

Abstract
The chromatin remodeling amine oxidase lysine-specific demethylase 1 (LSD1) has become an attractive target for the design of specific inhibitors with therapeutic potential. We, and others, have described LSD1 inhibitors that have potential as antitumor agents. Many of the currently known LSD1 inhibitors are poor drug candidates, or are structurally based on the tranylcypromine backbone, thus increasing the potential for off-target effects mediated by other amine oxidases. We now describe a series of potent LSD1 inhibitors based on a novel 1,2,4-triazole scaffold; these inhibitors show a high degree of specificity for LSD1 in vitro, and cause increases in cellular histone 3 dimethyllysine 4 (H3K4me2), a gene transcription activating mark. Importantly, these inhibitors are not toxic to mammalian cells in vitro, and thus they may show utility in the treatment of epigenetically-based diseases where cell death is not a desired endpoint
Figure 1. Structures of LSD1 inhibitors 1, verlindamycin 2, (bis)thioureas 3, amidoxime 4, cyclic peptide 5, N3-(2-chloro-6-phenoxybenzyl)-4H-1,2,4-triazole-3,5-diamine 6 and N3,N5-bis(2-methoxybenzyl)-1H-1,2,4-triazole-3,5-diamine 7.
INTRODUCTION
The dynamic interplay between histones and chromatin remodeling is critical for selective control of gene expression, and chromatin remodeling enzymes have now become attractive therapeutic targets for multiple diseases with an epigenetic basis. A number of post-translational histone modifications are known to control gene expression, including methylation, ubiquitination, sumoylation, ADP-ribosylation and acetylation of histone lysine or arginine residues, ADP-ribosylation of glutamate residues and phosphorylation of histone serine residues.1 The FAD-dependent amine oxidase lysine-specific demethylase 1 (LSD1, also known as KDM1A or BHC110) bound to the co-repressor CoREST, preferentially demethylates the mono- and dimethylated forms of the activating mark histone 3 lysine 4 (H3K4) to repress gene transcription.2, 3 Under some conditions (e.g. when bound to the androgen receptor), LSD1 also demethylates the mono- and dimethylated forms of the deactivating chromatin mark H3K9.4 Because LSD1 is overexpressed in a number of human cancers (acute myeloid leukemia, neuroblastoma, retinoblastoma, prostate cancer, breast cancer, lung cancer and bladder cancer),5–8 the protein has emerged as an important target for the development of specific inhibitors as a new class of antitumor agents.9 Importantly, LSD1 is now regarded as an emerging drug target for diseases other than cancer, such as neurological disease,10, 11 blood disorders,12, 13 viral infection,14 diabetes15, 16 and fibrosis.17 As such, there is a need for potent epigenetic modulators that do not cause overt cytotoxicity.
To date, a number of small molecule inhibitors of LSD1 have been described, as shown in Figure 1. Effective LSD1 inhibitors include tranylcypromine-based analogues3, 18–20 such as 1,21 oligoamines such as verlindamycin 222 and related isosteric ureas and thioureas related to 3,23, 24 small-molecule amidoximes such as 4,25 and peptide based LSD1 inhibitors such as 5.26, 27 Many of the most potent and selective LSD1 inhibitors are structurally based on the clinically used antidepressant tranylcypromine, which has an IC50 value of 20.7 μM for LSD1. Because tranylcypromine is a potent inhibitor of monoamine oxidases and other flavin-dependent amine oxidases, there is a potential for undesired off-target effects in tranylcypromine-based LSD1 inhibitors. In addition, compounds built on a tranylcypromine scaffold rely on covalent, irreversible adduct formation with FAD to inactivate the enzyme. Herein we describe a novel scaffold for a new series of reversible, competitive inhibitors of LSD1, the 3,5-diaminotriazole moiety. The 3,5-diaminotriazole scaffold was used to produce histone demethylase inhibitors exhibiting increased potency for LSD1 (IC50 1–2 μM), higher specificity when compared to monoamine oxidase A and B (IC50 values > 100 μM), and reduced cytotoxicity. These analogues have great therapeutic potential for treatment of cancer, and importantly, for use in other epigenetically driven disorders where cytotoxicity is not a desired endpoint.
Figure 1.

Structures of LSD1 inhibitors 1, verlindamycin 2, (bis)thioureas 3, amidoxime 4, cyclic peptide 5, N3-(2-chloro-6-phenoxybenzyl)-4H-1,2,4-triazole-3,5-diamine 6 and N3, N5-bis(2-methoxybenzyl)-1H-1,2,4-triazole-3,5-diamine 7.
RESULTS
Virtual screen for novel LSD1 inhibitors
Potential new scaffolds for small-molecule LSD1 inhibitors were identified through a virtual screen of the Maybridge Hitfinder 5 compound library, as previously described.25 The crystal structure of LSD1/CoREST (PDB 2V1D) was prepared using PrepWizard, and SiteMap was then used to assess efficient binding within the LSD1 histone-binding pocket. Lowest energy conformers of 3D compounds were determined and docked in the LSD1 active site using Glide. A total of 10 hits were identified with Glide scores lower than −7.5 kcal/mol. The synthesis and biological evaluation of other lead compounds identified in this screen have been previously published.25 In the present study, two hits from the virtual screen, compounds 6 and 7 (Figure 1), were identified, and these compounds, as well as related analogues 8–20, were purchased and evaluated (Table 1).
Table 1.
Structures, cLogP and LSD1 inhibitory activity for 3,5-diaminotriazoles 6–20. Each data point is the average of 3 determinations ± standard error of the mean.
| Structure | Cmpd. | cLogP | Residual % LSD1 Activity ([I] 10μM) |
|---|---|---|---|

|
6 | 3.73 | 15.1 ± 4.7 |

|
7 | 3.58 | 25.1 ± 1.9 |

|
8 | 4.46 | 72.3 ± 9.0 |

|
9 | 4.46 | 69.4 ± 6.4 |

|
10 | 3.21 | 97.8 ± 6.9 |

|
11 | 2.90 | 103.1 ± 7.8 |

|
12 | 2.49 | 95.4 ± 6.9 |

|
13 | 3.648 | 81.9 ± 8.2 |

|
14 | 3.53 | 93.1 ± 7.5 |

|
15 | 3.21 | 69.1 ± 7.7 |

|
16 | 3.08 | 103 ± 12.7 |

|
17 | 3.03 | 80.1 ± 8.4 |

|
18 | 2.62 | 77.9 ± 7.3 |

|
19 | 1.27 | 69.2 ± 5.7 |

|
20 | 1.795 | 79.2 ± 5.0 |
In vitro activity against recombinant LSD1/CoREST
The ability of compounds 6–20 to inhibit purified recombinant LSD1 was measured using a commercially available fluorescence-based assay kit (Cayman Chemicals #700120). Initially, all compounds were tested at a concentration of 10μMin phosphate-buffered saline (PBS) containing <1% DMSO(Table 1). The screen was performed as suggested by the supplier and modified as previously described.23, 25 Compounds 6 and 7 were the most effective inhibitors of LSD1 (%LSD1 activity remaining 15.1 ± 4.7 and 25.1 ± 1.9, respectively). For comparison, tranylcypromine (TCP) and compound 2 reduced LSD1 activity to 72.6 and 15.8%, respectively. Compounds 6 and 7 were then subjected to titration analysis to determine the in vitro IC50 of each compound against LSD1 (Figure 2, Panel A). Compound 6 possessed an IC50 value of 1.19 μM while compound 7 had an IC50 value of 2.22 μM.
Figure 2. Potency, inhibition kinetics, binding and selectivity of 3,5-diaminotriazoles 6 and 7.

Panel A. Dose-response of LSD1 following treatment with compounds 6 and 7. Compound 6 IC50 = 1.19 μM; compound 7 IC50 = 2.22 μM Panel B. Competitive enzyme inhibition kinetics for LSD1 treated with compound 6 at 0, 0.375, 0.75, and 1 μM concentrations over a range of substrate concentrations between 0 and 100 μM. Ki for 6 = 2.2 μM Panel C. Isothermal calorimetry trace for LSD1 showing a release of heat upon titration with compound 6. Ka = 48.89 nM, R2=0.96 Panel D. MAO A and MAO B enzyme activity for 3,5-diaminotriazoles 6 and 7 compared to the known MAO inhibitor tranylcypromine (TCP). TCP IC50= 4.2 μM and 5.8 μM for MAO A and MAO B, respectively). Compounds 6 and 7 exhibited IC50 values > 100 μM for both MAO A and B. For Panels A. B and D, each data point is the average of 3 determination that in each case differed by 3% or less.
Compound 6 is a reversible, competitive inhibitor of LSD1
The fluorescence-based assay method described above was then used to determine Michaelis-Menten kinetics for 6, as shown in Figure 2, Panel B. In brief, compound 6 was incubated at 0, 0.375, 0.75 and 1.0 μM for 30 mins with 15 ng/μL of LSD1 at 37°C prior to addition of increasing amounts of the H3K4me2 peptide substrate (concentrations between 0 and 100 μM). Initial rates were determined by linear least-squares fit, and Km and Vmax values were determined using the GraphPad Prism 6 software package. The Vmax remained constant (30.62 ± 0.8 unit/min) indicating competitive inhibition, and the Ki for 6 was determined to be 2.20 μM.
To assure that 3,5-diaminotriazoles were bound to LSD1 with 1:1 stoichiometry, nanoisothermal titration calorimetry (ITC) was performed (Figure 2, Panel C) using compound 6 and purified LSD1 (Enzo Life Sciences, #BML-SE544-0050). Titration of compound 6 to LSD1 resulted in an independent binding isotherm signifying significant heat release on binding of 6 to LSD1. The R2 of heat released and the molar ratio was found to be 0.96.
Amine oxidase selectivity of 3,5-diaminotriazoles
LSD1 and other amine oxidases, such as the monoamine oxidases (MAO), use the cofactor FAD to reoxidize molecular oxygen to produce H2O2. Tranylcypromine and derivatives often show significant off-target efficacy against MAO/A and MAO/B. A commercial recombinant luminescent assay (Promega Corporation, #V1401) was performed as described by the manufacturer to assess the ability of 3,5-diaminotriazole 6 and 7 to inhibit monoamine oxidase (MAO) A and B isoforms. Compounds 6 and 7 were diluted in 1% DMSO prior to measuring the activity of MAO A and B. Compounds 6 and 7 both exhibited IC50 values greater than 100 μM against both MAO isoforms, while tranylcypromine inhibited MAO A and B with IC50 values of 4.2 μM and 5.8 μM, respectively (Figure 2D).
Molecular modeling studies for compound 6
In silico molecular modeling (GOLD software package, version 5.1, Cambridge Crystollagraphic Data Center, Cambridge, UK) was performed to predict key residues interacting with compound 6 (Figure 3) and compound 7 (data not shown). The LSD1 active site (PDB #3ZMT, LSD1-CoREST in complex with a peptide with the sequence PRSFLV) was defined as a sphere enclosing residues within 10Å around the crystallographic peptide ligand. Prior to energy minimization, proteins were protonated and the pH was set to 7.4. Initial screens focused on using the PRSFLV peptide for defining potential key interaction sites. The 3D inhibitor structures were energy minimized using the MM94x force field for 1000 iterations and a convergence value of 0.001 kcal/mol/Å as the termination criterion. Initial docking results yielded 60 poses of each structure bound in the active site of LSD1. The top 5 poses that yielded the lowest E-score were chosen for further analysis. The best fit for binding was analyzed for interacting residues. Key interactions with compound 6 include two hydrogen bonds with aspartate 555 and another hydrogen bond with the carbonyl of alanine 539. In addition, the compound participates in pi-stacking with the flavin ring of the FAD cofactor within 2.98Å. Thus, compound 6 shows close association with the active site and effectively prohibits substrate binding. A 2-dimensional rendition of the binding of 6 to LSD1/CoREST appears in the supplement, Figure S1.
Figure 3.

In silico analysis of compound 6 in the LSD1/CoREST catalytic site (PDB file 3ZMT). The aromatic portion of the o-phenoxy substituent lies 2.98Å from the FAD cofactor.
Cytotoxicity, Cellular Effects, and Global H3K4me2 Levels
Calu-6 human lung adenocarcinoma cells were purchased from ATCC (HTB-56), and cultured in EMEM growth medium containing 10% (v/v) fetal bovine serum and 5% penicillin and streptomycin. Cultures were grown at 37°C in a humidified environment containing 5% CO2. The cells were plated and maintained in a clear bottom, 96 well plate and seeded at 1,000 cells/well. After attachment, the cells were exposed to varying concentrations of the drug, while being kept in a humidified environment for 48 or 72 hours. Compounds were dissolved in DMSO and then further diluted with culture medium. The cells were exposed to DMSO concentrations of less than 1%, and 1% DMSO was used as a negative control for cell growth. The known LSD1 inhibitor verlindamycin 2 was used as a positive control. Cells were fixed with 4% PFA, permeabilized with 1% Triton-X100 and stained with DAPI at 1:1000 with in PBS. As shown in Figure 4, compounds 6 and 7 were not cytotoxic to cultured Calu-6 lung tumor cells (IC50 values > 100 μM), while 2 was cytotoxic with an IC50 value of 5 μM. TCP was also not cytotoxic in the Calu-6 cell line at concentrations up to 100 μM, and in fact did not cause significant cytotoxicity at much higher concentrations (Figure S2).
Figure 4. Cell viability and compound cytotoxicity in Calu-6.
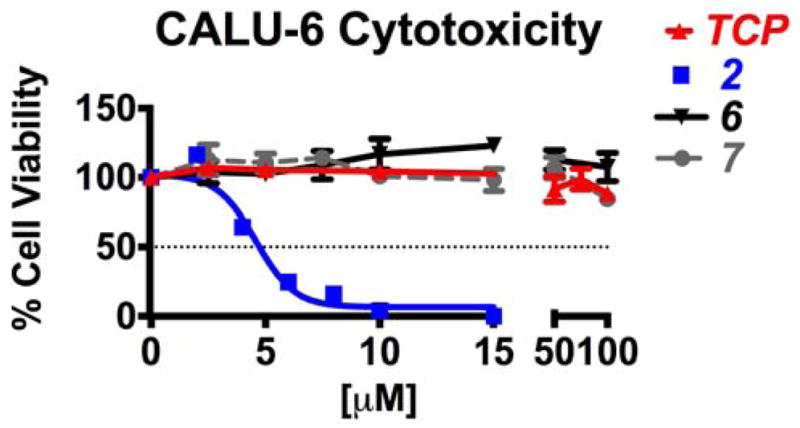
Cells were seeded at 3×103 cells/well and treated 48–72 hrs with increasing concentrations of TCP, 2, 6 or 7. Cell viability was determined by colormetric CellTiter 96 Aqueous MTS (Promega, #G3580).
The cytotoxicity of 6 and 7 was examined in 5 additional cell lines including CA46 Burkitt’s lymphoma, the PC3 human prostate cancer cell line, the PANC1 human pancreatic cell line, the MDA-MB-231 estrogen receptor-negative cell line and the MCF-10A human breast epithelial cell line. LSD1 has been shown to be overexpressed in the PC3, PANC1 and MDA-MB-231 cell lines. Compounds 6 and 7 produced no significant cytotoxicity in the CA46 and MCF-10A lines, although 6 had an IC50 of 52 μM in the MCF-10A line. In the PC3 line, both 6 and 7 exhibited IC50 values near 74 μM, compared to 2, which produced 85% growth inhibition at 8 μM. In the PANC1 and MDA-MB-231 lines, 7 was not an effective growth inhibitor, with IC50 values of 80 and 55 μM, respectively. Interestingly, compound 6 was an effective growth inhibitor in the PANC1 and MDA-MB-231 cell lines, exhibiting IC50 values of 19 and 12 μM, respectively. The mechanism underlying this activity remains to be determined; however, it can be generally stated that 6 and 7 produce little cytotoxicity in multiple cell lines in vitro.
To measure the cellular effects of 3,5-diaminotriazoles 6 and 7, treated cells from the cytotoxicity assay above were stained for immunofluorescence (IF) imaging using the appropriate fluorescently labelled secondary antibodies. In a 96-well plate, 5% BSA was added to specific wells and the plate was allowed to stand for 2 hours. The plate was then incubated at 4°C overnight with the primary antibody for H3K4me2 (Cell Signaling #2139S) diluted in 1% bovine serum albumin (BSA). Fluorescent secondary antibodies were then added to each well at 1:500 dilutions in 1% BSA for 2 hours. Cells were washed, suspended in PBS and viewed for intensity of signal per cell (Hermes WiScan, Idea Biomedical). The imaging system is able to generate 10–40x images as well as quantify average intensity on a per-well basis, eliminating any bias towards IF staining. Compounds 6 and 7 at both 1 and 10 μM developed green fluorescence in the nucleus at 48 hours that was comparable to the fluorescence promoted by 30 μM tranylcypromine (Figure 5), indicating a significant increase in H3K4me2 levels. The H3K4me2 levels were quantified and graphed as cell count vs. average intensity (RFU) as shown in the histogram (Figure 5). The mean population intensity (MPI) ± SEM for the vehicle was 1.44×104 ± 71 RFU. By way of comparison, the MPI for 30 μM TCP was 1.86×104 ± 136 RFU (1.3-fold increase), for 10 μM compound 6 1.82×104 ± 105 RFU (1.26-fold increase), and for 10 μM compound 7 1.91×104 ± 124 RFU (1.33-fold increase). Thus, compounds 6 and 7 at 1.0 μM were as effective as 30 μM TCP at increasing H3K4me2 levels in Calu-6 cells in vitro.
Figure 5. Global methylation changes by immunofluorescent staining.

Calu-6 human lung adenocarcinoma cells were plated at 1×103 cells/well and treated with 30μM TCP, 1- or 10μM compound 4, and 1- or 10μM compound 5 for 48hrs. Cells were stained for nucleus (DAPI), F-actin (Alexa Fluor 594 Phalloidin), and dimethyl-H3K4 (Alexa Fluor 488 Secondary Antibody). Fluorescent intensity on cell-by-cell basis was obtained by Hermes WiScan (IDEA Biomedical) and graphed as a frequency distribution histogram of relative cell count at specific intensities. Two-way ANOVA; *** P-value < 0.001, representative of 3 experiments with n=4–6 wells each.
Synthesis of 3,5-diaminotriazole analogues
In order to produce additional analogues in the 3,5-diaminotriazole library, a synthesis of 6 was developed. as shown in Scheme 1. Condensation of phenol 21 and 2-cyano-3-chlorofluorobenzene 22 (K2CO3, microwave, 190°C, 6 min) resulted in the phenoxyphenyl intermediate 23. The cyano group in compound 23 was then reduced (LiALH4) to afford primary amine 24, which was reacted with 25 (microwave, 40°C, 5 min) to yield 26. Intermediate 26 was then treated with hydrazine (microwave, 90°C, 10 min) to produce the desired 3,5-diaminotriazole 6. This route was used to synthesize the previously unreported 3,5-diaminotriazoles of the general structure shown in Scheme 1, compounds 27–42 (Table 2). Each compound was evaluated as an inhibitor of recombinant LSD1 as described above at a concentration of 10 μM (Figure 6). Six compounds (6, 7, 32, 35, 37 and 39) were more effective LSD1 inhibitors at 10 μM than the known LSD1 inhibitor 2, while 11 of the 16 compounds measured inhibited LSD1 by 50% or more. The small number of analogues shown in Table 2 are insufficient for the development of structure/activity relationships, however, the activity associated with 6, 32, 35, 37 and 39 suggests that activity is retained with the addition of small electron releasing groups to the phenoxy aromatic ring at positions R1–R4. Compounds 37 and 39, which have LSD1 inhibitory activity comparable to 6, have been selected for further study, and are being subjected to the bioevaluation studies described above. In addition, we are continuing to synthesize analogues in this series for the purpose of refining a structure/activity model for 3,5-diaminotriazole-based LSD1 inhibitors. These results will be reported in a subsequent publication along with the complete biological characterization of 37 and 39.
Scheme 1.

Table 2.
Structures, cLogP and LSD1 residual activity for 3,5-diaminotriazoles 2, 6, 7 and 27–42 at 10 μM. Each data point is the average of 3 determinations ± standard error of the mean. Compounds 34 and 36 were insoluble and thus were not screened for LSD1 inhibition. TCP = tranylcypromine

| |||||||
|---|---|---|---|---|---|---|---|
| # | X | R1 | R2 | R3 | R4 | cLogP | % LSD1 Activity (10μM) |
| TCP | -- | -- | -- | -- | -- | 64.2 ± 6.1 | |
| 2 | -- | -- | -- | -- | -- | 33.9 ± 6.7 | |
| 6 | -- | H | H | H | H | 3.73 | 15.1 ± 4.7 |
| 7 | -- | -- | -- | -- | -- | 3.58 | 25.1 ± 1.9 |
| 27 | O | H | H | CF3O | H | 5.47 | 61.4 ± 5.6 |
| 28 | O | H | H | CH3 | H | 4.94 | 40.7 ± 6.1 |
| 29 | O | (CH3)2CH | H | H | CH3 | 5.97 | 43.7 ± 3.2 |
| 30 | O | H | CH3O | H | H | 4.36 | 55.4 ± 5.1 |
| 31 | O | H | H | (CH3)3C | H | 6.27 | 41.8 ± 8.8 |
| 32 | O | H | CH3 | H | CH3 | 5.44 | 32.1 ± 8.8 |
| 33 | O | H | CF3 | H | CF3 | 6.21 | 55.1 ± 4.6 |
| 34 | O | H | H | S-CF3 | H | 6.11 | insolb. |
| 35 | O | CF3 | H | Br | H | 6.19 | 32.2 ± 5.4 |
| 36 | O | H | H | S-CH3 | H | 6.11 | insolb. |
| 37 | O | CH3O | H | CH3 | H | 4.51 | 15.3 ± 3.2 |
| 38 | O | H | CH3O | H | CH3O | 4.45 | 36.7 ± 8.6 |
| 39 | O | CH3 | CH3 | H | H | 5.39 | 19.4 ± 1.9 |
| 40 | O | H | O-CH2-O | H | 4.41 | 34.2 ± 6.4 | |
| 41 | S | H | H | H | H | 4.74 | 39.2 ± 7.9 |
| 42 | S | H | CH3O | CH3O | H | 4.40 | 46.8 ± 7.4 |
Figure 6. LSD1 inhibition assay.

Percent LSD1 activity remaining after treatment with 3,5-diaminotriazoles 6, 7, 27–33, 35, 37–42 at 10 μM. Each data point is the average of 3 determinations ± SEM (see Table 2).
Discussion
Newly identified roles for epigenetic modulation involving LSD1 continue to emerge, both in cancer and in other disease states, and thus, has become a promising target for therapeutic intervention. The TCP-based LSD1 inhibitors are the most advanced chemical class with respect to drug development, and the first clinical trial for a tranylcypromine-based LSD1 inhibitor for treatment of acute myeloid leukemia began earlier this year.28 TCP is a moderately potent, irreversible inhibitor of LSD1 and continues to provide a popular scaffold for the design of clinically relevant LSD1 inhibitors. However, it is challenging to design a TCP-based LSD1 inhibitor for clinical use that is specific, has low toxicity, and is devoid of the many biological responses to TCP itself. Thus, these analogues have the potential to produce off-target effects mediated through other flavin-dependent amine oxidase enzymes.29 As such, there is a continuing need to identify novel small-molecule scaffolds for inhibitors of LSD1 that can be used to design highly specific LSD1 inhibitors. The 1,2,4-triazole moiety described in this manuscript could be used as such a scaffold, and the preliminary studies described herein support the contention that potent, non-toxic LSD1-specific inhibitors can be designed in this chemical class.
During the preparation of this manuscript, a series of 1,2,3-triazole-based LSD1 inhibitors were described.30–32 In these analogues, the triazole ring system is derived from click chemistry used in their preparation, and is regioisomeric to the 1,2,4-triazoles described in this manuscript. Although the 1,2,3-triazole moiety contributes to binding to LSD1, the activity of the 1,2,3-triazoles depends on a dithiocarbamate moiety, and the compounds act as irreversible inactivators of LSD1.32 The compounds we have described in this report do not require activation and/or covalent attachment to the FAD cofactor for inhibition of LSD1, and our enzymatic kinetic studies have revealed that these compounds act as potent competitive inhibitors (Figure 2B). Thus, compounds 6 and 7 are among the first reversible small molecule compounds that possess selectivity for LSD1 over MAO A and B. Compounds 6 and 7 also produce the desired epigenetic effect, namely a significant increase in H3K4me2 levels, indicating that they enter mammalian cells and are active within the cell nucleus. The low level of toxicity to mammalian cells produced by 6 and 7 demonstrate that the 1,2,4-triazole scaffold can be used to produce LSD1 inhibitors that can be used in non-cancer disease states. As shown in Table 2, our early hit-to-lead studies suggest that we will be able to identify more potent analogues in the series. As such, the design, synthesis and evaluation of additional analogues related tp 6 and 7 is an ongoing concern in our laboratories.
Conclusions
In summary, we have identified the 3,5-diamino-1,2,4-triazole nucleus as a novel scaffold for the design of nontoxic, reversible small molecule inhibitors of LSD1 that do not contain a tranylcypromine backbone. To our knowledge, compound 6 is one of the first small molecule, reversible LSD1 inhibitors that is active at low micromolar concentrations. There is a high probability that hit-to-lead and lead optimization studies will lead to more potent inhibitors that have high selectivity for LSD1, improved pharmacokinetic parameters and minimal off-target effects, and these studies are currently underway.
Supplementary Material
Acknowledgments
The research described in this manuscript was supported by NIH/NCI grant 5 RO1 CA149095 (PMW).
Footnotes
Electronic Supplementary Information (ESI) available: complete experimental section, including synthetic details and a description of all biological procedures.
Notes and references
- 1.Andreoli F, Del Rio A. Drug Disc Today. 2014 doi: 10.1016/j.drudis.2014.05.005. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 2.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Cell. 2004;119:941. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 3.Suzuki T, Miyata N. J Med Chem. 2011;54:8236. doi: 10.1021/jm201048w. [DOI] [PubMed] [Google Scholar]
- 4.Varier RA, Timmers HT. Biochim Biophys Acta. 2011;1815:75. doi: 10.1016/j.bbcan.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 5.Hayami S, Kelly JD, Cho HS, Yoshimatsu M, Unoki M, Tsunoda T, Field HI, Neal DE, Yamaue H, Ponder BA, Nakamura Y, Hamamoto R. Int’l J Cancer. 2011;128:574. doi: 10.1002/ijc.25349. [DOI] [PubMed] [Google Scholar]
- 6.Lim S, Janzer A, Becker A, Zimmer A, Schule R, Buettner R, Kirfel J. Carcinogenesis. 2010;31:512. doi: 10.1093/carcin/bgp324. [DOI] [PubMed] [Google Scholar]
- 7.Rotili D, Mai A. Genes & Cancer. 2011;2:663. doi: 10.1177/1947601911417976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schulte JH, Lim S, Schramm A, Friedrichs N, Koster J, Versteeg R, Ora I, Pajtler K, Klein-Hitpass L, Kuhfittig-Kulle S, Metzger E, Schule R, Eggert A, Buettner R, Kirfel J. Cancer Res. 2009;69:2065. doi: 10.1158/0008-5472.CAN-08-1735. [DOI] [PubMed] [Google Scholar]
- 9.Stavropoulos P, Hoelz A. Exp Op Ther Targets. 2007;11:809. doi: 10.1517/14728222.11.6.809. [DOI] [PubMed] [Google Scholar]
- 10.Gottesfeld JM, Pandolfo M. Future Neurol. 2009;4:775. doi: 10.2217/fnl.09.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lakowski B, Roelens I, Jacob S. J Mol Neurosci. 2006;29:227. doi: 10.1385/JMN:29:3:227. [DOI] [PubMed] [Google Scholar]
- 12.Ginder GD. Transl Res. 2014 doi: 10.1016/j.trsl.2014.05.002. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi L, Cui S, Engel JD, Tanabe O. Nat Med. 2013;19:291. doi: 10.1038/nm.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liang Y, Quenelle D, Vogel JL, Mascaro C, Ortega A, Kristie TM. mBio. 2013;4:e00558. doi: 10.1128/mBio.00558-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mishra M, Zhong Q, Kowluru RA. Free Rad Biol Med. 2014 doi: 10.1016/j.freeradbiomed.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pan D, Mao C, Wang YX. PLoS ONE. 2013;8:e66294. doi: 10.1371/journal.pone.0066294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang IV, Schwartz DA. Transl Res. 2014 doi: 10.1016/j.trsl.2014.03.011. epub ahead of print. [DOI] [PMC free article] [PubMed]
- 18.Guibourt N, Ortega-Munoz A, Castro-Palomino Laria J. 20120004262. US Patent Application. Phenylcyclopropylamine derivatives and their medical use. 2012.
- 19.Mimasu S, Umezawa N, Sato S, Higuchi T, Umehara T, Yokoyama S. Biochemistry. 2010;49:6494. doi: 10.1021/bi100299r. [DOI] [PubMed] [Google Scholar]
- 20.Ortega-Munoz A, Castro-Palomino Laria J, Fyfe MCT. Lysine-specific demethylase 1 inhibitors and their use. 2011035941A1. WO. 2011 Mar 31;
- 21.Neelamegam R, Ricq EL, Malvaez M, Patnaik D, Norton S, Carlin SM, Hill IT, Wood MA, Haggarty SJ, Hooker JM. ACS Chem Neurosci. 2012;3:120. doi: 10.1021/cn200104y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang Y, Greene E, Stewart TM, Goodwin AC, Baylin SB, Woster PM, Casero RA. Proc Nat’l Acad Sci USA. 2007;104:8023. doi: 10.1073/pnas.0700720104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharma S, Wu Y, Steinbergs N, Crowley M, Hanson A, Casero RAJ, Woster P. J Med Chem. 2010;53:5197. doi: 10.1021/jm100217a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharma SK, Hazeldine S, Crowley ML, Hanson A, Beattie R, Varghese S, Sennanayake TMD, Hirata A, Hirata F, Huang Y, Wu Y, Steinbergs N, Murray-Stewart T, Bytheway I, Casero J, RA, Woster PM. Med Chem Comm. 2012;3:14. doi: 10.1039/C1MD00220A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hazeldine S, Pachaiyappan B, Steinbergs N, Nowotarski S, Hanson AS, Casero RA, Jr, Woster PM. J Med Chem. 2012;55:7378. doi: 10.1021/jm3002845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forneris F, Binda C, Adamo A, Battaglioli E, Mattevi A. J Biol Chem. 2007;282:20070. doi: 10.1074/jbc.C700100200. [DOI] [PubMed] [Google Scholar]
- 27.Kumarasinghe IR, Woster PM. ACS Med Chem Lett. 2014;5:29. doi: 10.1021/ml4002997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.http://www.oryzon.com/files/NdP2014/PRESS_RELEASE_01-2014def.pdf.
- 29.Khan MN, Suzuki T, Miyata N. Med Res Rev. 2013;33:873. doi: 10.1002/med.21269. [DOI] [PubMed] [Google Scholar]
- 30.Duan YC, Ma YC, Zhang E, Shi XJ, Wang MM, Ye XW, Liu HM. Eur J Med Chem. 2013;62:11. doi: 10.1016/j.ejmech.2012.12.046. [DOI] [PubMed] [Google Scholar]
- 31.Duan YC, Zheng YC, Li XC, Wang MM, Ye XW, Guan YY, Liu GZ, Zheng JX, Liu HM. Eur J Med Chem. 2013;64:99. doi: 10.1016/j.ejmech.2013.03.058. [DOI] [PubMed] [Google Scholar]
- 32.Zheng YC, Duan YC, Ma JL, Xu RM, Zi X, Lv WL, Wang MM, Ye XW, Zhu S, Mobley D, Zhu YY, Wang JW, Li JF, Wang ZR, Zhao W, Liu HM. J Med Chem. 2013;56:8543. doi: 10.1021/jm401002r. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.