

Abstract
Over the past several years, mitochondrial dysfunction has been linked to an increasing number of human illnesses, making mitochondrial proteins (MPs) an ever more appealing target for therapeutic intervention. With 20% of the mitochondrial proteome (312 of an estimated 1500 MPs) having known interactions with small molecules, MPs appear to be highly targetable. Yet, despite these targeted proteins functioning in a range of biological processes (including induction of apoptosis, calcium homeostasis, and metabolism), very few of the compounds targeting MPs find clinical use. Recent work has greatly expanded the number of proteins known to localize to the mitochondria and has generated a considerable increase in MP 3D structures available in public databases, allowing experimental screening and in silico prediction of mitochondrial drug targets on an unprecedented scale. Here, we summarize the current literature on clinically active drugs that target MPs, with a focus on how existing drug targets are distributed across biochemical pathways and organelle substructures. Also, we examine current strategies for mitochondrial drug discovery, focusing on genetic, proteomic, and chemogenomic assays, and relevant model systems. As cell models and screening techniques improve, MPs appear poised to emerge as relevant targets for a wide range of complex human diseases, an eventuality that can be expedited through systematic analysis of MP function.
Keywords: Drug−protein interactions, human disease, mitochondria, model system, network, pharmacological target, protein complex, pathways, small molecules, systems biology
1. Introduction
Mitochondria are essential organelles responsible for diverse functions, including ATP production, ion homeostasis, and the initiation of apoptosis.1−3 These functions are exercised not only within the mitochondria itself but also through its interaction with other organelles such as the endoplasmic reticulum (ER),4,5 for example, in coordinating interorganellar tethering and lipid and Ca2+ exchange. While ancestral mitochondria had distinct genomes, over evolutionary time the majority of proteins required for mitochondrial function have been transferred to the nuclear genome and are imported via mitochondrial localization signals.6
Estimates suggest that as many as 1 in 5000 individuals suffer from an illness with mitochondrial etiology,7 making the mitochondria an appealing pharmacological target. These illnesses include Parkinson’s disease (PD), Alzheimer’s disease (AD), and amyotrophic lateral sclerosis (ALS).8,9 Mitochondrial protein (MP) dysfunction has also been linked to schizophrenia and autism,10,11 cancer,12 and metabolic disorders.13 For example, mutations in the mitochondrially encoded superoxide dismutase SOD1, which functions to protect mitochondria from oxidative damage, have been linked to the progression of ALS;14 NADH dehydrogenase 4, to Leber hereditary optic neuropathy;5PARKIN, to the familial form of PD;6 and Krebs tricarboxylic-acid cycle enzymes, to oncogenesis.7 These are just few examples of disease-associated mutations that affect mitochondrial function; for a more detailed summary of the role of various MPs in disease, readers are urged to consult any of several recently published reviews.2,3,15
Currently, there are 327 mitochondria-targeted small molecules (as annotated in Drug Bank16 and various literature sources, including Wagner et al.;17 see Table S1), suggesting that targeting the mitochondria is an effective avenue for therapeutic modulation. These include the triaminopyridine flupirtine, a nonopioid analgesic drug with mitochondria-dependent antioxidant and free radical scavenging activity that has been shown to be effective against ischemic neuronal damage, apoptosis, and age-associated brain disorders.18−20 Similarly, 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors such as the commercially available statins (e.g., atorvastatin and simvastatin) have been shown to exhibit neuroprotective effects against the pathologies of PD, AD, traumatic brain injury, secondary progressive multiple sclerosis, and several other neuropathological conditions.21−24 Other bioactive molecules targeting mitochondria (for more detail, see Table S1) and the mechanisms underlying their efficacy have been reviewed elsewhere.15,25−28
While our compendium of currently reported small molecules (Table S1) serves as a useful resource for gross indication of targeted mitochondrial pathways and processes, determining whether the physiological effect of individual drugs is elicited exclusively within the mitochondria is, in many cases, unknown and requires further in-depth experimentation that is beyond the scope of this review. Given the involvement of MPs in a myriad of essential functions, it is possible that the listed small molecules either diffuse out of the mitochondria to improve the efficacy of their action on extramitochondrial targets,29,30 or interact with related extramitochondrial pathways. For this reason, certain mitochondria-targeting small molecules, such as adenosine triphosphate or dimethyl sulfoxide (Table S1), are unsuitable for therapeutic intervention, as they are prone to broader effects on cellular metabolism. Because mitochondrial protein interactions are incompletely characterized at present,31 further examination of mitochondrial pathways, specifically as they integrate with extramitochondrial processes, may help to anticipate these extramitochondrial effects.
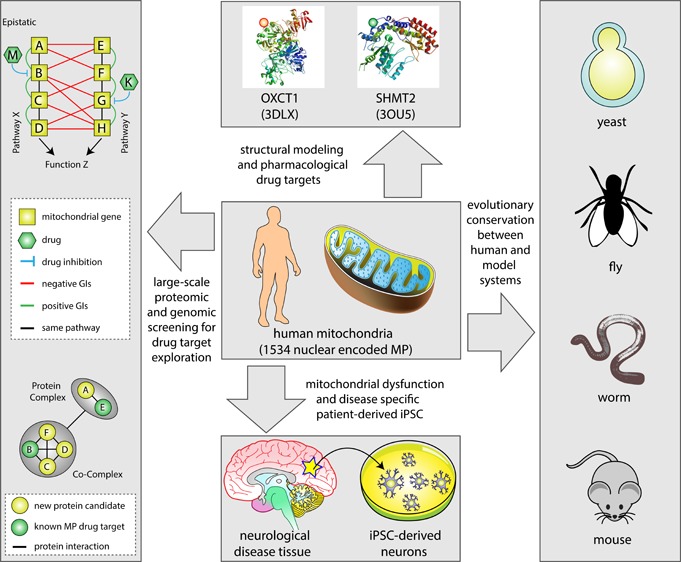
In this review, we examine MPs targeted by current therapeutics, presenting a literature-compiled census of known drug interactions for 1534 human MPs. We also discuss the high-throughput discovery of drugs that target nuclear-encoded MPs from a network pharmacology standpoint. We discuss how such an approach can accelerate the pace of drug discovery by identifying candidates that may be further studied to derive more specific mechanistic details using standard pharmacological methods. Finally, we outline how shifting experimental platforms from model organisms to human cell lines can accelerate compound discovery and drug target identification and how the application of developing technologies and improvements in three-dimensional (3D) structural analysis could spur future pharmacological discovery.
2. Systematic Characterization of the Mitochondrial Proteome
While the biochemical and structural characteristics of the mitochondria have been well-known for decades, the emergence of high-throughput proteomic screening efforts around the turn of the millennium has greatly accelerated the pace of characterizing MPs, pathways, and complexes. For example, in the model yeast Saccharomyces cerevisiae, more than 400 additional MPs have been identified (1200 total; up from ∼750)32,33 during the past decade, and in human, more than 450 have been identified (∼1100 total; up from 615)34,35 (Figure 1A). Representative discoveries, shown in Figure 1B, have informed our understanding of basic mitochondrial biology, and modern proteomics and integrative systems biology have helped to address how MP dysfunction can lead to human diseases.
Figure 1.

Composition of mitochondria and timeline of important discoveries. (A) Human MPs show a steady increase in the number of annotated genes over time. (B) Key milestones and discoveries in mitochondrial research. (C) Subcellular localization of MPs, including the inner and outer mitochondrial membrane (IMM and OMM, respectively), intermembrane space (IMS), and matrix, with highly organized cristae structures forming from the inner membrane into the matrix. These subcompartments contain many unique MPs. (D, E) Functional grouping (D) and overlap of our compiled MP catalogue with that from a recently published study (E) of MPs in human heart function.41
Based on published proteomics studies, the total number of MPs in humans is estimated to approach 1500.34,36 To generate a census of currently described MPs, we systematically annotated human proteins as being mitochondrial on the basis of experimentally supported database annotations, manual curation of the peer-reviewed literature, and a hand-picked set of well-established and widely used subcellular localization databases (Table S1). While our analysis allowed us to define a comprehensive catalogue of 1534 putative nuclear-encoded human MPs, establishing localization of these proteins within the mitochondria has been more challenging, as only 48% (750 of 1534) of these MPs have been experimentally identified to specifically localize to a mitochondrial subcompartment in mammalian cells (147 inner membrane, 107 outer membrane, 23 intermembrane space, 441 mitochondrial matrix, 1 cristae, and 21 mitochondrial ribosome proteins). 34% of MPs (526 of 1534) have been found to localize to both mitochondria and other compartments,37,38 leaving 268 proteins with unclear submitochondrial localization (Figure 1C and Table S1). This suggests that more experimentation, such as that being conducted for the ongoing Human Protein Atlas39 project, is required to understand organellar functionality comprehensively.40 Additionally, 373 human MPs are of unknown function and thus provide an opportunity for novel functional discovery (Figure 1D and Table S1).
As an independent assessment to measure the quality of our annotation efforts, we compared the MP target index to a recently published study focusing on annotating MPs expressed in human heart tissue.41 While our analysis captured about 74% of the current mitochondrial annotations (573 of the 775 MPs reviewed in UniProt; Figure 1E), the remainder of the annotations we report are new and well-supported by other sources of information (Table S1), providing a valuable resource for future studies on mitochondrial biology.
Finally, we examined protein abundance for the 1534 human MPs using a recently published large-scale human proteome draft map, in which protein abundance was measured for 16 570 annotated human proteins in 24 histologically healthy tissue samples.42 Our analysis indicated that nearly 43% (619 of 1433 detected) of the human MPs are constitutively expressed in all tissues, versus ∼13% (1991 of 15 134) of non-MPs (Figure 2A,B and Table S2). Thus, while there appears to be considerable tissue-specific variation in MP expression (consistent with previous reports43,44), we feel that this set of proteins represents a constitutive current estimate of MP composition.
Figure 2.

Disease association for human MPs. (A, B) Heat map showing expression of 93% of the human MPs (1433 of 1534) from our target index (A) and their distribution in tissue expression as compared against non-MPs (B) that are expressed in at least one of the 24 histologically healthy tissue samples, extracted from a recently published large-scale human proteome draft map.42 The bold text in the zoomed-in view of the inset indicates proteins constitutively expressed in the majority of human tissues examined and enriched specifically for processes encoding for programmed cell death (p-value ≤ 1.3 × 10–6; significance computed using Fisher’s exact test.). (C) Distribution of disease associations of human MPs. In the case of MPs associated with multiple diseases, assignment was made only to one disease type (see Table S1 for details).
3. Mitochondrial Dysfunction in Disease Pathologies
Establishing an accurate and complete list of mammalian MPs, as well as characterizing submitochondrial localization, is complicated by the fact that MP localization can be tissue- or condition-specific.43,45,46 Thus, both the experimental model and survey conditions play substantial roles in the elucidation of MP function.47 Fortunately, tissue lineages are being established to model specific effects of mitochondrial dysfunction. For example, familial PD, AD, and other neurodegenerative disorders caused by the impairment of mitochondrial processes, including oxidative stress and quality control factors (e.g., PINK1 and PARKIN), can be modeled in vitro using disease-specific patient-derived induced pluripotent stem cells (iPSC) to elucidate unknown disease mechanisms.48 Reprogramming of differentiated somatic cells provides an avenue for exploring various neurological diseases and allows for patient stratification, which will inform specific molecular disease etiology.48,49 Small molecules can then be screened for efficacy in specific molecular disease states.48 This has been well-demonstrated in two recent publications50,51 wherein the drug targets identified in a yeast model of α-synuclein toxicity (a small lipid binding protein involved in several neurodegernative disorders, including PD) led to the identification of early pathogenic phenotypes in iPS neurons derived from patients harboring an α-synuclein mutation.
Next, to clarify our 1534 compiled human MPs as being either causal or associated with mitochondrial diseases, we collected large sets of disease association data from databases (e.g., CORUM, GAD, OMIM, and CGP), and literature sources (from both full-text articles and abstracts), including our previous study.3 This effort resulted in 514 disease-linked and 9 disease-causing MPs, of which 210 were deemed to be both disease-linked and disease-causing (Table S1 in the Supporting Information). Strikingly, ∼63% (465 of 733) of the human MPs with disease evidence are associated with cancer, metabolic, and neurological disorders (Figure 2C and Table S1). These disorders can arise either from mutations in a mitochondrial gene (whether encoded by the nuclear or mitochondrial genome52) or from the indirect disruption of a mitochondria-dependent metabolic or homeostatic processes.53−55
Disruptions in mitochondrial function that have been subsequently linked to disease can be broadly classified as those that result in the increased generation of reactive oxygen species (ROS), the dyshomeostasis of calcium buffering and storage, or the disruption of ATP production,30 although alterations in ATP production and calcium homeostasis are inherently linked. For example, damage to the mitochondrial membranes affects the ability of mitochondria to generate ATP, which, in turn, leads to a lack of efficient pumping of calcium outside the cell or into other intracellular calcium stores (such as the ER).56 Excessive intracellular calcium leads to the formation of so-called mitochondrial permeability transition pores that bypass both mitochondrial membranes.57 This further disrupts mitochondrial function, ultimately causing swelling, rupture, and activation of apoptosis through the release of cytochrome C from the mitochondrial intermembrane space into the cytosol, resulting in apoptosis observed in ischemia/reperfusion injury.56
Modifications in mitochondrial function have also been linked to cancer through several lines of evidence. For example, a shift in mitochondrial metabolism from primarily oxidative phosphorylation (OXPHOS) to a near sole dependence on glycolysis, a phenomenon termed the Warburg effect, is a long-known hallmark of cancer cells.58,59 As a result of being less dependent on OXPHOS, cancerous cells are able to survive in microenvironments with poor blood supply, such as those frequently encountered in rapidly growing tumors. Additionally, increased production of reactive oxygen and nitrogen species alters cell signaling in a manner that promotes cancer cell survival.60 This can occur via inhibition of mitogen-activated protein kinase phosphatases and through numerous transcription factors, including ETS1 (V-Ets Avian Erythroblastosis Virus E26 oncogene homologue 1), which controls the differentiation, survival, and proliferation of lymphoid cells.61 Excessive production of reactive oxygen and nitrogen species, as well as oxidative and nitrosative stress, are also major contributing factors to AD and Type 2 Diabetes Mellitus.62−67
Finally, remodelling of mitochondrial structures through fusion (joining of mitochondria involving the coordinated activity of both outer and inner mitochondrial membranes) has contributed greatly to the pathogenesis of human disorders, especially in neurodegeneration.68 Alternately, mitochondrial fission (or division) rarely results in human disease, principally due to the essentiality of this process for cell survival.26 This is evidenced by the known embryonic lethality of fission mediator DRP1 knockout in mice.69
4. Pharmacological Targeting of Mitochondria
Of the 1534 compiled human MPs, 312 are known targets of one or more existing small molecules (Figure 3A and Table S1). This represents almost 20% of the human mitochondrial proteome, significantly more than the ∼5% of targeted non-MPs (p ≤ 2.2 × 10–16). As mitochondria are key sites for the production of ATP, it is not surprising that the bulk of mitochondrial drug targets, almost 200, are involved in energy metabolism (Figure 3B). The remaining targets are widely distributed across a variety of biological processes (e.g., mitochondrial transport, respiration, transcription, and genome maintenance; Figure 3B), reflecting the importance of mitochondria in diverse cellular functions.
Figure 3.

Small molecules targeting MPs and their associations to protein complexes and pathways. (A) Fraction of mitochondrial and non-MPs that are potential drug targets; p-value was computed using Fisher’s exact test. (B) Functional grouping of MP drug targets as annotated by Gene Ontology bioprocess terms. (C) Gene expression profiles of various mitochondrial physiological activities measured for each of the 56 MPs with disease evidence against 125 distinct chemical perturbations compiled from the study of Wagner et al.17 The physiological measurements were performed for mitochondrial oxidative damage (MitOX), nuclear oxidative damage (NucOX), gene expression-based high-throughput screening (GE-HTS), cytochrome C activity (CytC), reactive oxygen species (ROS), mitochondrial membrane potential (MMP), and MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide], a measure of mitochondrial dehydrogenase activity. The expression values are represented as z-scores; score details are shown in Table S3.
When grouped according to broad disease classes, the gene expression profiles of MPs involved in various physiological activities targeted by small molecules (from the study of Wagner et al.17) are largely distributed to metabolic, neurological, or cancer-related illnesses, consistent with mitochondrial function in energy production and apoptosis (Figure 3C and Table S3). The most prominent category of mitochondria-targeted small molecules are antioxidants or free radical scavengers. While antioxidant therapy has been typically limited due to difficulties in accessing the mitochondria, recent advances have allowed mitochondrial targeting of such molecules as the lipophilic cation-associated drug MitoQ, the antioxidant Szeto–Schiller peptides, or drugs encapsulated in dequalinium-containing liposomes.71−73
Another category of pharmacological agents exploit the fact that mitochondria regulate the apoptotic cascade. One such group of drugs (including ABT-737, A-385358, and gossypol) reduce the ability of cancer cells to resist chemotherapeutic agents by inhibiting the sequestration of pro-apoptotic proteins (such as BCL2) by antiapoptotic proteins (such as BAK).27 Additionally, as mentioned above, the Warburg effect in cancer cells makes glycolysis an attractive pharmacological target. For instance, phloretin or 2-deoxy-d-glucose interferes with glucose uptake and has shown promising anticancer activity.74 Currently, there are several inhibitors of glycolysis that enhance cancer cell sensitivity to chemotherapeutic agents and are undergoing preclinical trials.27 Likewise, mitochondrial metabolic modulators are undergoing clinical trials for improvement of ischemic heart disease and cardiomyopathy.75
Increasing the NAD+/NADH ratio directly, as is done by nicotinamide, enhances mitophagy (mitochondrial degradation) and the subsequent removal of dysfunctional mitochondria.76 This and other effects on mitochondrial function make nicotinamide useful in treating a range of disorders, including AD.77 Indirect increase of the NAD+/NADH ratio activates AMP-activated protein kinase (AMPK), as is seen with resveratrol.78 While resveratrol has limited health benefits, a synthetic activator of AMPK, 5-amino-1-β-d-ribofuranosyl-imidazole-4-carboxamide (AICAR), corrected cytochrome c-oxidase deficiency in a knockout mouse model, improving motor performance.79 Similarly, direct activation of the downstream effectors of NAD+/NADH, namely, the sirtuin family of histone deacetylases, results in enhanced mitophagy.80 It is unclear whether mitochondrial dysfunction is the cause81 or the result of metabolic syndromes;82,83 however, improvement of mitochondrial function apparently results in an overall improvement of patient condition. Compounds that enhance a specific step in the electron transport chain can also help to improve mitochondrial function.84 For example, methylene blue, which enhances cytochrome C oxidase and mitochondrial respiration, has been used to counteract respiratory defects associated with AD.85
Several nuclear proteins that increase the expression of mitochondrial metabolic enzymes and enhance mitochondrial function are reported to ameliorate metabolic disorders upon drug-based stimulation. These include the retinoic acid receptor,86 peroxisome proliferator-activated receptors,87 estrogen-related receptors,88 and mitochondrial transcription factor A.89 Some of these transcription factors are already targets of clinically used drugs.90
Recently, impaired mitophagy was implicated in PD,91 and chemical inhibitors like nicotinamide that can enhance removal of defective mitochondria may slow the progression of neurodegenerative disorders.92 A genome-wide high-throughput RNA interference (RNAi) screen identified a diverse set of genes that affect mitophagy rates (e.g., TOMM7, HSPA1L, BAG4, and SIAH3),93 suggesting the existence of multiple pathways that influence this process. Compounds that inhibit mitochondrial fission94 and fusion95 were also shown to attenuate apoptosis in models of neurodegenerative disorders. These include MDIVI-1, which blocks outer membrane permeabilization via inhibition of DNM1 and which is currently undergoing clinical evaluation.94,96
The ability of FDA approved drugs to be repurposed for mitochondrial diseases has also been an area of intense interest. For example, cyclosporine A (CsA), a clinical immunosuppressant used to prevent rejection of transplanted organs, has been used for years to inhibit mitochondrial permeability of the transition pore through its molecular target cyclophilin D.97 CsA was shown to reduce the size of myocardial infarctions in humans98 and is also being tested for its ability to ameliorate neuronal damage after stroke,99 with both diseases involving mitochondrial dysfunction through extra-mitochondrial pathology. CsA analogues that specifically act on mitochondrial cyclophilin D, but not on cytosolic isoforms, would have the added advantage of lacking immunosuppressant side effects.100
However, currently there are few clinically approved drugs that directly influence disease pathologies that are linked to MPs compiled in this study, whereas others may be palliative and treat only symptoms and not the cause of the actual disease. Furthermore, many of the clinical drugs that affect MP function lack the specificity to allow them to be employed for clinical intervention in mitochondrial diseases. This is largely due to the inaccessibility of the inner mitochondrial membrane to small molecules.101 However, as our understanding of mitochondrial pathways is still incomplete,102 emerging methods aimed at systematically understanding mitochondrial gene function may yet uncover new avenues for therapeutic modulation.
5. Identification of Potential Pharmacological Targets through Systematic Screening
The recent prevalence of large-scale genomic and proteomic data sets has facilitated a paradigm shift in therapeutic design towards an approach that integrates pathway and systems data and that is increasingly tailored to individual patients.103 In examining large-scale protein–protein interaction (PPI) data, interacting proteins have been found to be more likely to share similar fitness profiles across tested drugs (i.e., deletion of genes encoding physically connected subunits of a protein complex result in similar sensitivities to structurally diverse compounds, as evidenced by correlated fitness similarity profiles),104,105 making protein complex members likely drug co-targets (Figure 4A).
Figure 4.

Fitness profiling and interaction networks in mitochondrial drug target discovery.(A) Fitness profiles of interacting proteins complex members sharing phenotypic responses (I) and subnetworks of physically connected disease-linked MP complex subunits (II), targeted by small molecules. (B) Subnetworks depicting the physical connectivity, either directly (left) or within a complex (right), between a new and known MP drug target. (C) Illustration of several possible co-fitness profiles of single gene deletion mutant strains grown in the presence small molecules targeting related mitochondrial processes or pathways. (D) Epistatic interactions for two redundant pathways (X and Y). In pathway X, gene B that exists with A, C, and D in a linear pathway is inhibited by the drug M, whereas gene G in pathway Y (which is in a pathway with E, F, and H) is inhibited by drug K. Here, the additional pathway information provided by genetic interaction (GI) mapping enabled the discovery that drugs M and K inhibit parallel pathways, which may suggest, for example, combination drug therapies involving both M and K.
Conversely, members of functionally related disease- or non-disease-linked MPs can be used to predict relevant complexes or pathways targeted by a drug (Figure 4A). For example, loss-of-function of any one of the three functionally and physically connected voltage-dependent anion channel (VDAC) MP members VDAC1–3, associated with a variety of diseases (e.g., ALS, PD, AD, Huntington’s disease (HD), and cardiomyopathies106,107) has been shown to cause sensitivity to the small molecule dihydroxyaluminum.104,108−110 Similarly, methylmalonic acidemia (MMA) is a hallmark of genetic metabolic disorders and is caused by mutations in genes (MMAA, MMAB, MMAC, and MUT) involved in the translocation of cobalamin (vitamin B12) into the mitochondria. These proteins are all targeted by hydroxocobalamin and cyanocobalamin. Also, physical interactions have been observed between the human GTPase MMAA (methylmalonic aciduria type A) and MUT (mthylmalonyl-CoA mutase),111 and while these MPs are not expected to have comparable binding affinities for the same compounds, it seems likely that members of this pathway may be targeted to achieve the same physiological effect (Figure 4A).
Physical (i.e., protein–protein) interaction data can provide information on protein complex composition, thus identifying potential additional drug targets through guilt-by-association (Figure 4B).112 Moreover, drugs often function by perturbing physical interactions between proteins. For example, melatonin has been shown to interfere with the apoptotic pathway by impeding the dimerization of the pro-apoptotic protein BAX (the interacting partner of BCL2).113 Also, the small molecule 10058-F4, known to bind to the C-terminal domain of MYC, inhibits a family of transcriptional factors including c-MYC, MYCN, and MAX. The subsequent disruption of the c-MYC/MAX and MYCN/MAX heterodimeric interactions leads to protein degradation, apoptosis, and lipid formation in neuroblastoma cells.114,115 Some drug treatments use the opposing strategy of enhancing physical associations. For example, the free radical scavenger edaravone, which has neuroprotective effects, causes an increase in binding stability between the pro-apoptotic protein BAD and the conserved regulatory protein 14-3-3 in response to oxidative stress, thereby reducing apoptosis.116
Large-scale protein interaction data has been generated mostly using scalable platforms such as yeast two-hybrid,117,118 protein–fragment complementation,119 and affinity purification–mass spectrometry.31,120−123 Currently, due to difficulties in accessing MPs using standard laboratory conditions, their interactions are considered to be under-surveyed. To improve coverage in MP interaction screens, the currently employed practices such as growth in standard fermentative culturing conditions (YPD media) and growth of culture to saturation (which induces mitophagy) will likely need to be revisited, as they tend to inhibit MP gene expression.
Genetic interaction screening presents a useful alternative for definition of complexes. Of specific relevance is the identification of drug targets through systematic competitive growth of gene deletion mutant strains in the presence of a given small molecule and subsequent identification of sensitive strains (i.e., haploinsufficiency profiling and homozygous deletion profiling, or HIP–HOP; Figure 4C).124,125 HIP screening in yeast has revealed that gene deletions in mitochondrial translation confer sensitivity to tigecycline, which led to the discovery of tigecycline’s anti-leukemic action.126
Similarly, the enzyme kynurenine 3-monooxygenease (KMO), which is targeted by the small molecules lanthellamide A and UPF648,127,128 was identified as a potential therapeutic target for HD based on loss-of-function yeast screens.129 KMO inhibition has also recently been observed to improve neurodegenerative conditions in mouse models.130 Also, yeast genetic screens assaying growth of loss-of-function alleles under high temperature and using glycerol as a carbon source identified the antibacterial chlorhexidine as a potential candidate for ameliorating mitochondrial dysfunction.131 This was later confirmed in mammalian tissues.131
As HOP profiling depends on an epistatic relationship between the drug target and a second gene with a loss of function allele, characterization of the yeast genetic interaction network has been highly useful in deciphering drug target pathways. Synthetic genetic array (SGA) screening132,133 has allowed near-comprehensive surveys of genetic interactions regardless of protein localization and thus is particularly useful in charting mitochondrial gene function (Figure 4D). Briefly, SGA quantifies the growth of thousands of yeast deletion strains, systematically identifying instances wherein double mutant strains grow either more or less than expected based on the growth of constitutive single deletion strains. Deviation from expected growth rate in the double mutant strain is indicative of a synergistic genetic relationship between the two genes. Resulting genetic interaction profiles have been used in combination with HOP data to identify novel drug targets,133 although this approach has not yet been specifically applied to mitochondrial genes.
Alternately, recent large-scale screening efforts in human cell lines have monitored fluorescent indicators of mitochondrial function to examine process-specific perturbation effects. For example, large-scale chemical screening at various stages of apoptotic processes in Jurkat cells identified compound (norgestrel and diclofenac)-specific profiles that activated mitochondrial annexin binding.134 The use of recently developed large-scale RNAi-mediated epistatic or chemogenomic screens through short hairpin RNA (shRNA), small interfering RNA (siRNA), and endoribonuclease-prepared siRNA (esiRNA) plasmid libraries in mammalian cells135−139 have raised the possibility of high-throughput assays of functional interactions, as has been done in yeast.133,140,141
Recently, an assessment of ATP production in various RNAi-mediated knockdowns revealed adenylate kinase to be a potential target for therapeutic intervention to counter electron transport chain or bioenergetic disruptions characteristic of cancers and neurodegenerative diseases.142 Similarly, analysis of PARKIN (a component of the E3 ubiquitin ligase complex associated with PD) in RNAi-treated HeLa cells using high-content microscopy has revealed regulators associated with mitochondrial damage.93 A similar screen in Drosophila melanogaster tissues identified a novel regulator of calcium transport, LETM1,143 whereas a Caenorhabditis elegans RNAi screen combined with the mitotoxic drug antimycin has identified additional genes important for mitochondrial protection.144
While RNAi may present an attractive approach for the systematic survey of mitochondrial gene function and chemogenomic analysis, off-target effects, uneven or limited gene coverage, and imperfect suppression of the target gene may obscure interpretation.145−147 The recent advent of RNA-guided CRISPRs (clustered regularly interspaced short palindrome repeats) for targeted gene disruption148,149 offers a promising strategy for gene deletion assays in mammalian cells. However, as with RNAi, potential off-target effects of CRISPRs would present a limitation to large-scale screening. More recent adaptations, such as the use of truncated sgRNAs (short or single-guide RNAs),150 seek to limit these off-target effects.
6. Interpreting Target Association Data
Although much of the large-scale protein and genetic interaction data generated over the past decade has come from model organisms such as yeast, fly, and worm,141 the high conservation of MPs and complexes (Figure 5A,B and Table S4) allows these results to be particularly transferable to humans through cross-species orthologue mapping. This strategy has been reported widely by us31,151 and others152−157 to inform human protein function.
Figure 5.

Human MP and complex conservation across species. (A) Venn diagram showing the overlap of 1534 human MPs with four other eukaryotes. The numbers in parentheses show the extent of human MP conservation in other species. (B) Evolutionary conservation map showing 119 (of the 1788) curated human protein complexes containing at least one drug-targeted MP in additional model species. As an example, the conserved ESR1–SP1 complex in the bottom inset highlights ESR1, as 32 drugs are known to target this MP. Node size is proportional to the number of subunits comprising the complex, and the colored wedges are sized according to the proportion of the human complex containing an MP drug target conserved in yeast, fly, worm, and mouse. The fraction of conserved MP drug complex subunits across species is shown as a bar graph. Edges in the network graph indicate significant PPIs (|z-score ≥ 1.96| versus random permutation; p-value ≤0.05) compiled from BioGRID (ver. 3.2.111), whereas the edge width reflects the degree of PPI connectivity between conserved complex subunits.
These lower eukaryotic models are not only generally more rapid and cost-effective for small molecule screening than their mammalian counterparts, but also can be vital when the phenotypic effect of certain mutations cannot be experimentally tested directly in disease tissues.157 For example, ample literature evidence supports the complex formation of an E3 ubiquitin ligase (PARKIN), a mitochondrial kinase (PINK1), and a protein of unknown function (DJ-1) based on their functional similarity in preserving mitochondrial integrity and promoting ubiquitination of Parkin substrates in human brain lysates.158 Knockout of both PARKIN and PINK1 in fly has drastic phenotypic effects due to mitochondrial damage, causing muscle degeneration, male infertility, and the loss of dopaminergic neurons.159,160
However, despite the utility of such highly tractable model organisms for identifying fundamental pathways and processes (Figure 6), they are inevitably limited in terms of modeling specific human disease states. For example, while neurotransmitter systems in fly mediate many behaviors (i.e., learning and memory) that are conserved in humans,157 the fly brain has no substantia nigra, which is pertinent to understanding how clinical features mediated by dopaminergic neuron loss in Parkinson’s disease correlate with behavioral phenotypes.157 Likewise, while essential molecular mechanisms underlying tumorigenesis and metastasis can be probed in fly, it is not feasible to model many types of malignancies that are common in humans, such as those related to specific tissues (e.g., prostate, ovarian, or breast cancer).157 Since cellular and molecular processes can vary between model species and humans, careful consideration of the model system is required when designing screens and interpreting data. Orthology mapping is therefore best suited as a means to generate new testable hypothesis or to lend ancillary support to an existing one.
Figure 6.

Relationship between the scale of research in tractable model systems and its relevance to clinical application. At each research level, the model organisms in use, methodologies available, and assays typically conducted are listed.
Mammalian in vivo models offer a more faithful modeling of disease but at the cost of scalability (Figure 6). For example, mouse models that closely replicate specific forms of mitochondria-associated neurodegenerative diseases are constantly being generated, such as SOD1-deficient mice for ALS161 or mice that model PD.162 However, these disease models lack the throughput needed for lead compound discovery.
7. Structural Modeling of Drug Targets for Therapeutic Discovery
In silico prediction of small molecule–protein interactions potentially provides an efficient alternative to experimental screening.163−167 This is particularly relevant for MPs, as they can be difficult to assay experimentally. For example, docking analysis of mitochondrial monoamine oxidase B facilitated the design of novel inhibitors of the protein.168 In addition to predicting novel drug binding interactions, in silico screening has been useful in the discovery of potential therapeutic effects for existing small molecules169 and in the optimization and development of small molecule agonists or antagonists from previously identified interactions.170 In silico target prediction typically depends largely on characterized 3D protein structures, which have increased dramatically for MPs over the past decade (Figure 7A,B and Table S1).
Figure 7.

Human MP structures and their relationships to small molecule inhibitors. (A) Bar graph showing the number of MPs with solved 3D structures (compiled from public databases) over time. (B) Timeline showing representative 3D MP structures (with year of publication) along with known small molecule inhibitors (denoted by dots). (C) Number of solved structures per human MP. Multiple solved 3D structures for the same human MP can arise in separate research publications. (D) Comparison of MPs with known targeted drugs and with solved 3D structures. (E, F) Fraction of human MPs targeted by small molecules conserved across eukaryotic species (E) and with known small molecule targets (F) by the number of solved 3D structures.
Approximately 37% of MPs have at least one solved 3D structure, as compiled by the Protein Data Bank (Figure 7C). However, as protein docking analysis has centered around identifying small molecule binding mechanisms, drug targets make up a significantly higher proportion (162 of 575; 28%) of solved 3D structures, with half (162/312) of all known mitochondrial drug targets having solved 3D structures (Figure 7D). Furthermore, a higher fraction of MP drug targets are conserved within other model species (Figure 7E) and have multiple solved structures when compared to non-drug-targeted MPs (Figure 7F), suggesting an intense research focus on MP–small molecule docking.
For those proteins that fail to crystallize properly for structural analysis, potentially due to low yield or protein instability, the high conservation of MPs (Figure 5A) provides relevant alternatives from other model organisms. For example, when the mammalian neurodegeneration-associated protein kynurenine 3-monooxygenease failed to crystallize, the homologous yeast protein provided the crystal structure of both the protein and the protein–small molecule (UPF 648) binding complex.128 Notably, this example may be somewhat unique, as use of alternative model organisms may not improve acquisition of high-quality crystals. This is especially true for membrane-bound MPs, where obtaining concentrated pure solution is often demanding and requires appropriate crystallization and solubilization conditions.50 Expressing a mutated form or altering the surface properties of the protein51 and improving the solubility of the native proteins in a different host52 can ease some of the associated problems, but this remains a slow process. Consequently, there is a pressing need to predict the structure of proteins in an informed way, which may involve the detection of homologues of known 3D structures using template-based structural homology modeling and fold recognition.53
In the absence of a solved structure, prospective drug targets can also be proposed based on amino acid conservation. For example, the structure of DRP1, modeled using similarity to dynamin, was queried for potential antagonists using automated docking analysis, finding multiple candidates with potential antiapoptotic effects.171 Compound activity can also be estimated through protein secondary structure172 and examination of protein interaction173 or expression174 data. For example, small molecules can be designed to specifically target interface sites between interacting protein pairs, to inhibit or stabilize important protein–protein interactions, or to disrupt or strengthen a protein complex175,176 (for a more in-depth review on protein–protein interaction interface targeting, see Duran-Frigola et al.176). As well, gene network expression changes characteristic of disease states can be identified using systematic transcriptional analysis,177−179 an approach useful for mitochondrial targets.174 Thus, in silico screening, in combination with dedicated experimental efforts aimed at elucidating mitochondrial gene function on a large scale,132 presents an attractive combination for future drug targeting studies.
8. Conclusions and Future Perspectives
The majority of drugs discovered to date that target MPs have been revealed through either target-based screens,180 systematic competitive growth assays,140 or phenotype-based screens.181 While our curation efforts have generated a list of 327 compounds targeting 312 unique MPs, future discovery of novel therapeutics can benefit from a greater systematic understanding of mitochondrial disease etiology and of mitochondrial gene function. Yeast, worm, and fly are scalable, easily manipulated systems that have been invaluable in assaying the molecular functions of thousands of genes, many of which are directly conserved across eukaryotes.124,151 However, these organisms are limited in terms of modeling tissue-dependent diseases in humans. The specific cellular environment of a disease state may play a significant role in disease progression, and understanding these effects will be critical in determining how a drug restores health to a dysfunctional mitochondrial network. This is particularly true for diseases that exhibit tissue specificity, such as PD,48 which requires specialized differentiated tissue culture types to examine.
Recent advances in mammalian cell lines, specifically using high-content screening of mitochondrial reporters, have made it possible to perform targeted lead compound discovery at high throughput. However, delivery of drugs in vivo still presents a substantial hurdle, as more rigorous optimization procedures are required with respect to testing drug–target specificity and selectivity in preclinical models to avoid clinical trial failure. Future combinations of existing drug information with structural modeling, gene network analysis, expression profiling, and gene deletion/knockdown assay in multiple model organisms, holds the promise to provide more effective and more easily deliverable therapeutics.
Acknowledgments
We thank members of the Babu laboratory for helpful comments and discussions. This research was supported by grants from the Canadian Institutes of Health Research (MOP nos. 125952 and 132191) to Z.Z. and M.B. and a National Institutes of Health grant (R01GM106019) to M.B. M.J. and J.V. are supported by the Saskatchewan Health Research Foundation Postdoctoral Fellowship.
Glossary
Abbreviations
- AD
Alzheimer’s disease
- ALS
amyotrophic lateral sclerosis
- CRISPR
clustered regularly interspaced short palindrome repeats
- CsA
cyclosporine A
- ER
endoplasmic reticulum
- HD
Huntington’s disease
- HIP
haploinsufficiency profile
- HOP
homozygous deletion profile
- iPSC
induced pluripotent stem cells
- MP
mitochondrial protein
- OXPHOS
oxidative phosphorylation
- PPI
protein–protein interaction
- PD
Parkinson’s disease
- RNAi
RNA interference
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- SGA
synthetic genetic array
- sgRNA
short or single-guide RNA
- shRNA
small hairpin RNA
- siRNA
silencing RNA
- VDAC
voltage-dependent anion channel
Supporting Information Available
Human MP index (including gene annotation, biological process, subcellular localization, and disease association) and known small molecule inhibitors are listed in Table S1. Protein composition of MPs from this study mapped against human proteins measured in 24 histologically healthy tissue samples (compiled from a recently published large-scale human proteome draft map42) are shown in Table S2. Bioactive compounds targeting MPs linked with disease (sheet 1) and the gene expression profiles (sheet 2) of various physiological effects of MPs against chemical perturbations (compiled from the study of Wagner et al.17) are shown in Table S3. Compilation of human protein complexes containing MPs that are known drug targets (sheet 1) and conservation of these MPs to other eukaryotic species (sheets 2 and 3) are indicated in Table S4. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
⊥ R.H.M., M.J., K.J., and G.M. contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Chacinska A.; Koehler C. M.; Milenkovic D.; Lithgow T.; Pfanner N. Importing mitochondrial proteins: machineries and mechanisms. Cell 2009, 138, 628–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari J.; Suomalainen A. Mitochondria: in sickness and in health. Cell 2012, 148, 1145–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlasblom J.; Jin K.; Kassir S.; Babu M. Exploring mitochondrial system properties of neurodegenerative diseases through interactome mapping. J. Proteomics 2014, 100, 8–24. [DOI] [PubMed] [Google Scholar]
- Hoppins S. The regulation of mitochondrial dynamics. Curr. Opin. Cell Biol. 2014, 29C, 46–52. [DOI] [PubMed] [Google Scholar]
- Kornmann B. The molecular hug between the ER and the mitochondria. Curr. Opin. Cell Biol. 2013, 25, 443–8. [DOI] [PubMed] [Google Scholar]
- Schmidt O.; Pfanner N.; Meisinger C. Mitochondrial protein import: from proteomics to functional mechanisms. Nat. Rev. Mol. Cell Biol. 2010, 11, 655–67. [DOI] [PubMed] [Google Scholar]
- Schaefer A. M.; Taylor R. W.; Turnbull D. M.; Chinnery P. F. The epidemiology of mitochondrial disorders—past, present and future. Biochim. Biophys. Acta 2004, 1659, 115–20. [DOI] [PubMed] [Google Scholar]
- Trushina E.; McMurray C. T. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 2007, 145, 1233–48. [DOI] [PubMed] [Google Scholar]
- Lin M. T.; Beal M. F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–95. [DOI] [PubMed] [Google Scholar]
- Rossignol D. A.; Frye R. E. Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol. Psychiatry 2012, 17, 290–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manji H.; Kato T.; Di Prospero N. A.; Ness S.; Beal M. F.; Krams M.; Chen G. Impaired mitochondrial function in psychiatric disorders. Nat. Rev. Neurosci 2012, 13, 293–307. [DOI] [PubMed] [Google Scholar]
- Wallace D. C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasrallah C. M.; Horvath T. L. Mitochondrial dynamics in the central regulation of metabolism. Nat. Rev. Endocrinol. 2014, 10, 650–658. [DOI] [PubMed] [Google Scholar]
- Vehvilainen P.; Koistinaho J.; Gundars G. Mechanisms of mutant SOD1 induced mitochondrial toxicity in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014, 8, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer G.; Horvath R.; Klopstock T.; Mootha V. K.; Suomalainen A.; Koene S.; Hirano M.; Zeviani M.; Bindoff L. A.; Yu-Wai-Man P.; Hanna M.; Carelli V.; McFarland R.; Majamaa K.; Turnbull D. M.; Smeitink J.; Chinnery P. F. New treatments for mitochondrial disease-no time to drop our standards. Nat. Rev. Neurol 2013, 9, 474–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S.; Knox C.; Guo A. C.; Cheng D.; Shrivastava S.; Tzur D.; Gautam B.; Hassanali M. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner B. K.; Kitami T.; Gilbert T. J.; Peck D.; Ramanathan A.; Schreiber S. L.; Golub T. R.; Mootha V. K. Large-scale chemical dissection of mitochondrial function. Nat. Biotechnol. 2008, 26, 343–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluter T.; Struy H.; Schonfeld P. Protection of mitochondrial integrity from oxidative stress by the triaminopyridine derivative flupirtine. FEBS Lett. 2000, 481, 42–6. [DOI] [PubMed] [Google Scholar]
- Perovic S.; Bohm M.; Meesters E.; Meinhardt A.; Pergande G.; Muller W. E. Pharmacological intervention in age-associated brain disorders by Flupirtine: Alzheimer’s and prion diseases. Mech. Ageing Dev. 1998, 101, 1–19. [DOI] [PubMed] [Google Scholar]
- Osborne N. N.; Schwarz M.; Pergande G. Protection of rabbit retina from ischemic injury by flupirtine. Invest. Ophthalmol. Visual Sci. 1996, 37, 274–80. [PubMed] [Google Scholar]
- Kumar A.; Sharma N.; Gupta A.; Kalonia H.; Mishra J. Neuroprotective potential of atorvastatin and simvastatin (HMG-CoA reductase inhibitors) against 6-hydroxydopamine (6-OHDA) induced Parkinson-like symptoms. Brain Res. 2012, 1471, 13–22. [DOI] [PubMed] [Google Scholar]
- Ramirez C.; Tercero I.; Pineda A.; Burgos J. S. Simvastatin is the statin that most efficiently protects against kainate-induced excitotoxicity and memory impairment. J. Alzheimer’s Dis. 2011, 24, 161–74. [DOI] [PubMed] [Google Scholar]
- Malkki H. Multiple sclerosis: could simvastatin slow down secondary progressive MS?. Nat. Rev. Neurol. 2014, 10, 241. [DOI] [PubMed] [Google Scholar]
- Wang H.; Lynch J. R.; Song P.; Yang H. J.; Yates R. B.; Mace B.; Warner D. S.; Guyton J. R.; Laskowitz D. T. Simvastatin and atorvastatin improve behavioral outcome, reduce hippocampal degeneration, and improve cerebral blood flow after experimental traumatic brain injury. Exp. Neurol. 2007, 206, 59–69. [DOI] [PubMed] [Google Scholar]
- Olszewska A.; Szewczyk A. Mitochondria as a pharmacological target: magnum overview. IUBMB Life 2013, 65, 273–81. [DOI] [PubMed] [Google Scholar]
- Andreux P. A.; Houtkooper R. H.; Auwerx J. Pharmacological approaches to restore mitochondrial function. Nat. Rev. Drug Discovery 2013, 12, 465–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulda S.; Galluzzi L.; Kroemer G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discovery 2010, 9, 447–64. [DOI] [PubMed] [Google Scholar]
- Eckert G. P.; Renner K.; Eckert S. H.; Eckmann J.; Hagl S.; Abdel-Kader R. M.; Kurz C.; Leuner K.; Muller W. E. Mitochondrial dysfunction—a pharmacological target in Alzheimer’s disease. Mol. Neurobiol. 2012, 46, 136–50. [DOI] [PubMed] [Google Scholar]
- Prime T. A.; Blaikie F. H.; Evans C.; Nadtochiy S. M.; James A. M.; Dahm C. C.; Vitturi D. A.; Patel R. P.; Hiley C. R.; Abakumova I.; Requejo R.; Chouchani E. T.; Hurd T. R.; Garvey J. F.; Taylor C. T.; Brookes P. S.; Smith R. A.; Murphy M. P. A mitochondria-targeted S-nitrosothiol modulates respiration, nitrosates thiols, and protects against ischemia-reperfusion injury. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 10764–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R. A.; Hartley R. C.; Cocheme H. M.; Murphy M. P. Mitochondrial pharmacology. Trends Pharmacol. Sci. 2012, 33, 341–52. [DOI] [PubMed] [Google Scholar]
- Babu M.; Vlasblom J.; Pu S.; Guo X.; Graham C.; Bean B. D. M.; Burston H. E.; Vizeacoumar F. J.; Snider J.; Phanse S.; Fong V.; Tam Y. Y. C.; Davey M.; Hnatshak O.; Bajaj N.; Chandran S.; Punna T.; Christopolous C.; Wong V.; Yu A.; Zhong G.; Li J.; Stagljar I.; Conibear E.; Wodak S. J.; Emili A.; Greenblatt J. F. Interaction landscape of membrane-protein complexes in Saccharomyces cerevisiae. Nature 2012, 489, 585–589. [DOI] [PubMed] [Google Scholar]
- Sickmann A.; Reinders J.; Wagner Y.; Joppich C.; Zahedi R.; Meyer H. E.; Schonfisch B.; Perschil I.; Chacinska A.; Guiard B.; Rehling P.; Pfanner N.; Meisinger C. The proteome of Saccharomyces cerevisiae mitochondria. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 13207–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess D. C.; Myers C. L.; Huttenhower C.; Hibbs M. A.; Hayes A. P.; Paw J.; Clore J. J.; Mendoza R. M.; Luis B. S.; Nislow C.; Giaever G.; Costanzo M.; Troyanskaya O. G.; Caudy A. A. Computationally driven, quantitative experiments discover genes required for mitochondrial biogenesis. PLoS Genet. 2009, 5, e1000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini D. J.; Rutter J. Hallmarks of a new era in mitochondrial biochemistry. Genes Dev. 2013, 27, 2615–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor S. W.; Fahy E.; Zhang B.; Glenn G. M.; Warnock D. E.; Wiley S.; Murphy A. N.; Gaucher S. P.; Capaldi R. A.; Gibson B. W.; Ghosh S. S. Characterization of the human heart mitochondrial proteome. Nat. Biotechnol. 2003, 21, 281–6. [DOI] [PubMed] [Google Scholar]
- Meisinger C.; Sickmann A.; Pfanner N. The mitochondrial proteome: from inventory to function. Cell 2008, 134, 22–4. [DOI] [PubMed] [Google Scholar]
- Hung V.; Zou P.; Rhee H. W.; Udeshi N. D.; Cracan V.; Svinkina T.; Carr S. A.; Mootha V. K.; Ting A. Y. Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol. Cell 2014, 55, 332–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Menachem R.; Tal M.; Shadur T.; Pines O. A third of the yeast mitochondrial proteome is dual localized: a question of evolution. Proteomics 2011, 11, 4468–76. [DOI] [PubMed] [Google Scholar]
- Berglund L.; Bjorling E.; Oksvold P.; Fagerberg L.; Asplund A.; Szigyarto C. A.; Persson A.; Ottosson J.; Wernerus H.; Nilsson P.; Lundberg E.; Sivertsson A.; Navani S.; Wester K.; Kampf C.; Hober S.; Ponten F.; Uhlen M. A genecentric Human Protein Atlas for expression profiles based on antibodies. Mol. Cell. Proteomics 2008, 7, 2019–27. [DOI] [PubMed] [Google Scholar]
- Scott M. S.; Calafell S. J.; Thomas D. Y.; Hallett M. T. Refining protein subcellular localization. PLoS Comput. Biol. 2005, 1, e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotz C.; Lin A. J.; Black C. M.; Zhang J.; Lau E.; Deng N.; Wang Y.; Zong N. C.; Choi J. H.; Xu T.; Liem D. A.; Korge P.; Weiss J. N.; Hermjakob H.; Yates J. R. III; Apweiler R.; Ping P. Characterization, design, and function of the mitochondrial proteome: from organs to organisms. J. Proteome Res. 2014, 13, 433–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M. S.; Pinto S. M.; Getnet D.; Nirujogi R. S.; Manda S. S.; Chaerkady R.; Madugundu A. K.; Kelkar D. S.; Isserlin R.; Jain S.; Thomas J. K.; Muthusamy B.; Leal-Rojas P.; Kumar P.; Sahasrabuddhe N. A.; Balakrishnan L.; Advani J.; George B.; Renuse S.; Selvan L. D.; Patil A. H.; Nanjappa V.; Radhakrishnan A.; Prasad S.; Subbannayya T.; Raju R.; Kumar M.; Sreenivasamurthy S. K.; Marimuthu A.; Sathe G. J.; Chavan S.; Datta K. K.; Subbannayya Y.; Sahu A.; Yelamanchi S. D.; Jayaram S.; Rajagopalan P.; Sharma J.; Murthy K. R.; Syed N.; Goel R.; Khan A. A.; Ahmad S.; Dey G.; Mudgal K.; Chatterjee A.; Huang T. C.; Zhong J.; Wu X.; Shaw P. G.; Freed D.; Zahari M. S.; Mukherjee K. K.; Shankar S.; Mahadevan A.; Lam H.; Mitchell C. J.; Shankar S. K.; Satishchandra P.; Schroeder J. T.; Sirdeshmukh R.; Maitra A.; Leach S. D.; Drake C. G.; Halushka M. K.; Prasad T. S.; Hruban R. H.; Kerr C. L.; Bader G. D.; Iacobuzio-Donahue C. A.; Gowda H.; Pandey A. A draft map of the human proteome. Nature 2014, 509, 575–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha V. K.; Bunkenborg J.; Olsen J. V.; Hjerrild M.; Wisniewski J. R.; Stahl E.; Bolouri M. S.; Ray H. N.; Sihag S.; Kamal M.; Patterson N.; Lander E. S.; Mann M. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell 2003, 115, 629–40. [DOI] [PubMed] [Google Scholar]
- Balaban R. S. The mitochondrial proteome: a dynamic functional program in tissues and disease states. Environ. Mol. Mutagen. 2010, 51, 352–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forner F.; Foster L. J.; Campanaro S.; Valle G.; Mann M. Quantitative proteomic comparison of rat mitochondria from muscle, heart, and liver. Mol. Cell. Proteomics 2006, 5, 608–19. [DOI] [PubMed] [Google Scholar]
- Mootha V. K.; Lepage P.; Miller K.; Bunkenborg J.; Reich M.; Hjerrild M.; Delmonte T.; Villeneuve A.; Sladek R.; Xu F.; Mitchell G. A.; Morin C.; Mann M.; Hudson T. J.; Robinson B.; Rioux J. D.; Lander E. S. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 605–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppen M.; Metodiev M. D.; Casari G.; Rugarli E. I.; Langer T. Variable and tissue-specific subunit composition of mitochondrial m-AAA protease complexes linked to hereditary spastic paraplegia. Mol. Cell. Biol. 2007, 27, 758–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaizumi Y.; Okano H. Modeling human neurological disorders with induced pluripotent stem cells. J. Neurochem. 2014, 129, 388–99. [DOI] [PubMed] [Google Scholar]
- Velasco I.; Salazar P.; Giorgetti A.; Ramos-Mejia V.; Castano J.; Romero-Moya D.; Menendez P. Concise review: generation of neurons from somatic cells of healthy individuals and neurological patients through induced pluripotency or direct conversion. Stem Cells 2014, 32, 2811–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardiff D. F.; Jui N. T.; Khurana V.; Tambe M. A.; Thompson M. L.; Chung C. Y.; Kamadurai H. B.; Kim H. T.; Lancaster A. K.; Caldwell K. A.; Caldwell G. A.; Rochet J. C.; Buchwald S. L.; Lindquist S. Yeast reveal a “druggable” Rsp5/Nedd4 network that ameliorates alpha-synuclein toxicity in neurons. Science 2013, 342, 979–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C. Y.; Khurana V.; Auluck P. K.; Tardiff D. F.; Mazzulli J. R.; Soldner F.; Baru V.; Lou Y.; Freyzon Y.; Cho S.; Mungenast A. E.; Muffat J.; Mitalipova M.; Pluth M. D.; Jui N. T.; Schule B.; Lippard S. J.; Tsai L. H.; Krainc D.; Buchwald S. L.; Jaenisch R.; Lindquist S. Identification and rescue of alpha-synuclein toxicity in Parkinson patient-derived neurons. Science 2013, 342, 983–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard J. V.; Schapira A. H. Mitochondrial respiratory chain disorders II: neurodegenerative disorders and nuclear gene defects. Lancet 2000, 355, 389–94. [DOI] [PubMed] [Google Scholar]
- Halestrap A. Biochemistry: a pore way to die. Nature 2005, 434, 578–9. [DOI] [PubMed] [Google Scholar]
- Murphy E.; Steenbergen C. What makes the mitochondria a killer? Can we condition them to be less destructive?. Biochim. Biophys. Acta 2011, 1813, 1302–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace D. C.; Fan W.; Procaccio V. Mitochondrial energetics and therapeutics. Ann. Rev. Pathol. 2010, 5, 297–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasters J. J.; Theruvath T. P.; Zhong Z.; Nieminen A. L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta 2009, 1787, 1395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchier-Hayes L.; Lartigue L.; Newmeyer D. D. Mitochondria: pharmacological manipulation of cell death. J. Clin. Invest. 2005, 115, 2640–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D.; Weinberg R. A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–74. [DOI] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science 1956, 123, 309–14. [DOI] [PubMed] [Google Scholar]
- Munoz-Pinedo C.; El Mjiyad N.; Ricci J. E. Cancer metabolism: current perspectives and future directions. Cell Death Dis. 2012, 3, e248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verschoor M. L.; Wilson L. A.; Singh G. Mechanisms associated with mitochondrial-generated reactive oxygen species in cancer. Can. J. Physiol. Pharmacol. 2010, 88, 204–19. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I.; Mattson M. P. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008, 31, 454–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straface E.; Matarrese P.; Gambardella L.; Vona R.; Sgadari A.; Silveri M. C.; Malorni W. Oxidative imbalance and cathepsin D changes as peripheral blood biomarkers of Alzheimer disease: a pilot study. FEBS Lett. 2005, 579, 2759–66. [DOI] [PubMed] [Google Scholar]
- Mastrocola R.; Restivo F.; Vercellinatto I.; Danni O.; Brignardello E.; Aragno M.; Boccuzzi G. Oxidative and nitrosative stress in brain mitochondria of diabetic rats. J. Endocrinol. 2005, 187, 37–44. [DOI] [PubMed] [Google Scholar]
- de la Torre J. C.; Stefano G. B. Evidence that Alzheimer’s disease is a microvascular disorder: the role of constitutive nitric oxide. Brain Res. Rev. 2000, 34, 119–36. [DOI] [PubMed] [Google Scholar]
- Moreira P. I.; Santos M. S.; Seica R.; Oliveira C. R. Brain mitochondrial dysfunction as a link between Alzheimer’s disease and diabetes. J. Neurol. Sci. 2007, 257, 206–14. [DOI] [PubMed] [Google Scholar]
- Zhao W. Q.; Townsend M. Insulin resistance and amyloidogenesis as common molecular foundation for type 2 diabetes and Alzheimer’s disease. Biochim. Biophys. Acta 2009, 1792, 482–96. [DOI] [PubMed] [Google Scholar]
- Knott A. B.; Perkins G.; Schwarzenbacher R.; Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 505–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N.; Nomura M.; Jofuku A.; Kato H.; Suzuki S. O.; Masuda K.; Otera H.; Nakanishi Y.; Nonaka I.; Goto Y.; Taguchi N.; Morinaga H.; Maeda M.; Takayanagi R.; Yokota S.; Mihara K. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 2009, 11, 958–66. [DOI] [PubMed] [Google Scholar]
- Chatr-Aryamontri A.; Breitkreutz B. J.; Heinicke S.; Boucher L.; Winter A.; Stark C.; Nixon J.; Ramage L.; Kolas N.; O’Donnell L.; Reguly T.; Breitkreutz A.; Sellam A.; Chen D.; Chang C.; Rust J.; Livstone M.; Oughtred R.; Dolinski K.; Tyers M. The BioGRID interaction database: 2013 update. Nucleic Acids Res. 2013, 41, D816–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gioscia-Ryan R. A.; LaRocca T. J.; Sindler A. L.; Zigler M. C.; Murphy M. P.; Seals D. R. Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. J. Physiol. 2014, 592, 2549–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R. A.; Hartley R. C.; Murphy M. P. Mitochondria-targeted small molecule therapeutics and probes. Antiox. Redox Signaling 2011, 15, 3021–38. [DOI] [PubMed] [Google Scholar]
- Szeto H. H.; Schiller P. W. Novel therapies targeting inner mitochondrial membrane—from discovery to clinical development. Pharm. Res. 2011, 28, 2669–79. [DOI] [PubMed] [Google Scholar]
- Maschek G.; Savaraj N.; Priebe W.; Braunschweiger P.; Hamilton K.; Tidmarsh G. F.; De Young L. R.; Lampidis T. J. 2-Deoxy-d-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Res. 2004, 64, 31–4. [DOI] [PubMed] [Google Scholar]
- Walters A. M.; Porter G. A. Jr.; Brookes P. S. Mitochondria as a drug target in ischemic heart disease and cardiomyopathy. Circ. Res. 2012, 111, 1222–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang S. Y.; Kang H. T.; Hwang E. S. Nicotinamide-induced mitophagy: event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J. Biol. Chem. 2012, 287, 19304–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green K. N.; Steffan J. S.; Martinez-Coria H.; Sun X.; Schreiber S. S.; Thompson L. M.; LaFerla F. M. Nicotinamide restores cognition in Alzheimer’s disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J. Neurosci. 2008, 28, 11500–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um J. H.; Park S. J.; Kang H.; Yang S.; Foretz M.; McBurney M. W.; Kim M. K.; Viollet B.; Chung J. H. AMP-activated protein kinase-deficient mice are resistant to the metabolic effects of resveratrol. Diabetes 2010, 59, 554–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viscomi C.; Bottani E.; Civiletto G.; Cerutti R.; Moggio M.; Fagiolari G.; Schon E. A.; Lamperti C.; Zeviani M. In vivo correction of COX deficiency by activation of the AMPK/PGC-1alpha axis. Cell Metab. 2011, 14, 80–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne J. C.; Lambert P. D.; Schenk S.; Carney D. P.; Smith J. J.; Gagne D. J.; Jin L.; Boss O.; Perni R. B.; Vu C. B.; Bemis J. E.; Xie R.; Disch J. S.; Ng P. Y.; Nunes J. J.; Lynch A. V.; Yang H.; Galonek H.; Israelian K.; Choy W.; Iffland A.; Lavu S.; Medvedik O.; Sinclair D. A.; Olefsky J. M.; Jirousek M. R.; Elliott P. J.; Westphal C. H. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 2007, 450, 712–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patti M. E.; Corvera S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr. Rev. 2010, 31, 364–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoukas M. A.; Farr O. M.; Mantzoros C. S. Advances in metabolism. Metab., Clin. Exp. 2013, 62, 1700–13. [DOI] [PubMed] [Google Scholar]
- Blake R.; Trounce I. A. Mitochondrial dysfunction and complications associated with diabetes. Biochim. Biophys. Acta 2014, 1840, 1404–12. [DOI] [PubMed] [Google Scholar]
- Murphy M. P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atamna H.; Kumar R. Protective role of methylene blue in Alzheimer’s disease via mitochondria and cytochrome c oxidase. J. Alzheimer’s Dis. 2010, 20, S439–52. [DOI] [PubMed] [Google Scholar]
- Chae S.; Ahn B. Y.; Byun K.; Cho Y. M.; Yu M. H.; Lee B.; Hwang D.; Park K. S. A systems approach for decoding mitochondrial retrograde signaling pathways. Sci. Signaling 2013, 6, rs4. [DOI] [PubMed] [Google Scholar]
- Knouff C.; Auwerx J. Peroxisome proliferator-activated receptor-gamma calls for activation in moderation: lessons from genetics and pharmacology. Endocr. Rev. 2004, 25, 899–918. [DOI] [PubMed] [Google Scholar]
- Schreiber S. N.; Emter R.; Hock M. B.; Knutti D.; Cardenas J.; Podvinec M.; Oakeley E. J.; Kralli A. The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 6472–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas R. R.; Khan S. M.; Smigrodzki R. M.; Onyango I. G.; Dennis J.; Khan O. M.; Portelli F. R.; Bennett J. P. Jr. RhTFAM treatment stimulates mitochondrial oxidative metabolism and improves memory in aged mice. Aging 2012, 4, 620–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csiszar A.; Labinskyy N.; Pinto J. T.; Ballabh P.; Zhang H.; Losonczy G.; Pearson K.; de Cabo R.; Pacher P.; Zhang C.; Ungvari Z. Resveratrol induces mitochondrial biogenesis in endothelial cells. Am. J. Physiol.: Heart Circ. Physiol. 2009, 297, H13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deas E.; Wood N. W.; Plun-Favreau H. Mitophagy and Parkinson’s disease: the PINK1-parkin link. Biochim. Biophys. Acta 2011, 1813, 623–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskovits A. Z.; Guarente L. Sirtuin deacetylases in neurodegenerative diseases of aging. Cell Res. 2013, 23, 746–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasson S. A.; Kane L. A.; Yamano K.; Huang C. H.; Sliter D. A.; Buehler E.; Wang C.; Heman-Ackah S. M.; Hessa T.; Guha R.; Martin S. E.; Youle R. J. High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature 2013, 504, 291–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy-Stone A.; Chipuk J. E.; Ingerman E.; Song C.; Yoo C.; Kuwana T.; Kurth M. J.; Shaw J. T.; Hinshaw J. E.; Green D. R.; Nunnari J. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 2008, 14, 193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue W.; Chen Z.; Liu H.; Yan C.; Chen M.; Feng D.; Yan C.; Wu H.; Du L.; Wang Y.; Liu J.; Huang X.; Xia L.; Liu L.; Wang X.; Jin H.; Wang J.; Song Z.; Hao X.; Chen Q. A small natural molecule promotes mitochondrial fusion through inhibition of the deubiquitinase USP30. Cell Res. 2014, 24, 482–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian W.; Wang J.; Roginskaya V.; McDermott L. A.; Edwards R. P.; Stolz D. B.; Llambi F.; Green D. R.; Van Houten B. Novel combination of mitochondrial division inhibitor 1 (mdivi-1) and platinum agents produces synergistic pro-apoptotic effect in drug resistant tumor cells. Oncotarget 2014, 5, 4180–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P.; Broekemeier K. M.; Pfeiffer D. R. Recent progress on regulation of the mitochondrial permeability transition pore; a cyclosporin-sensitive pore in the inner mitochondrial membrane. J. Bioenerg. Biomembr. 1994, 26, 509–17. [DOI] [PubMed] [Google Scholar]
- Gomez L.; Li B.; Mewton N.; Sanchez I.; Piot C.; Elbaz M.; Ovize M. Inhibition of mitochondrial permeability transition pore opening: translation to patients. Cardiovasc. Res. 2009, 83, 226–33. [DOI] [PubMed] [Google Scholar]
- Osman M. M.; Lulic D.; Glover L.; Stahl C. E.; Lau T.; van Loveren H.; Borlongan C. V. Cyclosporine-A as a neuroprotective agent against stroke: its translation from laboratory research to clinical application. Neuropeptides 2011, 45, 359–68. [DOI] [PubMed] [Google Scholar]
- Hansson M. J.; Mattiasson G.; Mansson R.; Karlsson J.; Keep M. F.; Waldmeier P.; Ruegg U. T.; Dumont J. M.; Besseghir K.; Elmer E. The nonimmunosuppressive cyclosporin analogs NIM811 and UNIL025 display nanomolar potencies on permeability transition in brain-derived mitochondria. J. Bioenerg. Biomembr. 2004, 36, 407–13. [DOI] [PubMed] [Google Scholar]
- Reichert A. S.; Neupert W. Contact sites between the outer and inner membrane of mitochondria—role in protein transport. Biochim. Biophys. Acta 2002, 1592, 41–9. [DOI] [PubMed] [Google Scholar]
- Calvo S. E.; Mootha V. K. The mitochondrial proteome and human disease. Annu. Rev. Genomics Hum. Genet. 2010, 11, 25–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loscalzo J.; Barabasi A. L. Systems biology and the future of medicine. Wiley Interdiscip. Rev.: Syst. Biol. Med. 2011, 3, 619–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillenmeyer M. E.; Ericson E.; Davis R. W.; Nislow C.; Koller D.; Giaever G. Systematic analysis of genome-wide fitness data in yeast reveals novel gene function and drug action. Genome Biol. 2010, 11, R30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S.; Kim D. Inference of protein complex activities from chemical-genetic profile and its applications: predicting drug-target pathways. PLoS Comput. Biol. 2008, 4, e1000162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viola H. M.; Adams A. M.; Davies S. M.; Fletcher S.; Filipovska A.; Hool L. C. Impaired functional communication between the L-type calcium channel and mitochondria contributes to metabolic inhibition in the mdx heart. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, E2905–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao V. K.; Carlson E. A.; Yan S. S. Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim. Biophys. Acta 2014, 1842, 1267–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D. W.; Colombini M. Inhibition by aluminum hydroxide of the voltage-dependent closure of the mitochondrial channel, VDAC. Biochim. Biophys. Acta 1989, 991, 68–78. [DOI] [PubMed] [Google Scholar]
- Zhang D. W.; Colombini M. Group IIIA-metal hydroxides indirectly neutralize the voltage sensor of the voltage-dependent mitochondrial channel, VDAC, by interacting with a dynamic binding site. Biochim. Biophys. Acta 1990, 1025, 127–34. [DOI] [PubMed] [Google Scholar]
- Fraser H. B.; Plotkin J. B. Using protein complexes to predict phenotypic effects of gene mutation. Genome Biol. 2007, 8, R252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froese D. S.; Kochan G.; Muniz J. R.; Wu X.; Gileadi C.; Ugochukwu E.; Krysztofinska E.; Gravel R. A.; Oppermann U.; Yue W. W. Structures of the human GTPase MMAA and vitamin B12-dependent methylmalonyl-CoA mutase and insight into their complex formation. J. Biol. Chem. 2010, 285, 38204–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuaje F. Drug interaction networks: an introduction to translational and clinical applications. Cardiovasc. Res. 2013, 97, 631–41. [DOI] [PubMed] [Google Scholar]
- Radogna F.; Cristofanon S.; Paternoster L.; D’Alessio M.; De Nicola M.; Cerella C.; Dicato M.; Diederich M.; Ghibelli L. Melatonin antagonizes the intrinsic pathway of apoptosis via mitochondrial targeting of Bcl-2. J. Pineal Res. 2008, 44, 316–25. [DOI] [PubMed] [Google Scholar]
- Muller I.; Larsson K.; Frenzel A.; Oliynyk G.; Zirath H.; Prochownik E. V.; Westwood N. J.; Henriksson M. A. Targeting of the MYCN protein with small molecule c-MYC inhibitors. PLoS One 2014, 9, e97285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zirath H.; Frenzel A.; Oliynyk G.; Segerstrom L.; Westermark U. K.; Larsson K.; Munksgaard Persson M.; Hultenby K.; Lehtio J.; Einvik C.; Pahlman S.; Kogner P.; Jakobsson P. J.; Henriksson M. A. MYC inhibition induces metabolic changes leading to accumulation of lipid droplets in tumor cells. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 10258–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J.; Zhang N.; Yin G.; Zhang Z.; Cheng G.; Qian W.; Long H.; Cai W. Edaravone protects cortical neurons from apoptosis by inhibiting the translocation of BAX and Increasing the interaction between 14-3-3 and p-BAD. Int. J. Neurosci. 2012, 122, 665–74. [DOI] [PubMed] [Google Scholar]
- Yu H.; Braun P.; Yildirim M. A.; Lemmens I.; Venkatesan K.; Sahalie J.; Hirozane-Kishikawa T.; Gebreab F.; Li N.; Simonis N.; Hao T.; Rual J. F.; Dricot A.; Vazquez A.; Murray R. R.; Simon C.; Tardivo L.; Tam S.; Svrzikapa N.; Fan C.; de Smet A. S.; Motyl A.; Hudson M. E.; Park J.; Xin X.; Cusick M. E.; Moore T.; Boone C.; Snyder M.; Roth F. P.; Barabasi A. L.; Tavernier J.; Hill D. E.; Vidal M. High-quality binary protein interaction map of the yeast interactome network. Science 2008, 322, 104–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider J.; Hanif A.; Lee M. E.; Jin K.; Yu A. R.; Graham C.; Chuk M.; Damjanovic D.; Wierzbicka M.; Tang P.; Balderes D.; Wong V.; Jessulat M.; Darowski K. D.; San Luis B. J.; Shevelev I.; Sturley S. L.; Boone C.; Greenblatt J. F.; Zhang Z.; Paumi C. M.; Babu M.; Park H. O.; Michaelis S.; Stagljar I. Mapping the functional yeast ABC transporter interactome. Nat. Chem. Biol. 2013, 9, 565–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarassov K.; Messier V.; Landry C. R.; Radinovic S.; Serna Molina M. M.; Shames I.; Malitskaya Y.; Vogel J.; Bussey H.; Michnick S. W. An in vivo map of the yeast protein interactome. Science 2008, 320, 1465–70. [DOI] [PubMed] [Google Scholar]
- Krogan N. J.; Cagney G.; Yu H.; Zhong G.; Guo X.; Ignatchenko A.; Li J.; Pu S.; Datta N.; Tikuisis A. P.; Punna T.; Peregrin-Alvarez J. M.; Shales M.; Zhang X.; Davey M.; Robinson M. D.; Paccanaro A.; Bray J. E.; Sheung A.; Beattie B.; Richards D. P.; Canadien V.; Lalev A.; Mena F.; Wong P.; Starostine A.; Canete M. M.; Vlasblom J.; Wu S.; Orsi C.; Collins S. R.; Chandran S.; Haw R.; Rilstone J. J.; Gandi K.; Thompson N. J.; Musso G.; St. Onge P.; Ghanny S.; Lam M. H.; Butland G.; Altaf-Ul A. M.; Kanaya S.; Shilatifard A.; O’Shea E.; Weissman J. S.; Ingles C. J.; Hughes T. R.; Parkinson J.; Gerstein M.; Wodak S. J.; Emili A.; Greenblatt J. F. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 2006, 440, 637–43. [DOI] [PubMed] [Google Scholar]
- Gavin A. C.; Aloy P.; Grandi P.; Krause R.; Boesche M.; Marzioch M.; Rau C.; Jensen L. J.; Bastuck S.; Dumpelfeld B.; Edelmann A.; Heurtier M. A.; Hoffman V.; Hoefert C.; Klein K.; Hudak M.; Michon A. M.; Schelder M.; Schirle M.; Remor M.; Rudi T.; Hooper S.; Bauer A.; Bouwmeester T.; Casari G.; Drewes G.; Neubauer G.; Rick J. M.; Kuster B.; Bork P.; Russell R. B.; Superti-Furga G. Proteome survey reveals modularity of the yeast cell machinery. Nature 2006, 440, 631–6. [DOI] [PubMed] [Google Scholar]
- Mak A. B.; Ni Z.; Hewel J. A.; Chen G. I.; Zhong G.; Karamboulas K.; Blakely K.; Smiley S.; Marcon E.; Roudeva D.; Li J.; Olsen J. B.; Wan C.; Punna T.; Isserlin R.; Chetyrkin S.; Gingras A. C.; Emili A.; Greenblatt J.; Moffat J. A lentiviral functional proteomics approach identifies chromatin remodeling complexes important for the induction of pluripotency. Mol. Cell. Proteomics 2010, 9, 811–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcon E.; Ni Z.; Pu S.; Turinsky A. L.; Trimble S. S.; Olsen J. B.; Silverman-Gavrila R.; Silverman-Gavrila L.; Phanse S.; Guo H.; Zhong G.; Guo X.; Young P.; Bailey S.; Roudeva D.; Zhao D.; Hewel J.; Li J.; Graslund S.; Paduch M.; Kossiakoff A. A.; Lupien M.; Emili A.; Wodak S. J.; Greenblatt J. Human-chromatin-related protein interactions identify a demethylase complex required for chromosome segregation. Cell Rep. 2014, 8, 297–310. [DOI] [PubMed] [Google Scholar]
- Kapitzky L.; Beltrao P.; Berens T. J.; Gassner N.; Zhou C.; Wuster A.; Wu J.; Babu M. M.; Elledge S. J.; Toczyski D.; Lokey R. S.; Krogan N. J. Cross-species chemogenomic profiling reveals evolutionarily conserved drug mode of action. Mol. Syst. Biol. 2010, 6, 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillenmeyer M. E.; Fung E.; Wildenhain J.; Pierce S. E.; Hoon S.; Lee W.; Proctor M.; St. Onge R. P.; Tyers M.; Koller D.; Altman R. B.; Davis R. W.; Nislow C.; Giaever G. The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science 2008, 320, 362–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrtic M.; Sriskanthadevan S.; Jhas B.; Gebbia M.; Wang X.; Wang Z.; Hurren R.; Jitkova Y.; Gronda M.; Maclean N.; Lai C. K.; Eberhard Y.; Bartoszko J.; Spagnuolo P.; Rutledge A. C.; Datti A.; Ketela T.; Moffat J.; Robinson B. H.; Cameron J. H.; Wrana J.; Eaves C. J.; Minden M. D.; Wang J. C.; Dick J. E.; Humphries K.; Nislow C.; Giaever G.; Schimmer A. D. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 2011, 20, 674–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y.; Bowden B. F.; Kapoor V.; Ianthellamide A. A selective kynurenine-3-hydroxylase inhibitor from the Australian marine sponge Ianthella quadrangulata. Bioorg. Med. Chem. Lett. 2012, 22, 3398–401. [DOI] [PubMed] [Google Scholar]
- Amaral M.; Levy C.; Heyes D. J.; Lafite P.; Outeiro T. F.; Giorgini F.; Leys D.; Scrutton N. S. Structural basis of kynurenine 3-monooxygenase inhibition. Nature 2013, 496, 382–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgini F.; Guidetti P.; Nguyen Q.; Bennett S. C.; Muchowski P. J. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat. Genet. 2005, 37, 526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwilling D.; Huang S. Y.; Sathyasaikumar K. V.; Notarangelo F. M.; Guidetti P.; Wu H. Q.; Lee J.; Truong J.; Andrews-Zwilling Y.; Hsieh E. W.; Louie J. Y.; Wu T.; Scearce-Levie K.; Patrick C.; Adame A.; Giorgini F.; Moussaoui S.; Laue G.; Rassoulpour A.; Flik G.; Huang Y.; Muchowski J. M.; Masliah E.; Schwarcz R.; Muchowski P. J. Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration. Cell 2011, 145, 863–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couplan E.; Aiyar R. S.; Kucharczyk R.; Kabala A.; Ezkurdia N.; Gagneur J.; St. Onge R. P.; Salin B.; Soubigou F.; Le Cann M.; Steinmetz L. M.; di Rago J. P.; Blondel M. A yeast-based assay identifies drugs active against human mitochondrial disorders. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 11989–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppins S.; Collins S. R.; Cassidy-Stone A.; Hummel E.; Devay R. M.; Lackner L. L.; Westermann B.; Schuldiner M.; Weissman J. S.; Nunnari J. A mitochondrial-focused genetic interaction map reveals a scaffold-like complex required for inner membrane organization in mitochondria. J. Cell Biol. 2011, 195, 323–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M.; Baryshnikova A.; Bellay J.; Kim Y.; Spear E. D.; Sevier C. S.; Ding H.; Koh J. L.; Toufighi K.; Mostafavi S.; Prinz J.; St. Onge R. P.; VanderSluis B.; Makhnevych T.; Vizeacoumar F. J.; Alizadeh S.; Bahr S.; Brost R. L.; Chen Y.; Cokol M.; Deshpande R.; Li Z.; Lin Z. Y.; Liang W.; Marback M.; Paw J.; San Luis B. J.; Shuteriqi E.; Tong A. H.; van Dyk N.; Wallace I. M.; Whitney J. A.; Weirauch M. T.; Zhong G.; Zhu H.; Houry W. A.; Brudno M.; Ragibizadeh S.; Papp B.; Pal C.; Roth F. P.; Giaever G.; Nislow C.; Troyanskaya O. G.; Bussey H.; Bader G. D.; Gingras A. C.; Morris Q. D.; Kim P. M.; Kaiser C. A.; Myers C. L.; Andrews B. J.; Boone C. The genetic landscape of a cell. Science 2010, 327, 425–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luu Y. K.; Rana P.; Duensing T. D.; Black C.; Will Y. Profiling of toxicity and identification of distinct apoptosis profiles using a 384-well high-throughput flow cytometry screening platform. J. Biomol. Screening 2012, 17, 806–12. [DOI] [PubMed] [Google Scholar]
- Roguev A.; Talbot D.; Negri G. L.; Shales M.; Cagney G.; Bandyopadhyay S.; Panning B.; Krogan N. J. Quantitative genetic-interaction mapping in mammalian cells. Nat. Methods 2013, 10, 432–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizeacoumar F. J.; Arnold R.; Vizeacoumar F. S.; Chandrashekhar M.; Buzina A.; Young J. T.; Kwan J. H.; Sayad A.; Mero P.; Lawo S.; Tanaka H.; Brown K. R.; Baryshnikova A.; Mak A. B.; Fedyshyn Y.; Wang Y.; Brito G. C.; Kasimer D.; Makhnevych T.; Ketela T.; Datti A.; Babu M.; Emili A.; Pelletier L.; Wrana J.; Wainberg Z.; Kim P. M.; Rottapel R.; O’Brien C. A.; Andrews B.; Boone C.; Moffat J. A negative genetic interaction map in isogenic cancer cell lines reveals cancer cell vulnerabilities. Mol. Syst. Biol. 2013, 9, 696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketela T.; Heisler L. E.; Brown K. R.; Ammar R.; Kasimer D.; Surendra A.; Ericson E.; Blakely K.; Karamboulas D.; Smith A. M.; Durbic T.; Arnoldo A.; Cheung-Ong K.; Koh J. L.; Gopal S.; Cowley G. S.; Yang X.; Grenier J. K.; Giaever G.; Root D. E.; Moffat J.; Nislow C. A comprehensive platform for highly multiplexed mammalian functional genetic screens. BMC Genomics 2011, 12, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aza-Blanc P.; Cooper C. L.; Wagner K.; Batalov S.; Deveraux Q. L.; Cooke M. P. Identification of modulators of TRAIL-induced apoptosis via RNAi-based phenotypic screening. Mol. Cell 2003, 12, 627–37. [DOI] [PubMed] [Google Scholar]
- Bassik M. C.; Kampmann M.; Lebbink R. J.; Wang S.; Hein M. Y.; Poser I.; Weibezahn J.; Horlbeck M. A.; Chen S.; Mann M.; Hyman A. A.; Leproust E. M.; McManus M. T.; Weissman J. S. A systematic mammalian genetic interaction map reveals pathways underlying ricin susceptibility. Cell 2013, 152, 909–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A. Y.; St. Onge R. P.; Proctor M. J.; Wallace I. M.; Nile A. H.; Spagnuolo P. A.; Jitkova Y.; Gronda M.; Wu Y.; Kim M. K.; Cheung-Ong K.; Torres N. P.; Spear E. D.; Han M. K.; Schlecht U.; Suresh S.; Duby G.; Heisler L. E.; Surendra A.; Fung E.; Urbanus M. L.; Gebbia M.; Lissina E.; Miranda M.; Chiang J. H.; Aparicio A. M.; Zeghouf M.; Davis R. W.; Cherfils J.; Boutry M.; Kaiser C. A.; Cummins C. L.; Trimble W. S.; Brown G. W.; Schimmer A. D.; Bankaitis V. A.; Nislow C.; Bader G. D.; Giaever G. Mapping the cellular response to small molecules using chemogenomic fitness signatures. Science 2014, 344, 208–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan C. J.; Cimermancic P.; Szpiech Z. A.; Sali A.; Hernandez R. D.; Krogan N. J. High-resolution network biology: connecting sequence with function. Nat. Rev. Genet. 2013, 14, 865–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanning N. J.; Looyenga B. D.; Kauffman A. L.; Niemi N. M.; Sudderth J.; DeBerardinis R. J.; MacKeigan J. P. A mitochondrial RNAi screen defines cellular bioenergetic determinants and identifies an adenylate kinase as a key regulator of ATP levels. Cell Rep. 2014, 7, 907–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D.; Zhao L.; Clapham D. E. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 2009, 326, 144–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Samuel B. S.; Breen P. C.; Ruvkun G. Caenorhabditis elegans pathways that surveil and defend mitochondria. Nature 2014, 508, 406–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker M.; Samsonova A. A.; Kwon Y.; Flockhart I.; Mohr S. E.; Perrimon N. False negative rates in Drosophila cell-based RNAi screens: a case study. BMC Genomics 2011, 12, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echeverri C. J.; Beachy P. A.; Baum B.; Boutros M.; Buchholz F.; Chanda S. K.; Downward J.; Ellenberg J.; Fraser A. G.; Hacohen N.; Hahn W. C.; Jackson A. L.; Kiger A.; Linsley P. S.; Lum L.; Ma Y.; Mathey-Prevot B.; Root D. E.; Sabatini D. M.; Taipale J.; Perrimon N.; Bernards R. Minimizing the risk of reporting false positives in large-scale RNAi screens. Nat. Methods 2006, 3, 777–9. [DOI] [PubMed] [Google Scholar]
- Kaelin W. G. Jr. Molecular biology. Use and abuse of RNAi to study mammalian gene function. Science 2012, 337, 421–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O.; Sanjana N. E.; Hartenian E.; Shi X.; Scott D. A.; Mikkelsen T. S.; Heckl D.; Ebert B. L.; Root D. E.; Doench J. G.; Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T.; Wei J. J.; Sabatini D. M.; Lander E. S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y.; Sander J. D.; Reyon D.; Cascio V. M.; Joung J. K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havugimana P. C.; Hart G. T.; Nepusz T.; Yang H.; Turinsky A. L.; Li Z.; Wang P. I.; Boutz D. R.; Fong V.; Phanse S.; Babu M.; Craig S. A.; Hu P.; Wan C.; Vlasblom J.; Dar V.-N.; Bezginov A.; Clark G. W.; Wu G. C.; Wodak S. J.; Tillier E. R. M.; Paccanaro A.; Marcotte E. M.; Emili A. A census of human soluble protein complexes. Cell 2012, 150, 1068–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker T.; Sharan R. Protein networks in disease. Genome Res. 2008, 18, 644–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande R.; Asiedu M. K.; Klebig M.; Sutor S.; Kuzmin E.; Nelson J.; Piotrowski J.; Shin S. H.; Yoshida M.; Costanzo M.; Boone C.; Wigle D. A.; Myers C. L. A comparative genomic approach for identifying synthetic lethal interactions in human cancer. Cancer Res. 2013, 73, 6128–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guruharsha K. G.; Rual J. F.; Zhai B.; Mintseris J.; Vaidya P.; Vaidya N.; Beekman C.; Wong C.; Rhee D. Y.; Cenaj O.; McKillip E.; Shah S.; Stapleton M.; Wan K. H.; Yu C.; Parsa B.; Carlson J. W.; Chen X.; Kapadia B.; VijayRaghavan K.; Gygi S. P.; Celniker S. E.; Obar R. A.; Artavanis-Tsakonas S. A protein complex network of Drosophila melanogaster. Cell 2011, 147, 690–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter H.; Hofree M.; Ideker T. Genotype to phenotype via network analysis. Curr. Opin. Genet. Dev. 2013, 23, 611–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi T. K.; Zhong J.; Mathivanan S.; Karthick L.; Chandrika K. N.; Mohan S. S.; Sharma S.; Pinkert S.; Nagaraju S.; Periaswamy B.; Mishra G.; Nandakumar K.; Shen B.; Deshpande N.; Nayak R.; Sarker M.; Boeke J. D.; Parmigiani G.; Schultz J.; Bader J. S.; Pandey A. Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nat. Genet. 2006, 38, 285–93. [DOI] [PubMed] [Google Scholar]
- Pandey U. B.; Nichols C. D. Human disease models in Drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol. Rev. 2011, 63, 411–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H.; Wang D.; Chen L.; Choo Y. S.; Ma H.; Tang C.; Xia K.; Jiang W.; Ronai Z.; Zhuang X.; Zhang Z. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J. Clin. Invest. 2009, 119, 650–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark I. E.; Dodson M. W.; Jiang C.; Cao J. H.; Huh J. R.; Seol J. H.; Yoo S. J.; Hay B. A.; Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–6. [DOI] [PubMed] [Google Scholar]
- Park J.; Lee S. B.; Lee S.; Kim Y.; Song S.; Kim S.; Bae E.; Kim J.; Shong M.; Kim J. M.; Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006, 441, 1157–61. [DOI] [PubMed] [Google Scholar]
- Sabado J.; Casanovas A.; Tarabal O.; Hereu M.; Piedrafita L.; Caldero J.; Esquerda J. E. Accumulation of Misfolded SOD1 in Dorsal Root Ganglion Degenerating Proprioceptive Sensory Neurons of Transgenic Mice with Amyotrophic Lateral Sclerosis. BioMed Res. Int. 2014, 2014, 852163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G.; Zhang Z.; Bao Q.; Zhang Z.; Zhou L.; Jiang J.; Li S. Protective effect of chinonin in MPTP-induced C57BL/6 mouse model of Parkinson’s disease. Biol. Pharm. Bull. 2014, 37, 1301–7. [DOI] [PubMed] [Google Scholar]
- Martinez-Jimenez F.; Papadatos G.; Yang L.; Wallace I. M.; Kumar V.; Pieper U.; Sali A.; Brown J. R.; Overington J. P.; Marti-Renom M. A. Target prediction for an open access set of compounds active against Mycobacterium tuberculosis. PLoS Comput. Biol. 2013, 9, e1003253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perot S.; Regad L.; Reynes C.; Sperandio O.; Miteva M. A.; Villoutreix B. O.; Camproux A. C. Insights into an original pocket-ligand pair classification: a promising tool for ligand profile prediction. PLoS One 2013, 8, e63730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozalbes R.; Mosulen S.; Orti L.; Rodriguez-Diaz J.; Carbajo R. J.; Melnyk P.; Pineda-Lucena A. Hit identification of novel heparanase inhibitors by structure- and ligand-based approaches. Bioorg. Med. Chem. 2013, 21, 1944–51. [DOI] [PubMed] [Google Scholar]
- Pang Y. P. In silico drug discovery: solving the “target-rich and lead-poor” imbalance using the genome-to-drug-lead paradigm. Clin. Pharmacol. Ther. 2007, 81, 30–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M. B. Seeing is believing: the impact of structural genomics on antimicrobial drug discovery. Nat. Rev. Microbiol. 2004, 2, 739–46. [DOI] [PubMed] [Google Scholar]
- Kare P.; Bhat J.; Sobhia M. E. Structure-based design and analysis of MAO-B inhibitors for Parkinson’s disease: using in silico approaches. Mol. Diversity 2013, 17, 111–22. [DOI] [PubMed] [Google Scholar]
- Li Y. Y.; An J.; Jones S. J. A computational approach to finding novel targets for existing drugs. PLoS Comput. Biol. 2011, 7, e1002139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girrbach M.; Meliciani I.; Waterkotte B.; Berthold S.; Oster A.; Brurein F.; Strunk T.; Wadhwani P.; Berensmeier S.; Wenzel W.; Schmitz K. A fluorescence polarization assay for the experimental validation of an in silico model of the chemokine CXCL8 binding to receptor-derived peptides. Phys. Chem. Chemi. Phys. 2014, 16, 8036–43. [DOI] [PubMed] [Google Scholar]
- Hanumanthappa P.; Rajanikant G. K. In silico identification of potential dynamin-related protein 1 antagonists: implications for diseases involving mitochondrial dysfunction. Comb. Chem. High Throughput Screening 2014, 17, 25–34. [DOI] [PubMed] [Google Scholar]
- Keiser M. J.; Roth B. L.; Armbruster B. N.; Ernsberger P.; Irwin J. J.; Shoichet B. K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [DOI] [PubMed] [Google Scholar]
- Yeh S. H.; Yeh H. Y.; Soo V. W. A network flow approach to predict drug targets from microarray data, disease genes and interactome network - case study on prostate cancer. J. Clin. Bioinf. 2012, 2, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y. A.; Wu J.; Zhang L.; Zhang Q.; Su D. M.; He P.; Wang B. D.; Li H.; Webster M. J.; Rennert O. M.; Ursano R. J. Dysregulated mitochondrial genes and networks with drug targets in postmortem brain of patients with posttraumatic stress disorder (PTSD) revealed by human mitochondria-focused cDNA microarrays. Int. J. Biol. Sci. 2008, 4, 223–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanzoni A.; Soler-Lopez M.; Aloy P. A network medicine approach to human disease. FEBS Lett. 2009, 583, 1759–65. [DOI] [PubMed] [Google Scholar]
- Duran-Frigola M.; Mosca R.; Aloy P. Structural systems pharmacology: the role of 3D structures in next-generation drug development. Chem. Biol. 2013, 20, 674–84. [DOI] [PubMed] [Google Scholar]
- Marton M. J.; DeRisi J. L.; Bennett H. A.; Iyer V. R.; Meyer M. R.; Roberts C. J.; Stoughton R.; Burchard J.; Slade D.; Dai H.; Bassett D. E. Jr.; Hartwell L. H.; Brown P. O.; Friend S. H. Drug target validation and identification of secondary drug target effects using DNA microarrays. Nat. Med. 1998, 4, 1293–301. [DOI] [PubMed] [Google Scholar]
- Hughes T. R.; Marton M. J.; Jones A. R.; Roberts C. J.; Stoughton R.; Armour C. D.; Bennett H. A.; Coffey E.; Dai H.; He Y. D.; Kidd M. J.; King A. M.; Meyer M. R.; Slade D.; Lum P. Y.; Stepaniants S. B.; Shoemaker D. D.; Gachotte D.; Chakraburtty K.; Simon J.; Bard M.; Friend S. H. Functional discovery via a compendium of expression profiles. Cell 2000, 102, 109–26. [DOI] [PubMed] [Google Scholar]
- Lamb J.; Crawford E. D.; Peck D.; Modell J. W.; Blat I. C.; Wrobel M. J.; Lerner J.; Brunet J. P.; Subramanian A.; Ross K. N.; Reich M.; Hieronymus H.; Wei G.; Armstrong S. A.; Haggarty S. J.; Clemons P. A.; Wei R.; Carr S. A.; Lander E. S.; Golub T. R. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–35. [DOI] [PubMed] [Google Scholar]
- Hawley S. A.; Fullerton M. D.; Ross F. A.; Schertzer J. D.; Chevtzoff C.; Walker K. J.; Peggie M. W.; Zibrova D.; Green K. A.; Mustard K. J.; Kemp B. E.; Sakamoto K.; Steinberg G. R.; Hardie D. G. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 2012, 336, 918–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeson C. C.; Beeson G. C.; Schnellmann R. G. A high-throughput respirometric assay for mitochondrial biogenesis and toxicity. Anal. Biochem. 2010, 404, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.