

Abstract
STAT3 is well corroborated preclinically as a cancer therapeutic target, but tractable translational strategies for its blockade by small molecule inhibitors have remained elusive. In this study, we report the development of a novel class of bifunctional STAT3 inhibitors, based on conjugation of a diarylidenyl-piperidone (DAP) backbone to an N-hydroxypyrroline (−NOH) group, which exhibits minimal toxicity against normal cells and good oral bioavailability. Molecular modeling studies of this class suggested direct interaction with the STAT3 DNA binding domain. In particular, the DAP compound HO-3867 selectively inhibited STAT3 phosphorylation, transcription, and DNA binding without affecting the expression of other active STATs. HO-3867 exhibited minimal toxicity toward noncancerous cells and tissues but induced apoptosis in ovarian cancer cells. Pharmacologic analysis revealed greater bioabsorption and bioavailability of the active (cytotoxic) metabolites in cancer cells compared with normal cells. The selective cytotoxicity of HO-3867 seemed to be multifaceted, eliciting differential activation of the Akt pathway in normal versus cancer cells. RNAi attenuation experiments confirmed the requirement of STAT3 for HO-3867–mediated apoptosis in ovarian cancer cells. In vivo testing showed that HO-3867 could block xenograft tumor growth without toxic side effects. Furthermore, in primary human ovarian cancer cells isolated from patient ascites, HO-3867 inhibited cell migration/invasion and survival. Our results offer preclinical proof-of-concept for HO-3867 as a selective STAT3 inhibitor to treat ovarian cancer and other solid tumors where STAT3 is widely upregulated.
Introduction
The toxic side effects that standard anticancer drugs exert on healthy tissues and normal cells present obstacles in cancer treatment. These effects lead to dose reductions, treatment delays, and even the discontinuation of therapy. “Targeted therapy” is a relatively modern term commonly used to describe new agents, including small molecules and monoclonal antibodies, specifically designed to take advantage molecular pathways involved in the pathophysiology to be treated. A secondary goal of such developments is to limit the negative side effects these molecules exert on normal tissues. Unfortunately, even new therapeutics cause significant adverse effects on normal tissues, leading to toxicity. Thus, development of safe and targeted anticancer therapies that selectively kill cancer cells while sparing the surrounding healthy tissues is essential.
We identified a novel class of bifunctional compounds based on a diarylidenyl-piperidone (DAP) backbone conjugated to an N-hydroxypyrroline (−NOH; a nitroxide precursor) group for their inhibitory property on STAT3 activation (1, 2). Previous work found that DAP compounds exhibit more toxicity toward cancer cells when compared with noncancerous cells. However, the mechanism behind this selectivity was not fully elucidated. We hypothesize that the addition of the −NOH moiety functions as a modulator of cytotoxicity, imparting antioxidant protection to noncancerous tissues, while allowing the compound to maintain toxicity toward cancer cells. Two compounds, HO-3867 (possessing the −NOH moiety) and H-4073 (lacking the −NOH moiety) with the same DAP backbone were used to expound upon this concept (1, 3-5). In this study, we confirm the selective toxicity of HO-3867 as compared with H-4073. The mechanism of this selectivity is a differential interaction of HO-3867 and H-4073 with STAT3 in normal and cancer cells, with no impact upon other STATs. In addition, both compounds exhibit greater uptake and bioavailability in cancer cells when compared with normal cells. HO-3867 demonstrated anticancer efficacy in both a xenograft model and cisplatin-sensitive and cisplatin-resistant primary ovarian cancer cell populations. These results support the translational potential for HO-3867 as a safe, targeted compound for the treatment of ovarian cancer.
Materials and Methods
Transfection of cells with pBOS-H2BGFP vector and chromatin aberration assay
A2780 and CHO cells were plated 1 day before and were ~80% confluent on the day of transfection. Cells were transfected with pBOS-H2BGFP vector DNA (1 μg/μL; BD Pharmingen) using Fugene 6 transfection reagent according to manufacturer’s instructions.
STAT3 DNA-binding assays
After treatment with HO-3867 for 24 hours, a Nuclear Extract Kit (Clontech Inc.) was used to prepare cell nuclear extracts following the manufacturer’s protocol (6, 7).
STAT3 knockdown experiments
For downregulation of STAT3 in SKOV3 cells, a lentiviral system (pGIPZ) with a set of different short hairpin RNAs (shRNA) was used (Thermo Scientific). A scrambled shRNA was used as a control.
Statistical analysis
Results are expressed as mean ± SE. Comparisons between groups were made by the Student t test and ANOVA as appropriate. The significance level was set at P ≤ 0.05. Prism Graph Pad was used for all statistical calculations.
Results
DAPs target STAT3
A new class of DAP compounds was synthesized by linking two diarylidene groups with a piperidone group; an antioxidant-promoting N-hydroxypyrroline moiety was attached to the piperidone N-terminal of the base compound H-4073 to obtain HO-3867 (Fig. 1A). Treatment of SKOV3 cells with different concentrations of HO-3867 and H-4073 resulted in significantly decreased expression of pSTAT3 (Fig. 1B). DAP compounds exhibited the highest binding affinity for a pocket located on the DNA-binding domain of the STAT3 molecule (Fig. 1C). This is in contrast to other known STAT3 inhibitors, many of which interact with STAT3 at the SH2 domain (8-10). In addition, HO-3867 interacts with both the STAT3 dimer and monomer structures (Fig. 1D); H-4073 docked with the STAT3 monomer is shown in Supplementary Fig. S1A.
Figure 1.

DAP compounds interact with STAT3. A, H-4073(top) is a 3,5-diarylidenyl-piperidone containing a para-fluorosubstitution on the phenyl groups. HO-3867 (bottom) contains an N-hydroxypyrroline moiety covalently linked to the N-terminus of the piperidone of H-4073. B, immunoblots show pSTAT3 is significantly decreased in a dose-dependent manner, with no change in total STAT3. C, H-4073 (left) and HO-3867 (right) docked to the STAT3 dimer. D, images of HO-3867 docked with the STAT3 monomer (left) and the dimer (right), with lowest calculated binding energies shown. Insets show the monomeric or dimeric structures highlighting the preferred binding site on the DNA-binding domain. E, immunoblot assay shows that HO-3867 did not inhibit other pSTATs. F, HO-3867 and H-4073 inhibit DNA-binding of STAT3, as indicated by ELISA assay results. G, purified human STAT3 protein (2 μg) was incubated with PBS or HO-3867 (10 μmol/L) for 2 hours. Spectra show molecular interaction between HO-3867 and STAT3. H, luciferase assay results show that HO-3867 specifically inhibits pSTAT3 and transcriptional activity with minimal impact on other pSTATs. I, STAT3 translocation assay images. Green fluorescence indicates pSTAT3, with DAPI used as a nuclear counterstain. Untreated controls showing the pSTAT3 expression and HO-3867 (10 μmol/L)–treated inhibition of pSTAT3; IL-6 (50 ng/mL) stimulated controls, showing nuclear translocation of pSTAT3; HO-3867 (10 μmol/L) –treated cells stimulated with IL-6, showing inhibition of pSTAT3 nuclear translocation.
Western blots demonstrate that HO-3867 does not impact expression of other pSTATs (Fig. 1E). Because in silico docking simulations suggested that the compounds interact with the DNA-binding domain of STAT3, we evaluated this in vitro. SKOV3 cells were treated with 10 μmol/L of H-4073 or HO-3867, and samples subjected to ELISA assay. DNA binding activity was measured, and both compounds inhibited STAT3 DNA binding activity when compared with control (Fig. 1F).
Additional mechanistic studies focused upon HO-3867, which was indicated to have the greater binding affinity than H-4073 for the STAT3 DNA-binding domain. The difference in binding affinity is believed to be because of the inclusion of the −NOH moiety in the HO-3867 structure. Purified STAT3 protein (2 μg) was incubated with HO-3867 (10 μmol/L) for 2 hours. STAT3 was immunoprecipitated, repeatedly washed to remove unbound HO-3867, and subjected to electron paramagnetic resonance (EPR) analysis at X-band (9.8 GHz). As shown in Fig. 1G, a clear triplet EPR signal, characteristic of nitroxide (−NO·) compounds, was obtained from human STAT3 incubated with HO-3867, suggesting DAP-STAT3 interaction. Figure 1H shows a reduction in luciferase expression when the cells were treated with HO-3867, whereas no change in STAT4 and STAT5 transcriptional activity was observed. Mechanistically, STAT3 nuclear translocation was examined using a previously reported method (10, 11). A2780 cells were treated for 3 hours with 10 μmol/L of HO-3867, after which STAT3 translocation was stimulated by adding IL-6 for 30 minutes. As shown in Fig. 1I, STAT3 nuclear translocation in the HO-3867–treated cells was inhibited (right) when compared IL-6–stimulated cells. The intensity of nuclear pSTAT3 staining was quantified from the green channel. Treatment with HO-3867 reduced overall pSTAT3 expression and nuclear expression of pSTAT3 when compared with control or IL-6 stimulated cells (Supplementary Fig. S1B). These results provide evidence that DAP compounds interact specifically with STAT3 and inhibit its binding to DNA by preventing dimerization, nuclear translocation and/or phosphorylation.
Effect of DAPs on cancer cells versus normal cells
Development of STAT3 inhibitors that are selectively cytotoxic to cancer cells while sparing normal cells would be a significant step forward in cancer therapeutics. We hypothesize that the −NOH moiety of HO-3867 serves as a modulator of cytotoxicity. H-4073, which possesses an identical backbone structure but lacks the −NOHantioxidant precursor group, was used for comparative purposes. Both DAP compounds inhibited proliferation in ovarian cancer cells in a dose- and time-dependent manner. However, HO-3867 exhibited minimal toxicity toward normal cells, whereas H-4073 was toxic to both cell types (Supplementary Fig. S2A and S2B). Clonogenic assays confirm that HO-3867 is less toxic to normal CHO cells when compared with H-4073 (22% vs. 92%, P < 0.05; Fig. 2A and B).
Figure 2.
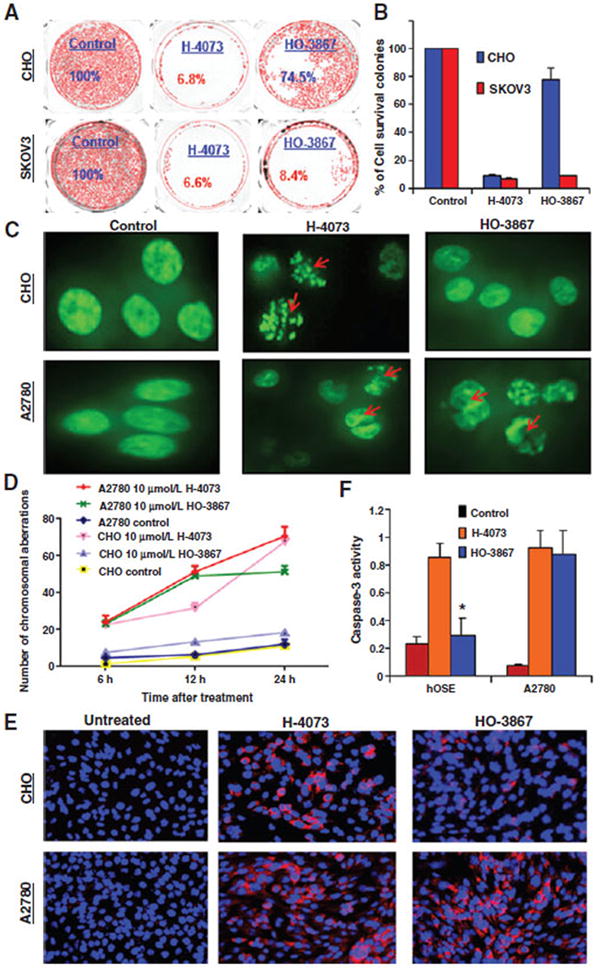
Selective cytotoxicity of HO-3867 toward to cancer cells. A, clonogenic assay: SKOV3 and CHO cells were treated with 10 μmol/L of H-4073 or HO-3867 for 24 hours, after which the drug was removed and cells were observed for 72 hours for colony-forming ability. B, quantification of colony survival. C, A2780 and CHO cells engineered to express a GFP-Histone 2B fusion protein were treated with 10 μmol/L of H-4073 or HO-3867 at different time points, followed by immunofluorescence microscopy. Red arrows, individual cells that underwent abortive mitosis—with transient chromatin condensation or apoptosis. D, quantification of chromosomal aberration at 6, 12, and 24 hours following treatment reveals that H-4073 causes more chromosomal aberration than HO-3867 in CHO cells (P < 0.0001 at all-time points) but not in A2780 cells where a difference only developed at 24 hours (P = 0.9982, 0.9216, <0.0001). E, immunofluorescent staining using 8-hydroxyguanosine (8OHdG), after treating cells with HO-3867 or H-4073 for 6 hours. Before treatment neither CHO nor A2780 stained positive for 8OHdG. After treatment with H-4073, both cell lines showed similarity to 8OHdG. Conversely, HO-3867 treatment induced elevated 8OHdG in A2780 relative to CHO cells (red, 8OHdG; blue, DAPI nuclear stain). Magnification, ×200. F, HO-3867 induced more caspase-3 activity in A2780 than hOSE cells compared with H-4073 (*, P ≤ 0.0001, n = 3).
Successful transfection of the A2780 and CHO cells with GFP-Histone 2B fusion protein was confirmed by fluorescence-activated cell sorting (FACS; Supplementary Fig. S3). Transfected cells were treated with 10 μmol/L of HO-3867 or H-4073 for up to 24 hours, followed by immunofluorescence microscopy. In Fig. 2C, red arrows indicate individual cells that underwent abortive mitosis with transient chromatin condensation or apoptosis. Quantification of chromosomal aberration at different time points (means ± SEM, n = 100 cells/data point), confirms the differential action of HO-3867 on normal versus cancer cells, P < 0.0001 (Fig. 2D). Using flow cytometry, apoptosis was quantified in A2780, hOSE, CHO, and H9c2 cells treated with 10 μmol/L of HO-3867 or H-4073 for 24 hours. HO-3867 induced less apoptosis in hOSE, CHO, and H9c2 cells when compared with A2780 cells (12.3% vs. 55.8%, P < 0.0001), respectively, whereas H-4073 induces apoptosis in both cell types (63.6% vs. 48.5%, P < 0.0001; Supplementary Table S1 and Fig. S4A and S4B). When compared with other STAT3 inhibitors HO-3867 show similar toxicity to cancer cells, but decreased toxicity to normal cells (Supplementary Fig. S5). Furthermore, we evaluated oxidative stress in the cells using 8-hydroxyguanosine (8OHdG) after treating cells with HO-3867 or H-4073 for 6 hours. After treatment with H-4073, both cell lines showed similar of levels of 8OHdG. Conversely, HO-3867 treatment resulted in elevated 8OHdG staining in cancer cells relative to normal cells (Fig. 2E). Based on our previous report (2), which showed that DAP compounds induce apoptosis via activation of caspase-3 in cancer cells, we examined the expression level of caspase-3 activity in both hOSE and A2780 cells treated with 10 μmol/L of HO-3867 or H-4073. An increase in caspase-3 activity expression was found in HO-3867–treated ovarian cancer cells when compared with HO-3867–treated normal cells and untreated controls, but no difference was observed after H-4073 treatment (Fig. 2F). This suggests a differential involvement of caspase-3 in HO-3867–induced apoptosis. It is clear that although HO-3867 and H-4073 show similar toxicity toward cancer cells, H-4073 is significantly more toxic toward normal cell types.
Differential bioabsorption of DAPs in ovarian cancer cells versus normal cells
To evaluate the mechanism behind the differential toxicity exhibited by the 2 compounds, we used EPR spectroscopy, UV/Vis spectrophotometry, and liquid chromatography/mass spectrometry (LC/MS) with both CHO or hOSE cells and A2780 cells (3). For the EPR study, hOSE and A2780 cells were treated with HO-3867 at 10 μmol/L for 3 hours, and then collected for analysis. The N-hydroxypyrroline (−NOH) moiety is capable of undergoing a reversible, one-electron oxidation to its nitroxide form (−NO·), which is paramagnetic and detectable by EPR spectroscopy (12). A substantial amount of the nitroxide form of HO-3867 was detected in both cells using EPR, suggesting that HO-3867 rapidly entered the cells during the 3 hour exposure period (Fig. 3A).
Figure 3.

Cellular absorption of H-4073 and HO-3867 in ovarian cancer cells versus normal cells. A, EPR spectra obtained from A2780 and hOSE cells treated with 10 μmol/L of HO-3867 for 3 hours. B, the cellular bio-absorption of H-4073 and HO-3867 was quantified using UV/Vis spectrophotometry in hOSE and A2780 cells after 3 hours of treatment with 10 μmol/L of H-4073 or HO-3867. HO-3867 demonstrates lower bio-absorption in hOSE cells compared with H-4073 (P ≤ 0.05, n = 6). C, LC/MS was used to evaluate bio-absorption of HO-3867 and H-4073 in CHO and A2780 cells. HO-3867 reached maximum concentration of 0.0548 ± 0.0163 μmol/L/million cells after 1 hour and plateaued at 0.0352 ± 0.0056 μmol/L/million cells after 3 hours. No drug was detected in CHO cells. D, LC/MS output of HO-3867 in CHO and A2780 cells treated with 10 μmol/L of HO-3867 for 1 hour. E, response area of the M+4 metabolite formed in A2780 and CHO cell lines after treatment with 10 μmol/L of H-4073 and HO-3867 at different time points.
The difference in signal amplitude observed in Fig. 3A would, at first, seem counterintuitive. It would be expected that a lower concentration of the nitroxide form would be found in the cancer cells, which have a more reductive environment than the corresponding normal cells, thereby producing lower signal amplitude. To further evaluate this, we quantified the cellular uptake of both H-4073 and HO-3867 at the same dose (10 μmol/L) and time point in both cells using UV/Vis spectrophotometry. The cellular uptake of HO-3867 was 5-fold and H-4073 was 2-fold greater in cancer cells than in normal cells after 3 hours of incubation (Fig. 3B). Uptake of HO-3867 byCHO and A-2780 cell lines was also analyzed by a liquid chromatography and tandem mass spectrometry (LC-MS/MS) assay. Both cell lines were treated with 10 μmol/L of either HO-3867 or H-4073, and cells were harvested at the allocated time points of 0, 1, 3 and 6 hours. Drug concentration was measured and calculated against the calibration curve of HO-3867. A2780 cells showed significant uptake of HO-3867, but CHO cells had no detectable levels of HO-3867 (Fig. 3C) and lower levels of H-4073 when compared with A2780 cells (Supplementary Fig. S6A). These results help to explain the greater EPR signal amplitude from the cancer cells. Despite the more reductive environment, a greater concentration of HO-3867 molecules, in both forms, would be present in the cancer cells, thereby providing a greater signal when compared with the noncancerous cells.
Next, in vitro metabolism studies were completed. In both A2780 and CHO cell lines, the major metabolite observed following incubation was the M+4 metabolite (Supplementary Table S2). The concentration, as represented by the response area of the M+4 metabolite, is much higher in CHO in comparison to A2780 cells for both compounds (Fig. 3D and E). This suggests that CHO cells metabolize HO-3867 faster than H-4073, which may explain why no HO-3867 was detected in the CHO cells by EPR spectroscopy. Extracted ion chromatograms of metabolites following exposure to H-4073 or HO-3867 in both CHO and A2780 cells at different time points were obtained (Supplementary Fig. S6B–S6E).
Differential effect of HO-3867 on STAT3/Akt signaling pathway in cancer cells versus normal cells
The differential bio-absorption study results obtained using EPR and LC/MS do not fully explain the selective mechanism of action of HO-3867. To elucidate this further, we examined the effect of HO-3867 on the STAT3/Akt signaling pathway, specifically looking at STAT3 activation and STAT3-mediated genes. After treatment of A2780 and SKOV3 cells with 10 μmol/L of HO-3867, analysis showed decreased levels of p-STAT3, p-Akt Ser473, and Bcl-2, with a concomitant increase in p53 or p21. Conversely, hOSE and CHO cells showed no change in p-STAT3, p53, or Bcl-2 levels, but an increase in pAkt Ser473 (Fig. 4A and B). This suggests that activation of pAkt in normal cells protects them from apoptosis, whereas inhibition of pSTAT3 and pAkt in HO-3867-treated A2780 or SKOV3 cells, promotes apoptosis. Changes in phosphorylated Akt expression were confirmed by confocal microscopy, as shown in Fig. 4C. Conversely, treatment with H-4073 showed decreased levels of pSTAT3 and elevated cleaved-caspase-7 in both CHO and A2780 cells (Supplementary Fig. S7A).
Figure 4.
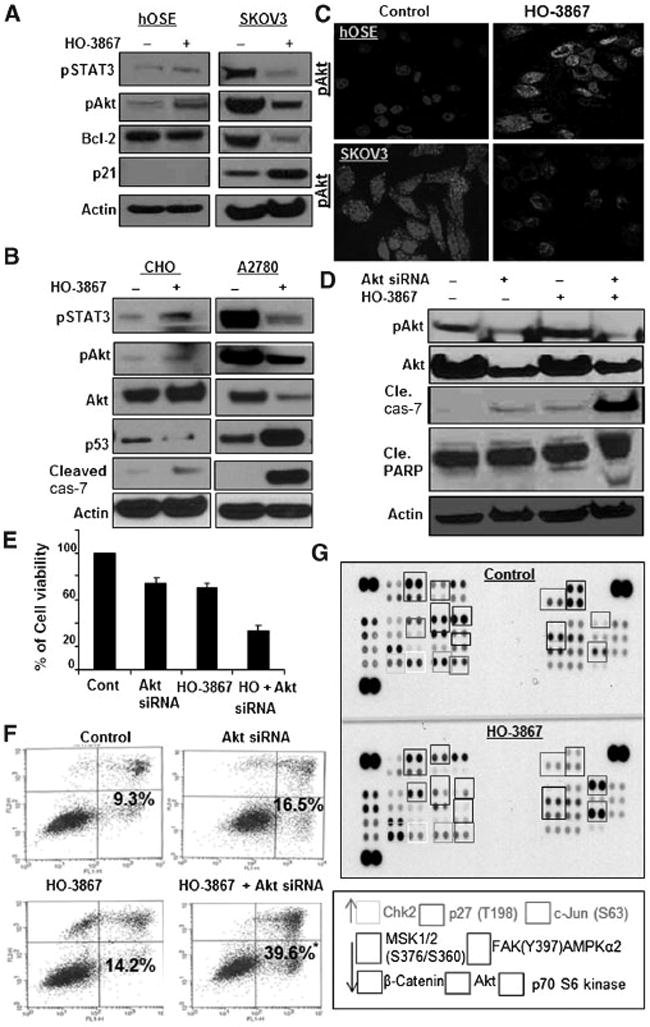
HO-3867 selectively inhibits STAT3 and Akt expression in cancer cells. A and B, immunoblot of hOSE, SKOV3, A2780, and CHO cells treated with 10 μmol/L of HO-3867 for 24 hours. C, confocal microscopy showing pAkt Ser473 expression in hOSE and SKOV3 cells before and after treatment with 10 μmol/L of HO-3867 for 24 hours. Treatment increased pAkt Ser473 in hOSE cells, but inhibited it in SKOV3 cells. D, Akt siRNA was used to silence Akt in CHO cells. Cells were then treated with 10 μmol/L of HO-3867 for 24 hours. Immunoblot analysis was used to evaluate Akt, pAkt Ser473, cleaved-caspase-7, and cleaved-PARP. E, cell viability was decreased in CHO cells transfected with Akt siRNA and treated with HO-3867 when compared with other groups (P < 0.05). F, Annexin V performed using CHO cells: control, Akt siRNA alone, 10 μmol/L of HO-3867 alone, and combination. Increased apoptosis was observed in CHO cells transfected with Akt siRNA and treated with 10 μmol/L of HO-3867 for 24 hours when compared with all other groups (*, P ≤ 0.05, n = 6). G, protein kinase array performed on SKOV3 cells with and without 10 μmol/L of HO-3867 treatment for 24 hours.
Akt siRNA reverses the selective cytotoxicity of HO-3867 in CHO cells
Akt siRNA was transfected into CHO cells for 24 hours to determine if the apparent selective activation of Akt is necessary for HO-3867–mediated cytotoxicity. Cells were treated with 10 μmol/L of HO-3867 for 24 hours. Western blot analysis confirmed transfection. In CHO cells transfected with Akt siRNA before treatment with HO-3867, lower levels of total Akt and pAkt were observed when compared with untransfected controls. This decrease in pAkt resulted in an increase in the apoptosis markers cleaved-caspase-7 and cleaved-PARP (Fig. 4D). Decreased viability and doubling of the number of cells undergoing apoptosis was observed in transfected cells treated with HO-3867 (Fig. 4E and F). This indicates that activation of Akt is essential in the reduced cytotoxicity ofHO-3867 in normal cells. Next, we sought to determine if the selective activation or inhibition of pAktwas because of alterations in kinase pathways induced by HO-3867. Akt kinase activity was evaluated after treatment with HO-3867, and activation or suppression of various kinases was noted in A2780 (Fig. 4G) but not CHO cells (Supplementary Fig. S7B). This suggests that the selective toxicity exhibited by HO-3867 is related to differential effects on the STAT3/Akt pathway in ovarian cancer cells. Further study is required in other cell lines to confirm the activation of Akt by HO-3867 in noncancerous cell lines.
Effect of HO-3867 treatment on STAT3 knockdown ovarian cancer cells
To investigate the requirement of STAT3 in HO-3867–mediated apoptosis, we knocked down STAT3 in ovarian cancer cells using shRNA. Western blot analysis confirmed knockdown. (Fig. 5A). STAT3 knockdown cells were treated with 10 μmol/L of HO-3867 for 24 hours, then cell viability and apoptosis were analyzed. Among SKOV3 STAT3 knockdown cells, viability was increased (Fig. 5B) with a 50% reduction of apoptosis following treatment with HO-3867 when compared with wild-type cells (Fig. 5C and D). These results confirm that HO-3867 inhibition of cell proliferation and induction of apoptosis is mediated primarily by targeting the STAT3 pathway.
Figure 5.
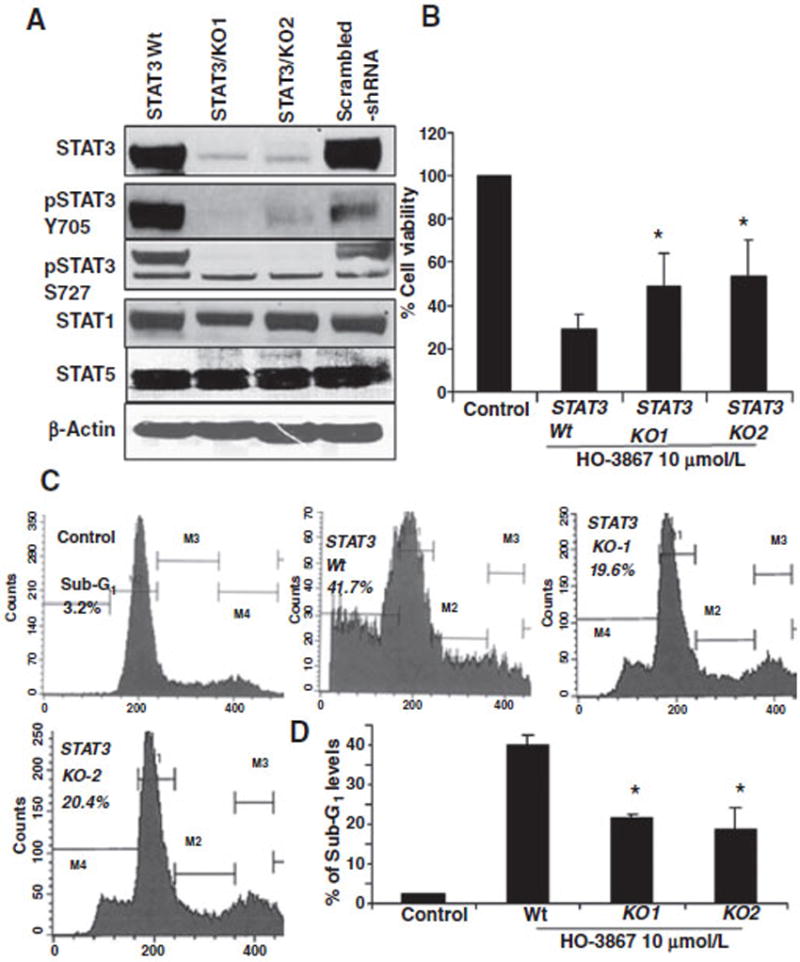
Knockdown STAT3 by s` transfection with shRNA decreased the apoptosis in HO-3867–treated ovarian cancer cells. A, lentiviral shRNA were used to knockdown STAT3 expression in SKOV3 cells using two different shRNA clones. Immunoblots confirm STAT3 knockdown. Wild-type and STAT3 knockdown cells were treated with 10 μmol/L of HO-3867 for 24 hours. B, cell viability was measured. Knockdown cells were less susceptible to the cytotoxic effects of HO-3867 than wild type (*, P ≤ 0.05, n = 5). C, cells were collected and fixed, then subjected to sub-G1 analysis (20,000 events in fixed gate). Intracellular propidium iodide fluorescence intensities are presented. D, quantification of sub-G1 levels; the sub-G1 population was significantly lower in knockout cell lines (*, P < 0.0001).
HO-3867 inhibits xenograft tumor growth without toxicity to other tissues in tumor-bearing mice
We evaluated the efficacy of HO-3867 in a chemotherapy-resistant human ovarian tumor model; mice were injected with A2780cDDP cells. After 28 days, the feed was supplemented with 2 concentrations of HO-3867 (50 or 100 ppm; Harlan-Teklad). Treated mice showed significant reductions in tumor mass, in a dose-dependent manner. Images of tumor bearing control and HO-3867-treated (100 ppm) mice are shown in Fig. 6A, with a graph showing the tumor weight at the time of tissue collection (Fig. 6B, top half). There was no decrease in body weight, a common side effect of traditional chemotherapy (Fig. 6B, bottom half). Excised tumor tissue from HO-3867 treated mice showed decreased levels of STAT3 Tyr705 and Bcl-2 levels, without affecting total STAT3 (Fig. 6C and Supplementary Fig. S8). To confirm uptake of HO-3867, tumor tissues and organs from HO-3867 treated and untreated mice were collected and prepared for EPR spectroscopic analysis as described previously (12). Clear, triplet signals indicating the presence of the −NO· radical (nitroxide) form of HO-3867 were obtained from liver, ovary, kidney, and spleen tissues. Brain, heart, and lung tissues seem to produce possible, but extremely weak signals (Supplementary Fig. S9). In addition, HO-3867 uptake by the tumor tissues was quantified using UV/Vis spectroscopy (Fig. 6D).
Figure 6.
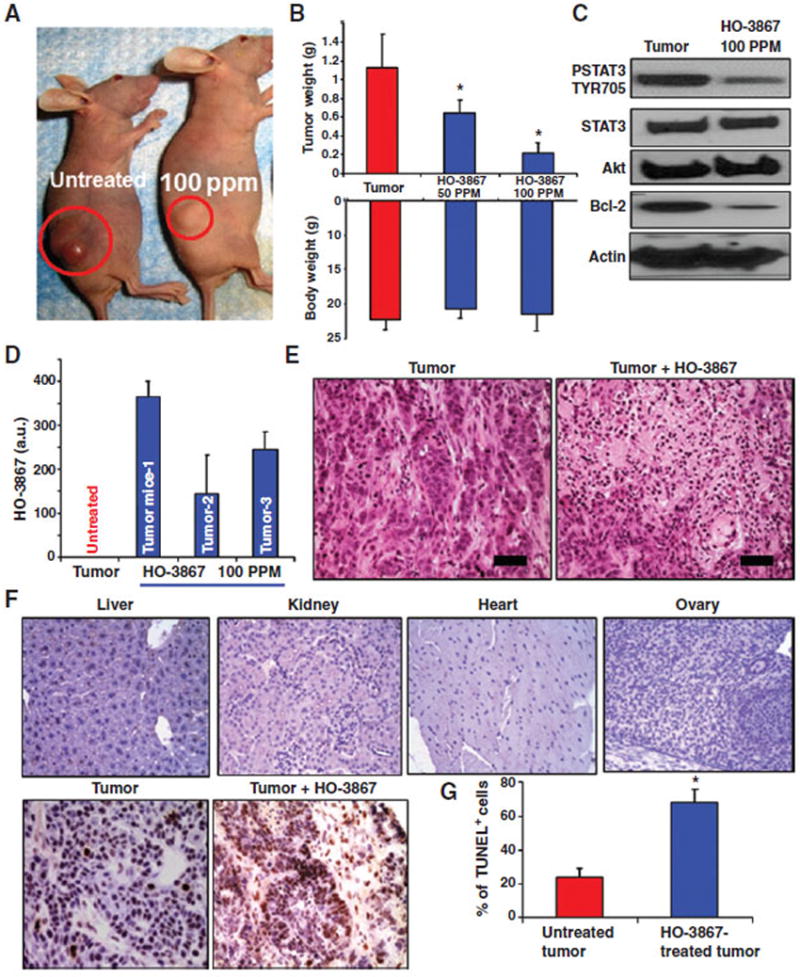
HO-3867 inhibits xenograft ovarian tumor growth. A, A2780 cells were injected into the bilateral flanks of nude mice. HO-3867 (50 or 100 ppm) was delivered in the feed to experimental groups. B, graphs showing the tumor weight and body weight in each group upon completion of the treatment period. Tumor weight was significantly reduced after treatment with either 50 (0.65 g ± 0.02) or 100 ppm (0.214 g ± 0.02) of HO-3867 versus untreated control (1.115 g ± 0.07, n = 7, mean ± SEM, P < 0.0001). Body weight was similar in all groups. C, immunoblot analysis for pSTAT3 Tyr705, Akt, and Bcl-2 using tissue lysates of xenograft tumors. D, HO-3867 levels in three tumor tissue samples measured using EPR. E, H&E staining of tumor with and without HO-3867 treatment (100 ppm); scale bar, 50 μm. F, TUNEL staining in various major organs and xenograft tumor. Apoptosis is present only in tumor specimens, not in the other organs of mice treated with HO-3867. G, quantification of TUNEL staining in tumor samples: control (21.46%) and HO-3867 treated (77.81%); *, P < 0.0001, n = 6.
The toxicity of HO-3867 in various organs and tumor tissues was evaluated using hematoxylin and eosin (H&E) and terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling (TUNEL) staining. H&E staining showed increased necrosis in tumors of HO-3867–treated mice when compared with control (Fig. 6E). No evidence of any cellular degeneration or inflammatory lesions was observed in mice supplemented with HO-3867 (Supplementary Fig. 10A). The results did not reveal increased apoptosis in the normal organ tissues of animals treated with HO-3867, however there was marked TUNEL-positive staining in tumor tissues treated with HO-3867 when compared with untreated controls (Fig. 6F and G and Supplementary Fig. S10B). These results confirm the efficacy of HO-3867 against chemoresistant ovarian tumors, with no apparent signs of toxicity toward normal tissues. The data show that HO-3867 inhibits ovarian cancer by targeting the STAT3 signaling pathway.
Ex vivo efficacy of HO-3867 on primary ovarian cancer cell populations
The efficacy of HO-3867 was evaluated in primary ovarian cancer populations using cell lines TR-127 and TR-182 (Dr. G. Mor, Yale University) derived from recurrent, chemotherapy-resistant tissues, and one collected from the ascites of a patient at The Ohio State University Wexner Medical Center. We found consistent expression of active STAT3 in primary cancer cell populations (Fig. 7A). Treatment with HO-3867 inhibited cell survival (Fig. 7B) and induced more cell death within 24 hours when compared with STATTIC and standard anticancer drugs (Supplementary Fig. S11A–S11C). Expression of STAT3, pAkt, and STAT3 target proteins cyclin D1, cyclin D2, and survivin was decreased after treatment with HO-3867 (Supplementary Fig. S12). Tumor cells in ascites are a major source of metastasis and disease recurrence in ovarian cancer patients. In vitro migration (Fig. 7C and D) and invasion (Fig. 7E and F) of these cells lines were inhibited by HO-3867. These results show that HO-3867 demonstrates anticancer efficacy in ovarian cancer cells lines, xenografts, and primary cell populations, suggesting the translational potential for HO-3867 as a safe, targeted compound for the treatment of ovarian cancer.
Figure 7.
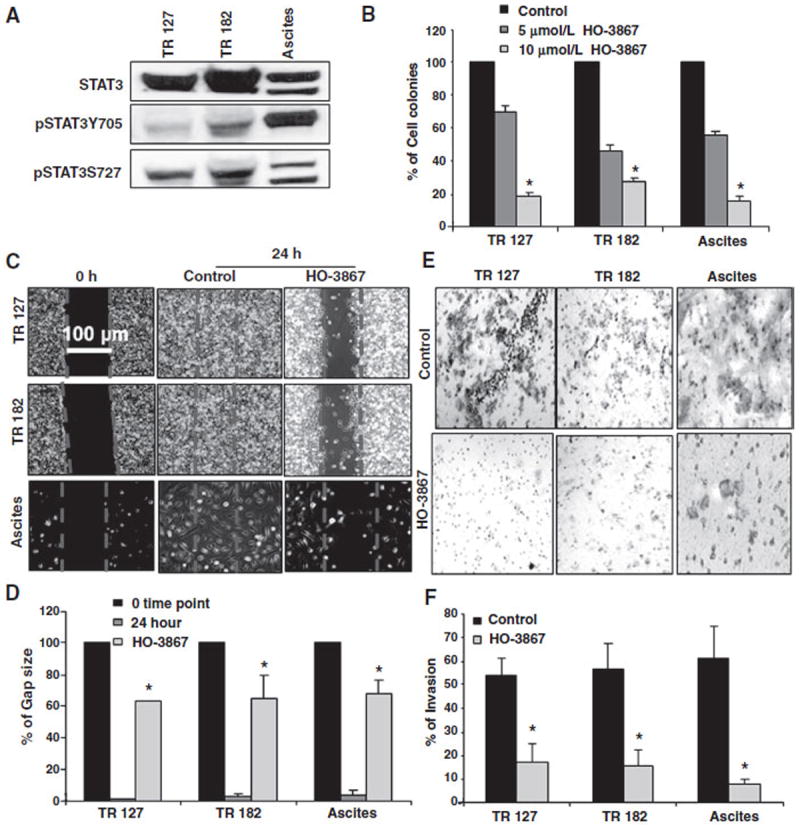
HO-3867 inhibits migration and invasion, and is cytotoxic to primary ovarian cancer cells. A, STAT3 expression in primary ovarian cell populations. B, three different primary ovarian cancer cell lines were treated with 5 or 10 μmol/L of HO-3867 for 24 hours and subjected to clonogenic assay. Quantification of clonogenic survival in cells showing decreased, dose-dependent survival in all cell lines when compared with control (n = 3, reported% ± SEM, P < 0.0001 all groups vs. control). C, cell migration/wound healing assay representative images using primary ovarian cell lines evaluated before and after 24 hours exposure to 10 μmol/L of HO-3867 with corresponding controls. D, the residual gap between the migrating cells from the opposing edges is expressed as a percentage of the initial, scraped area. HO-3867 significantly inhibits the reduction in gap size caused by cell migration (n =5, control, treated, mean ± SD; all *, P < 0.0001). E, representative images of the invasion assay results using a transwell Boyden chamber with primary ovarian cancer cell lines and corresponding controls. F, quantification of invasion assay. HO-3867 inhibits invasion versus control in all cell lines (n = 3, mean ± SD; *, P < 0.0001).
Discussion
Drug development is a long and arduous journey. Many compounds are developed and evaluated on a yearly basis to find a promising few that are suitable for human clinical trials. Our compound, HO-3867, shows great promise in preclinical evaluations for the treatment of ovarian cancer. It has shown toxicity toward recurrent primary ovarian cancer cell populations, excellent oral bio-absorption, and bio-availability in a xenograft model, as well as in vivo toxicity in platinum-resistant tumors. However, many compounds have these properties—the novel innovation that sets HO-3867 apart is its selective toxicity toward cancer cells while sparing normal cells in both in vitro and in vivo experiments by taking advantage of one of the underlying biomolecular causes of disease.
At present, only a few agents are known to possess the potential for selective/preferential elimination of cancer cells without affecting normal cells (13-15). Sanguinarine, an alkaloid derived from the root of Sanguinaria canadendid and used commonly in mouth washes and toothpaste is one such compound. In vitro data show that it displays toxicity toward human epidermoid carcinoma (A431) cells but not normal human epidermal keratinocytes (16).
In this study, wewere able to elucidate the mechanism behind the selective toxicity of HO-3867 and translate this to in vivo model showing little or no toxicity at clinically effective doses. The mechanism is multifaceted, but is based on the addition of the N-hydroxypyrroline (−NOH) moiety to H-4073, to obtain HO-3867. This moiety functions as a modulator of cytotoxicity, allowing the compound to exist in an equilibrium of oxidized (−NO·) and reduced forms (−NOH) depending on the cellular environment (17). In normal cells, the N-hydroxypyrroline group has the greater potential to oxidize, generating a nitroxide that imparts antioxidant protection to noncancerous tissues (17-20). In cancer cells and hypoxic tumors, the microenvironment is far more reductive in nature, inhibiting oxidation and reducing antioxidant protection to tumor cells (21-23).
The difference between oxidized and reduced forms of nitroxyl radicals has been well established using EPR (24-26). Previous (1) and current study from our lab revealed that after in vitro treatment with HO-3867, cancer cells have higher levels of reactive oxygen species (ROS) and elevated 8OHdG expression when compared with normal cells as measured by DHE and 8OHdG staining. This suggests that HO-3867 acts as an antioxidant in normal cells, but not in cancer cells. In this study, we show that the bio-absorption of HO-3867 is greater in cancer cells and more slowly metabolized, which likely contributes to the enhanced toxicity; allowing greater accumulation of ROS. This is in contrast to platinum-based chemotherapeutic agents, which promote ROS accumulation in both cancer and normal cells, leading to significant systemic toxicity. A previous study found that mice cotreated with cisplatin and HO-3867 did not have a reduction in body weight compared with control, whereas those treated with the same dose of cisplatin alone did lose weight during treatment (27). Given these findings it is possible that combining cisplatin with HO-3867 may protect against cisplatin-associated nephrotoxicity.
Another facet to the selective toxicity is a differential impact at the molecular level. HO-3867 preferentially inhibits phosphorylation of STAT3, and to a lesser extent pAkt, in cancer cells, but activates Akt expression in normal cells. This leads to selective cell killing of cancer cells while protecting normal cells. This is congruent with previous work from our lab, showing that cotreatment with HO-3867 and doxorubicin prevents development of doxorubicin-associated cardiomyopathy in mice through a similar mechanism (4). These data establish a critical and unique dependency on Akt activation for the protection of normal cells and inhibition of STAT3 activation for anticancer effect.
To our knowledge, this is the first systematic study showing the selective induction of apoptosis in ovarian cancer cells versus normal cells using an orally deliverable STAT3 inhibitor. Most importantly, we show that HO-3867 is highly effective in primary cancer cell populations, including cells known to be resistant to conventional chemotherapeutics. Viable treatment options for this latter cancer cell type are currently very limited or unavailable, suggesting potential translational application of HO-3867 for the safe treatment of ovarian cancer, including recurrent disease. However, additional studies are needed to confirm the selective cytotoxicity of HO-3867 via Akt signaling mechanisms in vivo. Safe and targeted inhibitors are an essential tool for future therapeutic intervention, particularly in the treatment of cancers. The present results provide a convincing argument for preclinical evaluation of HO-3867 in the treatment of ovarian cancer, including cancer that has developed resistance to conventional chemotherapeutic intervention.
Supplementary Material
Acknowledgments
The authors thank Drs. P. Goodfellow, J. Hays, and B. Kaur for their valuable insights, comments, and suggestions in the construction of this article; Dr. M. Phelps and Dr. Y. Ling of the Pharmacoanalytical Shared Resource for LC/MS analysis; and Dr. G. Mor of Yale University for providing the primary ovarian cancer cell populations. The authors also thank graduate students RossWanner, Meryl Sudhakar, and Priya Nalluri for the cell culture and basic assay help.
Grant Support
This work was supported by Ovarian Cancer Research Fund (OCRF) and Kaleidoscope of Hope foundation awards to K. Selvendiran and OSU CCC Internal grant to D.E. Cohn.
Footnotes
Authors’ Contributions
Conception and design: K.S. Rath, S.K. Naidu, G.A. McCann, K. Hideg, P. Kuppusamy, D.E. Cohn, K. Selvendiran
Development of methodology: S.K. Naidu, B.H. Hemant, G.A. McCann, V. Bravo, G. Leone, K. Hideg
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): K.S. Rath, S.K. Naidu, P. Lata, B.H. Hemant, G.A. McCann, B.J. Tierney, A.C. ElNaggar, K. Hideg
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): K.S. Rath, S.K. Naidu, P. Lata, B.H. Hemant, B.K. Rivera, A.C. ElNaggar, G. Leone, K. Hideg, D.E. Cohn, K. Selvendiran
Writing, review, and/or revision of the manuscript: K.S. Rath, S.K. Naidu, B.H. Hemant, B.K. Rivera, G.A. McCann, A.C. ElNaggar, P. Houghton, K. Hideg, P. Kuppusamy, D.E. Cohn, K. Selvendiran
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): B.H. Hemant, B.K. Rivera, G.A.McCann, B.J. Tierney, G. Leone, K. Hideg
Study supervision: K. Hideg, D.E. Cohn, K. Selvendiran
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
References
- 1.Selvendiran K, Ahmed S, Dayton A, Kuppusamy ML, Tazi M, Bratasz A, et al. Safe and targeted anticancer efficacy of a novel class of antioxidant-conjugated difluorodiarylidenyl piperidones: differential cytotoxicity in healthy and cancer cells. Free Radic Biol Med. 2010;48:1228–35. doi: 10.1016/j.freeradbiomed.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Selvendiran K, Tong L, Bratasz A, Kuppusamy ML, Ahmed S, Ravi Y, et al. Anticancer efficacy of a difluorodiarylidenyl piperidone (HO-3867) in human ovarian cancer cells and tumor xenografts. Mol Cancer Ther. 2010;9:1169–79. doi: 10.1158/1535-7163.MCT-09-1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Selvendiran K, Ahmed S, Dayton A, Ravi Y, Kuppusamy ML, Bratasz A, et al. HO-3867, a synthetic compound, inhibits the migration and invasion of ovarian carcinoma cells through downregulation of fatty acid synthase and focal adhesion kinase. Mol Cancer Res. 2010;8:1188–97. doi: 10.1158/1541-7786.MCR-10-0201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dayton A, Selvendiran K, Meduru S, Khan M, Kuppusamy ML, Naidu S, et al. Amelioration of doxorubicin-induced cardiotoxicity by an anticancer-antioxidant dual-function compound, HO-3867. J Pharmacol Exp Ther. 2011;339:350–7. doi: 10.1124/jpet.111.183681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Selvendiran K, Tong L, Vishwanath S, Bratasz A, Trigg NJ, Kutala VK, et al. EF24 induces G2/M arrest and apoptosis in cisplatin-resistant human ovarian cancer cells by increasing PTEN expression. J Biol Chem. 2007;282:28609–18. doi: 10.1074/jbc.M703796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bid HK, Kibler A, Phelps DA, Manap S, Xiao L, Lin J, et al. Development, characterization, and reversal of acquired resistance to the MEK1 inhibitor selumetinib (AZD6244) in an in vivo model of childhood astrocytoma. Clin Cancer Res. 2013;19:6716–29. doi: 10.1158/1078-0432.CCR-13-0842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bid HK, Roberts RD, Cam M, Audino A, Kurmasheva RT, Lin J, et al. ΔNp63 promotes pediatric neuroblastoma and osteosarcoma by regulating tumor angiogenesis. Cancer Res. 2014;74:320–9. doi: 10.1158/0008-5472.CAN-13-0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang X, Sun Y, Pireddu R, Yang H, Urlam MK, Lawrence HR, et al. A novel inhibitor of STAT3 homodimerization selectively suppresses STAT3 activity and malignant transformation. Cancer Res. 2013;73:1922–33. doi: 10.1158/0008-5472.CAN-12-3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao W, Jaganathan S, Turkson J. A cell-permeable Stat3 SH2 domain mimetic inhibits Stat3 activation and induces antitumor cell effects in vitro. J Biol Chem. 2010;285:35855–65. doi: 10.1074/jbc.M110.154088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X, Yue P, Page BD, Li T, Zhao W, Namanja AT, et al. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc Natl Acad Sci U S A. 2012;109:9623–8. doi: 10.1073/pnas.1121606109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Selvendiran K, Koga H, Ueno T, Yoshida T, Maeyama M, Torimura T, et al. Luteolin promotes degradation in signal transducer and activator of transcription 3 in human hepatoma cells: an implication for the antitumor potential of flavonoids. Cancer Res. 2006;66:4826–34. doi: 10.1158/0008-5472.CAN-05-4062. [DOI] [PubMed] [Google Scholar]
- 12.Dayton A, Selvendiran K, Kuppusamy ML, Rivera BK, Meduru S, Kalai T, et al. Cellular uptake, retention and bioabsorption of HO-3867, a fluorinated curcumin analog with potential antitumor properties. Cancer Biol Ther. 2010;10:1027–32. doi: 10.4161/cbt.10.10.13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahmad N, Feyes DK, Nieminen AL, Agarwal R, Mukhtar H. Green tea constituent epigallocatechin-3-gallate and induction of apoptosis and cell cycle arrest in human carcinoma cells. J Natl Cancer Inst. 1997;89:1881–6. doi: 10.1093/jnci/89.24.1881. [DOI] [PubMed] [Google Scholar]
- 14.Donnerstag B, Ohlenschlager G, Cinatl J, Amrani M, Hofmann D, Flindt S, et al. Reduced glutathione and S-acetylglutathione as selective apoptosis-inducing agents in cancer therapy. Cancer Lett. 1996;110:63–70. doi: 10.1016/s0304-3835(96)04461-8. [DOI] [PubMed] [Google Scholar]
- 15.Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol. 2011;7:428–30. doi: 10.1038/nchembio.573. [DOI] [PubMed] [Google Scholar]
- 16.Ahmad N, Gupta S, Husain MM, Heiskanen KM, Mukhtar H. Differential antiproliferative and apoptotic response of sanguinarine for cancer cells versus normal cells. Clin Cancer Res. 2000;6:1524–8. [PubMed] [Google Scholar]
- 17.Rath KS, McCann GA, Cohn DE, Rivera BK, Kuppusamy P, Selvendiran K. Safe and targeted anticancer therapy for ovarian cancer using a novel class of curcumin analogs. J Ovarian Res. 2013;6:35. doi: 10.1186/1757-2215-6-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vitolo JM, Cotrim AP, Sowers AL, Russo A, Wellner RB, Pillemer SR, et al. The stable nitroxide tempol facilitates salivary gland protection during head and neck irradiation in a mouse model. Clin Cancer Res. 2004;10:1807–12. doi: 10.1158/1078-0432.ccr-03-0194. [DOI] [PubMed] [Google Scholar]
- 19.Hahn SM, Krishna MC, DeLuca AM, Coffin D, Mitchell JB. Evaluation of the hydroxylamine Tempol-H as an in vivo radioprotector. Free Radic Biol Med. 2000;28:953–8. doi: 10.1016/s0891-5849(00)00176-3. [DOI] [PubMed] [Google Scholar]
- 20.Hahn SM, Krishna CM, Samuni A, DeGraff W, Cuscela DO, Johnstone P, et al. Potential use of nitroxides in radiation oncology. Cancer Res. 1994;54:2006s–10s. [PubMed] [Google Scholar]
- 21.Denko NC, Fontana LA, Hudson KM, Sutphin PD, Raychaudhuri S, Altman R, et al. Investigating hypoxic tumor physiology through gene expression patterns. Oncogene. 2003;22:5907–14. doi: 10.1038/sj.onc.1206703. [DOI] [PubMed] [Google Scholar]
- 22.Verrax J, Taper H, Buc Calderon P. Targeting cancer cells by an oxidant-based therapy. Curr Mol Pharmacol. 2008;1:80–92. [PubMed] [Google Scholar]
- 23.Giaccia AJ, Simon MC, Johnson R. The biology of hypoxia: the role of oxygen sensing in development, normal function, and disease. Genes Dev. 2004;18:2183–94. doi: 10.1101/gad.1243304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuppusamy P, Li H, Ilangovan G, Cardounel AJ, Zweier JL, Yamada K, et al. Noninvasive imaging of tumor redox status and its modification by tissue glutathione levels. Cancer Res. 2002;62:307–12. [PubMed] [Google Scholar]
- 25.Mitchell JB, Krishna MC, Kuppusamy P, Cook JA, Russo A. Protection against oxidative stress by nitroxides. Exp Biol Med. 2001;226:620–1. doi: 10.1177/153537020222600703. [DOI] [PubMed] [Google Scholar]
- 26.Bratasz A, Weir NM, Parinandi NL, Zweier JL, Sridhar R, Ignarro LJ, et al. Reversal to cisplatin sensitivity in recurrent human ovarian cancer cells by NCX-4016, a nitro derivative of aspirin. Proc Natl Acad Sci U S A. 2006;103:3914–9. doi: 10.1073/pnas.0511250103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Selvendiran K, Ahmed S, Dayton A, Kuppusamy ML, Rivera BK, Kalai T, et al. HO-3867, a curcumin analog, sensitizes cisplatin-resistant ovarian carcinoma, leading to therapeutic synergy through STAT3 inhibition. Cancer Biol Ther. 2011;12:837–45. doi: 10.4161/cbt.12.9.17713. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.