

Abstract
In this report, we demonstrate a rapid, simple, and green method for synthesizing silver-gold (Ag-Au) bimetallic nanoparticles (BNPs). We used a novel modification to the galvanic replacement reaction by suspending maltose coated silver nanoparticles (NPs) in ≈ 2% aqueous solution of EO100PO65EO100 (Pluronic F127) prior to HAuCl4 addition. The Pluronic F127 stabilizes the BNPs, imparts biocompatibility, and mitigates the toxicity issues associated with other surfactant stabilizers. BNPs with higher Au:Ag ratios and, subsequently, different morphologies were successfully synthesized by increasing the concentration of gold salt added to the Ag NP seeds. These BNPs have enhanced catalytic activities than typically reported for monometallic Au or Ag NPs (∼ 2–10 fold) of comparable sizes in the sodium borohydride reduction of 4-nitrophenol. The 4-nitrophenol reduction rates were highest for partially hollow BNP morphologies.
Keywords: bimetallic nanoparticles, triblock copolymer, tunable composition, 4-nitrophenol, catalytic activity
Introduction
Bimetallic nanoparticles (BNPs) have attracted considerable interest in recent years due to their unique catalytic, electronic, and optical properties.1-5 These properties are distinctly different from those of single component nanomaterials and can be optimized by controlling the size, shape, and metallic composition of the particles.6, 7 Several methods for synthesizing BNPs exist,8-10 including a growing volume of literature describing environmentally benign BNP synthesis methods.11, 12 These environmentally benign routes are categorized broadly into physical, biological, and chemical approaches. Physical methods of BNP synthesis include radiation-induced,13, 14 electrochemical,15, 16 sonochemical,17, 18 and spark generation19 techniques. Biological nanomaterial synthesis methods are more cost effective than physical methods and use plant extracts20-23 or microorganisms23-26 to produce the nanoparticles (NPs). These methods, however, can be hard to reproduce, difficult to implement on the large scale, and time consuming. Chemical synthesis methods use reducing agents such as ascorbate27 citrate,28 or borohydride 5 to facilitate the formation of the BNPs and are by far the most commonly used methods for synthesizing BNPs.
Ag-Au BNPs have been extensively studied 5, 8, 11, 29-35 due to their utility in catalysis,1, 36 plasmonics,37 sensors,38, 39 and surface enhanced Raman-scattering.40-42 Xia and coworkers synthesized 50 nm Ag-Au nanocages and boxes with excellent catalytic activity against 4-nitrophenol by the galvanic replacement reaction (GRR).43 Zhang and colleagues synthesized hollow ∼20 nm Ag-Au BNPs by first forming Ag NPs in a polyelectrode multilayer film via an ion-exchange/reducing agent process and then reacting these synthesized Ag NPs with HAuCl4.30 Fu et al. synthesized bimetallic nanowires by the GRR between HAuCl4 and Ag nanowires formed via the reduction of AgNO3 by vanadium oxide.31 Core-shell Ag-Au BNPs with high catalytic activity for glucose oxidation were synthesized by the co-reduction of AgClO4 and HAuCl4 with NaBH4 in poly(N-vinyl-2-pyrrolidone).32 Moreover, the similarities in lattice constants between Ag and Au permit for single-phase alloys of Ag-Au NPs with varying compositions.5, 33, 34
Ag-Au BNPs with very unique structures of high complexity have also been reported.29, 44-50 Mechanistic interpretations for their formation include sequential or simultaneous processes of GRR,29, 51, 52 nanoscale Kirkendall growth,48, 53 aggregation and coalescence,48 and Ostwald ripening.29, 52 Net growth or etching of specific facets occurs during the reaction depending on the competition between Au atom deposition and Ag atomic dissolution on these facets.29 Moreover, template hollowing can also be ascribed to the nanoscale Kirkendall effect,48, 54 where there is faster diffusion of the Ag+ ions compared to gold at the Ag/Au interfacial regions resulting in the formation and coalescence of void vacancies.
Because the differing Ag-Au BNP synthetic methods result in the formation of nanomaterials with differing physical properties, it is likely that these various preparations of Ag-Au BNPs will have differing catalytic properties. It is well established that the catalytic properties of BNPs can be influenced by their morphology,55, 56 composition,57, 58 and size.59, 60 Wu and colleagues synthesized hollow Ag-Au bimetallic nanospheres and nanoboxes and found that the morphology of the nanomaterials affected the catalytic activity more than composition.61 Endo and coworkers found that the 7–10 nm Ag-Au BNPs with an Au:Ag ratio of 3:1 had higher catalytic activity for the reduction of 4-nitrophenol compared to 7 - 10 nm Ag-Au BNPs with an Au:Ag ratio of 1:1 or 1:3.62 Petri and colleagues synthesized Ag-Au BNPs with varying structures and compositions and found that both composition and structure were important for optimizing the catalytic activity towards the reduction of 4-nitrophenol and 4-aminophenol.63 Menezes and coworkers synthesized Ag-Au BNPs of varying sizes and found that the smallest particles showed the highest turnover frequencies for catalyzing CO oxidation.64 Taken together, these studies demonstrate that the physical properties of Ag-Au BNPs greatly influence their catalytic activity.
While the field of BNP synthesis and catalysis is mature, 46, 47 reproducible descriptions of Ag-Au BNP synthesis with good size and shape control using triblock copolymers (TBPs) are sparse.65 TBPs can be used as reducing and stabilizing agents to synthesize monometallic Ag and Au NPs.65-69 Moreover, the correlation between morphology of TBP stabilized Ag-Au BNPs and their catalytic activity is yet to be established. Herein, we describe a simple, reproducible, and green method for the synthesis of Ag-Au alloy BNPs with good size control. In this method, maltose coated Ag NP seeds are exposed to Au3+ ions in EO100PO65EO100 (Pluronic F127) aqueous solutions. The Au3+ reduction occurs on the surface of the silver NPs. Furthermore, by adjusting the concentration of gold salt added to the Ag NP seeds, the Au to Ag ratio and subsequent morphology of the BNPs can be controlled. The resulting BNPs show enhanced catalytic activity relative to Pluronic F127 stabilized Ag and Pluronic F127 stabilized Au NPs in the borohydride reduction of 4-nitrophenol with the highest turnover frequencies achieved by the highly etched Ag-Au BNPs.
Experimental Section
Materials
The ammonium hydroxide (28-30%), D-(+)-Maltose monohydrate (≥99%), NaOH (≥98%), silver nitrate (≥99%), gold (III) chloride hydrate (HAuCl4.×H2O; 99.999% trace metals basis), Pluronic F127 (EO100PO65EO100 MW ≈ 12500; batch no. 119K0073), ascorbic acid (reagent grade), sodium borohydride (< 90%), 4-nitrophenol (<98 %; 4-NP), and 4-nitrothiophenol (80%; 4-NTP) were used as received (Sigma-Aldrich, Milwaukee, WI, USA). Milli-Q water (Millipore 18.2 MΩ cm) was used.
Synthesis of monometallic Ag nanoparticles
Maltose coated Ag NPs (≈ 22 nm) were prepared via the reduction of AgNO3 by maltose in an alkaline environment as previously described.70, 71 A solution of 2.0 × 10-2 M AgNO3 was prepared by dissolving 34 mg of AgNO3 in 10 mL water. A stock solution containing 1.0 × 10-2 M ammonium hydroxide and 1.0 × 10-2 M maltose was prepared by adding 28 μL of ammonium hydroxide (28% NH3) and 0.137g of D-maltose to 40 mL of water. To 10 mL of stock solution, 0.5 mL of 2.0 × 10-2 M AgNO3 was added. The pH of the solution was adjusted to ≈ 11.5 using 1 M NaOH. The clear solution was then incubated at 30 °C until the solution turned amber in color (≈5 min). The resulting D-maltose coated Ag NPs were centrifuged (10,000 × g, 15 minutes) and resuspended in ≈ 2% w/v Pluronic F127 to an optical density of 12 at λmax ≈ 400 nm. The concentrations of the Ag nanoparticle seeds and comprising Ag atoms at an absorbance of 12 were ≈ 1 nM (∼6 × 1011 particles/mL) and ≈ 3 × 10-4 M as determined by Mie calculations, respectively.72
Preparation of monometallic Au nanoparticles
Gold NPs (≈ 25 nm) were prepared as follows. Trisodium citrate solution (2 mL of 1% (w/v)) was added to 98 mL of H2O and the solution was heated to reflux (∼100 °C). Then, HAuCl4 (100 μL of 0.1 M) was added and the solution was further refluxed for 15 minutes prior to cooling to room temperature. These particles (≈12 – 15 nm) were further seeded (after removing a 5 mL aliquot for analysis for each seeding cycle) to obtain ≈25 nm NPs. For seeding, the Au NP solution (≈ 100 mL) was heated at 95 °C and 5 mL of trisodium citrate (15 mM), followed by HAuCl4 (100 μL of 0.02 M), were added to the solution. This solution was then heated to reflux (∼100 °C) for 30 minutes. This seeding was repeated twice to get ≈ 25 nm NPs. For catalysis experiments, the NPs were coated in F127 by centrifuging (10,000×g, 15 minutes) followed by suspension in ≈ 2% (w/v) aqueous Pluronic F127. In 4-nitrothiophenol adsorption experiments, the NPs were first suspended in 5% (w/v) F127 by dissolving 2.5 g in 50 mL NP aqueous solution and then centrifuged (10,000×g, 15 minutes) and resuspended in ≈ 2% (w/v) aqueous Pluronic F127.
Preparation of Ag-Au bimetallic nanoparticles
Ag-Au BNPs were prepared by the galvanic replacement reaction between maltose coated Ag nanoparticle seeds and HAuCl4 in ≈ 2% (w/v) aqueous Pluronic F127 solutions. Triblock copolymers are known reducing and capping agents but, in this case, the rate of heterogeneous Au3+ surface reduction on the Ag NPs is rapid compared to solution based Au3+ reduction to Au by the TBPs. Without the presence of TBPs, GRR will occur on the Ag-NPs but it is followed by NP aggregation. In contrast, TBP stabilized BNPs are colloidally stable at room temperature for a period of several weeks.
On the 1 mL scale, 0.1 M HAuCl4 (1 to 8 μL) was added to the F127 stabilized (≈ 2% (w/v)) Ag NP seed solution (1 mL, OD = 12 at λmax ≈ 400 nm) at 25 °C or 100 °C. The reactions were allowed to run for 30 minutes (the color change was instantaneous upon thorough mixing) before air cooling to room temperature (≈ 1 hour). Then, the solutions were centrifuged at 1,500 × g for 5 minutes to remove any AgCl precipitate.
Instrumentation
UV-Vis spectra were recorded using a Varian Cary 300 spectrophotometer equipped with a temperature controller. All UV-Vis measurements were made using a quartz cell (1 cm path length, 25 °C) unless otherwise indicated.
Digital transmission electron microscopy (TEM) was carried out on a Philips Tecnai 12 instrument operating at 80 kV fitted with a Gatan camera. Samples were prepared for electron microscopy measurements by centrifuging (10,000×g, 15 minutes) twice to remove excess reagents and suspended in water followed by dropping 5 to 10 μL of solution onto a carbon-coated Cu grid (Ted Pella 200 mesh) and allowing the samples to air dry.
Powder X-ray diffraction (XRD) of samples was performed on a Siemens D500 X-ray diffractometer using Cu-Kα radiation (λ =1.54184 Å) at 40 kV, 40 mA, a scanning rate of 58°/min, and a 2θ angle ranging from 30 to 80°. Samples were dried onto a silicon low background sample holder prior to analysis. Data were analyzed with Jade10 software (Materials Data Inc.) using the NIST ICSD-Min (2013) database and each spectra matched to a powder diffraction file (PDF). A Thermo NNS Energy-dispersive X-ray (EDX) analyzer attached to a Vega LSH scanning electron microscope was used to determine the composition of the BNPs. EDX measurements for each BNP were performed on at least five different regions on two different samples.
Particle sizes were determined using a Nicomp380 XLS Zeta Potential/Particle Sizer (PSS Nicomp, USA) equipped with a He-Ne laser wavelength of λ = 638 nm and a power output of 60 mW. All data were collected at 25 °C and at a scattering angle of 90° with a square acrylic cuvette (3 mL volume) containing a suspension of diffusible particles. Values of the refractive index n = 1.33, and viscosity, η = 8.9×10−4 Ns m−2 for water were assumed to be applicable to the solutions. Prior to measurements, all samples were centrifuged (8000 × g, 30 min) to remove excess surfactant and suspended in deionized water.
Atomic force microscopy (AFM) images were generated with a multimode 8 scanning probe microscope (Bruker, Santa Barbra CA) in the peak force tapping (k = 0.4 Nm-1, f = 70 kHz) mode as previously described. 68, 69
Catalytic Reduction of 4-nitrophenol by Ag-Au bimetallic nanoparticles
We used the reduction of 4-nitrophenol as a model system to quantify the catalytic activity of the F127 capped Ag-Au BNPs. The catalytic reduction of 4-nitrophenol is known to follow pseudo-first order decay kinetics.73 All reactions were performed in a standard 3 mL quartz cuvette with a 1 cm path length. Stock solutions of the 4-nitrophenolate anion and NaBH4 were prepared in pH 9 water on the day of the experiment. The absolute concentration of 4-nitrophenolate was determined from the absorbance at 400 nm (ε = 19200 M-1 cm-1).74 Fresh 0.015 M NaBH4 (5.7 mg in 10 mL water) was prepared 10 minutes before each experiment to minimize hydrolysis. Briefly, 1.0 mL of 0.015 M NaBH4 was added to 1.7 mL of 0.15 mM 4-nitrophenolate together with 300 μL of Ag-Au BNPs (as-prepared). The UV-Vis absorbance spectra were recorded at 4 s (399 – 401 nm) or 30 s (200 – 700 nm) intervals. Monometallic Ag and Au NPs (≈1 nM) were used as controls. The catalytic activity was quantified in terms of turnover frequency (TOF) with dimensions of time-1 (Supplemental for details). We estimated the NP surface areas from the 4-nitrothiophenol adsorption isotherms (Supplemental Figure 1). We did the experiments between 25 and 45 °C to determine activation energies calculated from the gradient of natural log of the reaction rate (s-1) against reciprocal temperature.
Results and Discussion
Characterization of Ag-Au nanoparticles
In this study, Ag-Au BNPs were synthesized by adding chloroauric acid (0.1 – 0.8 mM) to F127 stabilized, maltose coated, Ag NP seeds (≈ 22 nm and 1 nM; Supplemental Figure 2). TEM images indicate that there is a change in BNP morphology upon the addition of increasing concentrations of gold salt (Figure 1). Extensive pitting and hollowing is observed below 50% Au composition, transitioning into pseudo spherical morphologies above 80% Au atom incorporation. The Ag-Au BNPs are formed by the GRR (3Ag(s) + AuCl4-(aq) → Au(s) + 3AgCl(s) + Cl-(aq)) between Ag NPs and chloroaurate ions. This reaction occurs because the redox potential of gold (III) ions is higher than that of solid silver (∼ +0.8 V for Ag versus ∼ +1.5 V for Au3+). EDX analysis confirmed the presence of both silver and gold in the nanostructures (Supplemental Figure 3). The Au:Ag ratios were estimated as a function of [Au3+ added]/[3 × 10-4 M], where the BNPs synthesized with increasing chloroauric acid concentration having higher Au:Ag ratios (Supplemental Figure 4). Below 50% Au composition (4 × 10-4 M HAuCl4 added, Au:Ag < 1.5) GRR is observed (Figure 1 A-B, E-F). The darker electron contrast for ∼20 – 50% Au composition indicates heterogeneous electron scattering from the gold and silver. This suggests that the particles are either core-shell or have hollow interiors.
Figure 1.
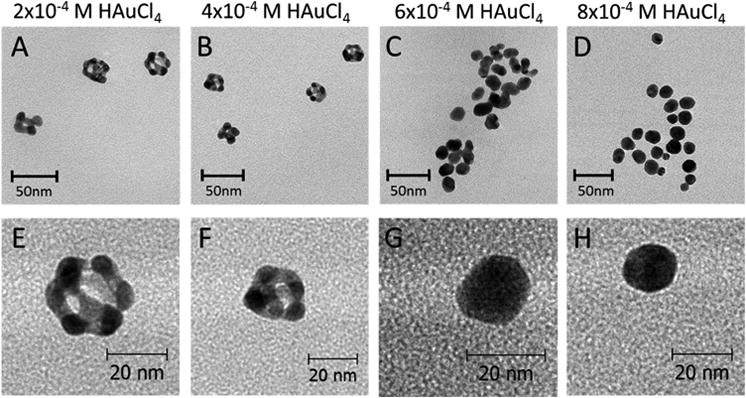
TEM images of Ag-Au nanoparticles. The nanoparticles were prepared by adding HAuCl4 (1 to 8 μL; 0.1 M) to 1 mL of ≈ 1 nM (OD ≈ 12, ≈ (22 ± 3) nm) Ag nanoparticles. Top panel: representative TEM images of Ag-Au NPs prepared at 100°C with (A) 2 × 10-4 M (≈ 20% Au), (B) 4 ×10-4 M (≈ 50% Au), (C) 6 × 10-4 M (≈ 90% Au), and (D) 8 ×10-4 M (≈ 93% Au) of HAuCl4. Bottom panel: high magnification TEM images of Ag-Au NPs prepared at 100°C with (E) 2 × 10-4 M (≈ 20% Au), (F) 4 × 10-4 M (≈ 50% Au), (G) 6 ×10-4 M (≈ 90% Au), and (H) 8 × 10-4 M (≈ 93% Au) of HAuCl4.
We also noted that the Ag-Au BNPs synthesized at 100 °C, compared to those prepared at 25 °C, were morphologically different and had a higher maximum gold content (Supplemental Figures 4 and 5). For example, when 6 × 10-4 M HAuCl4 (Au:Ag ≈ 2) was added, the BNPs prepared at 25 °C (∼60 percent gold) showed signs of “necklace chaining” while the BNPs prepared at 100 °C (∼80 percent gold) were dispersed. X-ray diffraction characterization at the 25 °C preparation had an additional (200) peak from AgCl (PDF#31-1238) at 32.0 ° that persisted after excess AgCl was removed by centrifugation. This peak quantification indicated ∼ 30% AgCl contamination on the formed BNPs when 1 to 5 × 10-4 M of HAuCl4 (Au:Ag ∼ 0.3 –1.7) was added to the Ag NPs at 25 °C. It is likely that this AgCl contamination contributes to the “necklace chaining” and lower maximum gold incorporation for the 25 °C preparation. It is well established that, during the formation of Au-Ag BNPs, oxidized silver species can exist as the sparingly soluble AgCl precipitate and in the form of stable solute complexes {[AgCln]1-n} at higher ligand concentrations.75, 76 The insoluble silver chloride precipitate can contaminate the surface of the nanoparticles, reduce the surface charge of the BNPs, and inhibit further Ag+ surface oxidation and further incorporation of Au. By heating the Ag NP seed solutions to ∼100 °C prior to adding the gold salt followed by centrifugation, we found that the AgCl contamination can be effectively avoided (Figure 2). Powder XRD of samples prepared at 25 and 100 °C confirmed the presence of metallic Ag with intensity maxima at 2-theta diffraction angles of 38.2°, 44.2°, 64.4°, and 77.5° (these correspond to characteristic diffractions from (111), (200), (220) and (310) planes of Ag (PDF# 4-783)). Unfortunately, we were not able to determine the Ag:Au elemental ratio of the BNPs by X-ray diffraction because the lattice constants for Ag and Au are comparable. EDX instead was used to quantify elemental composition.
Figure 2.
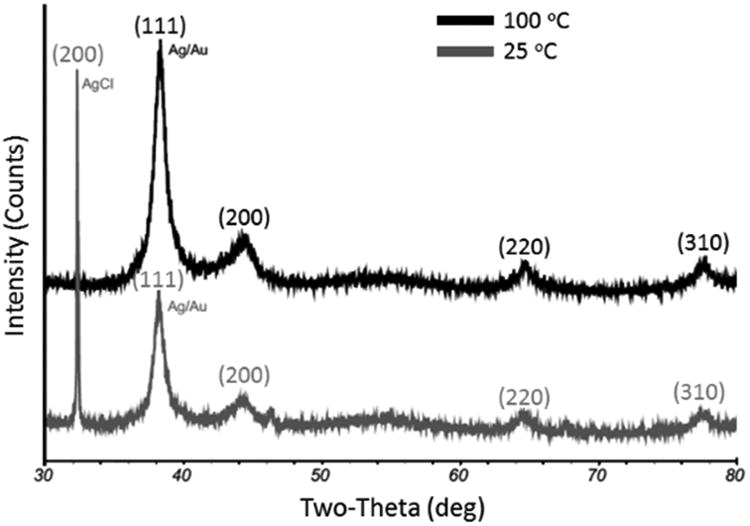
Representative XRD patterns of the Ag-Au nanoparticles. The nanoparticles were prepared by adding 1 × 10-4 M HAuCl4 (1 μL; 0.1 M) to 1 mL of ≈ 1 nM (OD ≈ 12) 25 nm Ag NP seeds at 25 or 100 °C. Samples were centrifuged at 1500 × g for 5 minutes to remove any AgCl precipitate prior XRD characterization. The 25 °C preparation has a (200) Bragg peak from AgCl.
The size and population distributions of the Ag-Au BNPs prepared at 100 °C were determined by TEM, AFM, and DLS. These data show one dominant population with nominal sizes of ∼10 – 25 nm for TEM (Supplemental Figure 4), 25-35 nm for AFM (Supplemental Figure 6), and 55 – 70 nm for DLS (Supplemental Figure 7). The larger NP sizes, as determined by AFM and DLS, are indicative of the block copolymer overlayer. From the BNP size distribution, we can assume that the concentrations of as-prepared BNPs are the same as the starting Ag NP seed concentrations. This is no longer true when Au3+ is added in large molar excess because additional gold nuclei are formed by TBP facilitated reduction in the solution, resulting in a bimodal size population distribution (Supplemental Figure 8).68, 69
Based on the TEM analysis, the dissolution of Ag and the deposition of Au are not uniform on the NP surface (Figure 1, Supplemental Figure 4). Under our experimental conditions, Au deposition will probably occur initially (Au:Ag < 1) and preferentially on the high-energy {110} and {100} facets.52 Consequently, the preferential gold deposition/reduction on these high-energy facets will inhibit the oxidation of Ag from them. The uncoated low energy {111} facets become sites for pitting, void formation, and dealloying,77 where silver behaves as a sacrificial anode. Such growth behavior also suggests that, although the Ag atoms drive the Au3+ reduction, the deposition of Au atoms is a self-seeding process. This is ultimately responsible for the formation of the hollow structures observed when the BNPs are prepared at a low concentration of Au3+. There is likely alloying at the interfacial regions resulting in an overall reduction in the number of atoms in the structure.48 When greater concentrations of HAuCl4 are added (Au:Ag > 1), the faster kinetics for the Au growth presumably decreases the surface energy differentiation between {111} and {100}/{110} facets and results in more uniform BNPs (Figure 1 G - H). At Au:Ag > 1, reduced Au atoms also fill void vacancies (dealloying) and replace some of Ag scaffolding by Au. This concentration is critical for the formation of core-shell structure of the BNPs.
With increasing HAuCl4 concentration, the localized surface plasmon peaks (LSPR) peak maxima shift from ≈ 400 to ≈ 525 nm (Figure 3). This change in LSPR maxima is non-linear, rapidly changing from ≈ 440 to ≈ 510 nm, between 0.3 to 0.7 mM HAuCl4 (Au: Ag ≈ 1–2) added. This suggests there is an Au3+ concentration threshold for promoting surface GRR.
Figure 3.
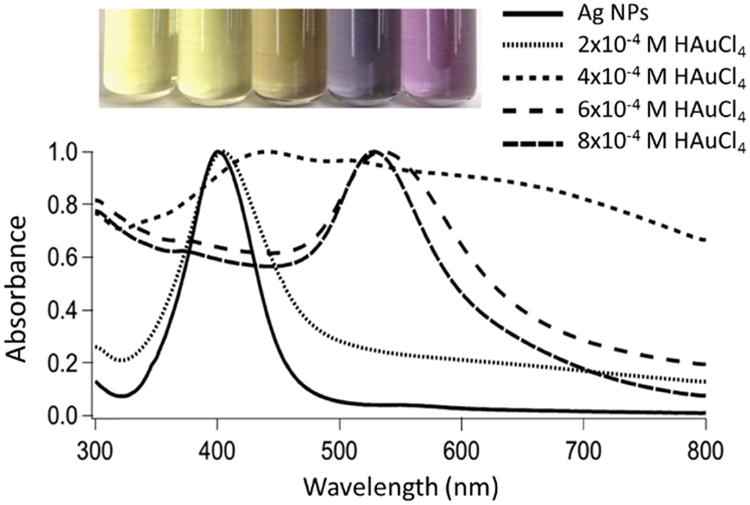
Representative optical images and UV-vis absorption spectra of Ag-Au BNPs with increasing Au fractional composition as determined by EDX. All spectra are normalized to their localized surface plasmon peak maxima. The nanoparticles were prepared by heating 1 mL of ≈ 1 nM (OD ≈ 12) 22 nm Ag nanoparticles to ∼ 100 °C and adding HAuCl4 (1 to 8 μL; 0.1 M) to the hot solution. The inset is a digital photograph of Ag and Ag-Au (made with 2 ×10-4 M, 4 × 10-4 M, 6 × 10-4 M, and 8 × 10-4 M of HAuCl4) nanoparticles. For the UV-vis absorption spectra, the black solid line at 400 nm represents Ag nanoparticles and the dotted lines correspond to Ag-Au nanoparticles made with 2 × 10-4 M, 4 × 10-4 M, 6× 10-4 M, and 8 × 10-4 M of HAuCl4, respectively.
Bimetallic nanoparticle catalytic efficiency
One of the applications of BNPs is their ability to enhance catalytic activity and selectivity as a result of their structural effects on electronic states.78-80 We evaluated the catalytic behavior of the synthesized Ag-Au BNPs using the reduction of 4-nitrophenol (4-NP) by NaBH4 as a model reaction. Kinetic data were analyzed in terms of the Langmuir-Hinshelwood model assuming that the NaBH4 is in molar excess and the reaction rate is dependent only on the 4-NP concentration.73,81 Under our experimental conditions, as-prepared NPs ranging from 100% Ag to ≈ 95% Au incorporation (Au:Ag ≈ 2.5) were used. Moreover, the comparable nominal size determinations enable a meaningful comparison of the effects of BNP composition and shape on catalytic activity.
The time evolution for the reduction of 4-NP by NaBH4 in the absence and presence of nanoparticles was monitored by UV-Vis spectroscopy both at 400 nm and between 700 and 200 nm (Figure 4). The reduction of 4-NP became significant in the presence of nanoparticles as evidenced by the decrease in the absorbance at 400 nm (4-NP) and the concomitant growth of the 300 nm 4-aminophenolate peak. Clearly 4-NP reduction is facilitated by the presence of a NP catalyst. Representative absorbance spectra indicate that the rates of degradation in the presence of a NP catalyst are in the order Ag < Au < Ag-Au (Figure 4 A – D). Observed lag times before 4-NP degradation are assigned to adsorbate induced surface restructuring and were shortest for the most active NPs (Figure 4 E).73
Figure 4.
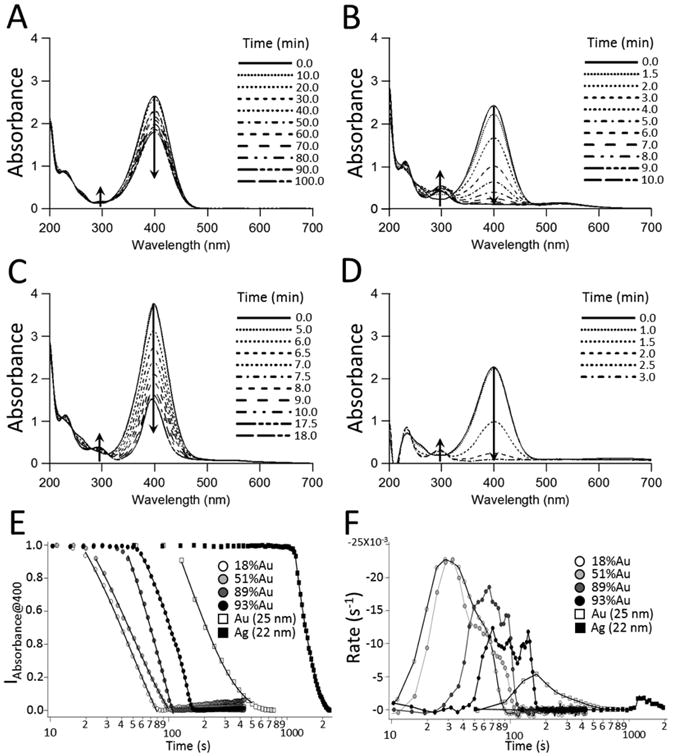
The reduction of 4–nitrophenol. The time evolution for the reduction of 1.5 × 10-4 M (1.7 mL; 0.15 mM) 4–nitrophenol by 5 × 10-3 M (1.0 mL; 0.015 M) NaBH4 determined from UV-vis spectroscopy at 4 s (399 – 401 nm) or 30 s (200 – 700 nm) intervals. (A) Borohydride only, (B) Au NPs (300 μL) (≈ 25 nm), (C) Ag NPs (300 μL) (≈ 22 nm), and (D) Ag-Au NPs (300 μL) roughly 50% Au and ≈ 22 nm. (E) The absorbance spectra at 400 nm for the reduction of 1.5 × 10-4 M 4-NP by ∼ 5 × 10-3 M NaBH4 in the presence of Ag-Au NPs (≈ 18, 51, 89, and 93% Au) Au NPs (≈ 25 nm), and Ag NPs (≈ 22 nm). (F) The time evolution in the TOF for Ag-Au NPs (≈ 18, 51, 89, and 93% Au) Au NPs (≈ 25 nm), and Ag NPs (≈ 22 nm). All reactions were run at 25 °C.
We determined the NP surface-to-volume (S/V) ratios from the 4-nitrothiophenol (4-NTP) adsorption isotherms. Figure 5A shows the adsorption isotherms of Au, Ag and Ag-Au BNPs (20% – 95% Au) where Cads and Ceq are the adsorption and the equilibrium solution concentrations of 4-NTP, respectively. Assuming a Langmuir isotherm model, the 4-NTP surface-to-volume ratios were estimated from 1/(slope) of the plots of Ceq/Cads against Cads [S/V = 1/slope (mol/L) × Avogadro's number (6 × 1023 4-NTP/mol) × 4-NTP surface area (1.87 × 10-19 m2)] (Figure 5B, Supplemental Figure 1, Table 1). For the monometallic NPs and Ag NP seeds prior to making the BNPs concentrations, the starting concentration were ∼ 1 mM. We note that the 4-NTP adsorption concentration (Cads) is proportional to the NP concentration. The adsorption saturation concentrations cluster for Ag and Ag-Au BNPs (20% – 93% Au). In contrast, Au and Ag-Au BNPs (∼95% Au) have the lowest and highest adsorption saturation concentrations, respectively. This can be understood from morphological and NP number density considerations. The Ag-Au BNPs (20% – 95% Au) concentrations were comparable to the silver NP seeds. But the subtle variations in the saturation adsorption concentrations in Ag-Au BNPs (∼20 – 50% Au) are likely from the variations in surface areas, which is confirmed by the larger S/V ratios of the Ag-Au BNPs with ∼20% Au (Table 1). We note that 4-NTP addition to the citrated Au NPs always resulted in some precipitation. The anomalously low Au saturation concentration is probably from partial Au NP aggregation due to thiol exchange with the surface citrate. For Ag-Au BNPs (≈ 95% Au), the large molar excess of HAuCl4 (Au(III)/Ag(I) > 3) will result in Ag NP dissolution, and the formation of smaller NP seeds. The concentration of the NPs will be higher than the initial Ag seeds. These smaller NP seeds will likely form nucleation sites seeding the growth in an aggregative type mechanism.68, 69
Figure 5.
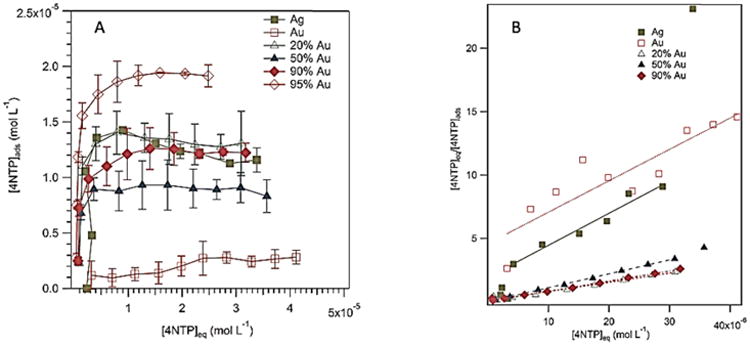
Adsorption isotherms of 4-nitrothiophenol on the nanoparticles at 25 °C. Each data point is the average of three independent experiments. The volume of NP added was 50 μL in a 1 mL total volume 4-NTP aqueous solution. For each experiment, the concentrations of the NPs were fixed while 4-NTP concentrations were varied (0.002 – 0.05 mM). The error bars are the standard deviations. (A) Adsorption isotherms of Ag, citrated Au, and Ag-Au NPs. (B) Langmuir Plots of Ceq/Cads against Ceq of Ag, citrated Au, and Ag-Au NPs. The reciprocal of the slopes gives monolayer 4-nitrothiophenol coverages.
Table 1.
Kinetic parameters for 4-nitrophenol reduction.a
| Au% (EDX) | Size (nm) | kapp(s-1) | TOF (s-1) | Ea (kJ mol-1) | tof°(s-1) | S/V (m2/L) |
|---|---|---|---|---|---|---|
| 0 | 22 ± 3 (TEM) | 5.75 ± 0.10 × 10-3 | 3.70 ± 0.10 × 10-3 | 63 ± 8 | 3.31 ± 0.30 × 10-4 | 0.45 ± 0.07 |
| 18 ± 4 | 22 ± 4 (TEM) | 1.89 ± 0.02 × 10-2 | 2.42 ± 0.20 × 10-2 | 56 ± 7 | 2.78 ± 0.42 × 10-3 | 1.55 ± 0.20 |
| 51 ± 3 | 21 ± 4 (TEM) | 2.45 ± 0.49 × 10-2 | 2.64 ± 0.52 × 10-2 | 51 ± 2 | 3.65 ± 0.72 × 10-3 | 1.02 ± 0.17 |
| 89 ± 1 | 14 ± 1 (TEM) | 1.35 ± 0.70 × 10-2 | 1.93 ± 0.09 × 10-2 | 70 ± 8 | 1.32 ± 0.12 × 10-3 | 1.41 ± 0.16 |
| 93 ± 1 | 16 ± 2 (TEM) | 5.09 ± 0.69 × 10-3 | 1.05 ± 0.27 × 10-2 | 64 ± 7 | 8.93 ± 0.30 × 10-4 | 2.23 ± 0.14 |
| 100 | 24 ± 4 (AFM) | 6.23 ± 0.35 × 10-3 | 7.10 ± 2.4 x 10-3 (citrated) | 28 ± 4 (citrated)b(50) | 2.29 ± 0.37 × 10-3 (citrated)b1.03 ± 0.2 × 10-3 | 0.45 ± 0.24 |
At 298.15 K.
Using activation energy based on literature values for gold.83
kapp: apparent rate constant.
TOF: turnover frequency.
Ea: activation energy of the TOF against 1/temperature.
TOF°: standard TOF at a concentration of [C] equal 1 M, temperature 273.13 K and 1 bar pressure.
S/V: surface-to-volume ratios.
Values are reported as mean ± SD.
We used TOFs to quantify the relative catalytic efficiencies of the NPs. This allows for a meaningful comparison with published reports of catalytic activity of Ag, Au and Ag-Au nanomaterials. Moreover, TOFs are independent of the reaction order as they are a measure of the reaction rate. In contrast, the assumption that the rate constants are first-order may not be always be valid.82 However, we can assume that the reaction order is ≈ 1 as the rate constants and TOFs are in good agreement (Table 1). These TOFs were measured above the saturation limit of the catalyst (∼ 10-11 M), and are a true measure of the TOF (Supplemental Figure 9). The BNPs represent superior catalytic performance compared to the monometallic NPs (Figure 4F). BNPs of Au:Ag ratios between 0.5 (20% Au) and 1.5 (50% Au) have the highest TOFs (Table 1) even when the TOFs are normalized by estimated surface areas. We note that the TOFs and activation energies for citrated NPs will be artificially high and low, because the surface citrate can act as a reductant in the system.43
The Ag-Au BNPs TOFs at 25 °C of ∼ 10-2 s-1 are comparable to the values obtained by BNPs derived from pueria starch12 and partially hollow nanorods,84 but is much higher than pure Ag NPs with a TOF (25 °C) of 10-3 s-1. BNPs above 60 Au% content have catalytic efficiencies comparable to the monometallic Au NPs. Assuming an activation energy of ∼ 50 kJ mol-1, based on known literature values for cetyltrimethylammonium bromide coated Au NPs,83 gives a standard TOF° of ∼ 10-3 s-1 extrapolated from room temperature, which is ∼ 3 to 4 fold lower than the BNPs. Further, the efficiency of the BNPs is about ∼10 fold higher compared to Ag NPs. If the BNPs TOFs ° are normalized to S/V, the relative enhancements against citrated Au and Ag NPs are ∼ 2 and ∼ 5 fold higher, respectively. Indeed, the confounding effect of the surface citrate on catalytic activity is demonstrated by the observation that the catalytic performance of BNPs comprising ∼20 – 50% Au is 4–fold (8–fold when normalized to S/V) higher than those comprising over 90% Au. Overall, there is a correlation of higher TOFs with partially hollow BNPs shapes than with Au:Ag molar ratios or size.
Higher catalytic efficiencies of the BNPs can be rationalized by structural and electronic effects. Structural effects such as partial hollowing act like “nano-reactor cages” and facilitate the confinement of the reactants inside the NP voids. Surface areas inside the voids, along with higher densities of lower coordinated binding (kinks, edges, and higher energy surfaces {100}) are known to facilitate higher catalytic activity and selectivity.86 Enhanced catalytic activities for BNPs may also be related to additional Ag/Au interfaces.85 Electronic effects, like stronger adsorbate binding to surfaces, may also be responsible for the enhanced catalytic activity of the BNPs. In our study, TEM confirms that the sizes and morphologies for all bimetallic compositions are broadly similar, suggesting that the enhancements observed in the catalytic activities of 20 – 50% BNPs are not explained exclusively by size, shape, or surface area differences. Slater and coworkers, in their EDX characterization of Ag-Au NPs within scanning transmission electron microscope (STEM), found that the catalytic performance for a specific bulk composition will depend upon the surface ratio of the Au to Ag.87 Notably, the best catalytic performance occurred near the Au composition (∼ 18%) where the NPs changed from Au surface segregation to a homogeneously alloyed composition.87 This homogenous surface alloying may also occur at 20 – 50% Au:Ag compositions under our experimental conditions, which are responsible for the high value of TOFs. Surface segregation of Ag and Au will modify the surface electron density and hence adsorbate binding. Because the d-band controls the bonding interactions, the low lying d-bands below the Fermi level of Ag and Au allow more antibonding states compared to earlier transition metals resulting in weak chemisorption.88, 89 When disparate metals are alloyed, charge rearranges to equilibrate the different Fermi levels.89 The greater electronegativity of Au with respect to Ag will facilitate charge transfer from Ag to Au. The decreasing of the electron density at Ag will improve adsorbate binding and reduce the activation barrier, while the increasing of the electron density on the Au will benefit for electron transfer to 4-NP. This charge transfer mechanism has been established by XPS measurements.1
We recognize that the homogenous surface segregation (alloying), optimal at 20% Au composition, will be important for the catalytic activity.87 As the Au composition changes between 20 – 50%, the morphology and surface area of our BNPs does not vary significantly, where the additional Au atoms are likely replacing the Ag atoms in the center of the BNPs. Consequently, the surface alloy composition will be approximately constant for the 20 – 50% Au composition range where the addition of 18 % Au is sufficient for achieving high reactivity. This explains why the measured TOF values are similar for the 20 – 50% Au in the BNPs.
Conclusions
We have demonstrated a novel, facile, and green method for the rapid and reproducible synthesis of Ag-Au BNPs using TBPs. Of particular interest are the partially hollow nanostructures that form when the GRR between Ag seeds and HAuCl4 is performed at 100 °C and the Ag:Au ratio is < 2. These synthesized BNPs were shown to enhance the reduction of 4–nitrophenol by NaBH4 when compared to both Ag seeds and Au NPs. This enhanced catalytic activity is ascribed to greater surface areas containing higher energy facets and Ag/Au interfacial regions where homogenous surface segregation (alloying) occurs that have excess electron density compared to monometallic NPs. This interpretation is consistent with the observation that the rate of 4–nitrophenol reduction was highest with partially hollow BNPs in the 20 – 50% Au range than with monometallic Ag or Au NPs.
The catalytic activity of the BNPs used in this study are larger (∼ 2–10 fold) than typically reported for monometallic Au or Ag NPs of comparable sizes.90 Furthermore, the methodology described is general and can be applied to TBPs or other polymers with equivalent reducing groups. Our synthesis of BNP structures allows us to optimize the catalytic activity at an atomic level for specific applications. This method provides a general framework for the synthesis cage like and pseudo-spherical BNPs that can be used in multiple fields e.g., antimicrobials,91 biomedical imaging,92 and nanocatalysis.93
Supplementary Material
Acknowledgments
This work was supported by the Loma Linda University. Access to the TEM was provided by the Central Facility for Advanced Microscopy and Microanalysis (CFAMM) at the University of California Riverside (UCR). Research support for (M.H) was provided by the National Institute of Health Disparities and Minority Health of the National Institutes of Health under award number P20MD006988. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Electronic Supplementary Information (ESI) available: details of turnover frequency calculations, surface area calculations, material characterizations (TEM, AFM, EDX, and DLS) are provided.
References
- 1.Zhang H, Haba M, Okumura M, Akita T, Hashimoto S, Toshima N. Langmuir. 2013;29:10330–10339. doi: 10.1021/la401878g. [DOI] [PubMed] [Google Scholar]
- 2.Mizukoshi Y, Fujimoto T, Nagata Y, Oshima R, Maeda Y. J Phys Chem B. 2000;104:6028–6032. [Google Scholar]
- 3.Toshima N, Yonezawa T. New J Chem. 1998;22:1179–1201. [Google Scholar]
- 4.Harikumar KR, Ghosh S, Rao CNR. J Phys Chem A. 1997;101:536–540. [Google Scholar]
- 5.Mallin MP, Murphy CJ. Nano Letters. 2002;2:1235–1237. [Google Scholar]
- 6.Teo BK, Keating K, Kao YH. J Am Chem Soc. 1987;109:3494–3495. [Google Scholar]
- 7.Schmid G, West H, Mehles H, Lehnert A. Inorg Chem. 1997;36:891–895. [Google Scholar]
- 8.Zhang Q, Xie J, Liang J, Lee JY. Adv Funct Mater. 2009;19:1387–1398. [Google Scholar]
- 9.Banerjee M, Sharma S, Chattopadhyay A, Ghosh SS. Nanoscale. 2011;3:5120–5125. doi: 10.1039/c1nr10703h. [DOI] [PubMed] [Google Scholar]
- 10.Liu X, Wang D, Li Y. Nano Today. 2012;7:448–466. [Google Scholar]
- 11.Zhang Q, Lee JY, Yang J, Boothroyd C, Zhang J. Nanotechnology. 2007;18:245605. [Google Scholar]
- 12.Xia B, He F, Li L. Langmuir. 2013;29:4901–4907. doi: 10.1021/la400355u. [DOI] [PubMed] [Google Scholar]
- 13.Treguer M, de Cointet C, Remita H, Khatouri J, Mostafavi M, Amblard J, Belloni J, de Keyzer R. J Phys Chem B. 1998;102:4310–4321. [Google Scholar]
- 14.Roy K, Lahiri S. Anal Chem. 2008;80:7504–7507. doi: 10.1021/ac800593u. [DOI] [PubMed] [Google Scholar]
- 15.Yancey DF, Carino EV, Crooks RM. J Am Chem Soc. 2010;132:10988–10989. doi: 10.1021/ja104677z. [DOI] [PubMed] [Google Scholar]
- 16.Yang J, Deng S, Lei J, Ju H, Gunasekaran S. Biosens Bioelectron. 2011;29:159–166. doi: 10.1016/j.bios.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 17.Anandan S, Grieser F, Ashokkumar M. J Phys Chem C. 2008;112:15102–15105. [Google Scholar]
- 18.Kumar PSS, Manivel A, Anandan S, Zhou M, Grieser F, Ashokkumar M. Colloids and Surfaces A: Physicochemical and Engineering Aspects. 2010;356:140–144. [Google Scholar]
- 19.Byeon JH, Park JH, Hwang J. J Aerosol Sci. 2008;39:888–896. [Google Scholar]
- 20.AbdelHamid AA, Al-Ghobashy MA, Fawzy M, Mohamed MB, Abdel-Mottaleb MMSA. ACS Sustainable Chemistry & Engineering. 2013;1:1520–1529. [Google Scholar]
- 21.Smuleac V, Varma R, Sikdar S, Bhattacharyya D. J Membr Sci. 2011;379:131–137. doi: 10.1016/j.memsci.2011.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhan G, Huang J, Du M, Abdul-Rauf I, Ma Y, Li Q. Mater Lett. 2011;65:2989–2991. [Google Scholar]
- 23.Sawle BD, Salimath B, Deshpande R, Bedre MD, Prabhakar BK, Venkataraman A. Science and Technology of Advanced Materials. 2008;9:035012. doi: 10.1088/1468-6996/9/3/035012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Senapati S, Ahmad A, Khan MI, Sastry M, Kumar R. Small. 2005;1:517–520. doi: 10.1002/smll.200400053. [DOI] [PubMed] [Google Scholar]
- 25.Zheng D, Hu C, Gan T, Dang X, Hu S. Sensors and Actuators B: Chemical. 2010;148:247–252. [Google Scholar]
- 26.Govindaraju K, Basha S, Kumar V, Singaravelu G. J Mater Sci. 2008;43:5115–5122. [Google Scholar]
- 27.Nadagouda MN, Varma RS. Cryst Growth Des. 2007;7:2582–2587. [Google Scholar]
- 28.Kim K, Kim KL, Shin KS. J Phys Chem C. 2011;115:14844–14851. [Google Scholar]
- 29.Zhang Q, Xie J, Lee JY, Zhang J, Boothroyd C. Small. 2008;4:1067–1071. doi: 10.1002/smll.200701196. [DOI] [PubMed] [Google Scholar]
- 30.Zhang X, Zhang G, Zhang B, Su Z. Langmuir. 2013;29:6722–6727. doi: 10.1021/la400728k. [DOI] [PubMed] [Google Scholar]
- 31.Fu H, Yang X, Jiang X, Yu A. Langmuir. 2013;29:7134–7142. doi: 10.1021/la400753q. [DOI] [PubMed] [Google Scholar]
- 32.Zhang H, Okuni J, Toshima N. J Colloid Interface Sci. 2011;354:131. doi: 10.1016/j.jcis.2010.10.036. [DOI] [PubMed] [Google Scholar]
- 33.Okamoto H, Massalski TB ASM International. Phase diagrams of binary gold alloys. ASM International; Metals Park, Ohio: 1987. [Google Scholar]
- 34.Link S, Wang ZL, El-Sayed MA. J Phys Chem B. 1999;103:3529–3533. [Google Scholar]
- 35.Wang C, Yin H, Chan R, Peng S, Dai S, Sun S. Chem Mater. 2009;21:433–435. [Google Scholar]
- 36.Wang AQ, Chang CM, Mou CY. J Phys Chem B. 2005;109:18860–18867. doi: 10.1021/jp051530q. [DOI] [PubMed] [Google Scholar]
- 37.Pena-Rodriguez O, Pal U. Nanoscale Research Letters. 2011;6:279. doi: 10.1186/1556-276X-6-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jha R, Sharma AK. Journal of Optics A: Pure and Applied Optics. 2009;11:045502. [Google Scholar]
- 39.Sharma AK, Gupta BD. Nanotechnology. 2006;17:124. [Google Scholar]
- 40.Fan M, Lai FJ, Chou HL, Lu WT, Hwang BJ, Brolo AG. Chemical Science. 2013;4:509–515. [Google Scholar]
- 41.Yi Z, Chen S, Chen Y, Luo J, Wu W, Yi Y, Tang Y. Thin Solid Films. 2012;520:2701–2707. [Google Scholar]
- 42.Yin Y, Qiu T, Ma L, Lang X, Zhang Y, Huang G, Mei Y, Schmidt OG. J Phys Chem C. 2012;116:25504–25508. [Google Scholar]
- 43.Zeng J, Zhang Q, Chen J, Xia Y. Nano Letters. 2010;10:30–35. doi: 10.1021/nl903062e. [DOI] [PubMed] [Google Scholar]
- 44.Ma Y, Li W, Cho EC, Li Z, Yu T, Zeng J, Xie Z, Xia Y. ACS Nano. 2010;4:6725–6734. doi: 10.1021/nn102237c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen J, Yang M, Zhang Q, Cho EC, Cobley CM, Kim C, Glaus C, Wang LV, Welch MJ, Xia Y. Adv Funct Mater. 2010;20:3684–3694. doi: 10.1002/adfm.201001329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cortie MB, McDonagh AM. Chemical Reviews. 2011;111:3713–3735. doi: 10.1021/cr1002529. [DOI] [PubMed] [Google Scholar]
- 47.Ghosh Chaudhuri R, Paria S. Chemical Reviews. 2011;112:2373–2433. doi: 10.1021/cr100449n. [DOI] [PubMed] [Google Scholar]
- 48.González E, Arbiol J, Puntes VF. Science. 2011;334:1377–1380. doi: 10.1126/science.1212822. [DOI] [PubMed] [Google Scholar]
- 49.Sun YG, Xia YN. 2002;298:2176. doi: 10.1126/science.1077229. [DOI] [PubMed] [Google Scholar]
- 50.Hong X, Wang D, Cai S, Rong H, Li Y. J Am Chem Soc. 2012;134:18165–18168. doi: 10.1021/ja3076132. [DOI] [PubMed] [Google Scholar]
- 51.You L, Mao Y, Ge J. The Journal of Physical Chemistry C. 2012;116:10753–10759. [Google Scholar]
- 52.Gilroy K, Sundar A, Farzinpour P, Hughes R, Neretina S. Nano Research. 2014;7:365–379. [Google Scholar]
- 53.Yin Y, Rioux RM, Erdonmez CK, Hughes S, Somorjai GA, Alivisatos AP. Science. 2004;304:711–714. doi: 10.1126/science.1096566. [DOI] [PubMed] [Google Scholar]
- 54.Wang W, Dahl M, Yin Y. Chem Mater. 2012;25:1179–1189. [Google Scholar]
- 55.Mu R, Fu Q, Xu H, Zhang H, Huang Y, Jiang Z, Zhang S, Tan D, Bao X. Journal of the American Chemical Society. 2011;133:1978–1986. doi: 10.1021/ja109483a. [DOI] [PubMed] [Google Scholar]
- 56.Stephens IEL, Bondarenko AS, Perez-Alonso FJ, Calle-Vallejo F, Bech L, Johansson TP, Jepsen AK, Frydendal R, Knudsen BP, Rossmeisl J, Chorkendorff I. Journal of the American Chemical Society. 2011;133:5485–5491. doi: 10.1021/ja111690g. [DOI] [PubMed] [Google Scholar]
- 57.Zhang H, Jin M, Wang J, Li W, Camargo PHC, Kim MJ, Yang D, Xie Z, Xia Y. Journal of the American Chemical Society. 2011;133:6078–6089. doi: 10.1021/ja201156s. [DOI] [PubMed] [Google Scholar]
- 58.Yin M, Huang Y, Liang L, Liao J, Liu C, Xing W. Chemical Communications. 2011;47:8172–8174. doi: 10.1039/c1cc12561c. [DOI] [PubMed] [Google Scholar]
- 59.Mazumder V, Chi M, Mankin MN, Liu Y, Metin Ö, Sun D, More KL, Sun S. Nano Letters. 2012;12:1102–1106. doi: 10.1021/nl2045588. [DOI] [PubMed] [Google Scholar]
- 60.Park JY, Zhang Y, Joo SH, Jung Y, Somorjai GA. Catalysis Today. 2012;181:133–137. [Google Scholar]
- 61.Wu H, Wang P, He H, Jin Y. Nano Res. 2012;5:135–144. [Google Scholar]
- 62.Endo T, Yoshimura T, Esumi K. Journal of Colloid and Interface Science. 2005;286:602–609. doi: 10.1016/j.jcis.2005.01.057. [DOI] [PubMed] [Google Scholar]
- 63.Petri MV, Ando RA, Camargo PHC. Chemical Physics Letters. 2012;531:188–192. [Google Scholar]
- 64.Menezes WG, Zielasek V, Thiel K, Hartwig A, Bäumer M. Journal of Catalysis. 2013;299:222–231. [Google Scholar]
- 65.Sakai T, Alexandridis P. Chem Mater. 2006;18:2577–2583. [Google Scholar]
- 66.Sakai T, Alexandridis P. Langmuir. 2005;21:8019–8025. doi: 10.1021/la050756h. [DOI] [PubMed] [Google Scholar]
- 67.Chen S, Guo C, Hu GH, Wang J, Ma JH, Liang XF, Zheng L, Liu HZ. Langmuir. 2006;22:9704–9711. doi: 10.1021/la061093m. [DOI] [PubMed] [Google Scholar]
- 68.Sabir TS, Yan D, Milligan JR, Aruni AW, Nick KE, Ramon RH, Hughes JA, Chen Q, Kurti RS, Perry CC. J Phys Chem C. 2012;116:4431–4441. [Google Scholar]
- 69.Sabir TS, Rowland LK, Milligan JR, Yan D, Aruni AW, Chen Q, Boskovic DS, Kurti RS, Perry CC. Langmuir. 2013;29:3903–3911. doi: 10.1021/la400387h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kvitek L, Prucek R, Panacek A, Novotny R, Hrbac J, Zboril R. J Mater Chem. 2005;15:1099–1105. [Google Scholar]
- 71.Kvitek L, Panaček A, Soukupova J, Kolar M, Večerova R, Prucek R, Holecova M, Zboril R. J Phys Chem C. 2008;112:5825–5834. [Google Scholar]
- 72.Navarro JRG, Werts MHV. Analyst. 2013;138:583–592. doi: 10.1039/c2an36135c. [DOI] [PubMed] [Google Scholar]
- 73.Wunder S, Polzer F, Lu Y, Mei Y, Ballauff M. J Phys Chem C. 2010;114:8814–8820. [Google Scholar]
- 74.Kotronarou A, Mills G, Hoffmann MR. J Phys Chem. 1991;95:3630–3638. [Google Scholar]
- 75.Jonte JH, Martin DS. J Am Chem Soc. 1952;74:2052–2054. [Google Scholar]
- 76.Lu L, Burkey G, Halaciuga I, Goia DV. J Colloid Interface Sci. 2013;392:90–95. doi: 10.1016/j.jcis.2012.09.057. [DOI] [PubMed] [Google Scholar]
- 77.Xia X, Wang Y, Ruditskiy A, Xia Y. Adv Mater. 2013;25:6313–6333. doi: 10.1002/adma.201302820. [DOI] [PubMed] [Google Scholar]
- 78.Sinfelt JH. Acc Chem Res. 1987;20:134–139. [Google Scholar]
- 79.Tao F. Chem Soc Rev. 2012;41:7977–7979. doi: 10.1039/c2cs90093a. [DOI] [PubMed] [Google Scholar]
- 80.Pei Y, Zhou G, Luan N, Zong B, Qiao M, Tao F. Chem Soc Rev. 2012;41:8140–8162. doi: 10.1039/c2cs35182j. [DOI] [PubMed] [Google Scholar]
- 81.Wunder S, Lu Y, Albrecht M, Ballauff M. ACS Catal. 2011;1:908–916. [Google Scholar]
- 82.Li M, Chen G. Nanoscale. 2013;5:11919–11927. doi: 10.1039/c3nr03521b. [DOI] [PubMed] [Google Scholar]
- 83.Fenger R, Fertitta E, Kirmse H, Thunemann AF, Rademann K. Phys Chem Chem Phys. 2012;14:9343–9349. doi: 10.1039/c2cp40792b. [DOI] [PubMed] [Google Scholar]
- 84.Khalavka Y, Becker J, Sönnichsen C. J Am Chem Soc. 2009;131:1871–1875. doi: 10.1021/ja806766w. [DOI] [PubMed] [Google Scholar]
- 85.Huang J, Vongehr S, Tang S, Lu H, Shen J, Meng X. Langmuir. 2009;25:11890–11896. doi: 10.1021/la9015383. [DOI] [PubMed] [Google Scholar]
- 86.Linic S, Christopher P, Xin H, Marimuthu A. Acc Chem Res. 2013;46:1890–1899. doi: 10.1021/ar3002393. [DOI] [PubMed] [Google Scholar]
- 87.Slater TJA, Macedo A, Schroeder SLM, Burke MG, O'Brien P, Camargo PHC, Haigh SJ. Nano letters. 2014;14:1921–1926. doi: 10.1021/nl4047448. [DOI] [PubMed] [Google Scholar]
- 88.Hammer B, Norskov JK. Nature. 1995;376:238–240. [Google Scholar]
- 89.Pozun ZD, Rodenbusch SE, Keller E, Tran K, Tang W, Stevenson KJ, Henkelman G. J Phys Chem C. 2013;117:7598–7604. doi: 10.1021/jp312588u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Herves P, Perez-Lorenzo M, Liz-Marzan LM, Dzubiella J, Lu Y, Ballauff M. Chem Soc Rev. 2012;41:5577–5587. doi: 10.1039/c2cs35029g. [DOI] [PubMed] [Google Scholar]
- 91.Sharma VK, Yngard RA, Lin Y. Advances in Colloid and Interface Science. 2009;145:83–96. doi: 10.1016/j.cis.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 92.Wang Y, Liu Y, Luehmann H, Xia X, Wan D, Cutler C, Xia Y. Nano Letters. 2013;13:581–585. doi: 10.1021/nl304111v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mahmoud MA, O'Neil D, El-Sayed MA. Chem Mater. 2013;26:44–58. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.