

Abstract

With increasing efficiency, accuracy, and speed we can access complete genome sequences from thousands of infectious microorganisms; however, the ability to predict antigenic targets of the immune system based on amino acid sequence alone is still needed. Here we use a Leptospira interrogans microarray expressing 91% (3359) of all leptospiral predicted ORFs (3667) and make an empirical accounting of all antibody reactive antigens recognized in sera from naturally infected humans; 191 antigens elicited an IgM or IgG response, representing 5% of the whole proteome. We classified the reactive antigens into 26 annotated COGs (clusters of orthologous groups), 26 JCVI Mainrole annotations, and 11 computationally predicted proteomic features. Altogether, 14 significantly enriched categories were identified, which are associated with immune recognition including mass spectrometry evidence of in vitro expression and in vivo mRNA up-regulation. Together, this group of 14 enriched categories accounts for just 25% of the leptospiral proteome but contains 50% of the immunoreactive antigens. These findings are consistent with our previous studies of other Gram-negative bacteria. This genome-wide approach provides an empirical basis to predict and classify antibody reactive antigens based on structural, physical–chemical, and functional proteomic features and a framework for understanding the breadth and specificity of the immune response to L. interrogans.
Keywords: leptospirosis, protein microarray, enrichment analysis, antibody response
Introduction
Leptospirosis is an emerging zoonotic disease of global importance and is now the leading cause of hemorrhagic disease. Humans are usually infected following contact with the urine of reservoir animals via contaminated soil or water.1−3 While infection is asymptomatic in wild rodents and other reservoirs, in humans and other accidental hosts, it can cause severe clinical disease: hepato-renal failure (Weil’s disease), pulmonary hemorrhage syndrome (LPHS), and even death. LPHS and Weil’s disease have case fatalities of >50% and >10%, respectively.2,4,5 The development of disease complications is thought to depend on infectious dose or host immunity. Protective immunity during naturally acquired infection is associated with a humoral immune response, indicated by increased production of agglutinating antibodies following infection and animal passive transfer studies.6−10
A major hurdle in the development of effective vaccines and serodiagnostic against complex microorganisms that encode thousands of proteins is the identification of a limited number of antigens that will induce a protective immune response. The advent of high-throughput sequencing during the past decade has made possible an approach called “reverse vaccinology”,11 which takes advantage of bioinformatics to narrow down the number of potential vaccine antigen candidates. Some proteomic features possibly associated with antigenicity and vaccine efficacy, such as extracellular location, outer-membrane proteins, signal peptides, and B- and T-cell epitopes, can be computationally predicted based on the amino acid sequence alone, reducing the number of candidates to several hundred. Although bioinformatic approaches can be used to remove groups of antigens from consideration as vaccine and serodiagnostic candidates, it tends to produce large lists of potential candidates that require additional expensive and time-consuming laboratory investigations to further narrow down the candidates. Bioinformatic approaches to antigen prediction are also inherently oversimplistic, and bona fide antigens can be missed.
More recently, whole proteome microarray chips, developed in our lab and by others, have provided an empirical approach to interrogate the entire proteome of any microorganism and to efficiently determine antibody profiles associated with acute or chronic infection or with vaccine mediated protection from infection.12−14 Proteomic studies are a powerful tool for elucidating the molecular mechanisms involved in cellular functioning as well as disease development and progression. Protein microarray chips are particularly effective for the study of human serum samples, defining the antibody immune response against infectious agents on a proteomic scale and enabling the access to the complete antibody repertoire produced during an infection.15,16 Additionally, the protein array usually encompasses thousands of proteins, representing the entire encoded proteome. The individual proteins printed on these chips capture antibodies in serum from infected individuals, which can be quantified using a fluorescent secondary antibody. In this fashion, antibodies produced after infection can be identified, characterizing the type of infection. The objective of proteomic studies is to provide a more complete understanding of the immune response to infection, also allowing the identification of novel serodiagnostic and prognostic markers as well as potential subunit vaccines.14
The mechanism for host recognition remains unclear, and the ability to predict antigenicity in silico is imperfect. Usually only a relatively small subset of the proteome of infectious agents, such as L. interrogans, is recognized by the immune system, and little is known about the characteristics of these antigens.17 Consequently, our group has used proteome microarray data and enrichment analyses to identify proteomic features observed in the immunodominant and serodiagnostic antigen sets for more than 25 medically important agents and has successfully identified antigens that were later employed in a variety of diagnostic platforms.13,15,18,19 Among the organisms studied there are viruses, protozoa, helminthes, and bacteria.12,19−22 Here we have continued this iterative approach, first using a proteome microarray to identify the spectrum of immunoreactive L. interrogans antigens recognized in human leptospirosis cases and then classifying their reactivity according to annotated functional and computationally predicted features. These results inform us about the benefits and limitations of antigen prediction and provide a framework for future studies to improve predictive capability for serodominant antigens.
Material and Methods
Ethics Statement
The institutional review board committees of Yale University and Oswaldo Cruz Foundation approved the study protocol. Samples from infected patients came from the following projects: “Natural History of Urban Leptospirosis” (R01AI052473), “Disease Determinants of Urban Leptospirosis (U01AI088752), and “Ecoepidemiology of Leptospirosis” (R01TW009504). All participants provided written informed consent. After collection, a unique code identifier was assigned to each sample so that all samples were deidentified for researchers before their use.
Human Serum Samples
The study was conducted with a group of 90 laboratory-confirmed leptospirosis patients from the state of Bahia, Brazil, including 30 patients with mild clinical presentations, 31 patients with severe disease, and 30 patients who died due to leptospiral infection. Laboratory confirmation was defined based on the results of the microagglutination test (MAT) and according to the criteria of seroconversion or a four-fold rise in titer in patients with paired serum sample or a single titer of 1:800 in patients with only one serum sample. Sera samples from patients with severe leptospirosis were collected at three different time points and are designated as follows: (i) early acute sample, collected at patient admittance at the health care unit, (ii) late acute sample, collected 2 to 3 days after early acute sample collection, and (iii) convalescent sample, collected at least 14 days after the first sampling. For patients with mild leptospirosis, no late acute sample was collected; that is, only early acute and convalescent samples were provided. An early acute sample was collected from all deceased patients, but only five patients from this group survived through the late acute sampling. Samples were organized in groups, separated by clinical presentation (mild, severe, or deceased) and by time point (early acute, late acute, or convalescent) so that a total of 188 samples were categorized into seven groups.
Leptospira ORF Amplification and High-Throughput Cloning
The complete ORFeome of Leptospira interrogans serovar Copenhageni strain Fiocruz L1-130 was amplified by PCR and cloned into pXI vector using a high-throughput PCR recombination cloning method described elsewhere.20,23 The cloning strategy allows the expression of recombinant proteins containing an N-terminal hemaglutinin (HA) tag and a C-terminal poly histidine (His) tag. Genes larger than 3 kb were cloned as smaller segments as previously described, and the ligA and ligB genes (LIC10465 and LIC10464, respectively) were fragmented according to the repeated Big domains present in the structure of each protein (LigB Repeats 7–12, LigA Repeats 7–13, and LigA/B Repeats 1–6),24 which are recognized by human sera as previously described.20 All PCR product sizes were confirmed by gel electrophoresis before cloning. Recombinant plasmids were confirmed by PCR using the insert specific primers for amplification. After identifying the seroreactive antigens on the microarrays, the inserts in the corresponding plasmids were confirmed by nucleotide sequencing by the Sanger method.
Microarray Production and Probing
Microarray fabrication was performed as previously described.20,23 In brief, purified mini-preparations of DNA were used for expression in a 10 μL E. coli in-vitro-based transcription-translation (IVTT) reaction system (RTS Kit, Roche) for 16 h at 26 °C with shaking (300 rpm) according to the manufacturer’s instructions. Negative control reactions were those performed in the absence of DNA template (“NoDNA” controls). A protease inhibitor mixture (Complete, Roche) and Tween-20 (0.5% v/v final concentration) were added to the reactions to minimize protein degradation and improve protein solubilization. Unpurified supernatants were immediately printed onto nitrocellulose-coated glass FAST slides using an Omni Grid 100 microarray printer (Genomic Solutions) together with multiple negative control reactions and positive control spots of an IgG mix containing mouse, rat, and human IgG and IgM (Jackson Immuno Research).
Protein expression was verified by probing the array with monoclonal antipolyhistidine (Sigma-Aldrich) and antihemaglutinin (Roche Applied Science) diluted 1/400 in Protein Array Blocking Buffer (Whatman), as previously described.20 Probing with human sera samples was performed with samples diluted 1/100 in Protein Array Blocking Buffer (Whatman) supplemented with E. coli lysate 10 mg/mL (McLab) at a final concentration of 10% v/v and incubated 30 min at room temperature with constant mixing. Antibodies bound to E. coli proteins were removed by centrifugation prior to addition to the microarray. Arrays were blocked for 30 min with Protein Array Blocking Buffer and then incubated with diluted samples overnight at 4 °C with gentle rocking. Washes and incubation with conjugate antibodies were performed as previously described.20 Slides were scanned in a PerkinElmer ScanArray confocal laser and intensities were quantified using QuantArray package.
Genome Annotation and Computational Predictions
Enrichment analysis was performed based on Leptospira interrogans serovar Copenhageni L1-130 strain genome annotations available in the National Center for Biotechnology Information (NCBI) and John Craig Venter Institute (JCVI) databases. The antigens were classified according to annotated functional features and computationally predicted features. The clusters of orthologous groups (COG) information and the Mainrole Classification utilized can be found at NCBI and JCVI Web sites. The following programs were utilized for computational prediction: TMHMM v2.0 for trans-membrane domains prediction (http://www.cbs.dtu.dk/services/TMHMM/); SignalP v3.0 for signal peptide prediction (http://www.cbs.dtu.dk/services/SignalP/); and PSORTb v3.0 software for subcellular location prediction (http://www.psort.org/psortb/). Proteins with potentially biological importance, such as mass spectrometry (MS) evidence of in vitro expression25 or in vivo mRNA up-regulation,26 were also submitted to enrichment analysis.
Data Analysis
The reactivity was quantified using QuantArray software. Spot intensity raw data were obtained as the mean pixel signal intensity with automatic correction for spot-specific background. For each array, the average of control IVTT reactions (NoDNA controls) was subtracted from spot signal intensities to minimize background reactivity. Positive expression of proteins was determined by a signal intensity of 2.5 standard deviation (SD) above the mean of NoDNA control reactions for either the His or HA tags. The same cutoff was applied to identify the reactive proteins using the sera collection. Antigens were classified as seroreactive if (i) the average signal intensity of a sera group was above the established cutoff or (ii) at least 33% of the samples within a group showed individual signal intensity above the cutoff. Our rationale for using this second criterion was to include antigens that may not be strongly recognized by all of the patients but are recognized by a significant proportion of them. Enrichment statistical analysis was performed with Fisher’s exact test.12,19,22 When segments of fragmented proteins showed reactivity, enrichment analysis was performed considering the annotation/predictions of the original protein.
Results
Gene Amplification, Cloning, and Protein Expression
We amplified the 3667 predicted ORFs from L. interrogans serovar Copenhageni from genomic DNA and cloned them into the pXi expression vector, as full or partial length proteins, using the high-throughput recombination cloning method developed by our group.23 Cloning efficiency was ∼94%, and all cloned ORFs were expressed under a T7 promoter in the E. coli in vitro transcription/translation system. Microarray probing with anti-His and anti-HA antibodies revealed an expression level of ∼91% of all 3819 proteins and fragments printed on the array (3359 of 3667 total ORFs). A list of the proteins not represented on the microarrays is provided in Table S1 in the Supporting Information.
Overall Antibody Reactivity
We probed leptospiral protein arrays with a collection of 188 serum samples, composed of longitudinal samples from patients with mild and severe clinical presentations of leptospirosis, at different phases of the disease, as well as patients that died from acute leptospirosis infection. Analyzing longitudinal samples increased the likelihood of detecting an antigen with transient seroreactivity. In Figure 1, we show representative histograms of the number of reactive antigens selected from the convalescent time point for the severe patient group (n = 30) using the inclusion criteria (described in the Materials and Methods). For some antigens, the high average signal intensity is due to the strong reactivity of a few patients, as observed by the five orange dots below the orange dotted line in Figure 1B. We also observed that antigens with lower average signal intensity show positive reactivity in a few patients, as observed by the last 17 antigens in Figure 1A.
Figure 1.
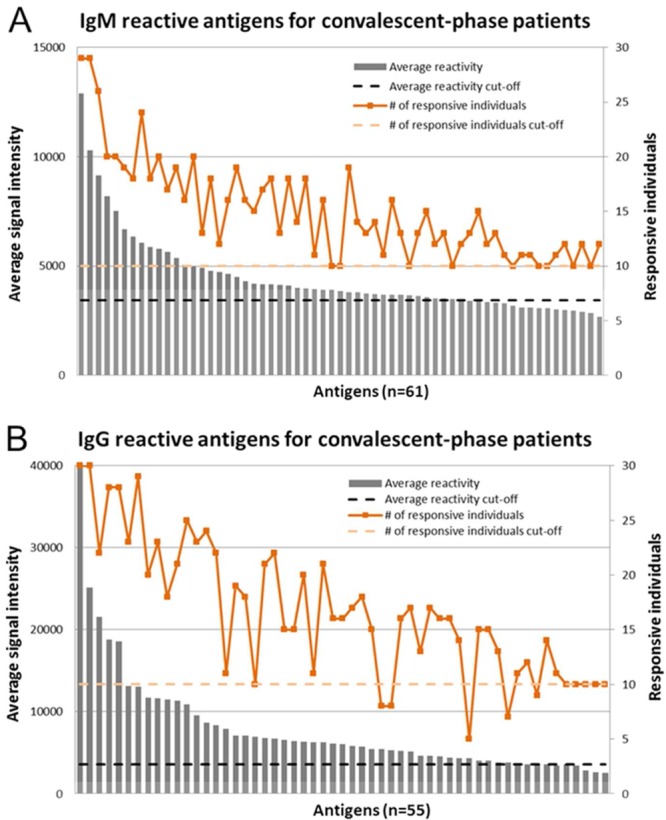
Representative histograms showing the criteria for selecting reactive antigens. An antigen was considered to be reactive if either the group average signal intensity or at least 33% of the samples within a group showed signal intensity above 2.5 standard deviations of the NoDNA control reactions. The histogram plots the average signal intensity (Y axis) and the number of responsive individuals (secondary Y axis) for each reactive antigen selected (X axis) for IgM (A) or IgG (B) probing of convalescent-phase samples from severe patients. Dotted lines correspond to the control reactions cutoff (black) or the minimum number of responsive individuals (orange) included using this criteria.
By applying these criteria to all groups of samples separately, we identified a total of 191 reactive antigens. Of these, 49 were reactive for both IgM and IgG antibodies, 61 were reactive only for IgM and, 81 were only for IgG (Figure 2). In this work, we investigated the proteomic features shared by antigens that are recognized by human sera because these features might increase their likelihood to be seroreactive. The aim is to identify all leptosprial antigens that are reactive in humans, without excluding cross-reactive antigens reactive in healthy unexposed individuals. A complete evaluation of the antibody immune response during naturally acquired leptospirosis compared with healthy controls, along with the analysis of new diagnostic and vaccine candidates, will be the focus of a separate study.
Figure 2.

Heat map showing the overall IgM and IgG reactivity detected for mild, severe, and deceased patients. Reactivity intensity is shown according to the colorized scale. Antigens are shown in rows, grouped as reactive for both IgM and IgG antibodies, or for IgM- or IgG-only antibodies; patients are in columns, organized from left to right by the increasing average reactivity detected for the reactive antigens. Samples from patients with mild leptospirosis are shown in green, patients with severe presentations are in yellow, and deceased patients are in blue.
Functional Enrichment Analysis
L. interrogans serovar Copenhageni proteins are annotated with NCBI Clusters of Orthologous Groups (COG) functional categories (Table S2 in the Supporting Information). Each COG is defined by a group of three or more proteins that are inferred to be orthologs, that is, they are direct evolutionary counterparts and represent a conserved functional category. Of the total 3667 leptospiral proteins, 1313 are not associated with COGs, whereas 500 proteins are associated with COGs but have not been assigned to a specific function, which are shown as general function prediction only (category R) or function unknown (category S). Additionally, some proteins have multiple category assignments. Using these definitions, we determined that there were 2416 COGs represented on the microarray.
Proteins with predicted COGs N (cell motility and secretion) and U (intracellular trafficking and secretion) were significantly enriched for both IgM and IgG reactivity, with significant fold increases of 4.4 (N) and 5.9 (U) for IgM and 3.8 (N) and 5.1 (U) for IgG, respectively (Table 1). We also found that proteins predicted as COG D (cell division and chromosome partitioning) were 4.5 times enriched in IgM responses. Interestingly, we determined that the IgG response to COGs S (unknown function) and V (defense mechanism) was 1.9- and 3.7-fold enriched, respectively, in IgG response, although these enrichments were less significant than COGs N and U. COG E (amino acid transport and metabolism) was lacking for IgG reactivity despite 4% of the L. interrogans ORFeome being classified in this category (Table 1).
Table 1. Enriched and Under-Represented Proteomic Features Identified for IgM or IgG Reactivity.
| IgM |
IgG |
|||
|---|---|---|---|---|
| categories | FoldEnrich | p value | FoldEnrich | p value |
| Enriched | ||||
| COG U - intracellular trafficking and secretiona | 5.9 | 1.49 × 10–7 | 5.1 | 9.85 × 10–7 |
| presence of signal peptidea | 2.3 | 5.00 × 10–7 | 2.4 | 1.92 × 10–9 |
| JCVI - cellular processesa | 3.7 | 4.11 × 10–6 | 3.0 | 1.27 × 10–4 |
| COG N - cell motility and secretiona | 4.4 | 1.12 × 10–5 | 3.8 | 5.67 × 10–5 |
| 1 trans-membrane domaina | 2.0 | 8.67 × 10–5 | 1.7 | 4.44 × 10–07 |
| JCVI - cell envelopea | 2.2 | 4.96 × 10–4 | 2.3 | 4.34 × 10–05 |
| mass spec positivea | 1.4 | 1.19 × 10–3 | 1.3 | 8.63 × 10–3 |
| outer membranea | 3.2 | 9.97 × 10–3 | 4.1 | 2.61 × 10–4 |
| up-regulated mRNA in vivoa | 2.5 | 1.48 × 10–2 | 2.9 | 1.25 × 10–3 |
| JCVI - protein fateb | 2.7 | 2.40 × 10–3 | 1.9 | 5.76 × 10–2 |
| COG D - cell division and chromosome partitioning | 4.5 | 2.67 × 10–2 | 1.3 | 5.46 × 10–1 |
| extracellularb | 1.6 | 4.37 × 10–1 | 2.7 | 2.22 × 10–2 |
| COG S - function unknownc | 0.8 | 8.32 × 10–1 | 1.9 | 2.64 × 10–2 |
| COG V - defense mechanismsc | 0.9 | 1.00 × 100 | 3.7 | 9.85 × 10–3 |
| Under-Represented | ||||
| lack of signal peptidea | 0.8 | 5.00 × 10–7 | 0.8 | 1.92 × 10–9 |
| 0 trans-membrane domaina | 0.8 | 8.21 × 10–5 | 0.4 | 1.50 × 10–9 |
| cytoplasmica | 0.8 | 1.94 × 10–2 | 0.6 | 1.31 × 10–5 |
| JCVI - energy metabolismb | 0.3 | 4.56 × 10–2 | 0.4 | 6.28 × 10–2 |
| COG E - amino acid transport and metabolismc | 0.2 | 8.94 × 10–2 | 0 | 6.17 × 10–3 |
| >1 trans-membrane domainsc | 1.2 | 4.08 × 10–1 | 0.3 | 1.47 × 10–2 |
Significant features for both IgM and IgG antibodies.
Significant features for IgM.
Significant features for IgG.
Leptospiral proteins are also annotated with JCVI Mainrole Classification functional categories (Table S3 in the Supporting Information). Each Mainrole category can be divided into specific subrole categories. A total of 305 leptospiral proteins are not classified in any category, whereas 1257 proteins are assigned to a category but have no specific function prediction, shown as unknown function or hypothetical protein. Because some proteins have multiple functional assignments, we identified 3212 proteins predicted with JCVI Mainrole Classification in total for the 3667 proteins represented on the chip.
Similar to the COG categories enriched in patient sera, we found that proteins predicted as Cell Envelope and Cellular Processes were significantly enriched for both IgM and IgG reactivity, with fold increases of 2.2 and 3.7 for IgM and 2.3 and 3.0 for IgG, respectively (Table 1). We observed that antigens classified in the Protein Fate category in IgM responses, which is related to protein folding, stabilization, and degradation, were enriched 2.7-fold. Similarly to the COGs classifications, we found that proteins categorized as enzymes involved in Energy Metabolism were under-represented in the IgM response, with a fold decrease of 0.3 (Table 1). Interestingly, the JCVI class of proteins that comprised most of the reactive antigens was the Hypothetical Proteins category, with 23 and 29% of IgM- and IgG-reactive antigens, respectively.
We also analyzed the enrichment of antigens using computationally predicted features to identify motifs that are enriched in the humoral immune response to Leptospira infection (Tables S4–S6 in the Supporting Information). As shown in Table 1, we found that proteins containing 1 transmembrane domain were highly significantly enriched for both IgM and IgG antibodies reactivity, with fold increases of 2.0 and 1.7, respectively. Indeed, we observed that proteins lacking transmembrane domains were an under-represented feature in both responses, with fold decreases of 0.8 (IgM) and 0.4 (IgG). Interestingly, we observed that proteins with more than one predicted transmembrane domain were under-represented in IgG responses (0.3 fold change). Conversely, we determined that proteins with predicted signal peptides were significantly enriched in both IgM and IgG responses, with fold increases of 2.3 and 2.4, respectively. Accordingly, outer membrane proteins were also significantly enriched in both responses (3.2 and 4.1 fold increase), and proteins predicted as cytoplasmic localization were significantly under-represented (0.8 and 0.6 fold change). Finally, prediction of subcellular localization as extracellular was enriched for IgG reactivity, with a fold increase of 2.7. Although the majority of the antibody reactive antigens lack both a signal peptide and a transmembrane domain or are classified as cytoplasmic by pSort, these groups account for a larger proportion of all of the computationally classified proteins in the L. interrogans proteome. Consequently, they are under-represented by the enrichment analysis.
Antibody Reactivity versus Previous in Vitro and in Vivo Studies
Another potential characteristic that could influence protein antigenicity is the in vivo expression level because leptospiral proteins expressed at higher levels have a higher probability of being recognized by the host immune system. Therefore, we interfaced our seroreactive data with two existing data sets: (i) MS data obtained from in vitro cultured bacteria and (ii) a transcriptome study that identified differentially expressed genes when the bacteria was grown in vivo in dialysis membrane chambers (DMCs) implanted in rats. More than 50% (102/191) of the seroreactive antigens identified in our study were identified in vitro by MS (Figure 3A), making this method an enriching feature for both IgM and IgG seroreactivity (Table 1). The up-regulated leptospiral genes identified in vivo were also enriched among the seroreactive antigens, with a fold change of 2.5 and 2.9 for IgM and IgG, respectively (Table 1).
Figure 3.

Correlation between the array seroreactivity and previously published studies. (A) Venn diagram of the leptospiral proteins identified by mass spectrometry when the bacteria were grown in vitro (red); proteins for which the corresponding mRNA was up-regulated when the bacteria were cultivated in dialysis membrane chambers in vivo (green); and antibody reactivity in leptospirosis patients detected by protein microarray (blue). (B) Scatter plots with the antibody reactivity and number of protein copies/cell detected by mass spectrometry. Array average signal intensity for IgM or IgG probing is plotted on the X axis; the number of protein copies per cell detected in vitro by mass spectrometry is shown on the Y axis.
Even though we observed a significant overlap between the number of seroreactive antigens identified in this study and the number of proteins detected in vitro by MS, there was no linear correlation between the number of protein copies per bacterium cell and the intensity of antibody reactivity detected on the protein arrays (Figure 3B). Few proteins had both high cellular concentrations and showed strong antibody response, as exemplified by LIC12966, LIC13050, LIC10191, and the Lig proteins (LIC10464 and LIC10465) in Figure 3B. Notably, >60% (67/108) of the up-regulated genes in vivo were not detected by MS under regular cultivation conditions, and only 15/108 up-regulated genes were also serodominant.
Discussion
A major component of the adaptive immune response to infection is the generation of protective and long-lasting humoral immunity, but factors governing selection of the antigens recognized by the immune system are largely unknown.17,19,27,28 It is not uncommon for viruses encoding a small number of proteins to elicit antibody responses against each structural protein. In contrast, for infectious bacteria or parasites that encode hundreds or thousands of proteins, only a subset of the proteome is recognized by the humoral immune response.14,16,19,27,29,28 Here we describe utilization of a Leptospira interrogans proteome microarray to determine empirically the entire antibody repertoire of infected individuals. This technique allows the identification of the types of proteomic structural, physical–chemical, and functional features that are recognized more frequently by the human immune system. We previously identified a set of serodiagnostic antigens from a partial L. interrogans proteome.20 An important difference between the present study and this previous report is that analysis of the complete proteome not only has allowed us to delineate additional serodiagnostic antigens but, more fundamentally, also has provided the basis for a rigorous, comprehensive, and quantitative determination of the protein characteristics of the entire set of the serodominant antigens on a genomic scale.
Our results represent a large-scale evaluation of L. interrogans proteins that are antigenic in the context of naturally acquired human infection. Our group has previously shown that >90% of the urban leptospirosis cases in Brazil are caused by L. interrogans serovar Copenhageni and has isolated strain L1-130 from an infected patient from the city of Salvador, Brazil.30,31 The homogeneity of pathogen exposure and the use of a bacterial strain isolated from our study site minimize possible errors due to the natural diversity of Leptospira strains. Additionally, the availability of strain L1-130 complete genome sequence makes it ideal for this proteomic study.
We identified 191 immunodominant protein antigens with either IgM or IgG recognition, which correspond to ∼6% of the leptospiral coding genome. This relatively small set of antigens represents the complete repertoire of antibodies that are generated during symptomatic leptospiral infections. Mounting an immune response against a limited set of antigens may have the advantage of minimizing energy consumption and avoiding an excessive innate inflammatory reaction or cross-reactive autoimmune responses while still controlling infection effectively.
We classified the annotated functional proteomic features that are associated with antigenicity in humans. Enrichment analysis found 14 protein features that were enriched in the seroreactive antigens, of which 9 had both IgM and IgG responses (Table 1). Interestingly, we identified features that were significantly enriched exclusively in IgM or in IgG responses. Accounting for these differences in enrichment categories between IgG and IgM antigen targets may be related to class switching from IgM to IgG and affinity maturation of the IgG. The most significant enriched or under-represented features, however, were the ones detected for both IgM and IgG antibodies.
Similar studies from our group with Brucella melitensis and Burkholderia pseudomallei corroborate the enriching features identified for Leptospira (Table 1).29,19 The most significant positive predictive features identified in this study, a signal peptide, 1 transmembrane domain, COG U (intracellular trafficking and secretion), and COG N (cell motility and secretion), were also enriched for B. melitensis. Accordingly, artemis-defined surface proteins (outer membrane, inner membrane, secreted, and surface structures) were enriched for B. pseudomallei. The enriched NCBI COG categories N and U are both related to protein secretion, and the externalization of such proteins can easily explain their accessibility to the host immune system. The presence of a signal peptide is a cellular tag for secretion or anchoring a protein on membranes, as we also found that proteins with one or more transmembrane domains, as well as outer-membrane subcellular location, were enriched. Cytoplasmic proteins were significantly under represented for all three pathogens. These studies demonstrate that antigens are not randomly recognized by the host immune response. Rather, the immune system focuses on specific types of antigens. Armed with this information and the increasing availability of genome sequences, we can begin to make generalizations to make a priori predictions about immunoreactivity of antigens based on amino acid sequence data alone.32
Sequence analysis and bioinformatics can be used to increase the odds of predicting immunoreactivity, but each has limitations that can be addressed by proteomic studies. For example, many reactive antigens cannot yet be predicted using bioinformatic methods because they do not contain features of an enrichment category, decreasing prediction sensitivity. In this study, we observed that 25% percent of the proteome is represented by enrichment categories, but only 12% of these proteins are seroreactive antigens. Furthermore, 50% of the reactive antigens identified here do not fall into any enrichment category and therefore cannot be predicted using this method. Use of the proteome microarray therefore allows the identification of a broader spectrum of reactive antigens that cannot be predicted by current methods; by selecting 25% of the proteome based on enriched categories, we would identify ∼50% of the immunoreactive antigens.
Proteomics is a powerful method for identifying seroreactive antigens but does have several limitations. For studies of this kind, we take advantage of bioinformatics tools to characterize the complete ORFeome of L. interrogans regarding proteomic features that help predict protein antigenicity in humans. Our findings are dependent on computational predictions and genome annotations. Even though different versions of the signal peptide predictor used here (SignalP v3 and v4) led to the same conclusions about enrichment (data not shown), we recognize that the use of different bioinformatics tools or possible errors and biases in the algorithms used can significantly alter the enrichment analysis. It is also possible that incorrect folding of the proteins printed on the microarrays may interfere with antigen–antibody recognition, either positively or negatively, and the accurate identification of immune-reactive proteins. Moreover, 9% of the leptospiral ORFeome were not represented on the microarrays, and we cannot affirm that those proteins are not seroreactive.
Our results correlate antigenicity with in vivo and in vitro expression of individual proteins allowing comparison of microarray data with data from previous protein expression studies. We found that >60% of the antigenic proteins were also detected by MS, indicating a large overlap in proteins expressed during both infection and in vitro growth. However, we identified no correlation between protein cellular concentration and seroreactivity intensity. Only 102 of the 1574 proteins identified by MS were found to be antigenic, suggesting that antigenicity involves structural features in addition to protein abundance. This discrepancy may be partially explained by differences between the level and identity of proteins in vitro and in vivo in the human host.
A similar argument can be made for the 93/108 differentially expressed leptospiral genes identified during cultivation in DMCs in rats that were not seroreactive on the array. For example, they identified eight up-regulated genes associated with virulence, of which four were identified as seroreactive in our study: LIC12631 and LIC12632 (hemolysins), LIC10465 (LigA), and LIC11219 (peroxiredoxin). Additionally, we identified 18 antigens associated with secretion (COG U) that could be involved in virulence. Of these, only LIC10053 was up-regulated in Leptospira in the rat reservoir. The profile of up-regulated leptospiral proteins may differ significantly between chronic carriage in an animal reservoir and the human host because of different host environments and immune responses. This lack of correlation between protein concentration and antigenicity supports the observation that up-regulated genes are not necessarily antigenic and that not all virulence factors elicit an antibody response.
Seroreactive antigens were selected based on a limited sera collection that comprised leptospirosis patients with different clinical outcomes from the city of Salvador in Bahia, Brazil. Expanding this collection or studying individuals from different areas, infected with different species or serovars, may provide an expanded set of reactive antigens. This study of specimens derived from one clinical site in Salvador involving exposure to one Leptospira interrogans strain has identified a reference set of antigens that are important in the humoral response to Leptospira infection in humans, including hypothetical proteins that would be missed by other bioinformatics approaches. Future studies comparing humoral responses among disease outcomes and against healthy, uninfected controls may identify prognostic markers and promising subunit vaccine candidates.
Acknowledgments
We acknowledge the institutions involved in this study and the Molecular and Cellular Biology Postgraduation Program of Oswaldo Cruz Institute, Fiocruz. Funding support: R01AI052473 - Natural History of Leptospirosis, R01TW009504 - Ecoepidemiology of Leptospirosis, U01AI088752 - Disease Determinants of Urban Leptospirosis.
Supporting Information Available
Table S1. Proteins not represented on the proteome array. Table S2. COGs enrichment table. Table S3. JCVI main role enrichment table. Table S4. Trans-membrane domain enrichment table. Table S5. Signal peptide enrichment table. Table S6. Subcellular localization enrichment table. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): P.L.F. receives income from Antigen Discovery, Inc., which is developing products related to the research described in this paper. The terms of this arrangement have been reviewed and approved by the University of California, Irvine in accordance with its conflict of interest policies.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Levett P. N. Leptospirosis. Clin. Microbiol. Rev. 2001, 142296–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride A. J.; Athanazio D. A.; Reis M. G.; Ko A. I. Leptospirosis. Curr. Opin. Infect. Dis. 2005, 185376–386. [DOI] [PubMed] [Google Scholar]
- Ko A. I.; Goarant C.; Picardeau M. Leptospira: the dawn of the molecular genetics era for an emerging zoonotic pathogen. Nat. Rev. Microbiol 2009, 710736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouveia E. L.; Metcalfe J.; de Carvalho A. L.; Aires T. S.; Villasboas-Bisneto J. C.; Queirroz A.; Santos A. C.; Salgado K.; Reis M. G.; Ko A. I. Leptospirosis-associated severe pulmonary hemorrhagic syndrome, Salvador, Brazil. Emerging Infect. Dis. 2008, 143505–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marotto P. C.; Nascimento C. M.; Eluf-Neto J.; Marotto M. S.; Andrade L.; Sztajnbok J.; Seguro A. C. Acute lung injury in leptospirosis: clinical and laboratory features, outcome, and factors associated with mortality. Clin. Infect. Dis. 1999, 2961561–1563. [DOI] [PubMed] [Google Scholar]
- Fraga T. R.; Barbosa A. S.; Isaac L. Leptospirosis: aspects of innate immunity, immunopathogenesis and immune evasion from the complement system. Scand. J. Immunol. 2011, 735408–419. [DOI] [PubMed] [Google Scholar]
- Maneewatch S.; Sakolvaree Y.; Saengjaruk P.; Srimanote P.; Tapchaisri P.; Tongtawe P.; Klaysing B.; Wongratanacheewin S.; Chongsa-Nguan M.; Chaicumpa W. Monoclonal antibodies to LipL32 protect against heterologous Leptospira spp. challenge. Hybridoma 2008, 276453–465. [DOI] [PubMed] [Google Scholar]
- Chapman A. J.; Everard C. O.; Faine S.; Adler B. Antigens recognized by the human immune response to severe leptospirosis in Barbados. Epidemiol. Infect. 1991, 1071143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi H. Etiology of Yellow Fever: X. Comparative Immunological Studies on Leptospira Icteroides and Leptospira IcterohaemoorrhagiaE. J. Exp. Med. 1920, 312135–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler B.; Faine S.; Muller H. K.; Green D. E. Maturation of humoral immune response determines the susceptibility of guinea-pigs to leptospirosis. Pathology 1980, 124529–538. [DOI] [PubMed] [Google Scholar]
- Pizza M.; Scarlato V.; Masignani V.; Giuliani M. M.; Aricò B.; Comanducci M.; Jennings G. T.; Baldi L.; Bartolini E.; Capecchi B.; Galeotti C. L.; Luzzi E.; Manetti R.; Marchetti E.; Mora M.; Nuti S.; Ratti G.; Santini L.; Savino S.; Scarselli M.; Storni E.; Zuo P.; Broeker M.; Hundt E.; Knapp B.; Blair E.; Mason T.; Tettelin H.; Hood D. W.; Jeffries A. C.; Saunders N. J.; Granoff D. M.; Venter J. C.; Moxon E. R.; Grandi G.; Rappuoli R. Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science 2000, 28754591816–1820. [DOI] [PubMed] [Google Scholar]
- Vigil A.; Ortega R.; Nakajima-Sasaki R.; Pablo J.; Molina D. M.; Chao C. C.; Chen H. W.; Ching W. M.; Felgner P. L. Genome-wide profiling of humoral immune response to Coxiella burnetii infection by protein microarray. Proteomics 2010, 10122259–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X.; Chun S.; Pablo J.; Felgner P.; Liang X.; Davies D. H. Failure of the smallpox vaccine to develop a skin lesion in vaccinia virus-naïve individuals is related to differences in antibody profiles before vaccination, not after. Clin. Vaccine Immunol. 2012, 193418–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigil A.; Chen C.; Jain A.; Nakajima-Sasaki R.; Jasinskas A.; Pablo J.; Hendrix L. R.; Samuel J. E.; Felgner P. L. Profiling the humoral immune response of acute and chronic Q fever by protein microarray. Mol. Cell. Proteomics 2011, 1010M110.006304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trieu A.; Kayala M. A.; Burk C.; Molina D. M.; Freilich D. A.; Richie T. L.; Baldi P.; Felgner P. L.; Doolan D. L. Sterile protective immunity to malaria is associated with a panel of novel P. falciparum antigens. Mol. Cell. Proteomics 2011, 109M111.007948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyles J. E.; Unal B.; Hartley M. G.; Newstead S. L.; Flick-Smith H.; Prior J. L.; Oyston P. C.; Randall A.; Mu Y.; Hirst S.; Molina D. M.; Davies D. H.; Milne T.; Griffin K. F.; Baldi P.; Titball R. W.; Felgner P. L. Immunodominant Francisella tularensis antigens identified using proteome microarray. Proteomics 2007, 7132172–2183. [DOI] [PubMed] [Google Scholar]
- Donati C.; Rappuoli R. Reverse vaccinology in the 21st century: improvements over the original design. Ann. N.Y. Acad. Sci. 2013, 1285, 115–132. [DOI] [PubMed] [Google Scholar]
- Barry A. E.; Trieu A.; Fowkes F. J.; Pablo J.; Kalantari-Dehaghi M.; Jasinskas A.; Tan X.; Kayala M. A.; Tavul L.; Siba P. M.; Day K. P.; Baldi P.; Felgner P. L.; Doolan D. L. The stability and complexity of antibody responses to the major surface antigen of Plasmodium falciparum are associated with age in a malaria endemic area. Mol. Cell. Proteomics 2011, 1011M111.008326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L.; Tan X.; Juarez S.; Villaverde H.; Pablo J.; Nakajima-Sasaki R.; Gotuzzo E.; Saito M.; Hermanson G.; Molina D.; Felgner S.; Morrow W. J.; Liang X.; Gilman R. H.; Davies D. H.; Tsolis R. M.; Vinetz J. M.; Felgner P. L. Systems biology approach predicts antibody signature associated with Brucella melitensis infection in humans. J. Proteome Res. 2011, 10104813–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessa-Aquino C.; Borges Rodrigues C.; Pablo J.; Sasaki R.; Jasinskas A.; Liang L.; Wunder E. A.; Ribeiro G. S.; Vigil A.; Galler R.; Molina D.; Liang X.; Reis M. G.; Ko A. I.; Medeiros M. A.; Felgner P. L. Identification of seroreactive proteins of Leptospira interrogans serovar copenhageni using a high-density protein microarray approach. PLoS Neglected Trop. Dis. 2013, 710e2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinne M.; Matsunaga J.; Haake D. A. Leptospiral outer membrane protein microarray, a novel approach to identification of host ligand-binding proteins. J. Bacteriol. 2012, 194226074–6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L.; Felgner P. L. Predicting antigenicity of proteins in a bacterial proteome; a protein microarray and naïve Bayes classification approach. Chem. Biodiversity 2012, 95977–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies D. H.; Liang X.; Hernandez J. E.; Randall A.; Hirst S.; Mu Y.; Romero K. M.; Nguyen T. T.; Kalantari-Dehaghi M.; Crotty S.; Baldi P.; Villarreal L. P.; Felgner P. L. Profiling the humoral immune response to infection by using proteome microarrays: high-throughput vaccine and diagnostic antigen discovery. Proc. Natl. Acad. Sci. U. S. A. 2005, 1023547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga J.; Barocchi M. A.; Croda J.; Young T. A.; Sanchez Y.; Siqueira I.; Bolin C. A.; Reis M. G.; Riley L. W.; Haake D. A.; Ko A. I. Pathogenic Leptospira species express surface-exposed proteins belonging to the bacterial immunoglobulin superfamily. Mol. Microbiol. 2003, 494929–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmström J.; Beck M.; Schmidt A.; Lange V.; Deutsch E. W.; Aebersold R. Proteome-wide cellular protein concentrations of the human pathogen Leptospira interrogans. Nature 2009, 4607256762–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caimano M. J.; Sivasankaran S. K.; Allard A.; Hurley D.; Hokamp K.; Grassmann A. A.; Hinton J. C.; Nally J. E. A model system for studying the transcriptomic and physiological changes associated with mammalian host-adaptation by Leptospira interrogans serovar Copenhageni. PLoS Pathog. 2014, 103e1004004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigil A.; Ortega R.; Jain A.; Nakajima-Sasaki R.; Tan X.; Chomel B. B.; Kasten R. W.; Koehler J. E.; Felgner P. L. Identification of the feline humoral immune response to Bartonella henselae infection by protein microarray. PLoS One 2010, 57e11447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour A. G.; Jasinskas A.; Kayala M. A.; Davies D. H.; Steere A. C.; Baldi P.; Felgner P. L. A genome-wide proteome array reveals a limited set of immunogens in natural infections of humans and white-footed mice with Borrelia burgdorferi. Infect. Immun. 2008, 7683374–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felgner P. L.; Kayala M. A.; Vigil A.; Burk C.; Nakajima-Sasaki R.; Pablo J.; Molina D. M.; Hirst S.; Chew J. S.; Wang D.; Tan G.; Duffield M.; Yang R.; Neel J.; Chantratita N.; Bancroft G.; Lertmemongkolchai G.; Davies D. H.; Baldi P.; Peacock S.; Titball R. W. A Burkholderia pseudomallei protein microarray reveals serodiagnostic and cross-reactive antigens. Proc. Natl. Acad. Sci. U. S. A. 2009, 1063213499–13504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko A. I.; Galvão Reis M.; Ribeiro Dourado C. M.; Johnson W. D.; Riley L. W. Urban epidemic of severe leptospirosis in Brazil. Salvador Leptospirosis Study Group. Lancet 1999, 3549181820–825. [DOI] [PubMed] [Google Scholar]
- de Faria M. T.; Calderwood M. S.; Athanazio D. A.; McBride A. J.; Hartskeerl R. A.; Pereira M. M.; Ko A. I.; Reis M. G. Carriage of Leptospira interrogans among domestic rats from an urban setting highly endemic for leptospirosis in Brazil. Acta Trop 2008, 10811–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnan C. N.; Zeller M.; Kayala M. A.; Vigil A.; Randall A.; Felgner P. L.; Baldi P. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics 2010, 26232936–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.