

Abstract
RATIONALE
Acyl-Coenzyme A (CoA) thioesters are the principal form of activated carboxylates in cells and tissues. They are employed as acyl carriers that facilitate the transfer of acyl groups to lipids and proteins. Quantification of medium-chain and long-chain acyl-CoAs represents a significant bioanalytical challenge because of their instability.
METHODS
Stable isotope dilution-liquid chromatography-selected reaction monitoring-mass spectrometry (LC-SRM/MS) provides the most specific and sensitive method for the analysis of CoA species. However, relevant heavy isotope standards are not available and they are challenging to prepare by chemical synthesis. Stable isotope labeling by essential nutrients in cell culture (SILEC) developed originally for the preparation of stable isotope labeled short-chain acyl-CoA thioester standards has now been extended to medium-chain and long-chain acyl-CoAs and used for LC-SRM/MS analyses.
RESULTS
Customized SILEC standards with > 98 % isotopic purity were prepared using mouse Hepa 1c1c7 cells cultured in pantothenic-free media fortified with [13C315N1]-pantothenic acid and selected fatty acids. A SILEC standard in combination with LC-SRM/MS was employed to quantify cellular concentrations of arachidonoyl-CoA (a representative long-chain acyl-CoA) in two human colon cancer cell lines. A panel of SILEC standards was also employed in combination LC-SRM/MS to quantify medium- and long-chain acyl-CoAs in mouse liver.
CONCLUSION
This new SILEC-based method in combination with LC-SRM/MS will make it possible to rigorously quantify medium- and long-chain acyl-CoAs in cells and tissues. The method will facilitate studies of medium- and long-chain acyl-CoA dehydrogenase deficiencies as well as studies on the role of medium- and long-chain acyl-CoAs in cellular metabolism.
INTRODUCTION
In prokaryotes and eukaryotes, coenzyme A (CoA) is an essential cofactor for biosynthetic and energetic metabolic pathways.1 Fatty acyl-CoAs are the precursors of sphingolipids, a major form of stored fatty acids 2,3 They are also involved in nuclear signaling,4 mitochondrial dysfunction,5 fatty acid catabolism,6 xenobiotic and biotransformations,7 post-translational protein modifications and gene regulation.3,4,8 As a result of these diverse functions, fatty acyl-CoAs have been implicated in obesity2,4, cardiovascular disease,9 diabetes mellitus,10–12 cancer,4 and a spectrum of genetic short, medium, and long-chain acyl-CoA disorders.6,13–16 Paradoxically, mammals are incapable of synthesizing the essential acyl-CoA precursor, pantothenic acid, which is also known as vitamin B5 (Figure 1).17 Previously, we have taken advantage of the essential requirement for pantothenic acid by replacing it in cell media with a [13C315N1]-pantothenic acid heavy isotope labeled analog, which is then incorporated into CoASH, and thus into any acyl-CoAs that are formed (Figure 1).18 We termed this methodology stable isotope labeling by essential nutrients in cell culture (SILEC),18,19 after the similarity to the stable isotope labeling by amino acids in cell culture (SILAC) methods employed for proteins.20 For acyl-CoA molecules, this imparts a +4 m/z shift to the internal standard, allowing distinction between the analyte and the internal standard in a mass spectrometer, without significant isotopic overlap or interference from biological matrices.19 The energy-rich thioester bond in acyl-CoAs, as compared with ordinary esters, is related primarily to the lack of resonance stabilization. As a result, thioesters are destabilized relative to the corresponding esters, making them more susceptible to hydrolysis. This means that CoA thioesters readily decompose in aqueous solutions and they are particularly labile under basic conditions.21 Consequently, there is a concern that selective losses of different CoA species could occur during complex bioanalytical procedures. Fortunately, the use of stable isotope analogs as internal standards corrects for such potential losses.22 Stable isotope analogs can also act as carriers for trace amounts of acyl-CoAs by preventing selective losses that can occur through binding active sites present on glassware and LC equipment.22 Therefore, stable isotope acyl-CoA analogs can improve the accuracy of assays for their corresponding endogenous acyl-CoAs present in biofluids particularly when only trace amounts of the endogenous acyl-CoAs are present.
Figure 1.

Biosynthesis of isotopically labeled medium- and long-chain acyl-CoAs and constant neutral loss of 507.2 Da from protonated acyl-CoA molecules.
A major limitation of our earlier SILEC studies was that the method was targeted to short-chain acyl-CoAs relevant to the Krebs cycle.18,19 The previous method inefficiently extracted naturally transient medium chain acyl-CoAs and more hydrophobic longer chain acyl-CoA species that are important for monitoring aberrant cellular metabolism. Our original SILEC method has now been modified so that a wider range of acyl-CoA species can be analyzed. In addition, the utility of the method has been established by quantifying the absolute levels of long-chain arachidonoyl-CoA in cell culture and acyl-CoA species in mouse liver tissue.
EXPERIMENTAL
Reagents
Arachidonoyl-CoA, 5-sulfosalicilic acid (SSA), ammonium formate solution, glacial acetic acid, and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO). Optima LC-MS grade methanol, acetonitrile (ACN), 2-propanol (IPA) and water were purchased from Fisher Scientific (Pittsburgh, PA). 2-(2-pyridyl) ethyl functionalized silica gel 100 mg/1 mL tubes were obtained from Supelco Analytical (Bellefonte, PA). F-12K media, DMEM, medium 200, low serum growth supplement (LSGS) kit, streptomycin, and penicillin were purchased from Invitrogen (Carlsbad, CA). Charcoal stripped fetal bovine serum (csFBS) was from Gemini Bioproducts (West Sacramento, CA). [13C315N1] calcium pantothenate was purchased from Isosciences (King of Prussia, PA). Peroxide free arachidonic acid (AA) was purchased from Cayman Chemical (Ann Arbor, MI). Human colorectal adenocarcinoma (LoVo) cells and human colon adenocarcinoma (HCA7) cells were obtained from ATCC (Manassas, VA). Livers from C57BL/6 mice were taken under University of Pennsylvania IACUC protocol # 803875.
Cell Culture
For SILEC labeling, Hepa1c1c7 cells were passaged at least 7 times in custom RPMI 1640 media without calcium pantothenate (AthenaES, Baltimore, MD) and containing 10% csFBS, 100 units/mL penicillin, 100 mg/L streptomycin, and 2 mg/L [13C315N1]-calcium pantothenate. 24 hours before extraction, “ultra-labeling” with media as above, but omitting csFBS, was performed in order to ensure optimal acyl-CoA stable isotope labeling. For unlabeled cell culture, HCA7 cells were grown in DMEM media with 2% FBS and 100 units/mL penicillin and 100 mg/L streptomycin. LoVo cells were grown in F-12K media with 2% FBS and 100 units/mL penicillin and 100 mg/L streptomycin.
Isolation of SILEC standards
Upon completion of labeling, Hepa1c1c7 cells were gently lifted, centrifuged at 500 x g for 5 minutes and resuspended in 750 μL/10cm2 plate of ACN:IPA (3:1; v/v). Samples were sonicated with a probe tip sonicator on ice 30 times for 0.5 s. 250 μL of 100 mM KH2PO4 (pH= 6.7) were added to the samples, then vortex-mixed and spun down for 10 min at 16,000 x g at 4°C. The supernatant was pooled and stored in −80°C until use.
Acyl-CoA analysis
Cells were gently lifted, centrifuged at 500 x g for 5 min and resuspended in 550 μL/10 cm2 plate of ACN:IPA (3:1; v/v) for extraction as described previously 23. The SILEC standard was added (200 μL) and samples were pulse sonicated with a probe tip sonicator on ice 30 times for 0.5 s. 250 μL of 100 mM KH2PO4 (pH= 6.7) were added to the samples, then vortex-mixed and spun down for 10 min at 16,000 x g at 4°C. The supernatant was transferred to a glass tube and acidified with 125 μL of glacial acetic acid. SPE columns were equilibrated with 1 mL of ACN:IPA:H2O:acetic acid (9:3:4:4; v/v) washing solvent. Samples were transferred to the columns, which ware washed two times with 1 mL of the washing solvent. The acyl-CoAs were then eluted by washing the columns twice with 500 μL methanol/250 mM ammonium formate (4:1; v/v) into glass tubes. After evaporation to dryness under nitrogen gas, the eluates were re-dissolved in 50 μL of H2O/ACN (70:30; v/v) containing 5 % SSA (w/v) and transferred to HPLC vials ready for LC-MS analysis.
Formation of arachidonyl-CoA in cell culture
HCA7 and LoVo cells were grown until 80% confluence as above. 25 μM arachidonic acid with 0.25% DMSO in DMEM media was added and at 30 min, 60 min, 90 min and 4 hours, the cells were gently scraped and collected in plastic tubes. Tubes were centrifuged for 5 min at 500 x g. Media was removed and the cell pellet was dissolved in 550 μL 3:1 ACN:IPA and 200 μL of the internal standard mix was added to each sample. The samples were then extracted and prepared as above.
Extraction of mixed length acyl-CoAs from mouse liver
Approximately 10 mg samples of frozen mouse liver were taken for analysis, and exact dry weight was determined for each liver. 750 μL of internal standard mix in 3:1 ACN:IPA was added to the liver samples. Samples were sonicated with a probe tip sonicator on ice 60 times for 0.5 s. Sonication produced a visually homogeneous mixture. Extraction proceeded as for the cells samples above, except that final liver acyl-CoAs were adjusted by weight of liver used for analysis.
Quantification of arachidonyl-CoA
Six standard solutions of arachidonyl-CoA in methanol were made corresponding to 20 pmol, 2 pmol, 0.2 pmol, 20 fmol, 2 fmol and 0 fmol on column. The standards were dried and then re-dissolved and spiked with internal standard as above. Samples were extracted using SPE columns as described for the mouse liver homogenate. Regression analyses of the standard curve and calculations of concentrations of arachidonoyl-CoA were performed with Excel.
LC-MS/MS
Samples were kept at 4°C in a Leap CTC autosampler (CTC Analytics, Switzerland) with 20 μL injections used for LC-MS analysis. Chromatographic separation was performed using a reversed phase Waters XBridge C18 column (2.1 × 150 mm, pore size 3 μm) on an Agilent 1100 HPLC system using a three solvent system: (A) 5 mM ammonium acetate in water, (B) 5 mM ammonium acetate in 95/5 ACN/water (v/v), and (C) 80/20/0.1 (v/v/v) ACN/water/formic acid, with a constant flow rate of 0.2 mL/min. Gradient elution was performed as follows: 2% B (isocratic) for 1.5 min, 2% to 20% (linear gradient) over 3.5 min, 20% to 100% B (linear gradient) B over 0.5 min, 100% B (isocratic) for 8 min, 100% C for 5 min, before equilibration at initial conditions for 5 min.
Samples were analyzed using an API 4000 triple quadrupole mass spectrometer (Applied Biosystems, Foster City, CA) in positive electrospray ionization (ESI) mode and data was analyzed using Analyst software as described previously 19. The mass spectrometer operating conditions were as follows: ion spray voltage (5.0 kV), compressed air as curtain gas (15 psi) and nitrogen as nebulizing gas (8 psi), heater (15 psi), and collision-induced dissociation (CID) gas (5 psi). The ESI probe temperature was 450°C, the declustering potential was 105 V, the entrance potential was 10 V, the collision energy was 45 eV, and the collision exit potential was 15 V. CoA thioesters were monitored using the selected reaction monitoring (SRM) transitions described in Table 1, which employed the major product ion resulting from the loss the phosphoadenosine diphosphate moiety (507.2 Da) as shown in Figure 1.
Table 1.
Medium– and long-chain acyl-CoAs with corresponding SILEC internal standards showing protonated molecules and product ions used for LC-SRM/MS analysis
| Acyl-CoA | MH+ (m/z) | Product ion (m/z) | SILEC MH+ (m/z) | SILEC product ion (m/z) |
|---|---|---|---|---|
| Butanoyl-CoA, C4:0 (butyryl-CoA) | 838.6 | 331.2 | 842.6 | 335.2 |
| Octanoyl-CoA, C8-0 (capryloyl-CoA) | 894.7 | 387.2 | 898.7 | 391.2 |
| Decanoyl-CoA, C10:0 (caproyl-CoA) | 922.8 | 415.3 | 926.8 | 419.3 |
| Dodecanoyl-CoA, C12:0 (lauroyl-CoA) | 950.8 | 443.3 | 954.8 | 447.3 |
| Tetradecanoyl-CoA, C14:0 (myristoyl-CoA) | 978.9 | 471.3 | 982.9 | 475.3 |
| Palmitoleoyl-CoA, C16:1 (9(Z)-hexadecenoyl-CoA) | 1004.9 | 497.3 | 1008.9 | 501.3 |
| Palmitoyl-CoA, C16:0 (hexadecanoyl-CoA) | 1006.9 | 499.3 | 1010.9 | 503.3 |
| Linoleoyl-CoA, C18:2 (9(Z),12(Z)-octadecadienoyl-CoA) | 1030.9 | 523.4 | 1034.9 | 527.4 |
| Oleoyl-CoA, C18:1 (9(Z)-octadecenoyl-CoA) | 1032.9 | 525.4 | 1036.9 | 529.4 |
| Stearoyl-CoA, C18:0 (octadecanoyl-CoA) | 1034.9 | 527.4 | 1038.9 | 531.4 |
| Arachidonyl-CoA, C20:4 (5(Z),8(Z),11(Z),14(Z)-eicosatetraenoyl-CoA) | 1055.0 | 547.4 | 1059.0 | 551.4 |
RESULTS
Mixed Acyl-CoA Extraction
Extracts of LoVo cells at 80% confluence were analyzed by the LC-MS method described above. The neutral loss scans of 507.2 Da are shown in Figure 2 for the mixed acyl-CoA solid phase extraction (Figure 2A), the short-chain acyl-CoA solid phase extraction (Figure 2B), and a biphasic liquid-liquid Folch extraction (Figure 2C). In our hands, the mixed extraction recovered the broadest range of acyl-CoA species as evidenced by the peaks corresponding to acyl-CoA species in the short, medium, and long chain range. The short-chain acyl-CoA extraction we have previously used gave intense CoASH and acetyl-CoA peaks, and overall higher signal intensity, but did not recover significant middle or long chain acyl-CoA species. Recovered species appeared less abundant in the Folch extract versus the mixed acyl-CoA SPE (Figure 2A, 2C).
Figure 2.

Neutral loss 507.2 Da scans of LoVo cells extracted by A) mixed acyl-CoA SPE B) Folch extraction C) short-chain acyl-CoA SPE.
Generation of a SILEC standard
Hepa1c1c7 mouse hepatocellular carcinoma cells were grown for 7 passages in SILEC media then “ultra-labeled” for 24 h in the same media with 0% FBS. The cells were harvested using the mixed acyl-CoA SPE method. Labeling for oleoyl-CoA was determined using integration of the peak area for the m/z 1032 → m/z 525 and m/z 1036 → m/z 529 transitions corresponding respectively to the unlabeled and labeled oleoyl-CoA (Figure 3), where labeling was over 98%. Commercial standards were analyzed together with SILEC internal standards for tetradecanoyl-, palmitoleoyl-, linoleolyl-, stearoyl-, and oleoyl-CoA (Table 1). A typical chromatogram for the long chain-fatty acyl-CoAs is shown in Figure 4. Importantly, the M2 isotopomers for overlapping isobaric species, such as oleoyl-CoA M2 and stearoyl-CoA, are shown to achieve baseline separation with the new LC method (Figure 4). Consistent with previous studies on short-chain acyl-CoAs,5,18,19 the distribution of acyl-CoA species was unequal, such that certain labeled molecules of interest might be of low abundance in the SILEC internal standard mixture for analytical use. To address this problem, the biosynthesis of acyl-CoAs in Hepa1c1c7 cells with various doses of fatty acid mixtures was conducted. A fatty acid mixture of sodium butyrate, sodium hexanoate, sodium octanoate, sodium decanoate, and sodium dodecanoate at 100 μM was found to be ideal, where above that concentration cellular toxicity was observed. A time course study revealed that a 3 h incubation produced the broadest distribution of acyl-CoA species (Figure 5).
Figure 3.
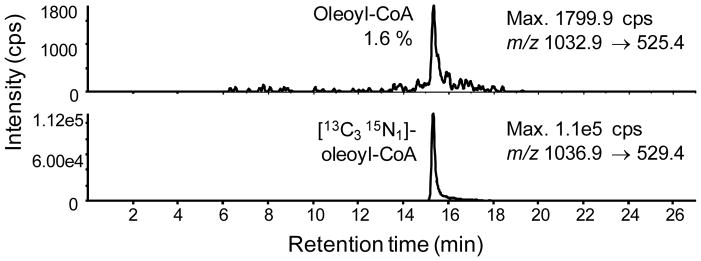
LC-SRM/MS chromatogram showing labeling efficiency of [13C315N1]-oleoyl-CoA extracted from Hepa1c1c7 SILEC extracts.
Figure 4.

LC-SRM/MS chromatograms of SILEC-derived long-chain acyl-CoAs analyzed with corresponding unlabeled authentic standard long-chain acyl-CoAs. Where overlap of isobaric ions was possible, clear chromatographic resolution was obtained (e.g. the M2 of oleoyl-CoA and M0 stearoyl-CoA).
Figure 5.

Constant neutral loss of 507.2 Da in m/z scans of [13C315N]-pantothenic acid derivatives. Hepa1c1c7 cells extracted at (A) baseline, (B) 30 min, (C) 3 h, (D) 6 h, (E) 12 h, (F) 24 h after treatment with sodium butyrate, sodium hexanoate, sodium octanoate, sodium decanoate, and sodium dodecanoate (100 μM of each). m/z 814 = [3C1315N]-acetyl-CoA; m/z 842 = [3C1315N]-butyryl-CoA; m/z 898 = [3C1315N]-octanoyl-CoA; m/z 926 = [3C1315N]-decanoyl-CoA; m/z 954 = [3C1315N]-lauroyl-CoA; m/z 982 = [3C1315N]-tetradecanoyl-CoA; m/z 1008 = [3C1315N]-palmitoleoyl-CoA; m/z 1010 = [3C1315N]-palmitoyl-CoA; m/z 1036 = oleoyl-CoA.
Time course for formation arachidonoyl-CoA in colon cancer cells
A special SILEC standard was generated by treating the SILEC Hepa1c1c7 cells with 25 μM arachidonic acid for one hour before extraction. This provided an enriched arachidonoyl-CoA internal standard, which made it possible to quantify endogenous arachidonoyl-CoA (Figure 6A) and illustrated the applicability of the SILEC method for targeted long chain acyl-CoA quantitation. LoVo and HCA-7 colon adenocarcinoma cells were treated with 25 μM arachidonic acid, and the cells were extracted and analyzed by our method at various time points. The resulting standard curve was linear over 4 orders of magnitude from 20 fmol to 20 pmol (y=1.3555x + 0.1267, R2=0.9978). Precision for analysis of low (0.60 fmol), middle (5 pmol) and high (15 pmol) quality control samples (n=5) were better than 15 %. Accuracies for analysis ranged were in the acceptable of between 85 % and 115 %. This made it possible to rigorously quantify the concentration range of arachidonoyl-CoA in the arachidonate-treated colon cancer cells. Interestingly, initial concentrations of arachidonoyl-CoA in the HCA-7 cells were almost two-fold higher (14. 9 pmol/106 cells) than the LoVo cells (8.9 pmol/106 cells). However, after the addition of arachidonic acid concentrations of arachidonyl-CoA increased to a maximum of 22.4 pmol/106 cells in both cell lines after 4 h (Figure 6B).
Figure 6.

(A) LC-SRM/MS analysis of endogenous arachidonoyl-CoA using the [3C1315N]-arachidonyl-CoA SILEC internal standard. (B) Absolute quantitation of the arachidonoyl-CoA in two different colon carcinoma cells lines from 0.5 to 4 h after the addition of 25 μM arachidonic acid. Analyses were performed in duplicate.
Tissue acyl-CoA quantitation
A set of three mouse liver tissue samples was analyzed by the new SILEC method, and absolute quantification of select acyl-CoA species was conducted. Standard curves were generated from 0.02 to 20 pmol for the acyl-CoAs. Regression lines for selected CoAs were as follows: butanoyl (y=1.77x+9.89e-5, R2=0.9907), octanoyl (4.07x+1.1e-5, R2=0.9974, decanoyl (7.44x+0.000226, R2=0.9994), dodecanoyl (12.1x+0.00171, R2=0.9919), tetradecanoyl (y=1.12x+0.000102, R2=0.9961), and palmitoleoyl (y=0.996x+8.82e-5, R2=0.9955). Precision for analysis of low (0.04 pmol), middle (5 pmol) and high (15 pmol) quality control samples (n=5) were better than 15 %. Accuracies for analysis were in the acceptable range from 85 % to 115 %. The relative abundance of the acyl-CoAs in the mouse liver samples ranged from 0.04 pmol/mg tissue for dodecanoyl-CoA to 0.52 pmol/mg tissue for butanoyl-CoA (Figure 7). This demonstrates the utility of the method for quantitative analysis of the medium- and long-chain acyl-CoA species that are present in tissue samples.
Figure 7.

Absolute quantitation of medium-chain acyl-CoA species from three mouse liver samples taken from different animals. Results are shown as means ± standard error of the mean.
CONCLUSIONS
Acyl-CoAs are important to cell bioenergetics and metabolism, and they play critical roles in lipid biosynthesis, cell signaling, post-translational modification of proteins, and metabolism of xenobiotics.5,7 Accurate and precise quantification of medium and long-chain acyl-CoAs has specific utility for the interrogation of a wide variety of inherited and acquired disorders of metabolism including medium- and long-chain acyl-coenzyme A dehydrogenase deficiencies.24–26 This is also necessary for elucidating the critical role of medium- and long-chain acyl-coenzyme A thioesters in numerous important cellular metabolic pathways.9,27–30 Long-chain acyl-CoAs act as a feedback regulators in bioenergetics and cellular carbon metabolism31 as well as regulating cardiac function at the level of ATP channels.32 Clearly, in order to establish the physiological relevance of altered levels of acyl-CoAs, the bioanalytical methodology that is employed must unequivocally determine the specific concentrations of these relatively unstable, structurally diverse molecules. The variety of individual chemical entities making up the acyl-CoA family poses a significant challenge to the analyst.33 LC-UV strategies for analysis of acyl-CoA species are insufficiently specific due to co-eluting peaks.34 Excellent LC-MS/MS strategies for the analysis of acyl-CoAs have been employed by other groups, including validated methods by Haynes et al.,3 Magnes, et al.,35 and Mauriala et al.36 However, these methods did not employ stable isotope internal standards, which limits their specificity, accuracy, and precision especially for complex biological matrices where interfering ion-suppressing molecules are likely to be present. Stable isotope analog internal standards overcome this problem by preventing selective suppression of the analyte signal.22
In the present study, our previous SILEC method for short-chain acyl-CoAs was expanded to include medium- and long-chain acyl-CoAs. A limitation of the method is that the cells have to be passaged up to 7 times in labeling media, which requires an initial time investment of approximately 3 weeks for most mammalian cell lines. However, once the labeled cell line has been established, expansion of the cells requires only 24 h/passage. Stable isotope labeled standards for the relevant low abundance endogenous acyl-CoA species were prepared by adding suitable fatty acid precursors to the Hepa 1c1c7 SILEC cell incubation. The utility of the method for medium-chain acyl-CoAs was demonstrated by quantifying medium- and long-chain acyl-CoAs present in mouse liver tissue. Long-chain acyl-CoAs could also be quantified as was demonstrated by monitoring the formation of arachidonyl-CoA in two human colon cancer cell lines in response to the addition of arachidonic acid. The SILEC method will be particularly useful for future studies on the quantification of acyl-CoAs derived from bioactive arachidonic acid metabolites such as 11- and 15-oxo-eicosatetraenoic acid37–39 to be conducted so that their role in intracellular signaling as well as the effects of non-steroidal anti-inflammatory drugs on acyl-CoA biosynthesis40,41 can be further elucidated.
Acknowledgments
We acknowledge NIH grants T32GM008076, T32ES019851, P30CA016520, U54HL117798, and P30ES013508.
References
- 1.Robishaw JD, Neely JR. Am J Physiol Endocrinol Metab. 1985;248:E1. doi: 10.1152/ajpendo.1985.248.1.E1. [DOI] [PubMed] [Google Scholar]
- 2.Leonhardt M, Langhans W. Physiol Behav. 2004;83:645. doi: 10.1016/j.physbeh.2004.07.033. [DOI] [PubMed] [Google Scholar]
- 3.Haynes CA, Allegood JC, Sims K, Wang EW, Sullards MC, Merrill AH., Jr J Lipid Res. 2008;49:1113. doi: 10.1194/jlr.D800001-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schroeder F, Petrescu AD, Huang H, Atshaves BP, McIntosh AL, Martin GG, Hostetler HA, Vespa A, Landrock D, Landrock KK, Payne HR, Kier AB. Lipids. 2008;43:1. doi: 10.1007/s11745-007-3111-z. [DOI] [PubMed] [Google Scholar]
- 5.Basu SS, Blair IA. Chemical Res Toxicol. 2011;24:1630. doi: 10.1021/tx200366j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lang TF. J Inherit Metab Dis. 2009;32:675. doi: 10.1007/s10545-009-1202-0. [DOI] [PubMed] [Google Scholar]
- 7.Tracy TS, Wirthwein DP, Hall SD. Drug Metab Dispos. 1993;21:114. [PubMed] [Google Scholar]
- 8.El-Husseini AED, Bredt DS. Nature Rev Neurosci. 2002;3:791. doi: 10.1038/nrn940. [DOI] [PubMed] [Google Scholar]
- 9.Heinonen TM. Expert Opin Investig Drugs. 2002;11:1519. doi: 10.1517/13543784.11.11.1519. [DOI] [PubMed] [Google Scholar]
- 10.Skibowska A, Raszeja-Specht A, Szutowicz A. Clin Chem Lab Med. 2003;41:1136. doi: 10.1515/CCLM.2003.176. [DOI] [PubMed] [Google Scholar]
- 11.Bavenholm PN, Kuhl J, Pigon J, Saha AK, Ruderman NB, Efendic S. J Clin Endocrinol Metab. 2003;88:82. doi: 10.1210/jc.2002-020330. [DOI] [PubMed] [Google Scholar]
- 12.Joyce M, Moore K, Thompson C, Fitzgerald P, Fennessy F, Kelly CJ, Bouchier-Hayes DJ. Eur J Vasc Endovasc Surg. 2004;27:432. doi: 10.1016/j.ejvs.2003.12.020. [DOI] [PubMed] [Google Scholar]
- 13.Gobin-Limballe S, Djouadi F, Aubey F, Olpin S, Andresen BS, Yamaguchi S, Mandel H, Fukao T, Ruiter JP, Wanders RJ, McAndrew R, Kim JJ, Bastin J. Am J Hum Genet. 2007;81:1133. doi: 10.1086/522375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilcken B. J Inherit Metab Dis. 2010;33:501. doi: 10.1007/s10545-009-9001-1. [DOI] [PubMed] [Google Scholar]
- 15.Kim SH, Park HD, Sohn YB, Park SW, Cho SY, Ji S, Kim SJ, Choi EW, Kim CH, Ko AR, Yeau S, Paik KH, Jin DK. Ann Clin Lab Sci. 2011;41:84. [PubMed] [Google Scholar]
- 16.Leoni V, Strittmatter L, Zorzi G, Zibordi F, Dusi S, Garavaglia B, Venco P, Caccia C, Souza AL, Deik A, Clish CB, Rimoldi M, Ciusani E, Bertini E, Nardocci N, Mootha VK, Tiranti V. Mol Gen Metab. 2012;105:463. doi: 10.1016/j.ymgme.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Webb ME, Smith AG, Abell C. Nat Prod Rep. 2004;21:695. doi: 10.1039/b316419p. [DOI] [PubMed] [Google Scholar]
- 18.Basu SS, Mesaros C, Gelhaus SL, Blair IA. Anal Chem. 2011;83:1363. doi: 10.1021/ac1027353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Basu SS, Blair IA. Nat Protoc. 2012;7:1. doi: 10.1038/nprot.2011.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Mol Cell Proteomics. 2002;1:376. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 21.Bishop JE, Hajra AK. Anal Biochem. 1980;106:344. doi: 10.1016/0003-2697(80)90531-x. [DOI] [PubMed] [Google Scholar]
- 22.Ciccimaro E, Blair IA. Bioanalysis. 2010;2:311. doi: 10.4155/bio.09.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Minkler PE, Kerner J, Ingalls ST, Hoppel CL. Anal Biochem. 2008;376:275. doi: 10.1016/j.ab.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iafolla AK, Thompson RJ, Jr, Roe CR. J Pediatr. 1994;124:409. doi: 10.1016/s0022-3476(94)70363-9. [DOI] [PubMed] [Google Scholar]
- 25.Hale DE, Batshaw ML, Coates PM, Frerman FE, Goodman SI, Singh I, Stanley CA. Pediatr Res. 1985;19:666. doi: 10.1203/00006450-198507000-00006. [DOI] [PubMed] [Google Scholar]
- 26.Mitchell GA, Gauthier N, Lesimple A, Wang SP, Mamer O, Qureshi I. Mol Genet Metab. 2008;94:4. doi: 10.1016/j.ymgme.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 27.Ellis JM, Frahm JL, Li LO, Coleman RA. Curr Opin Lipidol. 2010;21:212. doi: 10.1097/mol.0b013e32833884bb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ismail W, Gescher J. Appl Environ Microbiol. 2012;78:5043. doi: 10.1128/AEM.00633-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hunt MC, Tillander V, Alexson SE. Biochimie. 2014;98C:45. doi: 10.1016/j.biochi.2013.12.018. [DOI] [PubMed] [Google Scholar]
- 30.Darnell M, Weidolf L. Chem Res Toxicol. 2013;26:1139. doi: 10.1021/tx400183y. [DOI] [PubMed] [Google Scholar]
- 31.Eger-Neufeldt I, Teinzer A, Weiss L, Wieland O. Biochem Biophys Res Commun. 1965;19:43. [Google Scholar]
- 32.Liu GX, Hanley PJ, Ray J, Urgen D. Circ Res. 2001;88:918. doi: 10.1161/hh0901.089881. [DOI] [PubMed] [Google Scholar]
- 33.Haynes CA. Biochim Biophys Acta. 2011;1811:663. doi: 10.1016/j.bbalip.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 34.Deutsch J, Grange E, Rapoport SI, Purdon AD. Anal Biochem. 1994;220:321. doi: 10.1006/abio.1994.1344. [DOI] [PubMed] [Google Scholar]
- 35.Magnes C, Sinner FM, Regittnig W, Pieber TR. Anal Chem. 2005;77:2889. doi: 10.1021/ac048314i. [DOI] [PubMed] [Google Scholar]
- 36.Mauriala T, Herzig KH, Heinonen M, Idziak J, Auriola S. J Chromatogr B. 2004;808:263. doi: 10.1016/j.jchromb.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 37.Liu X, Zhang S, Arora JS, Snyder NW, Shah SJ, Blair IA. Chem Res Toxicol. 2011;24:2227. doi: 10.1021/tx200336f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Snyder NW, Revello SD, Liu X, Zhang S, Blair IA. J Lipid Res. 2013;54:3070. doi: 10.1194/jlr.M040741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wei C, Zhu P, Shah SJ, Blair IA. Mol Pharmacol. 2009;76:516. doi: 10.1124/mol.109.057489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferris JS, McCoy L, Neugut AI, Wrensch M, Lai R. Int J Cancer. 2012;131:E1031. doi: 10.1002/ijc.27536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kasuya F, Kazuhiro M, Tatsuya H, Nakamoto K, Tokuyama S, Masuyama T. J Enzyme Inhib Med Chem. 2013;28:223. doi: 10.3109/14756366.2011.636742. [DOI] [PubMed] [Google Scholar]