 2.2 | 10.2 ± 0.5 |
| Ganglioside sialic acid (μg/100mg dry weight) | 467 ± 14 | 443 ± 12 | 509 ± 22 | 507 ± 22 | 346 ± 14 | 325 ± 11 | 443 ± 13 | 432 ± 6 | 300 ± 17 | 294 ± 16 |
Values represent the mean±SE (n=3−4 independent samples). Asterisk indicates statistical significance where p<0.05 as determined by two-tailed t-test.
Table II.
Ganglioside distribution in different brain regions of Rett (Mecp2Bird and Mecp2308/y) micea
| Concentration (μg lipid/100 mg dry weight)b |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
Mecp2Bird (68 ± 2 days) |
MeCP2308/y (321 ± 13 days) |
|||||||||
| Cerebrum/Brainstem |
Hippocampus |
Cerebellum |
Cerebrum/Brainstem |
Cerebellum |
||||||
| +/y |
−/y |
+/y |
−/y |
+/y |
−/y |
+/y |
−/y |
+/y |
−/y |
|
| GM1 | 37.6 ± 1.9 | 35.2 ± 1.3 | 45.9 ± 1.5 | 41.2 ± 2.0 | 13.3 ± 0.4 | 12.2 ± 0.4 | 46.9 ± 3.4 | 51.3 ± 4.9 | 33.3 ± 4.6 | 33.5 ± 0.9 |
| GD3 | Trace | Trace | Trace | Trace | 9.0 ± 1.4 | 10.8 ± 0.6 | Trace | Trace | Trace | Trace |
| GD1a | 149.2 ± 4.1 | 126.9 ± 3.2 * | 176.8 ± 5.4 | 163.4 ± 5.5 | 44.2 ± 0.2 | 37.5 ± 2.7 | 130.9 ± 5.3 | 121.5 ± 5.6 | 42.4 ± 4.1 | 44.0 ± 2.3 |
| GT1a/LD1 | 24.0 ± 0.8 | 24.0 ± 1.1 | 31.4 ± 2.8 | 31.4 ± 1.9 | 33.3 ± 1.2 | 28.2 ± 1.6 | 25.0 ± 0.9 | 22.3 ± 0.8 | 27.7 ± 2.6 | 29.9 ± 2.2 |
| GD1b | 52.3 ± 2.4 | 53.9 ± 1.8 | 51.1 ± 2.3 | 51.8 ± 2.1 | 25.1 ± 2.1 | 26.0 ± 0.6 | 44.9 ± 1.4 | 43.4 ± 1.9 | 21.8 ± 1.8 | 18.5 ± 1.0 |
| GT1b | 140.4 ± 3.6 | 134.1 ± 4.7 | 147.2 ± 8.5 | 154.8 ± 8.4 | 140.3 ± 5.6 | 131.6 ± 2.0 | 134.8 ± 5.1 | 131.7 ± 1.9 | 112.0 ± 6.7 | 112.0 ± 7.5 |
| GQ1b | 64.3 ± 1.3 | 66.1 ± 1.5 | 56.5 ± 6.2 | 64.2 ± 2.5 | 76.9 ± 1.5 | 77.1 ± 1.6 | 60.4 ± 0.7 | 58.8 ± 1.2 | 63.2 ± 2.4 | 55.8 ± 3.6 |
Values represent the mean±SE (n=3−4 independent samples). Asterisk indicates statistical significance where p<0.01 (two-tailed t-test).
Determined from densitometric scanning of HPTLC plates, as shown in figure 1.
The qualitative and quantitative distribution of the individual neutral lipids and acidic lipids in the various brain regions of the two Mecp2 strains are shown in Figures 2 and 3 and Table 3. No statistically significant abnormalities for the content of any major neutral lipid or acidic lipid were found among the brain regions between the +/y and −/y mice in either of the two Mecp2 strains. Ceramide was present in only trace amounts in both the +/y and −/y mice of either strain and were not included in the analysis. In mice, cerebrosides are among the most sensitive biochemical markers for myelin content (15, 26). Glucocerebroside is a transient precursor for ganglioside biosynthesis and is present in only trace amounts in mammalian brain. The doublet band for cerebrosides shown on the HPTLC in Figure 2 comprises almost exclusively galactocerebroside. Our data on cerebrosides therefore reflect the content of galactocerebroside. Sulfatides and GM1 ganglioside, while not specific for myelin, are more concentrated in white matter than in gray matter (26, 38, 39). Our results show that neither cerebrosides, sulfatides, nor GM1 were significantly altered in the cerebrum/brainstem, hippocampus, or cerebellum of Rett mutant mice. Viewed collectively, these results indicate that neither the Mecp2tm1.1Bird male mice nor the Mecp2308/y male mice contain significant abnormalities in the content or in the distribution of the major neutral or acidic brain lipids.
Table III.
Neutal and acidic lipid distribution in different brain regions of Rett (Mecp2Bird and Mecp2308/y) micea
| Concentration (μg lipid/mg dry weight) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
Mecp2Bird (68 ± 2 days) |
Mecp2308/y (321 ± 13 days) |
|||||||||
| Cerebrum/Brainstem |
Hippocampus |
Cerebellum |
Cerebrum/Brainstem |
Cerebellum |
||||||
| Lipids | +/y |
−/y |
+/y |
−/y |
+/y |
−/y |
+/y |
−/y |
+/y |
−/y |
| Neutral b | ||||||||||
| Cholesterol | 93.5 ± 4.6 | 91.7 ± 5.1 | 71.3 ± 0.1 | 68.3 ± 0.2 | 46.2 ± 0.9 | 46.2 ± 1.1 | 123.6 ± 5.6 | 125.3 ± 4.4 | 43.6 ± 0.8 | 43.3 ± 0.5 |
| Cerebrosides | 40.1 ± 0.9 | 40.8 ± 1.9 | 19.6 ± 0.2 | 18.5 ± 0.1 | 24.3 ± 0.7 | 23.7 ± 0.7 | 51.9 ± 3.0 | 59.0 ± 5.5 | 34.7 ± 2.6 | 33.7 ± 1.9 |
| Phosphatidylethanolamine | 155.1 ± 2.9 | 152.0 ± 5.0 | 96.8 ± 0.1 | 93.7 ± 0.2 | 53.5 ± 1.2 | 54.0 ± 0.7 | 119.5 ± 1.3 | 119.2 ± 0.5 | 50.9 ± 2.1 | 50.0 ± 0.2 |
| Phosphatidylcholine | 77.2 ± 2.0 | 77.7 ± 0.3 | 78.9 ± 0.1 | 80.3 ± 0.3 | 44.8 ± 0.4 | 45.2 ± 0.6 | 78.8 ± 2.7 | 79.5 ± 4.4 | 43.6 ± 0.6 | 40.7 ± 2.9 |
| Sphingomyelin | 5.6 ± 0.4 | 5.6 ± 0.3 | 0.2 ± 0.1 | 0.1 ± 0.1 | 6.6 ± 0.2 | 6.8 ± 0.1 | 4.3 ± 0.4 | 4.2 ± 0.2 | 8.1 ± 0.2 | 8.2 ± 0.1 |
| Acidic c | ||||||||||
| Cardiolipin | 3.7 ± 0.2 | 3.9 ± 0.2 | 4.4 ± 0.1 | 4.4 ± 0.1 | 6.3 ± 0.3 | 6.8 ± 0.3 | 3.7 ± 0.1 | 3.7 ± 0.3 | 5.2 ± 0.3 | 5.6 ± 0.1 |
| Phosphatidic acid | Trace | Trace | 1.2 ± 0.3 | 3.0 ± 0.7 | 3.4 ± 0.3 | 2.2 ± 0.1 | Trace | Trace | 2.1 ± 0.2 | 2.2 ± 0.3 |
| Sulfatides | 8.4 ± 1.1 | 11.3 ± 2.6 | 4.1 ± 0.3 | 4.6 ± 0.1 | 6.7 ± 0.2 | 6.7 ± 0.3 | 11.4 ± 1.5 | 12.1 ± 1.6 | 11.5 ± 1.0 | 11.5 ± 1.1 |
| Phosphatidylserine | 18.6 ± 1.4 | 15.5 ± 3.7 | 15.7 ± 0.6 | 15.0 ± 0.5 | 20.3 ± 0.2 | 21.4 ± 0.2 | 18.4 ± 1.5 | 17.7 ± 4.1 | 21.8 ± 0.2 | 21.5 ± 0.7 |
| Phosphatidylinositol | 6.2 ± 0.1 | 6.1 ± 0.5 | 7.4 ± 0.1 | 7.6 ± 0.1 | 6.9 ± 0.4 | 7.3 ± 0.6 | 6.5 ± 0.1 | 6.7 ± 0.4 | 7.0 ± 0.3 | 7.1 ± 0.1 |
Discussion
Our findings showed that ganglioside GD1a was significantly lower in the cerebrum/brainstem of the −/y mice than in the +/y mice of the Mecp2tm1.1Bird strain. Interestingly, GD1a was also reduced in cerebral cortex of some Rett patients (10). The cerebrum/brainstem weighed less in the −/y mice than in the +/y mice of this mouse strain. Since GD1a is enriched in synaptic membranes (19, 40, 41), these findings suggest that cortical synapses are abnormal in the Mecp2tm1.1Bird male mice. These findings could be related to the small volumetric abnormalities found recently in the cerebral cortex of the Mecp2tm1.1Bird male mice (16). Although, recent studies showed reduced volume of hippocampus and cerebellum in the Mecp2tm1.1Bird male mice (16), we did not detect significantly reduced weights or lipid abnormalities in these brain regions. No significant abnormalities were found for the qualitative or quantitative distribution of other gangliosides or of major neutral and acidic lipids in cerebrum/brainstem, hippocampus or cerebellum of either the Mecp2tm1.1Bird or Mecp2308/y mutant male mice. We do not, however, exclude the possibility that subtle changes in lipid fatty acid molecular species might occur in the mutant mice. Nevertheless, our findings indicate that most of the behavioral and motor abnormalities previously reported in Rett mice are not associated with significant changes in content or compositions of the major brain lipids.
Previous studies showed elevations of ganglioside GD3 in temporal cortex, frontal cortex, and cerebellum of RS patients (9, 10). Ganglioside GD3 is enriched in reactive astrocytes and is increased in CNS diseases in association with neurodegeneration and gliosis (10, 19, 21, 42). The elevation of GD3 in the brains of RS patients is therefore indicative of neurodegeneration. The normal levels of GM1, GT1a/LD1, GT1b, GQ1b, and GD3 in the cerebrum/brainstem, hippocampus, and cerebellum of the Mecp2tm1.1Bird and the Mecp2308/y mutant male mice indicate that neurodegeneration is not a major phenotype in these mouse mutants in contrast to humans with the disease.
Galactocerebroside is enriched in mature myelin and is an excellent lipid marker for the content of CNS myelin (14, 26). Considerable ambiguity was found, however, for myelin and cerebroside content in cortex and cerebellum of RS patients (9, 10). The cerebroside deficiency reported in RS patients was observed only in temporal white matter, but was not observed in white matter from frontal cortex or from cerebellum (10). In contrast to temporal cortex white matter, cerebroside content was actually higher in Rett patients than in controls in cerebellum (9). The absence of differences for cerebrosides, sulfatides, and GM1 in the brains of the Mecp2tm1.1Bird and the Mecp2308/y mutant male mice indicates that myelin abnormalities are unlikely present in these mice. We were also unable to find changes in the content or distribution of phosphatidylcholine or cardiolipin, as was previously reported in the Mecp2tm1.1Bird male mice (17). Differences in lipid methodology could account for differences between our results and the previous findings of Viola et al. As we did not look specifically at plasmalogen content, we do not exclude the possibility that the Rett mice might contain subtle changes in brain plasmalogen content. In summary, our neurochemical findings show that, with the exception of GD1a, major lipid abnormalities are not present in the brains of the Mecp2tm1.1Bird or Mecp2308/y RS mice. Reduction of GD1a in the Mecp2tm1.1Bird mice suggests that these mice express abnormalities in cortical synapses.
Acknowledgements
This work was supported in part from NIH grant (NS055195), the Rett Syndrome Research Foundation (RSRF) and the Boston College Research Expense Fund. We thank Dr. Zhaolan Zhou and Dr. Michael Greenberg (Children's Hospital Boston, Harvard Medical School) for providing us with the Mecp2Bird mice used in our study. We also thank Nicholas C. Zimick and Christie L. Fritz for helping with the breeding and genotyping of the mice.
References
- 1.Shahbazian M, Young J, Yuva-Paylor L, et al. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002;35:243–254. doi: 10.1016/s0896-6273(02)00768-7. [DOI] [PubMed] [Google Scholar]
- 2.Hagberg B, Aicardi J, Dias K, et al. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett's syndrome: report of 35 cases. Ann Neurol. 1983;14:471–479. doi: 10.1002/ana.410140412. [DOI] [PubMed] [Google Scholar]
- 3.Percy AK. Rett syndrome. Current status and new vistas. Neurol Clin. 2002;20:1125–1141. doi: 10.1016/s0733-8619(02)00022-1. [DOI] [PubMed] [Google Scholar]
- 4.Amir RE, Van den Veyver IB, Wan M, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature genetics. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 5.Chahrour M, Jung SY, Shaw C, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guy J, Hendrich B, Holmes M, et al. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nature genetics. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- 7.Percy AK, Lane JB. Rett syndrome: clinical and molecular update. Curr Opin Pediatr. 2004;16:670–677. doi: 10.1097/01.mop.0000143693.59408.ce. [DOI] [PubMed] [Google Scholar]
- 8.Samaco RC, Nagarajan RP, Braunschweig D, et al. Multiple pathways regulate MeCP2 expression in normal brain development and exhibit defects in autism-spectrum disorders. Hum Mol Genet. 2004;13:629–639. doi: 10.1093/hmg/ddh063. [DOI] [PubMed] [Google Scholar]
- 9.Lekman A, Hagberg B, Svennerholm L. Altered cerebellar ganglioside pattern in Rett syndrome. Neurochem. Int. 1991;19:505–509. [Google Scholar]
- 10.Lekman AY, Hagberg BA, Svennerholm LT. Membrane cerebral lipids in Rett syndrome. Pediatric neurology. 1991;7:186–190. doi: 10.1016/0887-8994(91)90082-v. [DOI] [PubMed] [Google Scholar]
- 11.Lekman AY, Hagberg BA, Svennerholm LT. Cerebrospinal fluid gangliosides in patients with Rett syndrome and infantile neuronal ceroid lipofuscinosis. Eur J Paediatr Neurol. 1999;3:119–123. doi: 10.1016/s1090-3798(99)90099-5. [DOI] [PubMed] [Google Scholar]
- 12.Papadimitriou JM, Hockey A, Tan N, et al. Rett syndrome: abnormal membrane-bound lamellated inclusions in neurons and oligodendroglia. American journal of medical genetics. 1988;29:365–368. doi: 10.1002/ajmg.1320290216. [DOI] [PubMed] [Google Scholar]
- 13.Oldfors A, Sourander P, Armstrong DL, et al. Rett syndrome: cerebellar pathology. Pediatric neurology. 1990;6:310–314. doi: 10.1016/0887-8994(90)90022-s. [DOI] [PubMed] [Google Scholar]
- 14.Norton WT. Isolation and characterization of myelin. In: Morell P, editor. Myelin. Plenum; New York: 1977. pp. 161–199. [Google Scholar]
- 15.Muse ED, Jurevics H, Toews AD, et al. Parameters related to lipid metabolism as markers of myelination in mouse brain. J Neurochem. 2001;76:77–86. doi: 10.1046/j.1471-4159.2001.00015.x. [DOI] [PubMed] [Google Scholar]
- 16.Belichenko NP, Belichenko PV, Li HH, et al. Comparative study of brain morphology in Mecp2 mutant mouse models of Rett syndrome. The Journal of comparative neurology. 2008;508:184–195. doi: 10.1002/cne.21673. [DOI] [PubMed] [Google Scholar]
- 17.Viola A, Saywell V, Villard L, et al. Metabolic fingerprints of altered brain growth, osmoregulation and neurotransmission in a Rett syndrome model. PLoS ONE. 2007;2:e157. doi: 10.1371/journal.pone.0000157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Svennerholm L, Bostrom K, Fredman P, et al. Human brain gangliosides: developmental changes from early fetal stage to advanced age. Biochimica et biophysica acta. 1989;1005:109–117. doi: 10.1016/0005-2760(89)90175-6. [DOI] [PubMed] [Google Scholar]
- 19.Seyfried TN, Bernard DJ, Yu RK. Cellular distribution of gangliosides in the developing mouse cerebellum: analysis using the staggerer mutant. J Neurochem. 1984;43:1152–1162. doi: 10.1111/j.1471-4159.1984.tb12856.x. [DOI] [PubMed] [Google Scholar]
- 20.Seyfried TN, Yu RK. Cellular localization of gangliosides in the mouse cerebellum: analysis using neurological mutants. Advances in experimental medicine and biology. 1984;174:169–181. doi: 10.1007/978-1-4684-1200-0_15. [DOI] [PubMed] [Google Scholar]
- 21.Seyfried TN, Yu RK. Ganglioside GD3: structure, cellular distribution, and possible function. Mol. Cell. Biochem. 1985;68:3–10. doi: 10.1007/BF00219383. [DOI] [PubMed] [Google Scholar]
- 22.Seyfried TN, Miyazawa N, Yu RK. Cellular localization of gangliosides in the developing mouse cerebellum: analysis using the weaver mutant. J Neurochem. 1983;41:491–505. doi: 10.1111/j.1471-4159.1983.tb04767.x. [DOI] [PubMed] [Google Scholar]
- 23.Seyfried TN, Yu RK. Cerebellar ganglioside abnormalities in pcd mutant mice. Journal of neuroscience research. 1990;26:105–111. doi: 10.1002/jnr.490260113. [DOI] [PubMed] [Google Scholar]
- 24.Seyfried TN, Yu RK, Miyazawa N. Differential cellular enrichment of gangliosides in the mouse cerebellum: Analysis using neurological mutants. J. Neurochem. 1982;38:551–559. doi: 10.1111/j.1471-4159.1982.tb08662.x. [DOI] [PubMed] [Google Scholar]
- 25.Seyfried TN, Bernard D, Mayeda F, et al. Genetic analysis of cerebellar lipids in mice susceptible to audiogenic seizures. Exp. Neurol. 1984;84:590–595. doi: 10.1016/0014-4886(84)90206-1. [DOI] [PubMed] [Google Scholar]
- 26.Seyfried TN, Yu RK. Heterosis for brain myelin content in mice. Biochem. Genet. 1980;18:1229–1238. doi: 10.1007/BF00484350. [DOI] [PubMed] [Google Scholar]
- 27.Kiebish MA, Han X, Cheng H, et al. Lipidomic analysis and electron transport chain activities in C57BL/6J mouse brain mitochondria. J Neurochem. 2008;106:299–312. doi: 10.1111/j.1471-4159.2008.05383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seyfried TN, Glaser GH, Yu RK. Developmental analysis of regional brain growth and audiogenic seizures in mice. Genetics. 1978;88:S90. [Google Scholar]
- 29.Kasperzyk JL, El-Abbadi MM, Hauser EC, et al. N-butyldeoxygalactonojirimycin reduces neonatal brain ganglioside content in a mouse model of GM1 gangliosidosis. J Neurochem. 2004;89:645–653. doi: 10.1046/j.1471-4159.2004.02381.x. [DOI] [PubMed] [Google Scholar]
- 30.Baek RC, Kasperzyk JL, Platt FM, et al. N-butyldeoxygalactonojirimycin reduces brain ganglioside and GM2 content in neonatal Sandhoff disease mice. Neurochemistry international. 2008;52:1125–1133. doi: 10.1016/j.neuint.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 31.Macala LJ, Yu RK, Ando S. Analysis of brain lipids by high performance thin-layer chromatography and densitometry. Journal of lipid research. 1983;24:1243–1250. [PubMed] [Google Scholar]
- 32.Kasperzyk JL, d'Azzo A, Platt FM, et al. Substrate reduction reduces gangliosides in postnatal cerebrum-brainstem and cerebellum in GM1 gangliosidosis mice. Journal of lipid research. 2005;46:744–751. doi: 10.1194/jlr.M400411-JLR200. [DOI] [PubMed] [Google Scholar]
- 33.Folch J, Lees M, Sloane-Stanley GH. A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 34.Svennerholm L. Quantitative estimation of sialic acids II. A colorimetric resorcinol-hydrochloric acid method. Biochim. Biophys. Acta. 1957;24:604–611. doi: 10.1016/0006-3002(57)90254-8. [DOI] [PubMed] [Google Scholar]
- 35.Hauser EC, Kasperzyk JL, d'Azzo A, et al. Inheritance of lysosomal acid beta-galactosidase activity and gangliosides in crosses of DBA/2J and knockout mice. Biochemical genetics. 2004;42:241–257. doi: 10.1023/b:bigi.0000034429.55418.71. [DOI] [PubMed] [Google Scholar]
- 36.Ando S, Chang NC, Yu RK. High-performance thin-layer chromatography and densitometric determination of brain ganglioside compositions of several species. Anal. Biochem. 1978;89:437–450. doi: 10.1016/0003-2697(78)90373-1. [DOI] [PubMed] [Google Scholar]
- 37.Seyfried TN, Glaser GH, Yu RK. Cerebral, cerebellar, and brain stem gangliosides in mice susceptible to audiogenic seizures. J. Neurochem. 1978;31:21–27. doi: 10.1111/j.1471-4159.1978.tb12428.x. [DOI] [PubMed] [Google Scholar]
- 38.Sato C, Yu RK. Myelin galactolipid synthesis in different strains of mice. J Neurochem. 1987;49:1069–1074. doi: 10.1111/j.1471-4159.1987.tb09995.x. [DOI] [PubMed] [Google Scholar]
- 39.Morell P, Quarles RH. Myelin formation, structure and biochemistry. In: Siegel GJ, et al., editors. Basic Neurochemistry. Lippincott-Raven; New York: 1999. pp. 69–93. [Google Scholar]
- 40.Brigande JV, Wieraszko A, Albert MD, et al. Biochemical correlates of epilepsy in the E1 mouse: analysis of glial fibrillary acidic protein and gangliosides. J. Neurochem. 1992;58:752–760. doi: 10.1111/j.1471-4159.1992.tb09782.x. [DOI] [PubMed] [Google Scholar]
- 41.Yusuf HKM, Dickerson JWT. Disialoganglioside GD1a of rat brain subcellular particles during development. Biochem. J. 1978;174:655–657. doi: 10.1042/bj1740655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Levine SM, Seyfried TN, Yu RK, et al. Immunocytochemical localization of GD3 ganglioside to astrocytes in murine cerebellar mutants. Brain research. 1986;374:260–269. doi: 10.1016/0006-8993(86)90420-8. [DOI] [PubMed] [Google Scholar]