

The present study is the first intervention study in a well-established, translational mouse model for hyperlipidaemia and atherosclerosis showing that anacetrapib dose-dependently reduces atherosclerosis development and adds to the anti-atherogenic effects of atorvastatin. This effect is mainly ascribed to the reduction in non-HDL-C despite a remarkable increase in HDL-C and without affecting HDL functionality. In addition, anacetrapib improves lesion stability.
Keywords: Cholesteryl ester transfer protein, Non-HDL-cholesterol, HDL-cholesterol, HDL function, Atherosclerosis, Anacetrapib, Atorvastatin
Abstract
Background
The residual risk that remains after statin treatment supports the addition of other LDL-C-lowering agents and has stimulated the search for secondary treatment targets. Epidemiological studies propose HDL-C as a possible candidate. Cholesteryl ester transfer protein (CETP) transfers cholesteryl esters from atheroprotective HDL to atherogenic (V)LDL. The CETP inhibitor anacetrapib decreases (V)LDL-C by ∼15–40% and increases HDL-C by ∼40–140% in clinical trials. We evaluated the effects of a broad dose range of anacetrapib on atherosclerosis and HDL function, and examined possible additive/synergistic effects of anacetrapib on top of atorvastatin in APOE*3Leiden.CETP mice.
Methods and results
Mice were fed a diet without or with ascending dosages of anacetrapib (0.03; 0.3; 3; 30 mg/kg/day), atorvastatin (2.4 mg/kg/day) alone or in combination with anacetrapib (0.3 mg/kg/day) for 21 weeks. Anacetrapib dose-dependently reduced CETP activity (−59 to −100%, P < 0.001), thereby decreasing non-HDL-C (−24 to −45%, P < 0.001) and increasing HDL-C (+30 to +86%, P < 0.001). Anacetrapib dose-dependently reduced the atherosclerotic lesion area (−41 to −92%, P < 0.01) and severity, increased plaque stability index and added to the effects of atorvastatin by further decreasing lesion size (−95%, P < 0.001) and severity. Analysis of covariance showed that both anacetrapib (P < 0.05) and non-HDL-C (P < 0.001), but not HDL-C (P = 0.76), independently determined lesion size.
Conclusion
Anacetrapib dose-dependently reduces atherosclerosis, and adds to the anti-atherogenic effects of atorvastatin, which is mainly ascribed to a reduction in non-HDL-C. In addition, anacetrapib improves lesion stability.
Translational Perspective.
The present study is the first intervention study in a well-established, translational mouse model for hyperlipidaemia and atherosclerosis showing that anacetrapib dose-dependently reduces atherosclerosis development and adds to the anti-atherogenic effects of atorvastatin. This effect is mainly ascribed to the reduction in non-HDL-C despite a remarkable increase in HDL-C and without affecting HDL functionality. In addition, anacetrapib improves lesion stability.
Introduction
Intervention trials provide ample evidence that lowering of low-density lipoprotein-cholesterol (LDL-C) contributes to a reduction in cardiovascular (CV) risk.1–3 However, the residual risk that remains after statin treatment, as well as failure for some patients to reach recommended LDL-C targets despite statin treatment, support the addition of other LDL-C-lowering agents and also stimulate the search for secondary treatment targets.3,4 Prospective epidemiological studies propose high-density lipoprotein (HDL)-C as a potential target.5 Cholesteryl ester transfer protein (CETP) plays an important role in lipid metabolism by facilitating the transfer of cholesteryl esters from atheroprotective HDL to atherogenic (V)LDL in exchange for triglycerides (TG), and inhibition of CETP activity has been proposed as a therapeutic way to increase HDL-C levels.6–11
In mouse models for atherosclerosis, CETP expression aggravated atherosclerosis development.12,13 Most but not all studies in rabbits and mice showed that CETP inhibition reduced atherosclerosis development.14–19 However, torcetrapib failed to enhance the anti-atherogenic effects of atorvastatin and induced a pro-inflammatory, vulnerable plaque phenotype in APOE*3Leiden.CETP mice.19 In the large clinical outcome trial (ILLUMINATE), torcetrapib increased the risk of major CV events and mortality despite a 72% increase in HDL-C and a 25% reduction in LDL-C.20 The unexpected detrimental effects were ascribed to either an off-target blood pressure effect or the possible generation of dysfunctional HDL particles.20 The much less potent CETP inhibitor dalcetrapib increased HDL-C by 31–40% with a minimal reduction in LDL-C, but did not translate into clinical benefit and resulted in premature termination of the dal-OUTCOMES trial.21 Nonetheless, other CETP inhibitors are currently in clinical development. Among these, anacetrapib and evacetrapib have remarkable lipid-modulating abilities without the unwanted blood pressure effect as observed with torcetrapib.22 In phase II trials, anacetrapib (10–300 mg) decreased LDL-C by ∼15–40% and increased HDL-C by ∼40–140% and evacetrapib (30–500 mg) decreased LDL-C by ∼15–35% and increased HDL-C by ∼50–130%.23,24
To elucidate whether pharmacological CETP inhibition is anti-atherogenic and to what extent this is due to its LDL-C-lowering and HDL-C-raising abilities, we evaluated the effects of partial to full inhibition of CETP activity with a broad dose range of anacetrapib monotreatment on lipid modulation, atherosclerosis development, and HDL functionality in APOE*3Leiden.CETP mice. Secondly, to mimic clinical intervention trials where dyslipidaemic patients also receive statin treatment, we examined the possible additive/synergistic effects of anacetrapib on top of atorvastatin treatment in this well-established model for lipoprotein metabolism and atherosclerosis. These mice respond in a human-like manner to lipid-modulating interventions, including LDL-C-lowering19,25 and HDL-C-raising drugs.19,26,27 See Supplementary material online for more detailed information on the background of the APOE*3Leiden.CETP mice and their response to hypolipidaemic drugs.
Methods
Animals and diet
Female APOE*3Leiden.CETP transgenic mice13 were fed a semi-synthetic cholesterol-rich diet for a run-in period of 5 weeks (see Supplementary material online). Animals were matched based on body weight, total cholesterol (TC), TG, HDL-C and age (n = 15 per group) and received a control Western-type diet without or with incremental dosages of anacetrapib (0.03; 0.3; 3; and 30 mg/kg/day; Dalton Chemical Laboratories, Inc., Canada), atorvastatin (2.4 mg/kg/day) or a combination of atorvastatin (2.4 mg/kg/day) and anacetrapib (0.3 mg/kg/day) for a treatment period of 21 weeks. All animals were sacrificed by CO2 inhalation and hearts were isolated to assess atherosclerosis development. Animal experiments were approved by the Institutional Animal Care and Use Committee of The Netherlands Organization for Applied Research (TNO).
Plasma lipids, lipoprotein profile, endogenous cholesteryl ester transfer protein activity, cholesteryl ester transfer protein concentration, serum amyloid A and HDL functionality
Plasma TC, TG, and HDL-C were determined every 2–4 weeks and average TC, TG, and HDL-C levels were calculated by total exposure over number of weeks. To measure HDL-C, apoB-containing particles were precipitated from plasma with 20% polyethylene glycol in 200 mM glycine buffer (pH 10) and cholesterol was measured in the supernatant. The distribution of cholesterol over plasma lipoproteins was determined by fast-performance liquid chromatography (FPLC) as previously described.13
Plasma endogenous CETP activity and CETP concentration were determined as previously described.28 Serum amyloid A (SAA; Tridelta development, Co. Kildare, Ireland) was measured by using ELISA according to manufacturer's instructions.
Cultured arterial endothelial cells were incubated with HDL isolated from control- and anacetrapib-treated mice and pro-inflammatory cytokine-induced vascular cell adhesion molecule 1 (VCAM-1) expression and apoptotic cell death were assessed.
Atherosclerosis quantification
Cross-sections throughout the entire aortic root area were stained to assess atherosclerotic lesion area and severity. The lesions were classified into five categories: (i) early fatty streak, (ii) regular fatty streak, (iii) mild plaque, (iv) moderate plaque, and (v) severe plaque according to the American Heart Association classification and total lesion area, number of lesions, undiseased segments and lesion severity were determined as previously described.28,29 Lesion composition of the severe lesions (type IV–V) was assessed after immunostaining with anti-human alpha actin (1:800; Monosan, Uden, The Netherlands) for smooth muscle cells (SMC), and anti-mouse Mac-3 (1:50; BD Pharmingen, the Netherlands) for macrophages followed by sirius red staining for collagen. Necrotic area and cholesterol clefts, monocyte adhesion to the endothelium, and the calculation of plaque stability index were determined as previously described.28,29
Statistical analyses
Significance of differences between the groups was calculated non-parametrically using a Kruskal–Wallis test for independent samples, followed by a Mann–Whitney U test for independent samples. An analysis of covariance (ANCOVA) was performed to test for group differences in a lesion area with HDL-C and non-HDL-C exposure as covariates. To test whether collinearity was present between the explanatory variables, we calculated the variance inflation factor (VIF) and the condition index (CI). Values of VIF >5 and values of CI >10 were used as a cutoff for collinearity.30,31
SPSS 17.0 for Windows was used for statistical analysis. All groups were compared with the control group and the combination group was compared with the atorvastatin group. Bonferroni–Holm's method was used to determine the level of significance in the case of multiple comparisons. Values are presented as means ± SD. P-values <0.05 were considered statistically significant.
For the full descriptions of the used methods, please see the Supplementary material online.
Results
Anacetrapib, atorvastatin, and their combination decrease cholesteryl ester transfer protein activity despite an increase in cholesteryl ester transfer protein concentration
To assess the extent to which an ascending dose range of anacetrapib inhibits CETP, we measured CETP activity after 8 weeks of treatment and CETP concentration after 21 weeks of treatment (Table 1). Anacetrapib monotreatment (0.03; 0.3; 3; and 30 mg/kg/day) reduced CETP activity by −59 to −100% (P < 0.001) and increased plasma CETP concentration by +11% (NS) to +29% (P < 0.001). Both CETP activity and concentration were decreased by atorvastatin alone (−29 and −24%, P < 0.001) and in combination with 0.3 mg/kg/day anacetrapib (−84 and −23%, P < 0.001). Thus, adding anacetrapib to atorvastatin further reduced CETP activity (−78%, P < 0.001) without affecting CETP concentration when compared with atorvastatin.
Table 1.
Effect of anacetrapib, atorvastatin, and their combination on the cholesteryl ester transfer protein activity after 8 weeks of treatment and cholesteryl ester transfer protein concentration after 21 weeks of treatment
| Plasma CETP activity (nmol/mL/h) | Plasma CETP concentration (µg/mL) | |
|---|---|---|
| Control | 66.8 ± 10.1 | 15.7 ± 1.2 |
| 0.03 mg/kg/day anacetrapib | 27.1 ± 7.1*** (−59%) | 17.4 ± 3.1 (+11%) |
| 0.3 mg/kg/day anacetrapib | 6.3 ± 4.8*** (−91%) | 19.9 ± 2.4*** (+27%) |
| 3 mg/kg/day anacetrapib | 0.7 ± 3.3*** (−99%) | 20.2 ± 3.1*** (+29%) |
| 30 mg/kg/day anacetrapib | 0.0 ± 2.3*** (−100%) | 18.5 ± 4.1 (+18%) |
| Atorvastatin | 47.5 ± 8.2*** (−29%) | 11.9 ± 2.3*** (−24%) |
| Atorvastatin + 0.3 mg/kg/day anacetrapib | 10.5 ± 8.3***,### (−84%) | 12.1 ± 2.1*** (−23%) |
***P < 0.001 when compared with control.
###P < 0.001 when compared with atorvastatin. Data are presented as means ± SD (% inhibition or increase when compared with the control); n = 15 per group.
Anacetrapib alone and in combination with atorvastatin reduces plasma non-HDL-cholesterol and increases HDL-cholesterol
During the study, plasma lipids were measured every 2–4 weeks and average plasma TC (Figure 1A), TG (Figure 1B), non-HDL-C (Figure 1C), and HDL-C (Figure 1D) were calculated. In the control group, the Western-type diet resulted in an average plasma TC of 10.8 ± 1.1 mmol/L, TG of 1.8 ± 0.5 mmol/L, non-HDL-C of 9.5 ± 1.1 mmol/L and HDL-C of 1.2 ± 0.2 mmol/L. When compared with the control, anacetrapib monotreatment (0.03; 0.3; 3 and 30 mg/kg/day) decreased TC (−19; −25; −27, and −31%, P < 0.001 for all) mainly by decreasing non-HDL-C (−24; −36; −42, and −45%, P < 0.001 for all). In addition, anacetrapib monotreatment increased HDL-C (+30; +60; +86; and +86%, P < 0.001 for all) and decreased TG (−21%, P = 0.07; −22%, P = 0.06; −19%, NS and −27%, P < 0.01). Atorvastatin decreased TC (−33%, P < 0.001) by decreasing non-HDL-C (−37%, P < 0.001) and with no effect on HDL-C and TG. The combination treatment decreased TC (−48%, P < 0.001), non-HDL-C (−60%, P < 0.001) and TG (−33%, P < 0.01) and increased HDL-C (+56%, P < 0.001). Anacetrapib enhanced the lipid-modifying effects of atorvastatin with greater reductions in TC (−22%, P < 0.001), non-HDL-C (−36%, P < 0.001) and TG (−32%, P < 0.001) and a greater increase in HDL-C (+72%, P < 0.001) when comparing the combination treatment to atorvastatin monotreatment. Lipoprotein profiles confirmed the lipid-modifying effects of anacetrapib and revealed the formation of larger HDL particles, as observed previously19 after treatment with higher dosages of anacetrapib (3 and 30 mg/kg).
Figure 1.

Effect of anacetrapib, atorvastatin and their combination on total cholesterol, triglycerides, non-HDL-and HDL-cholesterol levels. Plasma total cholesterol (A), triglycerides (B) non-HDL-cholesterol (C) and HDL-cholesterol (D) were measured throughout the study and average levels were calculated. Lipoprotein profiles for cholesterol were assessed by FPLC lipoprotein separation to study effects of anacetrapib alone (E) and in combination with atorvastatin (F) after 18 weeks of treatment. **P < 0.01, ***P < 0.001 when compared with control; ###P < 0.001 when compared with atorvastatin. Data are presented as means ± SD (n = 15 per group).
Atorvastatin in combination with anacetrapib reduces atherosclerosis progression to a greater extent than atorvastatin alone
After 21 weeks of treatment, the effects of anacetrapib, atorvastatin and their combination on the progression of atherosclerosis were assessed in the aortic root area as illustrated by representative images in Figure 2. The number of lesions (Figure 3A), lesion area (Figure 3B), undiseased segments (Figure 3C), and lesion severity (Figure 3D) were assessed as previously described.28,29
Figure 2.
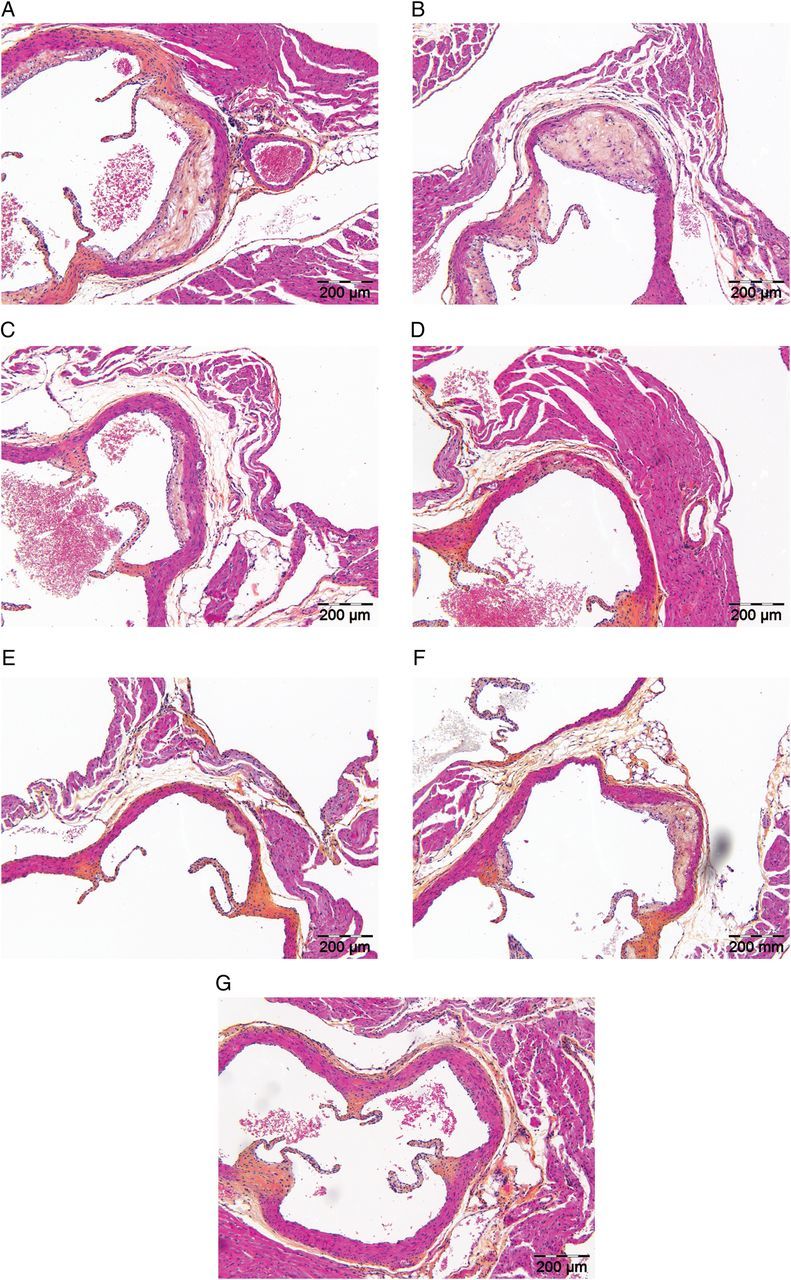
Effect of anacetrapib, atorvastatin, and their combination on plaque morphology. Representative images of haematoxylin–phloxine–saffron-stained atherosclerotic lesions in a cross-section of the aortic root area for the control group (A), 0.03 mg/kg/day anacetrapib (B), 0.3 mg/kg/day anacetrapib (C), 3 mg/kg/day anacetrapib (D), 30 mg/kg/day anacetrapib (E), atorvastatin group (F), and the combination group (G) after 21 weeks of treatment.
Figure 3.

Effect of anacetrapib, atorvastatin, and their combination on atherosclerosis development in the aortic root area. The number of lesions per cross-section (A), total lesion area per cross-section (B), the percentage undiseased segments (C), and lesion severity as a percentage of all lesions (D) were determined after 21 weeks of treatment. Lesion severity was classified as mild (type I–III) and severe (type IV–V) lesions. **P < 0.01, ***P < 0.001 when compared with control; #P < 0.05, ###P < 0.001 when compared with atorvastatin. Data are presented as means ± SD (n = 15 per group).
For the control group, 4.1 ± 0.6 lesions per cross-section developed with a total lesion area of 169 ± 51 × 103 µm2. Approximately 71% of these lesions were severe lesions (type IV–V) and only 5% of the segments were undiseased. Anacetrapib monotreatment (0.03; 0.3; 3; and 30 mg/kg/day) dose-dependently reduced the lesion area (−41%, P < 0.01; −72; −86; and −92%, P < 0.001 for all) and the number of lesions and improved lesion severity as indicated by less severe lesions (down to 15%, P < 0.001) and more undiseased segments (up to 46%, P < 0.001). Atorvastatin monotreatment reduced the total lesion area (−63%, P < 0.001) and improved lesion severity without affecting the number of lesions and undiseased segments. When compared with the control, the combination treatment further decreased the total lesion area (−95%, P < 0.001), the number of lesions (−41%, P < 0.01), and lesion severity and increased the percentage of undiseased segments. When compared with atorvastatin monotreatment, the combination treatment decreased the total lesion area (−87%, P < 0.001), the number of lesions (−34%, P = 0.06) and severity and increased the percentage undiseased segments to a greater extent, indicative of an additional effect of anacetrapib on top of the statin.
Anacetrapib, atorvastatin, and their combination improve lesion stability
In addition to atherosclerotic lesion size and severity, we assessed the number of monocytes adhering to the endothelium as a functional marker for vascular inflammation (Figure 4A). Adhering monocytes per cross-section in the control group (i.e. 4.1 ± 2.6) were reduced by the higher dosages of anacetrapib (−60%, P < 0.01 and −61%, P < 0.01), as well as by atorvastatin alone and in combination with anacetrapib (−48%, P < 0.05 and −78%, P < 0.001). When compared with atorvastatin, the combination treatment reduced the number of monocytes to a greater extent (−57%, P < 0.01). In addition, we analysed the composition of the severe lesions (type IV–V), since these lesions are considered to be most vulnerable and prone to rupture. All parameters of lesion composition were calculated per cross-section as absolute values and as a percentage of the lesion area (see Supplementary material online). To this end, collagen content (Figure 4B) and SMC content in the cap (Figure 4C) were considered as stabilization factors, and macrophage content (Figure 4D) and necrotic content (Figure 4E) were considered as destabilization factors. The severe lesions in the control group consisted of ∼54% collagen, 6% SMC in the cap, 10% macrophages and 4% necrosis. The lesion stability index for the control group presented as the ratio of stabilization to destabilization factors was 4.9 ± 2.0 (Figure 4F).
Figure 4.

Effect of anacetrapib, atorvastatin, and their combination on lesion composition. The number of monocytes adhering to the vascular endothelium per cross-section (A) was calculated. In the severe lesions (type IV and V), collagen content (B), and SMC content in the cap (C) were determined as stabilization factors and macrophage content (D) and necrotic content (E) were determined as destabilization factors, all as a percentage lesion area. The plaque stability index was calculated as the ratio of the stabilization factors to the destabilization factors (F). *P < 0.05, **P < 0.01, ***P < 0.001 when compared with control; ##P < 0.01 when compared with atorvastatin. Data are presented as means ± SD (n = 15 per group).
When corrected for the lesion area, the two higher dosages of anacetrapib (3 and 30 mg/kg/day) revealed a more stable plaque phenotype by increasing collagen content (+21%, P < 0.001 and +28%, P < 0.001; Figure 4B) and SMC content in the cap (+120%, P < 0.01 and +119%, P < 0.05) and by decreasing macrophage (−53%, P = 0.06 and −60%, P = 0.05) and necrotic (−73%, P < 0.001 and −46%, P < 0.05) content. This is reflected by an increase in lesion stability index in these two treatment groups (+427%, P < 0.001 and +366%, P < 0.01). Atorvastatin in combination with anacetrapib tended to increase the SMC content in the cap (+194%, P = 0.07) and decreased necrotic content (−96%, P < 0.05) with no effect on the lesion stability index. However, it should be noted that there were almost no lesions in the combination group and only two mice that received the combination treatment of anacetrapib and atorvastatin developed severe lesions.
Anacetrapib does not affect HDL function
To explore the contribution of the anacetrapib-induced increase in HDL-C to the reduction of atherosclerosis, we investigated the endothelial-vasoprotective properties of HDL, in particular with respect to anti-inflammatory and anti-apoptotic properties in cultured arterial endothelial cells. HDL isolated from anacetrapib-treated mice had no effect on pro-inflammatory cytokine-induced VCAM-1 expression (Figure 5A) or on apoptotic cell death (Figure 5B).
Figure 5.
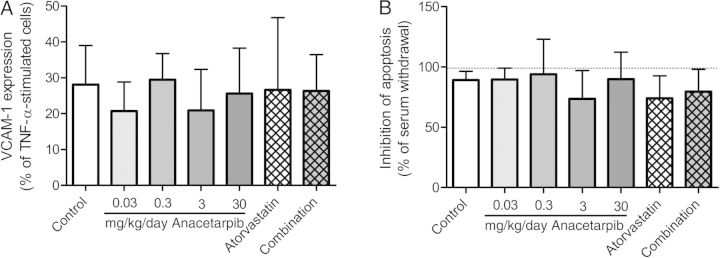
Effect of anacetrapib, atorvastatin, and their combination on endothelial-vasoprotective properties of HDL, in particular pro-inflammatory cytokine-induced VCAM-1 expression (A) and apoptotic cell death (B). Data are presented as means ± SD.
Anacetrapib reduces atherosclerosis progression primarily by reducing non-HDL-cholesterol exposure
We evaluated whether the effects of anacetrapib and atorvastatin on atherosclerosis development could be explained by either an increase in HDL-C or a decrease in non-HDL-C or both. The lesion area was normalized by cubic root transformation (lesion area(1/3)). Univariate regression analysis showed that the lesion area was predicted by TC (Figure 6A), mainly non-HDL-C (Figure 6B) and to a lesser extent by HDL-C (Figure 6C). Analysis of covariance showed that both anacetrapib treatment, at the dosages of 3 and 30 mg/kg/day (P < 0.05) and non-HDL-C (P < 0.001), but not HDL-C (P = 0.76), independently determined lesion size. Importantly, the variance inflation factors of HDL-C and non-HDL-C (VIF = 4.42 and 3.18, respectively) and the condition index (CI = 4.43) did not exceed the threshold for collinearity between the explanatory variables. Collectively, these data are compatible with a mechanism that anacetrapib mainly decreases atherosclerotic lesion development via a reduction of non-HDL-C with an additional effect by the compound itself at the higher doses (Figure 7).
Figure 6.
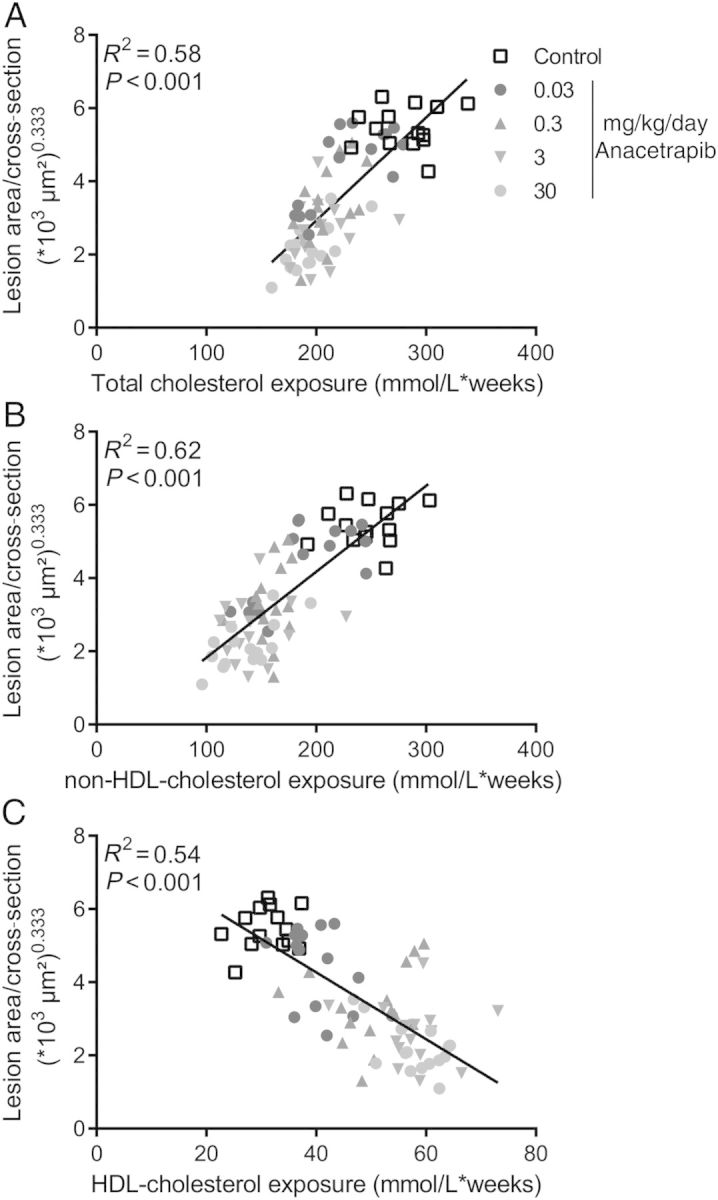
Correlation between plasma cholesterol exposure and lesion area. Linear regression analyses were performed on the cubic root of lesion area plotted against total cholesterol exposure (A), non-HDL-cholesterol exposure (B) and HDL-cholesterol exposure (C).
Figure 7.

Hypothetical scheme of factors contributing to the effect of anacetrapib on the atherosclerotic lesion area as suggested by statistical analyses. An analysis of covariance was performed to test for group differences in the lesion area with HDL-C and non-HDL-C exposure as covariates. HDL-C was not an independent predictor of the lesion area when non-HDL-C was included as covariate, suggesting that the effect of anacetrapib on atherosclerosis development was mainly mediated through the reduction of non-HDL-C. The higher dosages of anacetrapib (3 and 30 mg/kg/day) also revealed an effect on atherosclerosis that was independent of non-HDL-C, but this effect was not explained by the increase in HDL-C.
Anacetrapib slightly increased serum amyloid A as a marker of inflammation
To assess the effect of anacetrapib on general inflammatory status, we measured plasma SAA levels, a systemic inflammatory marker after 16 weeks of treatment (Figure 8). Plasma SAA levels in the control group were 1.6 ± 0.5 µg/mL. When compared with the control, 0.3 mg/kg/day anacetrapib tended to increase SAA levels (+37%, P = 0.07). No effects on body weight (gain) and food intake were noted with any of the treatments (data not shown).
Figure 8.

The effects of anacetrapib, atorvastatin, and their combination on plasma SAA levels were measured after 16 weeks of treatment. *P < 0.05 when compared with control. Data are presented as means ± SD (n = 15 per group).
Discussion
The present study is the first intervention study in a mouse model for atherosclerosis designed to investigate the effects of the CETP inhibitor anacetrapib alone and in combination with atorvastatin on the progression of atherosclerosis, lesion stability, and HDL function. In clinical trials, the effectiveness of novel treatment regimes in CVD is only being tested in patients on a statin background which makes this study unique in also evaluating the effects of anacetrapib monotreatment. In APOE*3Leiden.CETP mice, a broad dose range of anacetrapib dose-dependently reduced atherosclerosis development. This effect was mainly ascribed to the reduction in non-HDL-C despite a remarkable increase in HDL-C and without affecting HDL functionality. Anacetrapib improved lesion stability when given at a higher dose (3 and 30 mg/kg/day). In addition, a moderate dose of anacetrapib (0.3 mg/kg/day) added to the anti-atherogenic effects of atorvastatin.
In our study, incremental dosages of 0.03–30 mg/kg/day anacetrapib dose-dependently decreased CETP activity by >60%, decreased non-HDL-C by 24–45% and increased HDL-C by 30–86%. These lipid-altering effects are comparable with findings from phase I, II and III clinical trials. In phase I trials, an anacetrapib-induced reduction in CETP activity of >60% was accompanied by dose-dependent LDL-C-lowering and HDL-C-raising effects both in healthy subjects32 and in patients with dyslipidaemia.33 In line with our results, these studies also report an increase in CETP concentration possibly due to the formation of an inactive complex between CETP and HDL.34 In a phase II trial, 8 weeks of treatment with ascending dosages of anacetrapib monotreatment (10–300 mg/day) reduced LDL-C by 16–39% and increased HDL-C by 44–139%. Similar to our study, the addition of anacetrapib to atorvastatin produced incremental LDL-C reductions.23
The present study in APOE*3Leiden.CETP mice demonstrates that the total blockage of CETP does not reveal adverse effects on the clinical end-point when compared with partial blockage: anacetrapib dose-dependently reduced the progression of atherosclerosis and increased plaque stability where the anti-atherogenic effects of atorvastatin were enhanced in combination with a moderate dose of anacetrapib.
Inconsistent data have been reported on the effect of other CETP inhibitors on atherosclerosis development in animals expressing CETP. In rabbits, dalcetrapib reduced atherosclerosis in one study with no effect in another study.15,17 In contrast to the human situation,21 the reduction in atherosclerosis after dalcetrapib treatment was accompanied by a 40–50% decrease in non-HDL-C together with an increase in HDL-C.15 In mice, torcetrapib monotreatment decreased atherosclerosis.19 However, unlike the present study, these effects were not enhanced in combination with atorvastatin in the same mouse model.19 In rabbits, torcetrapib treatment decreased atherosclerosis where the aortic lesion area correlated with the TC/HDL-C ratio.18 This could suggest a possible anti-atherogenic role of increased HDL-C or other pleiotropic effects of HDL. However, in the APOE*3Leiden.CETP mouse model, statistical analyses revealed that HDL-C was not an independent predictor of the lesion area when non-HDL-C was included as covariate, suggesting that the effect of anacetrapib on atherosclerosis development was mainly mediated through the reduction of non-HDL-C. The higher dosages of anacetrapib (3 and 30 mg/kg/day) also revealed an effect on atherosclerosis that was independent of non-HDL-C, but this effect was not explained by the increase in HDL-C and could point to other hitherto unknown (off target) effects of anacetrapib.
Besides atherosclerotic lesion size, lesion composition should also be taken into consideration given that in the human situation, a vulnerable lesion consisting of more macrophages, a large necrotic core and a thin, collagen-poor, fibrous cap is more prone to rupture.35 Previously, our group showed that torcetrapib produced a pro-inflammatory, unstable plaque phenotype as seen by increased monocyte adherence to the vascular endothelium and consequently increased macrophage content of the lesions.19 In the present study, anacetrapib decreased monocyte adherence and improved lesion composition as shown by an increase in stabilization factors (collagen and SMC content) and a decrease in destabilization factors (macrophage and necrotic content). The inconsistencies can be ascribed to the off-target activation of the renin–angiotensin–aldosterone system (RAAS) and blood pressure effect of torcetrapib.36 Indeed, in our mouse model for atherosclerosis, torcetrapib also increased aldosterone levels in plasma.19
The large phase III DEFINE trial was designed to further assess the efficacy and tolerability of anacetrapib in statin-treated patients with or at risk for coronary heart disease.37 Anacetrapib (100 mg/day) decreased LDL-C by 40% and increased HDL-C by 138% with an acceptable safety profile and no indication for an increase in CV events. In fact, post hoc analyses suggested a reduction in CV end-points. These initial data provided a rationale for conducting a larger clinical end-point trial of pharmacological CETP inhibition despite the conflicting outcomes of genetic CETP deficiency and the ILLUMINATE trial.20 In view of the detrimental effects of torcetrapib in the ILLUMINATE trial, anacetrapib was thoroughly screened and revealed minimal side-effects without any indication for an off-target pressure effect.23,32,33
Despite the absence of reported side-effects of anacetrapib, there are some concerns about target-related side-effects due to formation of large buoyant cholesterol-rich HDL-2 particles after CETP inhibition,38,39 which may be dysfunctional with regard to their endothelial-vasoprotective effects and consequently their atheroprotective properties10,34,40,41 and that this may have contributed to the failure of torcetrapib.20 In the present study, we investigated the effects of HDL isolated from control and anacetrapib-treated mice on parameters of vascular inflammation and function. HDL from anacetrapib-treated mice did not suppress cytokine-induced adhesion molecule expression or cell apoptosis in endothelial cells. This is in line with results from recent studies where no differences were observed in the effect of HDL from control or anacetrapib-treated hamsters and humans on inflammatory markers (adhesion molecule expression, monocyte chemotactic protein-1 secretion, monocyte adhesion, NFkB activation, and cytokine mRNAs) in endothelial cells42 and macrophages.43 Importantly, although anacetrapib treatment did not improve the anti-inflammatory and anti-apoptotic effects of HDL, it also did not adversely affect these functions of HDL. In addition, we found no effect of anacetrapib on serum paraoxonase 1 activity and the aortic content of reactive oxygen species (data not shown). Formation of large cholesterol-rich HDL-2 particles in CETP-deficiency or after CETP inhibition has also been suggested to affect the cholesterol efflux capacity of these particles.38,39 Although we did not address this in the present study, data from literature consistently indicate that the cholesterol efflux capacity is not impaired but improved. HDL from CETP-deficient patients displayed enhanced ability to promote cholesterol efflux from macrophages in a ABCG1-dependent manner.38 In humans, anacetrapib-treated HDL showed increased ABCA1- and ABCG1-mediated cholesterol efflux capacity.43 Collectively, these data indicate that CETP inhibition does not result in formation of dysfunctional HDL with regard to its atheroprotective properties as assessed by ex vivo (cell) assays.
It should be noted that in the DEFINE trial, a non-significant 18% increase in C-reactive protein, a marker of inflammation, after anacetrapib treatment was found.37 In the present study, the inflammatory marker, SAA was slightly elevated after anacetrapib treatment, but this effect was alleviated when anacetrapib was given in combination with atorvastatin.
The effects of two other CETP inhibitors, DRL-17822 and TA-8995 (DEZ-001), as well as a vaccine against CETP, ATH03, are being tested in phase II clinical development. In large phase III clinical trials, the effects of 100 mg anacetrapib (REVEAL) and 130 mg evacetrapib (ACCELERATE) in patients on standard statin treatment on CV outcomes are currently being investigated and results are expected in 2016–17.44 The outcome of these trials will resolve the unanswered questions regarding possible beneficial effects of pharmacological CETP inhibition and may give additional insight into the HDL-hypothesis and the contribution of HDL and non-HDL to CV end-points.
Supplementary material
Supplementary material is available at European Heart Journal online.
Funding
This research was performed within the framework of CTMM, the Center for Translational Molecular Medicine (www.ctmm.nl), project PREDICCt (KOWVD, grant 01C-104), and supported by the Dutch Heart Foundation, Dutch Diabetes Research Foundation and Dutch Kidney Foundation and Netherlands Consortium for Systems Biology. We acknowledge the support from ‘the Netherlands CardioVascular Research Initiative: the Dutch Heart Foundation, Dutch Federation of University Medical Centers, the Netherlands Organization for Health Research and Development and the Royal Netherlands Academy of Sciences’ for the GENIUS project ‘Generating the best evidence-based pharmaceutical targets for atherosclerosis’ (CVON2011-19). PCNR is an Established Investigator of the Netherlands Heart Foundation (2009T038). Funding to pay the Open Access publication charges for this article was provided by TNO (Netherlands Organization for Applied Scientific Research TNO).
Conflict of interest: J.W.J. received research grants from and was speaker on (CME accredited) meetings sponsored by Amgen, Astellas, Astra-Zeneca, Biotronik, Boston Scientific, Daiichi Sankyo, Lilly, Genzyme, Medtronic, Merck-Schering-Plough, Pfizer, Orbus Neich, Novartis, Roche, Servier, Sanofi Aventis, the Netherlands Heart Foundation, the Interuniversity Cardiology Institute of the Netherlands and the European Community Framework KP7 Programme.
Acknowledgement
We would like to thank Erik Offerman (TNO), Wen Liang (TNO/LUMC) and Jasmin Manz (University of Zurich) for their excellent technical assistance.
References
- 1.Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, Kirby A, Sourjina T, Peto R, Collins R, Simes R Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–1278. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- 2.Mihaylova B, Emberson J, Blackwell L, Keech A, Simes J, Barnes EH, Voysey M, Gray A, Collins R, Baigent C Cholesterol Treatment Trialists’ (CTT) Collaborators. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet. 2012;380:581–590. doi: 10.1016/S0140-6736(12)60367-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, Peto R, Barnes EH, Keech A, Simes J, Collins R Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–1681. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davidson MH, Maki KC, Pearson TA, Pasternak RC, Deedwania PC, McKenney JM, Fonarow GC, Maron DJ, Ansell BJ, Clark LT, Ballantyne CM. Results of the National Cholesterol Education (NCEP) Program Evaluation ProjecT Utilizing Novel E-Technology (NEPTUNE) II survey and implications for treatment under the recent NCEP Writing Group recommendations. Am J Cardiol. 2005;96:556–563. doi: 10.1016/j.amjcard.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 5.Di Angelantonio E, Sarwar N, Perry P, Kaptoge S, Ray KK, Thompson A, Wood AM, Lewington S, Sattar N, Packard CJ, Collins R, Thompson SG, Danesh J Emerging Risk Factors Collaboration. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302:1993–2000. doi: 10.1001/jama.2009.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown ML, Inazu A, Hesler CB, Agellon LB, Mann C, Whitlock ME, Marcel YL, Milne RW, Koizumi J, Mabuchi H. Molecular basis of lipid transfer protein deficiency in a family with increased high-density lipoproteins. Nature. 1989;342:448–451. doi: 10.1038/342448a0. [DOI] [PubMed] [Google Scholar]
- 7.Boekholdt SM, Thompson JF. Natural genetic variation as a tool in understanding the role of CETP in lipid levels and disease. J Lipid Res. 2003;44:1080–1093. doi: 10.1194/jlr.R200018-JLR200. [DOI] [PubMed] [Google Scholar]
- 8.Inazu A, Brown ML, Hesler CB, Agellon LB, Koizumi J, Takata K, Maruhama Y, Mabuchi H, Tall AR. Increased high-density lipoprotein levels caused by a common cholesteryl-ester transfer protein gene mutation. N Engl J Med. 1990;323:1234–1238. doi: 10.1056/NEJM199011013231803. [DOI] [PubMed] [Google Scholar]
- 9.Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, Hindy G, Holm H, Ding EL, Johnson T, Schunkert H, Samani NJ, Clarke R, Hopewell JC, Thompson JF, Li M, Thorleifsson G, Newton-Cheh C, Musunuru K, Pirruccello JP, Saleheen D, Chen L, Stewart A, Schillert A, Thorsteinsdottir U, Thorgeirsson G, Anand S, Engert JC, Morgan T, Spertus J, Stoll M, Berger K, Martinelli N, Girelli D, McKeown PP, Patterson CC, Epstein SE, Devaney J, Burnett MS, Mooser V, Ripatti S, Surakka I, Nieminen MS, Sinisalo J, Lokki ML, Perola M, Havulinna A, de Faire U, Gigante B, Ingelsson E, Zeller T, Wild P, de Bakker PI, Klungel OH, Maitland-van der Zee AH, Peters BJ, de Boer A, Grobbee DE, Kamphuisen PW, Deneer VH, Elbers CC, Onland-Moret NC, Hofker MH, Wijmenga C, Verschuren WM, Boer JM, van der Schouw YT, Rasheed A, Frossard P, Demissie S, Willer C, Do R, Ordovas JM, Abecasis GR, Boehnke M, Mohlke KL, Daly MJ, Guiducci C, Burtt NP, Surti A, Gonzalez E, Purcell S, Gabriel S, Marrugat J, Peden J, Erdmann J, Diemert P, Willenborg C, Konig IR, Fischer M, Hengstenberg C, Ziegler A, Buysschaert I, Lambrechts D, Van de Werf F, Fox KA, El Mokhtari NE, Rubin D, Schrezenmeir J, Schreiber S, Schafer A, Danesh J, Blankenberg S, Roberts R, McPherson R, Watkins H, Hall AS, Overvad K, Rimm E, Boerwinkle E, Tybjaerg-Hansen A, Cupples LA, Reilly MP, Melander O, Mannucci PM, Ardissino D, Siscovick D, Elosua R, Stefansson K, O'Donnell CJ, Salomaa V, Rader DJ, Peltonen L, Schwartz SM, Altshuler D, Kathiresan S. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–580. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karalis I, Rensen PC, Jukema JW. Journey through cholesteryl ester transfer protein inhibition: from bench to bedside. Circ Cardiovasc Qual Outcomes. 2013;6:360–366. doi: 10.1161/CIRCOUTCOMES.111.000014. [DOI] [PubMed] [Google Scholar]
- 11.Barter PJ, Rye KA. Cholesteryl ester transfer protein inhibition as a strategy to reduce cardiovascular risk. J Lipid Res. 2012;53:1755–1766. doi: 10.1194/jlr.R024075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plump AS, Masucci-Magoulas L, Bruce C, Bisgaier CL, Breslow JL, Tall AR. Increased atherosclerosis in ApoE and LDL receptor gene knock-out mice as a result of human cholesteryl ester transfer protein transgene expression. Arterioscler Thromb Vasc Biol. 1999;19:1105–1110. doi: 10.1161/01.atv.19.4.1105. [DOI] [PubMed] [Google Scholar]
- 13.Westerterp M, van der Hoogt CC, de Haan W, Offerman EH, Dallinga-Thie GM, Jukema JW, Havekes LM, Rensen PC. Cholesteryl ester transfer protein decreases high-density lipoprotein and severely aggravates atherosclerosis in APOE*3-Leiden mice. Arterioscler Thromb Vasc Biol. 2006;26:2552–2559. doi: 10.1161/01.ATV.0000243925.65265.3c. [DOI] [PubMed] [Google Scholar]
- 14.Sugano M, Makino N, Sawada S, Otsuka S, Watanabe M, Okamoto H, Kamada M, Mizushima A. Effect of antisense oligonucleotides against cholesteryl ester transfer protein on the development of atherosclerosis in cholesterol-fed rabbits. J Biol Chem. 1998;273:5033–5036. doi: 10.1074/jbc.273.9.5033. [DOI] [PubMed] [Google Scholar]
- 15.Okamoto H, Yonemori F, Wakitani K, Minowa T, Maeda K, Shinkai H. A cholesteryl ester transfer protein inhibitor attenuates atherosclerosis in rabbits. Nature. 2000;406:203–207. doi: 10.1038/35018119. [DOI] [PubMed] [Google Scholar]
- 16.Rittershaus CW, Miller DP, Thomas LJ, Picard MD, Honan CM, Emmett CD, Pettey CL, Adari H, Hammond RA, Beattie DT, Callow AD, Marsh HC, Ryan US. Vaccine-induced antibodies inhibit CETP activity in vivo and reduce aortic lesions in a rabbit model of atherosclerosis. Arterioscler Thromb Vasc Biol. 2000;20:2106–2112. doi: 10.1161/01.atv.20.9.2106. [DOI] [PubMed] [Google Scholar]
- 17.Huang Z, Inazu A, Nohara A, Higashikata T, Mabuchi H. Cholesteryl ester transfer protein inhibitor (JTT-705) and the development of atherosclerosis in rabbits with severe hypercholesterolaemia. Clin Sci (Lond) 2002;103:587–594. doi: 10.1042/cs1030587. [DOI] [PubMed] [Google Scholar]
- 18.Morehouse LA, Sugarman ED, Bourassa PA, Sand TM, Zimetti F, Gao F, Rothblat GH, Milici AJ. Inhibition of CETP activity by torcetrapib reduces susceptibility to diet-induced atherosclerosis in New Zealand White rabbits. J Lipid Res. 2007;48:1263–1272. doi: 10.1194/jlr.M600332-JLR200. [DOI] [PubMed] [Google Scholar]
- 19.de Haan W, de Vries-van der Weij J, van der Hoorn JW, Gautier T, van der Hoogt CC, Westerterp M, Romijn JA, Jukema JW, Havekes LM, Princen HM, Rensen PC. Torcetrapib does not reduce atherosclerosis beyond atorvastatin and induces more proinflammatory lesions than atorvastatin. Circulation. 2008;117:2515–2522. doi: 10.1161/CIRCULATIONAHA.107.761965. [DOI] [PubMed] [Google Scholar]
- 20.Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, Lopez-Sendon J, Mosca L, Tardif JC, Waters DD, Shear CL, Revkin JH, Buhr KA, Fisher MR, Tall AR, Brewer B ILLUMINATE Investigators. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–2122. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- 21.Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, Chaitman BR, Holme IM, Kallend D, Leiter LA, Leitersdorf E, McMurray JJ, Mundl H, Nicholls SJ, Shah PK, Tardif JC, Wright RS dal-OUTCOMES Investigators. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367:2089–2099. doi: 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
- 22.Johns DG, Duffy J, Fisher T, Hubbard BK, Forrest MJ. On- and off-target pharmacology of torcetrapib: current understanding and implications for the structure activity relationships (SAR), discovery and development of cholesteryl ester-transfer protein (CETP) inhibitors. Drugs. 2012;72:491–507. doi: 10.2165/11599310-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 23.Bloomfield D, Carlson GL, Sapre A, Tribble D, McKenney JM, Littlejohn TW, 3rd, Sisk CM, Mitchel Y, Pasternak RC. Efficacy and safety of the cholesteryl ester transfer protein inhibitor anacetrapib as monotherapy and coadministered with atorvastatin in dyslipidemic patients. Am Heart J. 2009;157:352–360. doi: 10.1016/j.ahj.2008.09.022. e2. [DOI] [PubMed] [Google Scholar]
- 24.Nicholls SJ, Brewer HB, Kastelein JJ, Krueger KA, Wang MD, Shao M, Hu B, McErlean E, Nissen SE. Effects of the CETP inhibitor evacetrapib administered as monotherapy or in combination with statins on HDL and LDL cholesterol: a randomized controlled trial. JAMA. 2011;306:2099–2109. doi: 10.1001/jama.2011.1649. [DOI] [PubMed] [Google Scholar]
- 25.de Haan W, van der Hoogt CC, Westerterp M, Hoekstra M, Dallinga-Thie GM, Princen HM, Romijn JA, Jukema JW, Havekes LM, Rensen PC. Atorvastatin increases HDL cholesterol by reducing CETP expression in cholesterol-fed APOE*3-Leiden.CETP mice. Atherosclerosis. 2008;197:57–63. doi: 10.1016/j.atherosclerosis.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 26.van der Hoogt CC, de Haan W, Westerterp M, Hoekstra M, Dallinga-Thie GM, Romijn JA, Princen HM, Jukema JW, Havekes LM, Rensen PC. Fenofibrate increases HDL-cholesterol by reducing cholesteryl ester transfer protein expression. J Lipid Res. 2007;48:1763–1771. doi: 10.1194/jlr.M700108-JLR200. [DOI] [PubMed] [Google Scholar]
- 27.van der Hoorn JW, de Haan W, Berbee JF, Havekes LM, Jukema JW, Rensen PC, Princen HM. Niacin increases HDL by reducing hepatic expression and plasma levels of cholesteryl ester transfer protein in APOE*3Leiden.CETP mice. Arterioscler Thromb Vasc Biol. 2008;28:2016–2022. doi: 10.1161/ATVBAHA.108.171363. [DOI] [PubMed] [Google Scholar]
- 28.Kuhnast S, Louwe MC, Heemskerk MM, Pieterman EJ, van Klinken JB, van den Berg SA, Smit JW, Havekes LM, Rensen PC, van der Hoorn JW, Princen HM, Jukema JW. Niacin reduces atherosclerosis development in APOE*3Leiden.CETP mice mainly by reducing nonHDL-cholesterol. PLoS One. 2013;8:e66467. doi: 10.1371/journal.pone.0066467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuhnast S, van der Hoorn JW, van den Hoek AM, Havekes LM, Liau G, Jukema JW, Princen HM. Aliskiren inhibits atherosclerosis development and improves plaque stability in APOE*3Leiden.CETP transgenic mice with or without treatment with atorvastatin. J Hypertens. 2012;30:107–116. doi: 10.1097/HJH.0b013e32834ddd8e. [DOI] [PubMed] [Google Scholar]
- 30.Menard S. Applied Logistic Regression Analysis: Sage University Series on Quantitative Applications in the Social Sciences. Thousand Oaks, CA: Sage; 1995. [Google Scholar]
- 31.Besley DA. A guide to using the collinearity diagnostics. Comp Sci Econ Manage. 1991;4:33–50. [Google Scholar]
- 32.Krishna R, Bergman AJ, Jin B, Fallon M, Cote J, Van Hoydonck P, Laethem T, Gendrano IN, III, Van Dyck K, Hilliard D, Laterza O, Snyder K, Chavez-Eng C, Lutz R, Chen J, Bloomfield DM, De Smet M, Van Bortel LM, Gutierrez M, Al-Huniti N, Dykstra K, Gottesdiener KM, Wagner JA. Multiple-dose pharmacodynamics and pharmacokinetics of anacetrapib, a potent cholesteryl ester transfer protein (CETP) inhibitor, in healthy subjects. Clin Pharmacol Ther. 2008;84:679–683. doi: 10.1038/clpt.2008.109. [DOI] [PubMed] [Google Scholar]
- 33.Krishna R, Anderson MS, Bergman AJ, Jin B, Fallon M, Cote J, Rosko K, Chavez-Eng C, Lutz R, Bloomfield DM, Gutierrez M, Doherty J, Bieberdorf F, Chodakewitz J, Gottesdiener KM, Wagner JA. Effect of the cholesteryl ester transfer protein inhibitor, anacetrapib, on lipoproteins in patients with dyslipidaemia and on 24-h ambulatory blood pressure in healthy individuals: two double-blind, randomised placebo-controlled phase I studies. Lancet. 2007;370:1907–1914. doi: 10.1016/S0140-6736(07)61813-3. [DOI] [PubMed] [Google Scholar]
- 34.Gutstein DE, Krishna R, Johns D, Surks HK, Dansky HM, Shah S, Mitchel YB, Arena J, Wagner JA. Anacetrapib, a novel CETP inhibitor: pursuing a new approach to cardiovascular risk reduction. Clin Pharmacol Ther. 2012;91:109–122. doi: 10.1038/clpt.2011.271. [DOI] [PubMed] [Google Scholar]
- 35.Libby P. Mechanisms of acute coronary syndromes and their implications for therapy. N Engl J Med. 2013;368:2004–2013. doi: 10.1056/NEJMra1216063. [DOI] [PubMed] [Google Scholar]
- 36.Vergeer M, Bots ML, van Leuven SI, Basart DC, Sijbrands EJ, Evans GW, Grobbee DE, Visseren FL, Stalenhoef AF, Stroes ES, Kastelein JJ. Cholesteryl ester transfer protein inhibitor torcetrapib and off-target toxicity: a pooled analysis of the rating atherosclerotic disease change by imaging with a new CETP inhibitor (RADIANCE) trials. Circulation. 2008;118:2515–2522. doi: 10.1161/CIRCULATIONAHA.108.772665. [DOI] [PubMed] [Google Scholar]
- 37.Cannon CP, Shah S, Dansky HM, Davidson M, Brinton EA, Gotto AM, Stepanavage M, Liu SX, Gibbons P, Ashraf TB, Zafarino J, Mitchel Y, Barter P Determining the Efficacy and Tolerability Investigators. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med. 2010;363:2406–2415. doi: 10.1056/NEJMoa1009744. [DOI] [PubMed] [Google Scholar]
- 38.Matsuura F, Wang N, Chen W, Jiang XC, Tall AR. HDL from CETP-deficient subjects shows enhanced ability to promote cholesterol efflux from macrophages in an apoE- and ABCG1-dependent pathway. J Clin Invest. 2006;116:1435–1442. doi: 10.1172/JCI27602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krauss RM, Wojnooski K, Orr J, Geaney JC, Pinto CA, Liu Y, Wagner JA, Luk JM, Johnson-Levonas AO, Anderson MS, Dansky HM. Changes in lipoprotein subfraction concentration and composition in healthy individuals treated with the CETP inhibitor anacetrapib. J Lipid Res. 2012;53:540–547. doi: 10.1194/jlr.M018010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosenson RS, Brewer HB, Jr, Davidson WS, Fayad ZA, Fuster V, Goldstein J, Hellerstein M, Jiang XC, Phillips MC, Rader DJ, Remaley AT, Rothblat GH, Tall AR, Yvan-Charvet L. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 2012;125:1905–1919. doi: 10.1161/CIRCULATIONAHA.111.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ishigami M, Yamashita S, Sakai N, Arai T, Hirano K, Hiraoka H, Kameda-Takemura K, Matsuzawa Y. Large and cholesteryl ester-rich high-density lipoproteins in cholesteryl ester transfer protein (CETP) deficiency cannot protect macrophages from cholesterol accumulation induced by acetylated low-density lipoproteins. J Biochem. 1994;116:257–262. doi: 10.1093/oxfordjournals.jbchem.a124516. [DOI] [PubMed] [Google Scholar]
- 42.Han S, Levoci L, Fischer P, Wang SP, Gagen K, Chen Y, Xie D, Fisher T, Ehrhardt AG, Peier AM, Johns DG. Inhibition of cholesteryl ester transfer protein by anacetrapib does not impair the anti-inflammatory properties of high density lipoprotein. Biochim Biophys Acta. 2013;1831:825–833. doi: 10.1016/j.bbalip.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 43.Yvan-Charvet L, Kling J, Pagler T, Li H, Hubbard B, Fisher T, Sparrow CP, Taggart AK, Tall AR. Cholesterol efflux potential and antiinflammatory properties of high-density lipoprotein after treatment with niacin or anacetrapib. Arterioscler Thromb Vasc Biol. 2010;30:1430–1438. doi: 10.1161/ATVBAHA.110.207142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clinical Trials database. A service of the U.S. National Institutes of Health. Available from http://www.clinicaltrials.gov 9 January 2014.