

Abstract
We report the syntheses and electrochemical properties of nine new clusters ([LLnMnIV3O4(OAc)3(DMF)n]+ (Ln = La3+, Ce3+, Nd3+, Eu3+, Gd3+, Tb3+, Dy3+, Yb3+, and Lu3+, n = 2 or 3)) supported by a ligand (L3–) based on a 1,3,5-triarylbenzene motif appended with alkoxide and pyridine donors. All complexes were obtained by metal substitution of Ca2+ with lanthanides upon treatment of previously reported LMn3CaO4(OAc)3(THF) with Ln(OTf)3. Structural characterization confirmed that the clusters contain the [LnMn3O4] cubane motif. The effect of the redox-inactive centers on the electronic properties of the Mn3O4 cores was investigated by cyclic voltammetry. A linear correlation between the redox potential of the cluster and the ionic radii or pKa of the lanthanide metal ion was observed. Chemical reduction of the LMnIV3GdO4(OAc)3(DMF)2 cluster with decamethylferrocene, resulted in the formation of LGdMnIV2MnIIIO4(OAc)3(DMF)2, a rare example of mixed-valence [MMn3O4] cubane. The lanthanide-coordinated ligands can be substituted with other donors, including water, the biological substrate.
Short abstract
Mixed-oxide cubane clusters of manganese and lanthanides, [LnMnIV3O4], were synthesized as models of the oxygen evolving complex (OEC) of photosystem II. The redox potentials of these clusters show linear dependence with the lanthanide Lewis acidity, measured as radius of the ion or the pKa of the metal aquo complex. Rare examples of mixed-valent [GdMnIV2MnIIIO4] cluster and water coordination to the lanthanide in the [DyMnIV3O4] cubane provide models of the various states of the OEC.
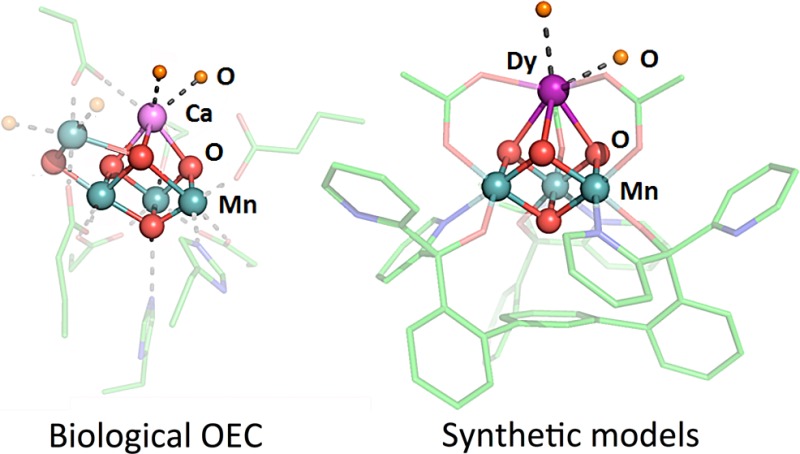
Biological water oxidation occurs in photosystem II (PSII) at the oxygen-evolving complex (OEC), an inorganic cluster displaying a Mn4CaOn moiety.1 Single crystal X-ray diffraction (XRD) studies have revealed that the OEC consists of a distorted Mn3CaO4 cubane bridged to the fourth Mn via oxide moieties.2 During catalysis, the OEC undergoes four electron transfer events, leading to a high oxidation state Mn cluster capable of promoting O–O bond formation and release of O2.3 Although its role remains under debate, the Ca2+ ion is instrumental for catalysis.4 Ca2+ has been proposed to affect the binding and activation of a water substrate as well as the proton coupled electron transfer and the redox properties of the cluster.5 More broadly, metals that are typically redox inactive at biologically relevant potentials affect the chemistry of a variety of redox processes,6 particularly involving transition metals, including dioxygen and peroxide activation,7 O- and H atom transfer,8 alkane oxidation,9 electron transfer rates, and reduction potentials.10 Crystallographic characterization of metal-oxo species involved in this chemistry that display both the redox inactive and active metals is rare.7a,10d Toward addressing the role of Ca2+ in PSII, synthetic models have been targeted,11 with small clusters displaying the biologically relevant CaMn3O4 cubane motif being recently reported.12 To facilitate structure–function studies that interrogate the effect of redox inactive metals in clusters, we have developed synthetic protocols for heterometallic models of the biological system that allow for the incorporation of diverse metals starting from trimetallic precursors and the generation of site-differentiated clusters.13 Studies of structurally related MMn3O4, MMn3O2, MFe3O(OH) clusters revealed a linear dependence of the reduction potential vs the pKa of the corresponding metal aqua ion as a measure of Lewis acidity.13a,13d,13e The significant slopes (70–100 mV/pKa unit) point to a role of the Lewis acid in tuning the cluster potential toward the appropriate level necessary for redox chemistry. Herein, we extend our synthetic, structural, and electrochemical studies to Mn cubanes displaying lanthanides, LnMn3O4, to evaluate how their systematic trends affect the properties of the clusters.
The lanthanides are an appealing choice in investigating systematic effects on the clusters’ physical properties given the monotonic change in radius and chemical properties.14 The preference of the lanthanides primarily for a single oxidation state simplifies the redox states available to the clusters.14 Additionally, lanthanides have been used as substitutes for Ca2+ in biological systems because of the similarities in their ionic radii and high coordination numbers.15
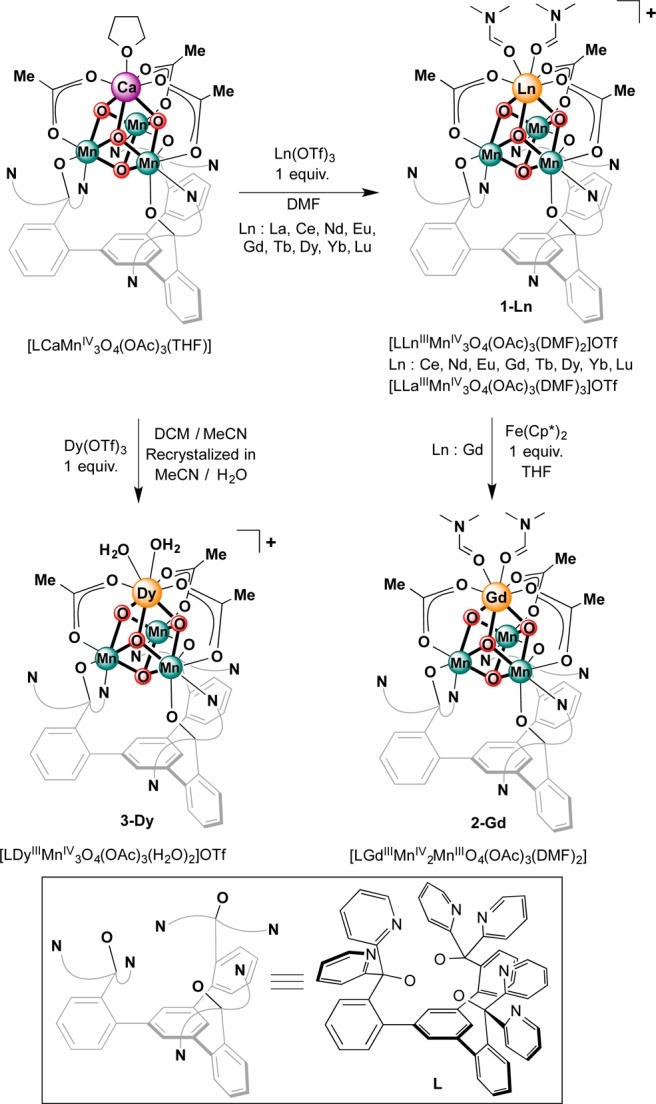
The [LnMn3O4] cubanes were targeted via a synthetic protocol previously employed for Zn2+, Sc3+, and Y3+. More Lewis acidic metals can substitute for Ca2+ in the Mn3CaO4 cubane.13d,13f The heteronuclear [LnMn3O4] complexes were synthesized by treatment of LCaMn3O4(OAc)3(THF) with Ln(OTf)3 (Ln = La3+, Ce3+, Nd3+, Eu3+, Gd3+, Tb3+, Dy3+, Yb3+, Lu3+) in dimethylformamide (DMF) (Scheme 1). The more Lewis acidic lanthanide ions readily substitute Ca2+, as supported by ESI-MS data indicating the incorporation of the respective lanthanide ion and loss of Ca2+. The 1H NMR spectra of the all resulting clusters are broad and paramagnetically shifted. Nevertheless, they show features similar to the starting material, in particular a broad peak upfield of −15 ppm (see the Supporting Information).
Scheme 1. Synthesis of Lanthanide-Mn-Oxide Clusters.
To confirm the formation of the cubane moiety, we performed single-crystal XRD studies for a subset of [LnMn3O4] complexes based on the size of the lanthanide. Clusters displaying large (La3+), intermediate (Gd3+), and small (Yb3+) lanthanides were selected for structural characterization in order to provide insight into the coordination environment as a function of the apical ion size. Dark brown crystals were grown by slow vapor diffusion of Et2O into a CH2Cl2/DMF (2:1 v/v) solution of the metal complexes, over the course of several days. [LYbIIIMnIV3O4(OAc)3(DMF3)](OTf) (1-Yb) and [LGdIIIMnIV3O4(OAc)3(DMF3)](OTf) (1-Gd) have very similar structures (see Figure 1), displaying eight-coordinate lanthanide ions: three bridging oxides, three bridging acetates, and two DMF ligands. In contrast, the larger La3+ ion (1-La) accommodates a ninth ligand in an additional DMF molecule. Structural parameters (Figure 2), reflect the change in average Ln-oxido bond distances from 2.57 Å (La3+) to 2.35 Å (Yb3+) with the size of the lanthanide. Consequently, the average Ln-Mn distance decreases from 3.45 Å (La3+) to 3.23 Å (Yb3+). The Mn-oxido distances remain essentially the same, independent of the nature of Ln.
Figure 1.
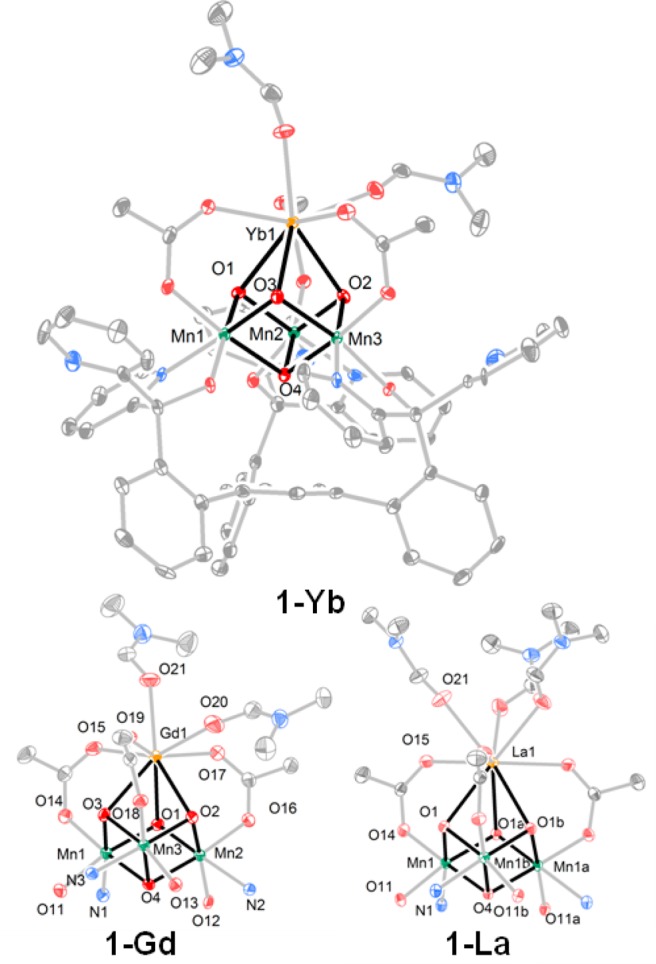
Solid-state structure of 1-Yb and the cubane core structures of 1-Gd and 1-La. Outer sphere solvents and anions and hydrogen atoms are omitted for clarity.
Figure 2.
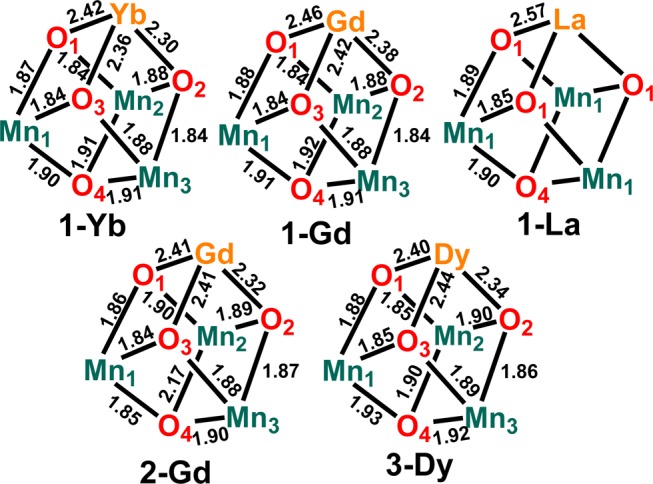
Metal-oxido distances of 1-Ln (Ln = La, Gd, and Yb), 2-Gd, and 3-Dy in Å.
With a series of nine 1-Ln complexes in hand, the effect of changing the nature of the lanthanide on the redox-properties of the clusters was studied by cyclic voltammetry (CV). The CVs of all complexes display a quasireversible wave assigned as the [MMnIV3O4]/[MMnIV2MnIIIO4] couple (Figure 3 and Figures S14–19 in the Supporting Information). The reduction potentials vary within the range of −0.35 (1-Lu) to −0.49 V (1-La) vs the ferrocene/ferrocenium couple (Fc/Fc+) (see Table S3 in the Supporting Information). Besides the quasi-reversible wave at −0.46 V assigned to [MMnIV3O4]/[MMnIV2MnIIIO4], the CV of 1-Ce exhibits an oxidation event at +0.62 V attributed to the oxidation from CeIII to CeIV.16 The two overlapping features for the reduction process at that potential suggest further chemical events following this oxidation.
Figure 3.
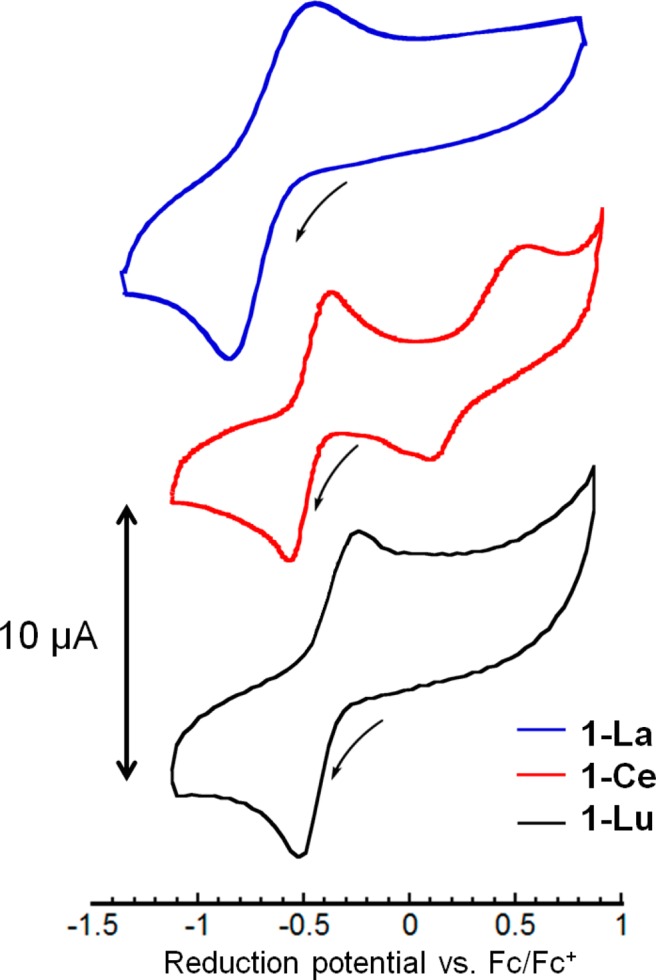
Cyclic voltammograms of 1-La, 1-Ce, and 1-Lu in 0.1 M NBu4PF6 solution in DMA at a scan rate of 100 mV/s. Potentials are referenced to Fc/Fc+.
A plot of the E1/2 values vs the pKa values of the lanthanide aqua ions, M(aqua)3+, revealed a linear correlation (Figure 4).17a We have previously reported similar linear E1/2 vs pKa correlations,13a,13d,13e but with wider variation in the Lewis acidity of the metal and, therefore, over a wider range of pKa and potential values. For the lanthanide series, the pKa variation is relatively narrow leading to a potential range of less than 150 mV. It is notable that even in this small range, the plot of redox potential vs pKa remains linear.18 When plotted with previously reported cubanes, the lanthanide series fits well on the line (Figure 4).
Figure 4.
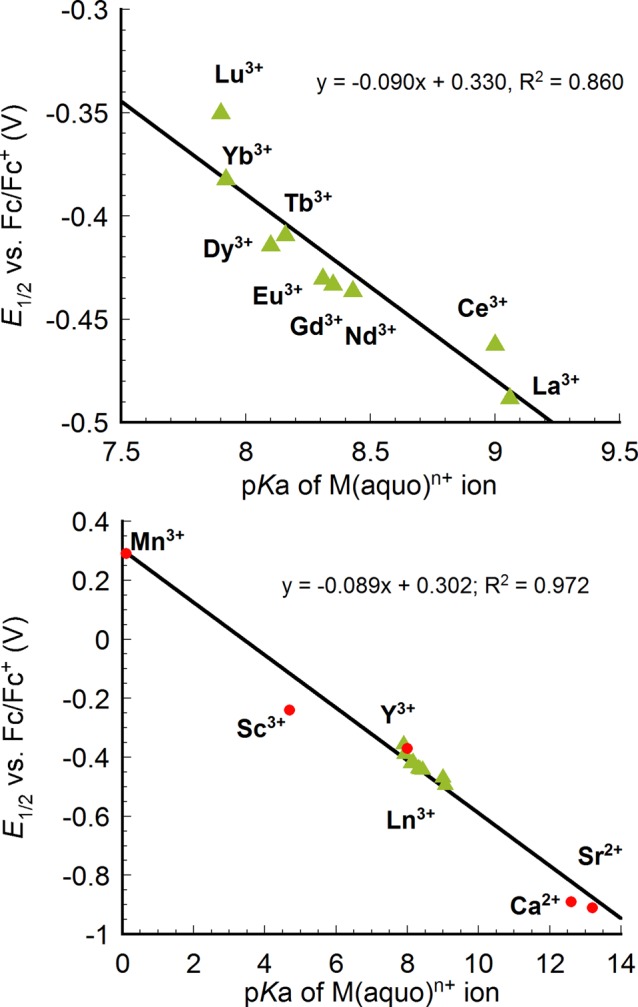
Plot of reduction potentials of [LnMn3O4] (green triangle(top and bottom)) and previously reported [MMn3O4] complexes (red circle(bottom)) vs pKa of the corresponding M(aqua)n+ ion as a measure of Lewis acidity. Potentials were referenced to Fc/Fc+.
With the expanded series of available M3+ cations in the composition of the [MMn3O4] cubanes, the redox potentials were plotted against the radius of M, including Mn3+, Sc3+, Y3+, and Ln3+ (Figure 5).17b Here as well, a linear correlation was observed between the potential and ionic radii of the rare earth ions. As expected, the reduction potential becomes more negative as the ionic radii increases and the Lewis acidity decreases. MnIII is well-removed from the line generated by the rare earth ions. This is likely a consequence of the different bonding character of these metals.14 The rare earth elements display primarily ionic interactions, therefore, the variation in radius is directly correlated to Lewis acidity giving linear dependence of redox potential vs both pKa and radius. MnIII is more covalent, and the change in radius does not track directly with its Lewis acidity. Therefore, the pKa of metal aquo complexes is favored as a measure of Lewis acidity as it incorporates a composite of effects including changes in the nature of bonding, radius, charge, and number of ligands.
Figure 5.
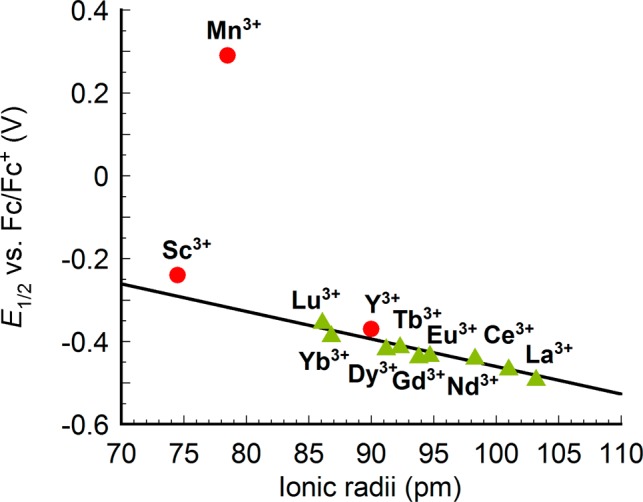
Plot of reduction potentials of [LnIIIMn3O4] complexes (green triangle) and other [MIIIMn3O4] complexes (red circle) vs ionic radii of the corresponding M(aqua)n+ ion.
Access to the above heterometallic clusters allows for further synthetic elaboration. To confirm the nature of the reduced species evidenced in the CV studies, we performed the chemical reduction of 1-Gd with decamethylferrocene. The reduced species, 2-Gd, was studied by XRD. Comparing the solid state structures of 1-Gd and 2-Gd (Figures 1, 6), the site of reduction is localized at Mn2 in 2-Gd. The MnIII center displays an elongation of the O4–Mn2–O16 axis due to population of a σ-antibonding orbital. The bond lengths along this axis (2.169(1) Å for Mn2–O4; 2.163(1) Å for Mn2–O16) are over 0.2 Å longer in comparison to the Mn–oxide bonds in 1-Gd (1.916(2)–1.941(2) Å) as well as the Mn–oxide bonds of Mn1 and Mn3 in 2-Gd (1.8473(9)–1.897(1) Å), which are assigned to MnIV centers. 2-Gd represents a rare example of mixed valent heterometallic cluster structurally related to the OEC. An analogous cluster, [ScMnIV2MnIIIO4], displays a similar localization of charge at Mn centers.13d Oxidation chemistry was also investigated with 1-Ce, by treatment with tris(p-tolyl)ammoniumyl triflate. A new species was generated (1H NMR spectroscopy) assigned as the [CeIVMnIV3] cluster, although attempts to grow single crystals for structural characterization have been unsuccessful to date.
Figure 6.
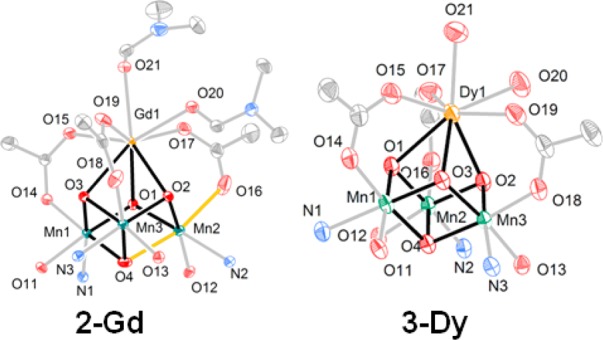
Solid-state structures of 2-Gd and 3-Dy. Only cubane core shown, for clarity. The elongated Mn–O bonds in 2-Gd are highlighted in orange.
In addition to redox chemistry, ligand substitution was investigated with these clusters. In particular, Ca2+-coordinated water ligands have been proposed to be involved in O–O bond formation and are observed in the crystal structure of PSII.2b,5b,5c In this context investigating the coordination of water and other ligands to cubane clusters is of interest. NMR experiments of 1-Gd in the presence of excess DMF show only one set of peaks for the ligand indicating fast exchange between bound and free DMF, that could not be frozen at low temperature (−40 °C) (see Figure S12 in the Supporting Information). Targeting water coordination, the Ca2+/Dy3+ substitution was performed in the absence of DMF. Crystallization in the presence of water afforded 3-Dy (Scheme 1) displaying water molecules bound to Dy3+ (Figure 6). The coordination of two water molecules to the lanthanide ion is reminiscent of the structure of the OEC.2b,13a In addition to modeling substrate binding as in the biological system, the isolation of 3-Dy also highlights the stability of the reported clusters in the presence of water, important for further mechanistic studies.
In summary, mixed metal oxide clusters of Mn and Ln displaying the cubane structural motif relevant to the OEC have been synthesized and studied. Nine complexes spanning the lanthanide series have been prepared in the redox state [LnMnIV3O4] via Ca2+ substitution with Ln3+ from a [CaMnIV3O4] precursor. A linear dependence was observed between the reduction potentials of these complexes and the pKa of the metal aquo, [Ln(H2O)n]3+, or the radius of Ln3+ indicating that the Lewis acidity of the nonmanganese ion tunes the potential of the cluster. A rare one-electron-reduced mixed valent cubane, [GdMnIV2MnIIIO4], was characterized by XRD and showed charge localization displayed by an axial distortion due to population of a σ-antibonding orbital. Reminiscent of binding of water molecules to Ca2+ in the biological system, a cluster displaying two water ligands bound to Dy3+ was structurally characterized and highlights the stability of the present models and their suitability for further mechanistic studies related to PSII.
Experimental Section
General Considerations
Unless otherwise specified, all compounds were manipulated using a glovebox or standard Schlenk line techniques with an N2 atmosphere. Anhydrous tetrahydrofuran (THF) was purchased from Aldrich in 18 L Pure-Pac containers. Anhydrous acetonitrile, benzene, dichloromethane, diethyl ether, and THF were purified by sparging with nitrogen for 15 min and then passing under nitrogen pressure through a column of activated A2 alumina (Zapp’s). Anhydrous N,N-dimethylformamide (DMF) was purchased from Aldrich and stored over molecular sieves. CD2Cl2 was purchased from Cambridge Isotope Laboratories, dried over calcium hydride, then degassed by three freeze–pump–thaw cycles and vacuum-transferred prior to use. 1H NMR spectra were recorded on a Varian 300 MHz instrument, with shifts reported relative to the residual solvent peak. 19F NMR spectra were recorded on a Varian 300 MHz instrument, with shifts reported relative to the internal lock signal. Elemental analyses were performed by Robertson Microlit Laboratories, NJ. All commercial chemicals were used as received. Ln(OTf)3 (Ln= La, Ce, Nd, Eu, Gd, Tb, Dy, Yb, Lu) were purchased from Strem. LMn3CaO4(OAc)3(THF) was prepared according to the previously published procedure.13f
Synthesis of [LLaMn3O4(OAc)3(DMF)3](OTf) (1-La) and [LLnMn3O4(OAc)3(DMF)2](OTf) (Ln: Ce (1-Ce), Nd (1-Nd), Eu (1-Eu), Gd (1-Gd), Tb (1-Tb), Dy (1-Dy), Yb (1-Yb), Lu (1-Lu))
A solution of Ln(OTf)3 (0.02 mmol) in DMF (2 mL) was added to LMn3CaO4(OAc)3(THF) (0.0274g, 0.02 mmol) solution in DMF (1 mL). The dark brown solution was stirred for an hour and then diethyl ether was added to precipitate the red-brown product. The precipitate was collected over Celite and extracted with CH2Cl2. The dark brown CH2Cl2 filtrate was concentrated in vacuo and crystallized from CH2Cl2/DMF/diethyl ether (1:1:5, v/v) to yield the product as red-brown crystals.
1-La: 1H NMR (CD2Cl2, 300 MHz): δ 16.6, 11.7, 11.1, 9.2, 7.9, 5.9, 5.3, 4.7, 3.0 1.2, 0.9, −18.5 ppm. 19F NMR (CD2Cl2, 282 MHz): δ −76.2 ppm. Yield: 88%. Anal. Calcd For C72H68F3LaMn3N9O19S: C, 49.24; H, 3.90; N, 7.18. Found: C, 49.52; H, 3.88; N, 7.03.
1-Nd: 1H NMR (CD2Cl2, 300 MHz): δ 16.3, 11.5, 10.5, 10.1, 9.5, 5.6, 5.3, 3.5, 1.3, 0.9, −19.0 ppm. 19F NMR (CD2Cl2, 282 MHz): δ −77.1 ppm. Yield: 86% Anal. Calcd For C70H62F3Mn3N8NdO18S: C, 49.42; H, 3.67; N, 6.59. Found: C, 49.37; H, 3.80; N, 5.61.
1-Ce: 1H NMR (CD2Cl2, 300 MHz): δ 18.3, 13.5, 13.1, 12.5, 11.5, 10.8, 7.3, 7.2, 3.3, 2.1, −17.0 ppm. 19F NMR (CD2Cl2, 282 MHz): δ −76.4 ppm. Yield: 41%. Anal. Calcd For C73H68CeCl6F3Mn3N8O18S: C, 44.92; H, 3.51; N, 5.74. Found: C, 44.83; H, 3.27; N, 6.12.
1-Eu: 1H NMR (CD2Cl2, 300 MHz): δ 15.9, 12.0, 11.7, 9.0, 6.8, 6.4, 5.4, 4.9, 4.0, 3.5, 2.7, 2.5, 1.7, 1.2, 1.0, 0.90, −20.46 ppm. 19F NMR (CD2Cl2, 282 MHz): δ −74.6 ppm. Yield: 95%. Anal. Calcd For C70H62EuF3Mn3N8O18S: C, 49.19; H, 3.66; N, 6.56. Found: C, 49.24; H, 3.72; N, 6.59.
1-Gd: 1H NMR (CD2Cl2, 300 MHz): δ 16.8, 11.9, 9.3, 6.0, 5.3, 3.0, 1.3, 1.2, −20.8 ppm. 19F NMR (CD2Cl2, 282 MHz): δ −77.2 ppm. Anal. Yield: 98%. Calcd For C70H62F3GdMn3N8O18S: C, 49.04; H, 3.65; N, 6.54. Found: C, 49.13; H, 3.71; N, 6.64.
1-Tb: 1H NMR (CD2Cl2, 300 MHz): δ 27.1, 17.9, 16.7, 15.3, 12.9, 9.0, 7.5, 6.7, 5.5, 3.6, 1.8, 1.4, 0.0, −1.4, −17.7 ppm. 19F NMR (CD2Cl2, 282 MHz): δ −81.2 ppm. Yield: 93%. Anal. Calcd For C70H62F3Mn3N8O18STb: C, 48.99; H, 3.64; N, 6.53. Found: C, 49.13; H, 3.58; N, 6.46.
1-Dy: 1H NMR (CD2Cl2, 300 MHz): δ 36.1, 21.1, 18.2, 17.3, 15.7, 10.4, 8.0, 5.3, 4.6, 3.4, 2.0, 1.3, −4.0, −16.6–42.0 ppm. 19F NMR (CD2Cl2, 282 MHz): δ −90.4 ppm. Yield: 92%. Anal. Calcd For C70H62DyF3Mn3N8O18S: C, 48.89; H, 3.63; N, 6.52. Found: C, 48.84; H, 3.70; N, 6.62.
1-Yb: 1H NMR (CD2Cl2, 300 MHz): δ 16.2, 14.5, 13.6, 9.3, 7.5, 5.6, 4.2, 3.7, 2.6, 1.7, 1.5, 1.2, −24.1 ppm. 19F NMR (CD2Cl2, 282 MHz): δ −78.4 ppm. Yield: 80%. Anal. Calcd For C73H69F3Mn3N9O19SYb: C, 48.62; H, 3.86; N, 6.99. Found: C, 48.49; H, 3.66; N, 7.31.
1-Lu: 1H NMR (CD2Cl2, 300 MHz): δ 14.2, 11.4, 11.0, 8.8, 7.3, 6.7, 5.5, 4.7, 3.7, 2.8, 2.2, 0.5, −23.4 ppm. 19F NMR (CD2Cl2, 282 MHz): δ −76.7 ppm. Yield: 42% Anal. Calcd For C70H62F3LuMn3N8O18S: C, 48.54; H, 3.61; N, 6.47. Found: C, 48.27; H, 3.86; N, 6.64.
Synthesis of 2-Gd
A solution of 1-Gd (0.037 g, 0.02 mmol) in THF (2 mL) was added to a decamethylferrocene (0.007 g, 0.02 mmol) solution in THF (2 mL). The dark brown solution was stirred overnight. A dark-brown precipitate was collected on a fritted glass funnel and washed with MeCN (4 mL) to remove the remaining [Cp*2Fe]+. The filter cake was washed with DCM, then dissolved in benzene/THF (1:1, v/v) and concentrated in vacuo. The resulting dark brown residue was recrystallized from DMF/benzene/diethyl ether (1:3:10, v/v) to yield the product as dark brown crystals. 1H NMR (C6D6, 300 MHz): δ 7.1, 3.5, 3.2,1.6, 1.4, 1.1 ppm. Yield: 49%. Anal. Calcd for C64H50Cl2GdMn3N6O13: C, 51.11; H, 3.35; N, 5.59. Found: C, 50.92; H, 3.30; N, 5.81.
Synthesis of 3-Dy
Performed in air, a solution of Dy(OTf)3 (0.02 mmol) in MeCN (1 mL) was added to [LMn3CaO4(OAc)3(THF)] (0.0274g, 0.02 mmol) in DCM (2 mL). Dark brown solution was stirred magnetically for overnight until the solution became homogeneous. A single-solvent system was tried but the reaction could not be completed because Dy(OTf)3 is poorly soluble in DCM and the CaMn3 cubane is poorly soluble in MeCN. The solution was filtered by Celite and the product was concentrated in vacuo. The product was recrystallized from H2O/MeCN/diethyl ether (1:10:30, v/v) to yield the product as red-brown crystals. 1H NMR (CD2Cl2, 300 MHz): δ 80.9, 44.5, 22.6, 17.6, 14.3, 11.7, 8.3, 6.2, 5.3, 2.0, 1.5, 1.1, 0.3, −18.5 ppm. Yield: 91%. Anal. Calcd for C69H63Cl6DyF3Mn3N9O19S: C, 43.09; H, 3.30; N, 5.10. Found: C, 42.82; H, 2.90; N, 5.17.
Synthesis of [LMn3CeO4(OAc)3(DMF)3](OTf)2
In glovebox, a solution of 1-Ce (75 mg, 0.042 mmol) in DCM (5 mL) was added with a DCM (5 mL) solution of tris(p-tolyl)ammoniumyl triflate (29.4 mg, 0.047 mmol), which was synthesized following procedure in the literature.19 Color of the solution turned from dark brown to dark orange brown. Mixture was stirred magnetically for 10 min and dried in vacuo. Diethyl ether was added to wash the mixture until filtrate cake becoming colorless and solid was collected over Celite. The filter cake was dissolved in DCM (∼5 mL) and the mixture was filtered through Celite. The filtrate was concentrated in vacuo to yield crystalline dark brown product. 1H NMR (CD2Cl2, 300 MHz): δ 11.8, 9.7, 6.0, 4.5, 3.2, 23.2 pm. Yield: 95%. Anal. Calcd For C76H73CeCl4F6Mn3N9O22S2: C, 43.69; H, 3.52; N, 6.03. Found: C, 43.97; H, 3.19; N, 6.30.
Acknowledgments
This work was supported by the California Institute of Technology and the NIH R01 GM102687A (T.A.). T.A. is a Sloan, Dreyfus, and Cottrell fellow. We thank Lawrence M. Henling for assistance with crystallography and Zhiji Han for assistance with the characterization of the Ce complexes. The Bruker KAPPA APEXII X-ray diffractometer was purchased via an NSF Chemistry Research Instrumentation award to Caltech (CHE-0639094).
Supporting Information Available
Experimental procedures and spectroscopic characterization, and crystallographic (CIF) data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a McEvoy J. P.; Brudvig G. W. Chem. Rev. 2006, 106, 4455. [DOI] [PubMed] [Google Scholar]; b Wydrzynski T. J.; Satoh K.. Photosystem II: The Light-Driven Water: Plastoquinone Oxidoreductase; Springer: Dordrecht, The Netherlands, 2005; Vol. 22. [Google Scholar]; c Yano J.; Yachandra V. Chem. Rev. 2014, 114, 4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ferreira K. N.; Iverson T. M.; Maghlaoui K.; Barber J.; Iwata S. Science 2004, 303, 1831. [DOI] [PubMed] [Google Scholar]; b Umena Y.; Kawakami K.; Shen J. R.; Kamiya N. Nature 2011, 473, 55. [DOI] [PubMed] [Google Scholar]
- a Kok B.; Forbush B.; Mcgloin M. Photochem. Photobiol. 1970, 11, 457. [DOI] [PubMed] [Google Scholar]; b Joliot P.; Barbieri G.; Chabaud R. Photochem. Photobiol. 1969, 10, 309. [Google Scholar]
- Yocum C. F. Coord. Chem. Rev. 2008, 252, 296. [Google Scholar]
- a Yachandra V. K.; Sauer K.; Klein M. P. Chem. Rev. 1996, 96, 2927. [DOI] [PubMed] [Google Scholar]; b Pecoraro V. L.; Baldwin M. J.; Caudle M. T.; Hsieh W. Y.; Law N. A. Pure Appl. Chem. 1998, 70, 925. [Google Scholar]; c Vrettos J. S.; Stone D. A.; Brudvig G. W. Biochemistry 2001, 40, 7937. [DOI] [PubMed] [Google Scholar]; d Riggs-Gelasco P. J.; Mei R.; Ghanotakis D. F.; Yocum C. F.; Penner-Hahn J. E. J. Am. Chem. Soc. 1996, 118, 2400. [Google Scholar]; e Lohmiller T.; Cox N.; Su J.-H.; Messinger J.; Lubitz W. J. Biol. Chem. 2012, 287, 24721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fukuzumi S.; Ohkubo K. Coord. Chem. Rev. 2010, 254, 372. [Google Scholar]; b Fukuzumi S. In Prog. Inorg. Chem.; John Wiley & Sons: New York, 2009; Vol. 56, p 49. [Google Scholar]
- a Park Y. J.; Ziller J. W.; Borovik A. S. J. Am. Chem. Soc. 2011, 133, 9258. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Park Y. J.; Cook S. A.; Sickerman N. S.; Sano Y.; Ziller J. W.; Borovik A. S. Chem. Sci. 2013, 4, 717. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Li F.; Van Heuvelen K. M.; Meier K. K.; Muenck E.; Que L. Jr. J. Am. Chem. Soc. 2013, 135, 10198. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lee Y. M.; Bang S.; Kim Y. M.; Cho J.; Hong S.; Nomura T.; Ogura T.; Troeppner O.; Ivanovic-Burmazovic I.; Sarangi R.; Fukuzumi S.; Nam W. Chem. Sci. 2013, 4, 3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Leeladee P.; Baglia R. A.; Prokop K. A.; Latifi R.; de Visser S. P.; Goldberg D. P. J. Am. Chem. Soc. 2012, 134, 10397. [DOI] [PubMed] [Google Scholar]; b Dong L.; Wang Y. J.; Lv Y. Z.; Chen Z. Q.; Mei F. M.; Xiong H.; Yin G. C. Inorg. Chem. 2013, 52, 5418. [DOI] [PubMed] [Google Scholar]
- Yiu S. M.; Man W. L.; Lau T. C. J. Am. Chem. Soc. 2008, 130, 10821. [DOI] [PubMed] [Google Scholar]
- a Morimoto Y.; Kotani H.; Park J.; Lee Y. M.; Nam W.; Fukuzumi S. J. Am. Chem. Soc. 2011, 133, 403. [DOI] [PubMed] [Google Scholar]; b Horwitz C. P.; Ciringh Y.; Weintraub S. T. Inorg. Chim. Acta 1999, 294, 133. [Google Scholar]; c Horwitz C. P.; Ciringh Y. Inorg. Chim. Acta 1994, 225, 191. [Google Scholar]; d Fukuzumi S.; Morimoto Y.; Kotani H.; Naumov P.; Lee Y. M.; Nam W. Nat. Chem. 2010, 2, 756. [DOI] [PubMed] [Google Scholar]; e Chen J.; Lee Y. M.; Davis K. M.; Wu X.; Seo M. S.; Cho K. B.; Yoon H.; Park Y. J.; Fukuzumi S.; Pushkar Y. N.; Nam W. J. Am. Chem. Soc. 2013, 135, 6388. [DOI] [PubMed] [Google Scholar]; f Yoon H.; Lee Y.-M.; Wu X.; Cho K.-B.; Sarangi R.; Nam W.; Fukuzumi S. J. Am. Chem. Soc. 2013, 135, 9186. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Fukuzumi S. Coord. Chem. Rev. 2013, 257, 1564. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Nam W.; Lee Y. M.; Fukuzumi S. Acc. Chem. Res. 2014, 47, 1146. [DOI] [PubMed] [Google Scholar]
- Tsui E. Y.; Kanady J. S.; Agapie T. Inorg. Chem. 2013, 52, 13833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kanady J. S.; Tsui E. Y.; Day M. W.; Agapie T. Science 2011, 333, 733. [DOI] [PubMed] [Google Scholar]; b Mukherjee S.; Stull J. A.; Yano J.; Stamatatos T. C.; Pringouri K.; Stich T. A.; Abboud K. A.; Britt R. D.; Yachandra V. K.; Christou G. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 2257. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kanady J. S.; Lin P.-H.; Carsch K. M.; Nielsen R. J.; Takase M. K.; Goddard W. A. III; Agapie T. J. Am. Chem. Soc. 2014, 136, 14373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tsui E. Y.; Tran R.; Yano J.; Agapie T. Nat. Chem. 2013, 5, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tsui E. Y.; Kanady J. S.; Day M. W.; Agapie T. Chem. Commun. 2011, 47, 4189. [DOI] [PubMed] [Google Scholar]; c Tsui E. Y.; Day M. W.; Agapie T. Angew. Chem., Int. Ed. 2011, 50, 1668. [DOI] [PubMed] [Google Scholar]; d Tsui E. Y.; Agapie T. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 10084. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Herbert D. E.; Lionetti D.; Rittle J.; Agapie T. J. Am. Chem. Soc. 2013, 135, 19075. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Kanady J. S.; Mendoza-Cortes J. L.; Tsui E. Y.; Nielsen R. J.; Goddard W. A.; Agapie T. J. Am. Chem. Soc. 2013, 135, 1073. [DOI] [PubMed] [Google Scholar]; g Kanady J. S.; Tran R.; Stull J. A.; Lu L.; Stich T. A.; Day M. W.; Yano J.; Britt R. D.; Agapie T. Chem. Sci. 2013, 4, 3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotton S.Lanthanide and Actinide Chemistry; John Wiley & Sons: Chichester, U.K., 2006. [Google Scholar]
- a Bakou A.; Buser C.; Dandulakis G.; Brudvig G.; Ghanotakis D. F. Biochim. Biophys. Acta 1992, 1099, 131. [Google Scholar]; b Bakou A.; Ghanotakis D. F. Biochim. Biophys. Acta 1993, 1141, 303. [Google Scholar]; c Lee C. I.; Lakshmi K. V.; Brudvig G. W. Biochemistry 2007, 46, 3211. [DOI] [PubMed] [Google Scholar]
- Piro N. A.; Robinson J. R.; Walsh P. J.; Schelter E. J. Coord. Chem. Rev. 2014, 260, 21. [Google Scholar]
- a Perrin D. D.Ionisation Constants of Inorganic Acids and Bases in Aqueous Solution; Pergamon: Oxford, U.K., 1982. Values of pKa reported under the same conditions (titration of 0.004−0.009 M Ln(ClO4)3 with 0.02 M Ba(OH)2; I = 0.3, NaClO4) were used for all ions except CeIII. [Google Scholar]; b Acta Cryst. 1976, A32, 751 [Google Scholar]
- One of the referees pointed out that the slope of the line in Figure 4 is close to unity if plotted on the same energy scale. We do not have an explanation for this behavior at this time.
- Barton D. H. R.; Haynes R. K.; Leclerc G.; Magnus P. D.; Menzies I. D. J. Chem. Soc., Perkin Trans. 1975, 1, 2055. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.