

Abstract
The p53 gene has been investigated for its role in epithelial ovarian cancer but data collected until now are contradictory. The evidence that p53 belongs with p63 and p73 to a family of transcription factors re-opened interest in this gene family.
Here, we used quantitative real time RT-PCR to determine expression levels of TAp53, TAp73 and their N-terminal splice variants in a cohort of 169 ovarian cancer patients with stage I and stage III disease. The TAp73 levels in stage III biopsies differed by 100-fold depending on the p53 status and overall survival appears to be significantly related to ΔNp73 expression. Kaplan–Meyer analyses did not suggest a correlation between overall survival and levels of TAp73, ΔNp73 or the ΔNp73/TAp73 ratio. In conclusion, these data suggest that at least in our patient cohort p53 and p73 expression levels are not correlated to malignant progression of ovarian cancer. They might, however, play a role in tumour initiation.
Keywords: p53, p73, Ovarian cancer, Splice variants
1. Introduction
p53 has long been considered a unique tumour suppressor, whose prominent position in cell cycle regulation, apoptosis and DNA repair has spurred extensive research into both its basic and clinical aspects.1,2 However, since this protein is at the crossroad of an extensive and complex network of stress response pathways, the role of wild-type p53 in oncogenesis extends far beyond our current knowledge. This is particularly evident in epithelial ovarian cancer (EOC) which is at present the most deadly cancer in women of western countries.3 Although platinum-based therapy improved EOC overall survival, more than 75% of patients are diagnosed at late stages of the disease and relapse after an initial response. This high rate of mortality is generally ascribed to a lack of molecular biomarkers for monitoring early disease onset and response to therapy.3 The role of p53 in EOC malignant transformation, clinical course and responsiveness to therapy is not completely clear and data are to some extent contradictory.4-6
p53 is the founding member of a gene family that includes p63 and p73, which share with p53 a high degree of structural similarities. This finding re-opened the debate on the role of these transcription factors in driving malignant transformation, in particular in EOC.7-9 However, structural similarities do not necessarily translate into functional similarity. p63 and p73 share a consensus DNA binding domain with p53 and in ectopic overexpression can act as a transcription factor on p53 target genes. However, they also have unique tissue distribution and functions during development and malignant transformation.10-12 The scenario is further complicated by the presence of alternative promoters and/or alternative splicing at the N-termini or C-termini, resulting in a plethora of different isoforms. Moreover, these isoforms can interact with each other in a dominant negative fashion. In addition, they might be endowed with unique transcriptional activity.13-17 Thus, analysis of a single isoform is not sufficient for drawing any conclusion on their role in human tumours. Instead, it is mandatory to study the relationship among the different isoforms, in particular between the full length isoforms (called TA, transactivation active), which are characterised by a p53 like anti-oncogenic activity, and their dominant negative ones (collectively called ΔTA), which are characterised by pro-oncogenic potential.16
The aim of this study was to assess a possible correlation between the expression levels of p53, p73 and their dominant negative isoforms and disease progression in a cohort of 169 EOC patients at an early and advanced stage of disease.
2. Materials and methods
2.1. Sample collection
Patient biopsies were collected during surgery at the department of Oncology and Gynecology, San Gerardo Hospital (Monza, Italy) from September 1992 to March 2004: 83 were diagnosed as stage I ovarian cancer and 86 as stage III. Their histopathologic features are summarised in Table 1. Stage and histological grade of the primary tumours were defined according to International Federation of Gynecology and Obstetrics (FIGO) staging. Fresh tumour tissues were obtained at the first laparotomy prior to any treatment. The tissues were freed from necrotic, haemorrhagic and connective tissues, minced and stored frozen at −80 °C in cryotubes until processed. Histopathological analysis revealed that tumour cells content accounted for more than 70% of tumour sample. The collection and use of tumour samples was approved by the local Scientific Ethical Committee, and patients gave their written consent.
Table 1.
Clinical parameters and p53 status according to stage, histotype and grading
| Clinical parameters
|
p53 status
|
|||
|---|---|---|---|---|
| Histopatological parameters | No. of cases | Wild-type | Mutated | ND |
| Stage I | 83 | |||
| a | 26 | 26 | 0 | 0 |
| b | 5 | 5 | 0 | 0 |
| c | 52 | 52 | 0 | 0 |
| Histotype | ||||
| Serous | 33 | 33 | 0 | 0 |
| Mucinous | 16 | 16 | 0 | 0 |
| Endometroid | 18 | 18 | 0 | 0 |
| Undifferentiated | 1 | 1 | 0 | 0 |
| Clear cell | 14 | 14 | 0 | 0 |
| Not available | 1 | 1 | 0 | 0 |
| Grade | ||||
| 1 | 17 | 17 | 0 | 0 |
| 2 | 18 | 18 | 0 | 0 |
| 3 | 33 | 33 | 0 | 0 |
| Bl | 15 | 15 | 0 | 0 |
| Stage III | 86 | |||
| a | 3 | 2 | 1 | 0 |
| b | 5 | 2 | 2 | 1 |
| c | 77 | 36 | 36 | 5 |
| Not available | 1 | 1 | 0 | 0 |
| Histotype | ||||
| Serous | 66 | 31 | 30 | 5 |
| Mucinous | 3 | 1 | 1 | 1 |
| Endometroid | 10 | 4 | 6 | 0 |
| Undifferentiated | 6 | 4 | 2 | 0 |
| Clear cell | 1 | 1 | 0 | 0 |
| Grade | ||||
| 1 | 5 | 4 | 0 | 1 |
| 2 | 18 | 8 | 8 | 2 |
| 3 | 58 | 25 | 31 | 2 |
| Bl | 5 | 4 | 0 | 1 |
| Not available | 0 | 0 | 0 | 0 |
| Total sample size | 169 | |||
ND, not detected.
2.2. Clinical data
Tumour histological classification was carried out according to the WHO system. A pathologist confirmed the diagnosis of ovarian cancer, the histological subtype and pathological stage (FIGO staging). Conventional clinical features including age and menopausal status were evaluated.
2.3. RNA isolation and cDNA preparation
From each frozen sample, a tumour fragment of approximately 30 mg was taken with a sterile and RNase-free scalpel. The division procedure lasted between 0.5 and 2 min, and included weighing of the aliquots to determine the amount of lysis solution needed for RNA isolation. Tissues were homogenised by ultraturrax at 4 °C and total RNA purified using the SV-Total RNA isolation system (Promega, Milan, Italy). Total RNA was measured by spectrophotometer. Aliquots were stored at −80 °C until use. Two hundred nanogram of total RNA was reverse transcribed in 20 μL of reaction mix with Archive Kit (Applied Biosystems, Foster City, USA) and random primers, and then stored at −80 °C until use.
2.4. Primer design
Primer pairs for TAp73, Ex2p73, Ex2/3p73 ΔNp73, ΔN′p73, 28 S and cyclophillin A (Cyclo A) were as previously described.18 For the other p53 family isoforms and housekeeping genes (glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and actin), optimal primer pairs were chosen spanning splice junctions, using PRIMER-3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). All primers were queried against the non-redundant Human Genome Database (National Center for Biotechnology Information). Specificity of primers was verified by detecting single bands of amplicons and sequencing the PCR products. Primers sequences are listed in Supplemental Table 1.
2.5. Real time RT-PCR
Absolute copy numbers for all isoforms were determined by real time RT-PCR (ABI-7900, Applied Biosystems, Monza, Italy) using the SYBR Green protocol (Applied Biosystems). Briefly, PCR reaction was performed in a 10 μL reaction mix with 2 μL of previously prepared cDNA (from 200 ng total RNA) and primer pairs at a final concentration of 200 nM. 384-well plates were prepared by automatic liquid handling (epMotion 5075, Eppendorf, Milan Italy). Annealing temperatures are reported in Supplemental Table 1. Melting curves for each amplicon were automatically generated to evaluate the specificity of the amplified products. The absolute copy number of target and housekeeping genes were calculated by interpolation using a standard curve generated from a serial dilution, ranging from 2 to 20 million copies, of a cDNA prepared from a plasmid expressing the specific transcript. Standard concentration was measured with Nanodrop (Celbio, Milan Italy) and gene copy numbers were determined according to the following formula: X(molecules/μl) = 6.023 × 1023/molecular weight × concentration of the plasmid(μg/μl) × 10−6.
2.6. Western blot
Total protein extracts were prepared by lysing tumour tissues in 50 mM Tris–HCl, pH 7.4, 250 mM NaCl, 0.1% Nonidet NP-40, 5 mM EDTA, 50 mM NaF with aprotinin, leupeptine and phenylmethyl-sulfonyl-fluoride (PMSF) as protease inhibitors, for 30 min on ice after sample homogenisation with an ultraturrax. Insoluble material was pelleted at 13,000g for 10 min at 4 °C and the protein concentration was determined by using Biorad kit (BioRad, Milan Italy). Total cellular proteins (100 μg) was separated on SDS–PAGE and transferred to a PVDF membrane. Immunoblotting was carried out with p73 monoclonal antibodies (GC-15 mAb, Oncogene Research, CA, USA). RAN, a highly conserved and ubiquitously expressed GTPase was used as housekeeping gene (clone 20, BD transduction laboratories, USA). Antibody binding was revealed by peroxidase labelled secondary antibodies and visualised using enhanced chemiluminescence (Amersham, Italy).
2.7. Mutational status of p53
The p53 mutational status in exons 4–9 was determined in tumour specimens. cDNA, obtained as previously described, was further PCR-amplified with the following primers: p53Fw 5′-GGGACAGCCAAGTCTGTGACT and p53Rw: 5′-CCTGGGCATCCTTGAGTT. Amplification was performed in a thermocycler (PTC-200 MJ, Biorad) with 33 cycles at 95 °C for 1′; 60 °C for 30″; 72 °C for 1′ and TAQ DNA polymerase with proof-reading activity (LA-Taq, Takara) and, Cambrex Milan, Italy). Sequencing was performed by ABI 3730 DNA sequencer.
2.8. Data and statistical analysis
Using the R 2.1.0 software (R Development Core Team 2005; R: A language and environment for statistical computing. R Foundation for Statistical Computing Vienna, Austria. URL http://www.R-project.org), we wrote a dedicated script based on ‘car’ and ‘stats’ R libraries to normalise the data and to plot the graphics. Four genes (actin, cyclophillin A, GAPDH and 28S) were chosen for data normalisation. A normalisation index was generated for each patient by computing the geometric mean of the four housekeeping genes. The reliability of the choice of those genes has been evaluated by determining the correlations among the genes and between each gene and the normalisation index, verifying that an acceptable correlation level exists. The calculated index for each patient was then used to normalise data of the target genes. Since the values of gene expression for both ovarian cancer stage I and ovarian cancer stage III displayed a non-normal distribution, all statistical analyses were performed using non-parametric tests.
We evaluated the correlation between each patient subset and also between full length and their relative splice variants using the Spearman correlation test. ΔTA and ΔN were analysed separately and also as a ratio. In the first case, we split the ΔTA and ΔN distribution in three groups defined by tertiles: patients who had expression range lower than the first tertile patients whose expression range was between first and second tertile and patients with expression range higher than the second tertile. Tertiles were calculated on patients who did not experience death at the end of the study.
The ratio between ΔN and TA isoforms of each target gene was analysed both as a continuous and as a dichotomous variable. The cut-off used to split the ratio into two categories was set to 1. Gaussian distribution of data was tested with the Shapiro–Wilk test for normality; as this test gave a non-significant result, a non-parametric analysis of variance (ANOVA) was used to evaluate the associations between ΔN/TA ratio and categorical variables such as stage, grade, residual tumour and histology.
Overall survival was classified as outcome measure and was defined as the length of time from the first surgery to the last follow-up date or death irrespective of the cause. Overall survival curves were plotted with the Kaplan–Meier method. The log-rank test and Cox proportional hazard models were used to compare time-to-event distributions between pre-defined groups. The estimates from the Cox regression model are presented as hazard ratios (HRs) and 95% confidence intervals (CIs).
Three different Cox multivariate models were built to study the prognostic effect of the ΔN/TA ratio, considered both as dichotomous and continuous variable: the first model analysed the whole sample of patients and included stage as covariate; the second model analysed stage I patients and included tumour grade, the third model analysed stage III patients and included the residual tumour as covariate.
Statistical significance was set at 0.05 and all tests were two-tailed. Analysis was performed using SAS software v9.0 (SAS Institute Inc, Cary, USA).
3. Results
A cohort of 169 patients with ovarian cancer biopsies procured at the time of diagnosis and a median follow-up of 6.8 years was selected for this study. Their histopathologic and clinical parameters are summarised in Table 1: 50% of patients had FIGO stage I disease and 50% stage III disease; 53% had a grade 3 tumour. The median age at the time of diagnosis was 53.6 years for the whole sample (median age of 49.4 years for stage I and 54.9 for stage III) and the median follow-up for the whole sample was 6.84 years (median age of 6.75 for stage I and 6.88 for stage III). The p53 mutational status was determined by sequencing the DNA binding region between exons 4 and 9. All stage I ovarian tumours harboured wild-type p53, while 45% of stage III (39 out of 86) harboured mutations (Table 1). Mutations were found more frequently in exon 5 (30%), followed by exons 7 and 8 (25%). For 7% of them (6 out 86), we were unable to obtain sequence. Kaplan–Meyer analysis showed that patients harbouring mut p53 showed a reduced overall survival compared to those harbouring wild-type (wt) p53 (p = 0.052, Supplemental Fig. 1). mRNA levels for TA, the ΔTA and the ΔNp73 isoforms were measured by real time RT-PCR in all 169 biopsies. Throughout this text, ΔTAp73 refers collectively to the three NH2-terminally truncated variants ΔNEx2p73, ΔNEx2/3p73 and ΔN′p73. ΔNp73 refers exclusively to the specific transactivation-deficient isoform that derives from the second alternative promoter. Absolute copy numbers per 200 ng total RNA for each isoform were normalised against 4 internal control housekeeping genes: cyclo A, actin B, GAPDH and 28S. Data validation for each specific real time RT-PCR was performed as previously described. We used this comprehensive data set to detect possible systematic expression changes in the TA/ΔN ratio of p53 and p73 family variants.
The box plots in Fig. 1 indicate in log scale the absolute mRNA copy number for full length TAp53, p53Δ40 and p53Δ133 (panel A) and for TAp73, ΔTAp73 and ΔNp73 (panel B), separated by FIGO stage. In stage III, p53 and its two main isoforms p53Δ40 and p53Δ133 had comparable levels of expression (median value of 9.4 × 10−3, 2.2 × 10−3 and 1.06 × 10−3 for full length p53, ΔN40 and ΔN133, respectively, see Table 2). No differences were evident between tumours with or without p53 mutations (Table 2).
Fig. 1.
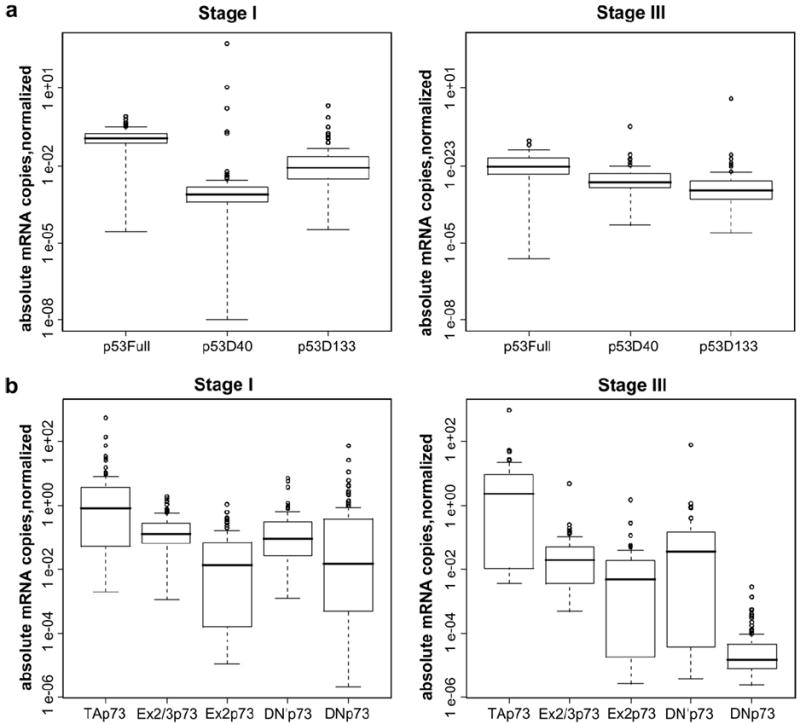
(a) Box plot diagrams showing the expression levels of p53, ΔN40 and Δ133 isoforms in 84 stage I (left side) and 86 stage III (right side) ovarian cancer biopsies. (b) Box plot diagrams showing the expression levels of TAp73, ΔTAp73 and ΔNp73 in 84 stage I (left) and 86 stage III (right) ovarian cancer biopsies. After normalisation of each sample to its own set of housekeeping genes, data are expressed as absolute copy numbers on a logarithmic scale. The line within the boxes indicates the median. The top edge of the boxes represents the 75th percentile, the bottom edge the 25th percentile. The range is shown as a vertical line. Outliers (circles) are defined as 1.5-fold above or below the 75th and 25th percentile values.
Table 2.
Median value of p53 and p73 expression
| Tumor stage | No. of samples | TAp53 | p53Δ40 | p53D133 |
|---|---|---|---|---|
| A | ||||
| Stage I | 83 | 1.15 × 10−1 | 7.2 × 10−4 | 8.2 × 10−3 |
| Stage III | 86 | 9.4 ×10−3 | 2.2 × 10−3 | 1.06 × 10−3 |
| p53wt | 41 | 5.57 × 10−3 | 1.52 × 10−3 | 9.7 × 10−4 |
| p53 mut | 39 | 1.33 × 10−2 | 3.35 × 10−3 | 1.08 × 10−3 |
| Tumor stage | No. of samples | TAp73 | Ex2.3p73 | Ex2p73 | ΔN′p73 | ΔNp73 |
|
| ||||||
| B | ||||||
| Stage I | 83 | 8.3 × 10−1 | 1.25 ×10−1 | 1.14 ×10−2 | 8.7 × 10−2 | 1.44 ×10−2 |
| Stage III | 86 | 2.34 | 1.94 ×10−2 | 4.8 ×10−3 | 3.4 × 10−2 | 1.4 ×10−5 |
| p53wt | 41 | 2.88 | 1.74 ×10−2 | 5.078 ×10−3 | 4.57 × 10−2 | 1.48 × 10−5 |
| p53 mut | 39 | 2.93 ×10−2 | 1.87 ×10−2 | 2.23 ×10−4 | 1.43 × 10−2 | 1.22 × 10−5 |
The median values of normalised copy numbers for the different p53 (A) and p73 (B) isoforms in the 169 ovarian cancer samples.
Stage I biopsies express 100-fold higher levels of TAp53 compared to stage III, while expression levels of p53ΔN40 and p53Δ133 were comparable between stage I and stage III (median value of 1.15 × 10−1, 7.2 × 10−4 and 8.2 × 10−3 for full length p53, p53ΔN40 and p53Δ133 respectively, Table 2). As already reported,18 our EOC cohort also showed that Ex2/3p73 and Ex2p73 remain largely unchanged. Concerning the expression levels of TAp73, ΔTAp73 and ΔNp73 (Fig 1B, right and left panels), specific differences were apparent. Within the ΔTAp73 isoforms, the median values are comparable between the two stages and even if there are substantial stage differences (Table 2), the box plots in Fig. 1 suggest that there is substantial overlap between stage I and stage III. When we subdivided stage III patients on the basis of their p53 status, we observed that samples lacking functional p53 had reduced levels of TAp73, whose values dropped 100-fold to a median of 2.93 × 10−2 in the p53-mutated subgroup. A different profile was observed for ΔNp73 expression, whose median value dropped a 1000-fold from 1.44 × 10−2 in stage I to 1.4 × 10−5 in stage III (Table 2). No differences were apparent between p53 wild-type or mutated subgroups.
Western blot analysis on a subset of patients showed that among the different C-terminal splice variants, ΔNp73β isoform represents the main form. Of note, its expression level is comparable between the two stages and consistent with the mRNA measurements in the same tumours. (Fig. 2). The α isoform was undetectable (Fig. 2). When we correlated expression levels of TAp73 with ΔTAp73 and ΔNp73 for each patient (Table 3), we found a significant correlation between the TA and the ΔNp73 forms in stage I (r = 0.97, p < 2.2e−16). Moreover, in stage III we found a good level of correlation for the isoforms encoded by the P1 promoter, but not for those encoded by the P2 promoter. Table 3 shows a level of correlation of 0.76 (p < 2.2e−16), 0.930 (p < 2.2e−16), 0.96 (p < 2.2e−16) for Ex2/3, Ex2 and ΔN′, respectively.
Fig. 2.
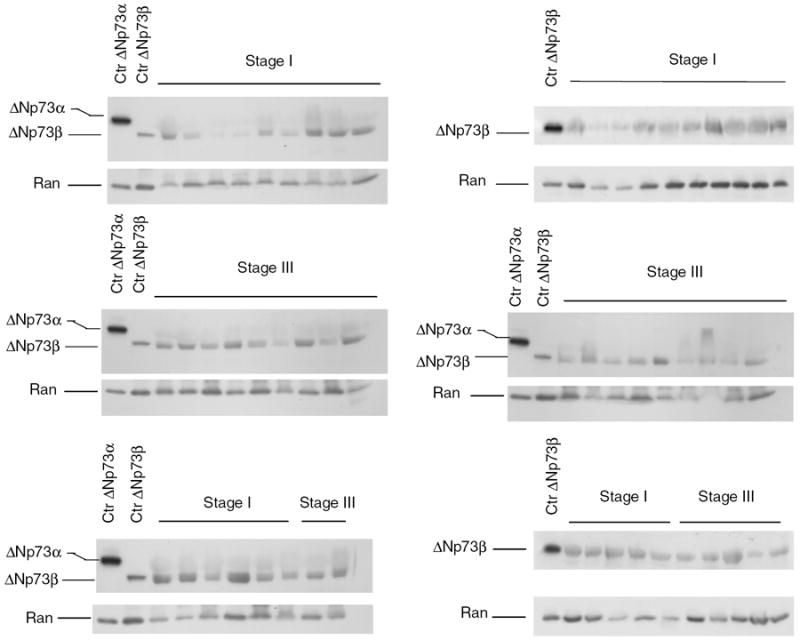
Western blot analysis of ΔNp73a and ΔNp73β isoforms in a subset of ovarian cancer patients from stage I and stage III. The ubiquitously expressed GTPase Ran was used as a loading control. Films were developed side by side.
Table 3.
Correlation between full length and transdominant isoforms of p53 and p73 in stage I and stage III ovarian cancer
| Stage I
|
Stage III
|
|||
|---|---|---|---|---|
| p53ΔN40 | p53 Δ133 | p53ΔN40 | p53 Δ133 | |
| p53full | ρ = 0.60 | ρ = 0.24 | ρ = 0.62 | ρ = 0.16 |
| p = 1.153e−9 | p = 0.0263 | p < 2.2e−16 | p = 0.1355 | |
| Stage I
|
Stage III
|
|||||||
| Ex2-3 | Ex2 | ΔN′ | ΔN | Ex2–3 | Ex2 | ΔN′ | ΔN | |
|
| ||||||||
| p73full | ρ = −0.03 | ρ = −0.6 | ρ = 0.24 | ρ = 0.97 | ρ = 0.76 | ρ = 0.93 | ρ = 0.96 | ρ = 0.25 |
| p = 0.7499 | p = 2.069e–9 | p = 0.02788 | p < 2.2e−16 | p < 2.2e−16 | p < 2.2e−16 | p < 2.2e−16 | p = 0.01741 | |
ρ is the Spearman correlation coefficient. Results are statistically relevant when p < 0.05.
We next attempted to correlate the expression levels of TAp73 with overall survival (OS). Tumour expression levels were subdivided into tertiles: the first tertile is defined as the lowest one-third; the second tertile as the middle one-third and the third tertile as the top one-third of the expression range. However, Kaplan–Meier curves (Fig. 3a–c) failed to show differences between patients with high expression of TAp73 compared to those with low expression. This was evident in the full cohort of 169 biopsies (p = 0.15, Fig. 3a) as well as in the two subsets of stage I and stage III patients (panels b and c, p-value 0.80 and 0.66, respectively).
Fig. 3.
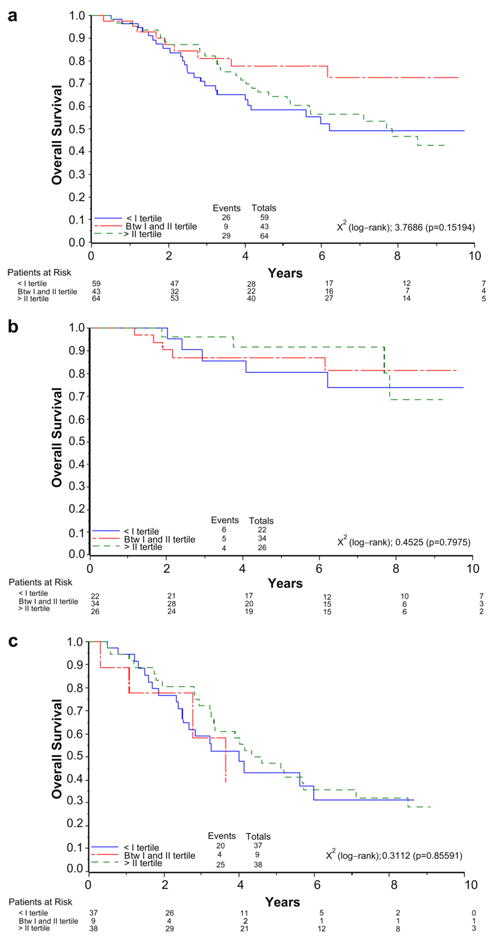
Kaplan–Meier survival curves in relation to TAp73 expression in all patients (a) and in stage I (b) and stage III (c) subpopulations.
When expression of ΔNp73 was analysed, we had an interesting trend (Fig. 4). In the whole cohort, a lower expression level of ΔNp73 strongly associates with poor survival (Fig. 4a, p = 0.00001). In fact, after three years follow-up, the OS for patients above the 2nd tertile was 92%, compared to those between the 1st and the 2nd tertile with 84% and for those within the 1st tertile with 64%. This difference became more consistent after 5 and 7 years of follow-up, when the OS was 89% above the 2nd tertile, 77% and 65% in the 1st and the 2nd tertile and fell to 44% or 36%, respectively, for patients within the 1st tertile. When we analysed stage I and stage III separately, we observed the same trend albeit only in stage III, although the difference due to the reduced number of samples was no longer significant (Fig. 4c). We calculated that at least 230 patients would be needed to confirm at a statistical significant level the trend seen in Fig. 4c.
Fig. 4.
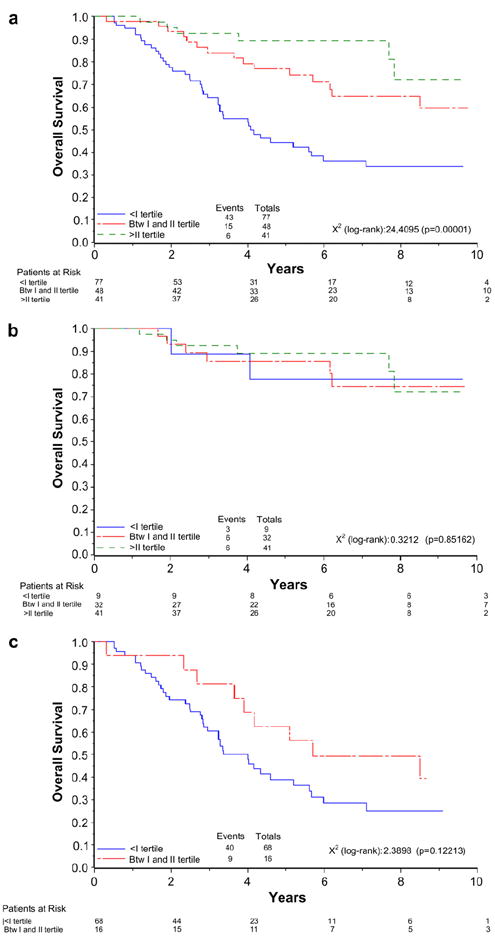
Kaplan–Meier survival curves in relation to ΔNp73 expression in all patients (a) and in stage I (b) and stage III (c) subpopulations.
Given the fact that the ΔNp73 protein acts in a dominant negative fashion on the TAp73 network, we tested the hypothesis that patients with a ΔNp73/TAp73 ratio different from 1 might have a different clinical outcome. The ΔNp73/TAp73 ratio was analysed as a dichotomous variable. However, as shown in Fig. 5, Kaplan–Meier curves showed that differences in the ΔNp73/TAp73 ratio did not affect OS, neither in the whole cohort nor when the two FIGO groups were considered separately.
Fig. 5.
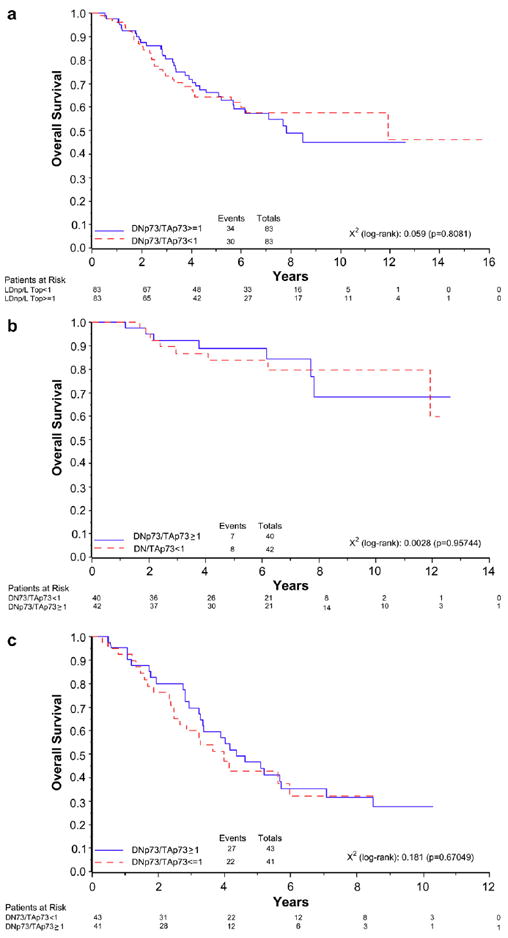
Kaplan–Meier survival curves in relation to the ratio between ΔNp73 and TAp73 expression in all patients (a) and in stage I (b) and stage III (c) subpopulations. Patients were divided into those with a ΔNp73/TAp73 ratio <1 and those with a ΔNp73/TAp73 ratio ≥1.
This result was confirmed by Cox analysis: the HR for mortality in stage I patients was 0.70 (95% CI: 0.25–2.00) in patients with a ratio >1 as compared to patients with a ratio <1, and 1.41 for stage III patients (95% CI: 0.74–2.71), respectively. Analysing the whole sample, we obtained a HR of 1.08 (95% CI: 0.66–1.77).
4. Discussion
In the present study, we measured the expression levels of TAp53, TAp73 and their different splice variants in 169 primary ovarian tumour biopsies with different histological and clinical features. We used a highly sensitive and specific quantitative RT-PCR, which amplified robustly and consistently TAp53, ΔNp40, ΔN133, TAp73, ΔTAp73 and ΔNp73 in all samples. One limiting factor of this experimental design is the lack of normal ovarian tissue to compare against. Also, this RT-PCR approach does not yield information on the carboxy termini of the different isoforms.
TAp53 alteration by itself has not clearly crystallised out as being a driving force for malignant progression in EOC. In line with data reported in the literature, in our cohort of patients we observed a trend relating p53 status and overall survival, although the relatively small sample size suggests that these data have to be interpreted with caution.19,20
Thus, molecular-prognostic analyses of ovarian carcinomas fail to provide a clear picture. The main reason for this situation may be that most studies were focused on p53 only and/or restricted to a small sample size with generally a high stage of malignancy. EOC is generally asymptomatic and diagnosis occurs late at a stage when the tumour has spread beyond the ovary or pelvis. Stage I accounts for less than 10% of diagnosed cases and is generally considered a rare disease.3
Over 15 years ago we started a project to bank ovarian tumour biopsies from patients with different grades and stages of malignancy. At present, we have stored more than 1000 human biopsies, 10% of which are stage I, and with a median follow-up of more than 5 years. This resource prompted us to reevaluate the role of some p53 isoforms in driving malignant progression of EOC by comparing, for the first time, their expression levels in a sizable cohort of stage I and stage III biopsies. A previous paper analysed the expression of TAp73 and its ΔNp73 variants in ovarian cancer biopsies and in healthy ovarian tissues with the aim to investigate if ΔNp73 can be considered a prognostic molecular marker.18 Our goal was to assess whether during EOC malignant progression defects in the tumour suppressor function of TAp53 and TAp73 could be ascribed to a deregulated expression of their N-terminally truncated isoforms.
Overall, the data presented in this study do not support a striking role for any of the N-terminally truncated isoforms in driving EOC malignant progression, although they might play a role in tumour initiation. In fact, our OS curves showed that patients with low levels of ΔNp73 mRNA are those with a trend for reduced overall survival, although a HR ratio close to 1 and non-significant confidence intervals indicate that more data are needed to assess whether this is an independent prognostic factor. However, despite this negative result in terms of clinical correlation, some points can now be discussed in more detail, particularly in light of the lack of knowledge about the complex genetic and epigenetic changes that occur during the development of EOC.
TAp53 is the main p53 isoform present in cells and its levels are regulated predominantly by post-translational mechanisms. The lack of commercially available antibodies to detect ΔN40 and Δ133 isoforms prompted us to measure mRNA expression. Our data are compatible with the notion that ΔN133 and ΔN40 isoforms are expressed at very low levels in cancer cells and represent a minority of total cellular p53.14,21 Moreover, in our dataset their expression is comparable between the two stages Thus, ΔN40 and ΔN133 levels do not seem to play an important role in modulating the transcriptional activity of p53 itself and the p53 family member TAp73.
Somewhat surprisingly, TAp73 levels in stage III biopsies differed by 100-fold, depending on their p53 status. Wtp53 tumours had higher TAp73 levels than mutant p53 tumours, a finding that is not easily explained (wtp53 has been shown to transactivate ΔNp73 from the P2 promoter but not the P1 promoter22). On the other hand, despite the drop in ΔNp73 mRNA levels in stage III, immunoblot analysis revealed that the total levels of ΔNp73 protein were comparable between the two stages. This can be explained on the basis that the ΔNp73 protein is encoded by two different promoters. ΔN′ mRNA, encoded by the P1 promoter, is the main isoform in stage III, rather than ΔNp73, encoded by the P2 promoter. This data are in agreement with a previous study in 100 tumour samples (80% were stage III), where the P1 promoter was mainly responsible for sustaining ΔNp73 levels.18 Thus, we speculate that an increase in ΔNp73 levels occurs as an early event during initiation of EOC malignant transformation. Alternatively, our finding that the main ΔNp73 protein is the β isoform is interesting in light of the different role that this specific isoform seems to play when ectopically expressed in cellular systems.23 Our unexpected finding that low levels of ΔNp73 tends to associate with worse survival could be related to the experimental evidence that high expression of the ΔNp73β isoform in some cellular context induces growth arrest.23 ΔNp73β could therefore act either as an oncogene due to its ability to counteract TAp53 and TAp73 activity, or as a growth suppressor via p53/p73 independent mechanisms.
In conclusion, although the negative conclusion with respect to mRNA levels and survival does not exclude a possible, yet difficult to asses, relationship at the protein levels, the robustness of the data we generated demonstrated the validity of our approach for translational research. The specificity and sensitivity of real time RT-PCR allowed to perform a high throughput analysis of a large cohort of human tumour samples and test the molecular models previously generated by in vitro experiments.
Supplementary Material
Acknowledgments
This work was partially supported by ‘Fondazione Cariplo’ and by FIRB MIUR (RBINO48RHA). The generous contributions of the Nerina and Mario Mattioli Foundation and of the Italian Association for Cancer Research were gratefully acknowledged. Eleonora Marrazzo is the recipient of a fellowship from the Italian Foundation for Cancer Research (FIRC).
Footnotes
Appendix A. Supplementary material
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.ejca.2007.10.011.
Conflict of interest statement
None declared.
References
- 1.Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006;6:909–23. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- 2.Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 3.Cannistra SA. Cancer of the ovary. N Engl J Med. 2004;351:2519–29. doi: 10.1056/NEJMra041842. [DOI] [PubMed] [Google Scholar]
- 4.Vikhanskaya F, Vignati S, Beccaglia P, Ottoboni C, Russo P, D’Incalci M, et al. Inactivation of p53 in a human ovarian cancer cell line increases the sensitivity to taxol by inducing G2 arrest and apoptosis. Exp Cell Res. 1998;241:96–101. doi: 10.1006/excr.1998.4018. [DOI] [PubMed] [Google Scholar]
- 5.Lavarino C, Delia D, Di Palma S, Zunino F, Pilotti S. p53 in drug resistance in ovarian cancer. Lancet. 1997;349:1556. doi: 10.1016/S0140-6736(05)62140-X. [DOI] [PubMed] [Google Scholar]
- 6.Perego P, Giarola M, Righetti SC, et al. Association between cisplatin resistance and mutation of p53 gene and reduced bax expression in ovarian carcinoma cell systems. Cancer Res. 1996;56:556–62. [PubMed] [Google Scholar]
- 7.Murray-Zmijewski F, Lane DP, Bourdon JC. p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ. 2006;13:962–72. doi: 10.1038/sj.cdd.4401914. [DOI] [PubMed] [Google Scholar]
- 8.Kaghad M, Bonnet H, Yang A, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90:809–19. doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- 9.Yang A, Kaghad M, Wang Y, et al. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell. 1998;2:305–16. doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]
- 10.Melino G, Lu X, Gasco M, Crook T, Knight RA. Functional regulation of p73 and p63: development and cancer. Trends Biochem Sci. 2003;28:663–70. doi: 10.1016/j.tibs.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 11.Moll UM, Erster S, Zaika A. p53, p63 and p73-solos, alliances and feuds among family members. Biochim Biophys Acta. 2001;1552:47–59. doi: 10.1016/s0304-419x(01)00036-1. [DOI] [PubMed] [Google Scholar]
- 12.Moll UM, Slade N. p63 and p73: roles in development and tumour formation. Mol Cancer Res. 2004;2:371–86. [PubMed] [Google Scholar]
- 13.Courtois S, de Fromentel CC, Hainaut P. p53 protein variants: structural and functional similarities with p63 and p73 isoforms. Oncogene. 2004;23:631–8. doi: 10.1038/sj.onc.1206929. [DOI] [PubMed] [Google Scholar]
- 14.Bourdon JC, Fernandes K, Murray-Zmijewski F, et al. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005;19:2122–37. doi: 10.1101/gad.1339905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rohaly G, Chemnitz J, Dehde S, et al. A novel human p53 isoform is an essential element of the ATR-intra-S phase checkpoint. Cell. 2005;122:21–32. doi: 10.1016/j.cell.2005.04.032. [DOI] [PubMed] [Google Scholar]
- 16.Melino G, De Laurenzi V, Vousden KH. p73: friend or foe in tumorigenesis. Nat Rev Cancer. 2002;2:605–15. doi: 10.1038/nrc861. [DOI] [PubMed] [Google Scholar]
- 17.Westfall MD, Pietenpol JA. p63: molecular complexity in development and cancer. Carcinogenesis. 2004;25:857–64. doi: 10.1093/carcin/bgh148. [DOI] [PubMed] [Google Scholar]
- 18.Concin N, Becker K, Slade N, et al. Transdominant {Delta}TAp73 isoforms are frequently up-regulated in ovarian cancer. Evidence for their role as epigenetic p53 inhibitors in vivo. Cancer Res. 2004;64:2449–60. doi: 10.1158/0008-5472.can-03-1060. [DOI] [PubMed] [Google Scholar]
- 19.Kmet LM, Cook LS, Magliocco AM. A review of p53 expression and mutation in human benign, low malignant potential, and invasive epithelial ovarian tumors. Cancer. 2003;97:389–404. doi: 10.1002/cncr.11064. [DOI] [PubMed] [Google Scholar]
- 20.Schuijer M, Berns EM. TP53 and ovarian cancer. Hum Mutat. 2003;21:285–91. doi: 10.1002/humu.10181. [DOI] [PubMed] [Google Scholar]
- 21.Boldrup L, Bourdon JC, Coates PJ, Sjostrom B, Nylander K. Expression of p53 isoforms in squamous cell carcinoma of the head and neck. Eur J Cancer. 2007;43:617–23. doi: 10.1016/j.ejca.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kartasheva NN, Contente A, Lenz-Stoppler C, Roth J, Dobbelstein M. p53 induces the expression of its antagonist p73 Delta N, establishing an autoregulatory feedback loop. Oncogene. 2002;21:4715–27. doi: 10.1038/sj.onc.1205584. [DOI] [PubMed] [Google Scholar]
- 23.Liu G, Nozell S, Xiao H, Chen X. DeltaNp73beta is active in transactivation and growth suppression. Mol Cell Biol. 2004;24:487–501. doi: 10.1128/MCB.24.2.487-501.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.