

Abstract
Influenza virus infections lead to numerous deaths and millions of hospitalizations each year. One challenge facing anti-influenza drug development is the heterogeneity of the circulating influenza viruses, which comprise several strains with variable susceptibility to antiviral drugs. For example, the wild-type (WT) influenza A viruses, such as the seasonal H1N1, tend to be sensitive to antiviral drugs, amantadine and rimantadine, while the S31N mutant viruses, such as the pandemic 2009 H1N1 (H1N1pdm09) and seasonal H3N2, are resistant to this class of drugs. Thus, drugs targeting both WT and the S31N mutant are highly desired. We report our design of a novel class of dual inhibitors along with their ion channel blockage and antiviral activities. The potency of the most active compound 11 in inhibiting WT and the S31N mutant influenza viruses is comparable with that of amantadine in inhibiting WT influenza virus. Solution NMR studies and molecular dynamics (MD) simulations of drug-M2 interactions supported our design hypothesis: namely, the dual inhibitor binds in the WT M2 channel with an aromatic group facing down toward the C-terminus, while the same drug binds in the S31N M2 channel with its aromatic group facing up toward the N-terminus. The flip-flop mode of drug binding correlates with the structure–activity relationship (SAR) and has paved the way for the next round of rational design of broad-spectrum antiviral drugs.
INTRODUCTION
Influenza virus infection poses a global health and economic challenge that has yet to be addressed.1 During an annual influenza season, an estimate of 35 000 people die due to influenza-related illnesses, which places influenza among top 10 leading causes of death in the U.S.2,3 What is more alarming is the emergence of highly pathogenic avian influenza (HPAI) strains, such as H5N1,4 and more recently the H7N9,5 which have much higher mortality rate than seasonal influenza strains. It has been shown that these HPAI strains need to acquire only one or a few mutations to become transmissible among humans, which raises the likelihood of the next influenza pandemic.6 There are currently two classes of approved anti-influenza drugs: M2 channel blockers (amantadine and rimantadine) and neuraminidase inhibitors (oseltamivir and zanamivir).7 Resistance to both classes of drugs raises great concern: resistance to M2 inhibitors is so prevalent that the Centers for Disease Control and Prevention (CDC) recommended discontinued use of this class of drugs, and resistance to the only orally bioavailable drug oseltamivir was dominant in the 2007–2008 influenza season.8,9 This leaves zanamivir as the last resort of treatment; however, the low bioavailability and its nasal route of administration limit its use in severely ill patients.10 Thus, there is a great need for next generation of orally bioavailable antiviral drugs.11 One challenge facing anti-influenza drug development is the heterogeneous makeup of the circulating influenza viruses, which comprise several influenza strains with different susceptibility to antiviral drugs. For example, among the influenza viruses in recent influenza seasons, the H1N1pdm09 and seasonal H3N2 strains are oseltamivir-susceptible and amantadine-resistant, while the seasonal H1N1 strains are mostly oseltamivir-resistant and amantadine-susceptible.12,13 Moreover, influenza viruses continue to evolve, and it is almost impossible to predict the drug susceptibility of a novel influenza strain.14,15 As an illustration, the H5N1 strains isolated from Vietnam, Thailand, and Cambodia have the characteristic S31N mutation, which confers amantadine resistance.16 However, strains isolated from other countries, such as China, Indonesia, Japan, and Korea, mostly carry the WT M2 and remain susceptible to amantadine. From the drug discovery standpoint, it would be ideal to develop broad-spectrum antiviral drugs that are active against multiple influenza virus strains, thus, circumventing the need of combination therapy which often has drug-drug interaction-related issues.17 Herein, we focus on M2 as a drug target and report the design of a novel class of M2 channel blockers that are active against both the WT and the S31N mutant.
Influenza A virus M2 protein (A/M2) is a virus-encoded proton channel that plays multiple roles during the viral replication cycle: in the early stage of virus uncoating, M2 facilitates unpacking of viral RNAs by acidifying the viral interior; in the late stage of viral replication, M2 equilibrates the pH across the Golgi apparatus in order to prevent premature conformational changes of another viral surface protein-hemagglutinin.18 M2 is a validated drug target of antiviral drugs, amantadine and rimantadine. However, mutations surrounding the drug binding site, such as S31N, V27A, A30T, and L26F, lead to escape of drug inhibition.19 In cell culture, a large number of drug-resistant M2 mutants readily emerged under amantadine drug selection pressure.20–22 A subset of these mutations was also observed in influenza-infected patients following treatment with amantadine.23 Reverse-engineered viruses harboring various pore-lining mutations were competent to replicate in mice,24 raising concerns that M2 might not be an ideal antiviral target. However, electrophysiological investigations showed that majority of the resulting amantadine-resistant M2 channels had shifts in their conductance characteristics that might render the viruses less fit.25,26 This is probably because even minor changes of one amino acid side chain lead to four changes in M2’s 4-fold symmetric homotetrameric pore. Indeed, many of these M2 mutations gave somewhat attenuated viruses that have a tendency to revert to the WT M2 in the absence of drug pressure and do not appear to be highly transmissible.22,27 Corroborating observations in cell cultures, large-scale sequencing of transmissible viruses from 1918 to 2008 have identified only three major amantadine-resistant M2 mutants, S31N, V27A, and L26F.28,29 Among these three drug resistant mutants, S31N is the predominant mutant and persists in more than 95% of currently circulating influenza viruses. Drug discovery efforts targeting these M2 mutants have long been hampered because of the lack of high-resolution structures, the constricted drug binding site, and the dynamic nature of this protein.30 Nevertheless, progress has been made in designing inhibitors targeting the drug-resistant mutants of M2 guided by MD simulations31–36 and NMR structures M2.37–40 The designed small molecule channel blockers inhibit V27A, L26F, and S31N with potencies greater than that of amantadine in inhibiting WT M2 in both electrophysiological assays and antiviral plaque reduction assays. These inhibitors not only represent excellent leads for further development, but also help stabilize the otherwise dynamic M2 protein, leading to the determination of the first high resolution solution NMR structure of the A/M2-S31N mutant.36 Herein, we describe the extension of these studies to design dual inhibitors targeting both WT and the S31N mutant. Such broadly specific antiviral drugs are superior to mutant-selective inhibitors: ideally, a single drug will be sufficient to combat multiple circulating influenza virus strains, thereby alleviating the need of combination therapy as required when treating human immunodeficiency virus (HIV) infections.41
MATERIALS AND METHODS
Materials
All starting materials for compound and peptide synthesis were purchased from commercial vendors and used without purification, unless otherwise stated. Reactions were carried out using HPLC grade solvents under N2 atmosphere. Compounds were purified by silica gel flash column chromatography and characterized by ESI-MS, 1H NMR, and 13C NMR.
Inhibitor Synthesis
Details about inhibitor synthesis procedures and characterization can be found in the Supporting Information.
Two-Electrode Voltage Clamp (TEVC) Assay
All compounds were initially tested in TEVC assay using Xenopus laevis frog oocytes microinjected with RNA expressing either the WT or the S31N mutant of M2 as described in a previous report.32 The potency of the inhibitors was expressed as the percentage inhibition of A/M2 current observed after 2 min of incubation with 100 μM compounds, and potent inhibitors were further tested at series dilutions to quantify their IC50 values.
Plaque Reduction Assay
The plaque reduction assays were performed according to a previou s report.35 WT M2-expressing A/Udorn/72 and M2-S31N-expressing A/WSN/33 were used to infect MDCK cells in the presence or absence of compounds to evaluate their antiviral activity. The EC50s were subsequently determined for potent dual inhibitors.
Molecular Dynamics (MD) Simulations
We used the 22–46 transmembrane segment of the tetrameric M2 protein (Udorn sequence) (M2-TM), modeled after the intermediate-pH structure of the 25–46 segments,42 with two histidines doubly protonated and the other two in the neutral state. We embedded each complex with compound 11 in an 8 × 8 nm2 1-palmitoyl-2-oleoylphospha-tidylcholine (POPC) bilayer, hydrated by a 150 mM KCl water solution. We used the CHARMM36,43,44 CGenFF,45 and TIP3P46 force fields for the treatment of M2-TM and lipids, 11, and water molecules, respectively. We optimized the partial charge of the bromine atom of 11 by MP2 calculations, as detailed in the Supporting Information. 11 was placed in the channel with the bromo-thiophene moiety facing H37 and V27 in the WT M2-TM and S31N mutant, respectively. We performed minimization and initial equilibration of water molecules, as well as relaxation of residues 22–24, with harmonic restraints on the protein backbone and on the bound 11. We gradually released these restraints over 50 ns of simulation followed by a 150 ns unrestrained run.
NMR Sample Preparation
HPLC purified peptides were mixed with deuterated n-dodecylphosphocholine (DPC) (Cambridge Isotope Laboratories) at 1:50 molar ratio in ethanol. Drug was added from the ethanol stock solution to the desired molar ratio. The resulting ethanol solution was mixed by vortexing, and the ethanol was removed by nitrogen purging. The sample was lyophilized overnight to remove residual ethanol. Next, aqueous buffer [10% D2O in H2O, 50 mM sodium phosphate (pH 6.8)] was added to the dried mixture and vortexed for 2 min, and the pH of the final sample was adjusted to the desired value with either NaOH or H3PO4. The final NMR sample conditions are the following: for S31N, 2 mM A/M2-S31N (19–49) (monomer), 20 mM 11, 100 mM deuterated DPC, and 50 mM sodium phosphate buffer in 10% D2O, 90% H2O, pH 6.8; for WT, 2 mM A/M2-WT (19–49) (monomer), 2.5 mM 11, 100 mM deuterated DPC (Cambridge Isotope Laboratories), and 50 mM sodium phosphate buffer in 10% D2O, 90% H2O, pH 7.5.
Peptide Synthesis and Purification
Isotopically labeled peptides used in this study, A/M2-WT (19–49) VASIGH and A/M2-S31N (19–49) VANIG, were synthesized using the optimized solid phase synthesis protocol as described previously.47 Peptides were purified by high performance liquid chromatography (HPLC) with a C4-Vydac reverse phase column. The purity and identify of the peptides were confirmed by analytical HPLC (>98% purity) and ESI-MS. For A/M2-WT (19–49) VASIGH, calcd MS: 1539.3 (+2), 1026.5 (+3). Obsd MS: 1539.3 (+2), 1026.5 (+3). For A/M2-S31N (19–49) VANIG, calcd MS: 1549.3 (+2H), 1033.2 (+3H). Obsd MS: 1549.3 (+2H), 1033.2 (+3H).
Solution NMR Experiments
All spectra were recorded with standard pulse sequences48,49 at 313 K on a Bruker 800 or 900 MHz spectrometers equipped with cryogenic probes. To obtain assignments, 2D 15N and 13C HSQC for both WT (19–49) VASIGH and S31N (19–49) VANIG M2 in the absence/presence of 11, and 2D 13C–(13C)–1H TOCSY experiments for both M2 samples in the presence of 11 were carried out. The 2D 13C–(13C)–1H TOCSY (70 ms) experiments were recorded with t1,max = 7 ms for the 13C dimension and t2,max = 81 ms for the 1H dimension with 32 scans. 2D 13C–(1H)–1H NOESY experiments with mixing time of 150 ms were carried out with t1,max = 7 ms for the 13C dimension and t2,max = 71 ms for the 1H dimension, 128 scans. The 1H carrier frequency was set to the water signal. Chemical shifts were referenced with respect to the residual water peak at 4.63 ppm, and 13C and 15N chemical shifts were referenced indirectly via gyromagnetic ratios. All spectra were processed and analyzed using the nmrPipe program.50 The time domain data of indirect 13C or 15N dimensions were extended once by linear prediction. The time domain data were multiplied by sine square bell window functions shifted by 90° and zero-filled once before Fourier transformation.
NOE Assignment and Model Calculation
The 2D 13C-edited NOESY allows observation of NOEs from the protons that are directly attached to the 13C atoms to any protons that are within approximately 5 Å, so both peptide–drug and peptide–peptide NOEs are present in a given experiment. Also, although the drug is not 13C labeled, intramolecular NOEs originated from its protons attached to 13C at natural abundance (1.1%) can be observed due to the fact that the drug is in large molar excess and the free drug has favorable relaxation properties. Thus, to assign peptide–drug NOEs we first focused on the isolated NOE peaks that could be assigned unambiguously; an intermediate model was generated once we were able to determine the drug orientation. With aid of the model, we continued to assign additional peptide–drug NOE peaks, even in overlapping areas, based on the network anchoring method.51–53 Note, however, that ambiguous NOEs have to be supported by other NOEs systematically in this procedure. Models generated using sparse NOEs could be misleading. The 1H–1H upper distance constraints between drug and peptide for structure modeling were extracted from the NOESY spectra and listed in Supporting Information Table S3. Models were calculated with those peptide–drug NOEs in addition to distance constraints and backbone dihedral angle constraints from the S31N-WJ332 (2) structure.36 The model structures were computed using XPLOR-NIH.54
RESULTS AND DISCUSSION
Rational Design of Dual Inhibitors Targeting WT and S31N
Previous SAR, structural studies, and MD simulations of drug–M2 interactions revealed a common mechanism of drug action: potent M2 inhibitors, regardless of whether they target WT, S31N, V27A, or L26F, all contain a positively charged ammonium, which presumably serves as a mimic of the conducting hydronium ion that forms water-mediated hydrogen bonds with backbone carbonyls of M2.36 In addition, channel blockers bind to M2 through hydrophobic interactions and 3-dimensional-shaped (3D-shaped) scaffolds are generally preferred over planar molecules.55,56,31,57 NMR structural studies of a high-affinity inhibitor of S31N revealed that this inhibitor bound to S31N in a different orientation than previously characterized inhibitors of the WT protein: drugs like amantadine (1) and rimantadine bind to WT M2 channel with their positively charged ammonium facing downward H37;47 while M2WJ332 (2) bound to the S31N mutant in the reverted orientation with their ammonium facing upward V27.35,36,58 The orientations of amantadine (1) and rimantadine in WT, and M2WJ332 (2) in M2-S31N, were modeled by MD simulations, and the orientations were confirmed by solution and solid-state NMR spectroscopies, and further corroborated by SAR studies.36,47,58 The observed drug-flipping phenomenon provides a rationale for design of dual inhibitors that target both WT and S31N in opposite orientations.
To design such dual inhibitors, we started with our earlier reported inhibitors that had moderate inhibition against both WT and S31N (Table 1).
Table 1.
Dual Inhibitors with Moderate Inhibition against Both WT and S31Na
| Substitution | Compound ID | %WT Inhibition* | %S31N Inhibition* | |
|---|---|---|---|---|

|
X = C, R = CH3, |
M2WJ369 (3) | 55 | 49 |
| X = N, R = CH3, |
M2WJ405 (4) | 41 | 60 | |
| X = N, R = H, |
M2WJ396 (5) | 60 | 52 |
Asterisk indicates that the values represent the mean of three independent measurements in TEVC assays. We typically see no more than 5% variation in the % inhibition on a given day, or 10% error for measurements made on different days with different batches of oocytes. All compounds were tested at 100 μM.
From the previous SAR studies,36 we noted that, as the R substitution on the five-membered heterocycles gets bulkier, the WT inhibition of the corresponding compounds decreases drastically. Therefore, to maintain the WT activity, we decided to examine compounds with small R substitutions (e.g., halide, methyl, methoxy). It is also known that a variety of hydrophobic scaffolds are tolerated for WT inhibition,31,57,59 while adamantane is the preferred moiety for S31N inhibition.35,36 Thus, we chose adamantane as the hydrophobic scaffold in order to retain S31N activity. With these two criteria in mind, we devised a general structure for the dual inhibitors as shown in Scheme 1. Diversity was introduced at the five-membered heterocycle rings. The linker between adamantane and the heterocycle was kept constant as an ammonium methylene, as it has been shown to be critical for S31N inhibition. On the basis of the binding mode of M2WJ332 (2) in S31N mutant as shown in Figure 1, the R group was expected to form hydrophobic interactions with the V27 side chain methyls.
Scheme 1.

Design of Dual Inhibitors Targeting Both WT and S31N
Figure 1.

Drug binding orientations in M2-WT and M2-S31N channels. (A) Solid state NMR structure of amantadine-bound WT A/M2 (PDB: 2KQT). Amantadine binds in the channel with its positively charged ammonium facing downward the C-terminal H37. (B) The solution NMR structure of M2WJ332 (2)-bound A/M2-S31N (PDB: 2LY0). M2WJ332 (2) binds with its positively charged ammonium facing upward the N-terminal V27. (C) Chemical structures of amantadine (1) and M2WJ332 (2).
Synthesis and Electrophysiological Testing of Designed Dual Inhibitors
The synthesis of the designed compounds is shown in the Supporting Information. Compounds were synthesized with highly efficient reductive amination and alkylation as described earlier.35,36 All compounds were initially tested in two-electrode voltage clamp assays at 100 μM concentration (Table 2). For the potent compounds, IC50’s were subsequently determined. As discussed previously,32,60 these measurements are made prior to the establishment of equilibrium due to very slow on and off rates for entry of the bulky drugs into the column and the problems of maintaining cells at low pH for extended periods. Hence the IC50 values determined by this procedure are significantly higher than the true binding constants. The most potent compound from the five-membered heterocyclic series was N-[(5-bromothiophen-2-yl)methyl]adamantan-1-amine (11), which showed 76% and 77% inhibition against S31N and WT M2, respectively. In comparison, the bromo-substituted thiazole and isoxazole analogues were less active (11 versus 8, 9, and 10). Chloro-substituted thiazole (6) had moderate activities against both WT and S31N. Interestingly, the chloro-substituted 1,2,4-thiadiazole analogue (7) showed improved potency against S31N, and retained its WT inhibition in comparison to 6. Methyl-substituted compounds were in general less active than their bromo- and chloro-substituted counterparts (15 versus 6 and 8). Methyl substitution slightly increased S31N inhibition, but decreased WT inhibition in the case of 1,2,4-isoxazole (21 versus 12). Compounds with hydrophilic amine substitution (16) and bulky tert-butyl substitution (24) were inactive against both WT and S31N.
Table 2.
Initial Screening of Dual Inhibitors Using the TEVC Assaysa
| R | Compound ID | %S31N inhibition* | % WT inhibition* | |
|---|---|---|---|---|
| Amantadine | 1 | 36 | 91 | |
|
|
|
6 | 42 | 56 |
|
|
7 | 64 | 54 | |
|
|
8 | 47 | 28 | |
|
|
9 | 38 | 36 | |
|
|
10 | 38 | 24 | |
|
|
11 | 76 | 77 | |
|
|
12 | 52 | 60 | |
|
|
13 | 6 | 66 | |
|
|
14 | 37 | 77 | |
|
|
15 | 28 | 22 | |
|
|
16 | 25 | 38 | |
|
|
17 | 36 | 57 | |
|
|
18 | 16 | 23 | |
|
|
19 | 49 | 55 | |
|
|
20 | 28 | 13 | |
|
|
21 | 60 | 41 | |
|
|
22 | 39 | 8 | |
|
|
23 | 33 | 8 | |
|
|
24 | 38 | 5 |
Asterisk indicates the values that represent the mean of three independent measurements in TEVC assays. We typically see no more than 5% variation in the % inhibition on a given day, or 10% error for measurements made on different days with different batches of oocytes. All compounds were tested at 100 μM.
Because the bromo-substituted thiophene 11 had the highest dual inhibition against both WT and S31N, we decided to further explore the SAR. As shown in Table 3, removal of the bromo substitution resulted in drastic decrease of S31N inhibition (11 versus 25), presumably due to the lack of favorable hydrophobic interactions with the V27 side chains. However, removal of the bromo substitution had no effect on the WT inhibition (11 versus 25). Other halides such as chloro and iodo were similarly tolerated in the same position, and the resulting compounds retained potent inhibition against both WT and S31N (11 versus 26 and 27). All potent compounds were also tested in plaque reduction assays. The EC50s of 5-halide-substituted adamantyl thiophene amines (11, 26, and 27) all fell in the single digit low micromolar range. Methyl-substituted thiophene (28) was 2-fold less active than its bromo analogue (11) in inhibiting both WT and S31N. Methoxy substitution at the same position also resulted in a less active compound (29). Compounds with ethyl (30) and cyclopropyl (31) substitutions showed drastic decrease in WT inhibition, while having little or no effect on their S31N inhibition. Collectively, the SAR correlates with the design rationale in which a bulky R group is not tolerated in WT inhibition. Moving the substitutions from the 5-position of the thiophene to the 4-position (32) or the 3-position (33) led to the loss of activity against both WT and S31N, suggesting the binding geometry of the drug is important. Introducing an extra methyl at the 4-position also decreased the activity (11 versus 34). Cellular cytotoxicity of the active dual inhibitors, 11, 26, 27, 28, and 29, were profiled using the 3-(4, 5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) assay.61 The CC50 values were in the range 145–196 μM. The selectivity index for the most potent dual inhibitors, 11 and 27, were >37, rendering them as promising candidates to be further pursued in the following in vivo mouse studies.
Table 3.
Effect of R Substitution on the Potency of Thiophene Inhibitorsa
| R | Compound ID | %S31N inhibition* | % WT(S31) inhibition* | A/WSN/33 (S3 IN) EC50 (μM) | A/Udorn/72 (S31) EC50 (μM) | CC50** (μM) | |
|---|---|---|---|---|---|---|---|
| Amantadine | 36 | 91 | 22.5 | 0.3 | >300 | ||

|
H | 25 | 29/N.T. | 78/N.T. | N.T. | N.T. | N.T. |
| Br | 11 | 76/N.T. | 77/N.T. | 1.8 | 4.6 | 123 | |
| CI | 26 | 72/N.T. | 91/N.T. | 3.5 | 3.8 | 133 | |
| 1 | 27 | 85/69 | 91/72 | 1.9 | 4.0 | 70 | |
| CH3 | 28 | 73/N.T. | 84/N.T. | 3.3 | 9.6 | 139 | |
| OCH3 | 29 | 68/N.T. | 86/N.T. | 3.9 | 6.8 | 186 | |
|
|
30 | 70/N.T. | 48/N.T. | N.T. | N.T. | N.T. | |
|
|
31 | 77/N.T. | 7/N.T. | N.T. | N.T. | N.T. | |

|

|
32 | 45/N.T. | 58/N.T. | N.T. | N.T. | N.T. |
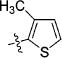
|
33 | 21/N.T. | 71/N.T. | N.T. | N.T. | N.T. | |

|
34 | 54/N.T. | 52/N.T. | N.T. | N.T. | N.T. |
One asterisk indicates the values represent the mean of three independent measurements in TEVC assays. We typically see no more than 5% variation in the percent inhibition on a given day, or 10% error for measurements made on different days with different batches of oocytes. All compounds were initially tested at 100 μM. The compounds that showed >80% inhibition at 100 μM were further tested at 30 μM. The data are presented as % inhibition at 100 μM/% inhibition at 30 μM. N.T. = not tested. Two asterisks indicate CC50 was measured using confluent monolayer Madin–Darby canine kidney (MDCK) epithelial cells. Cell viability was quantified after 72 h by MTT.61
NMR Studies of the Dual Inhibitor 11 Binding to WT and S31N M2
Our design was based on the idea that dual inhibitors bind to WT and S31N in opposite orientations. To experimentally validate this hypothesis of flip-flop drug binding, we recorded solution NMR NOESY spectra to determine drug-M2-TM interactions. Synthetic peptides with 15N–13C–labeled amino acids at selected positions were used for this study in order to simplify the spectrum and obtain unambiguous drug-M2-TM NOEs. The channel-forming transmembrane peptide corresponding to the pore of WT M2 is denoted WT (19–49) VASIGH, meaning residues at V27, A30, S31, I33, G34, and H37 are uniformly 15N–13C–labeled. For the S31N (19–49) VANIG peptide, residues at V27, A30, N31, I33, and G34 are uniformly 15N–13C–labeled. The chemical shifts for the complexes of WT VASIGH and S31N VANIG in the presence of 11 were very similar to those observed for the amantadine-bound WT M2 TM47 and the M2WJ332 (2)-bound S31N,36 respectively. Assignments were obtained by direct comparison and confirmed by 2D C(C)H-TOCSY, and assignments of I33 and V27 are labeled in Supporting Information Figure S3. Two-dimensional 13C-edited NOESY experiments with 150 ms mixing time were used to identify peptide–drug NOEs for S31N (19–49) VANIG peptide (Figure 2A) and the WT (19–49) VASIGH peptide (Figure 2B).
Figure 2.
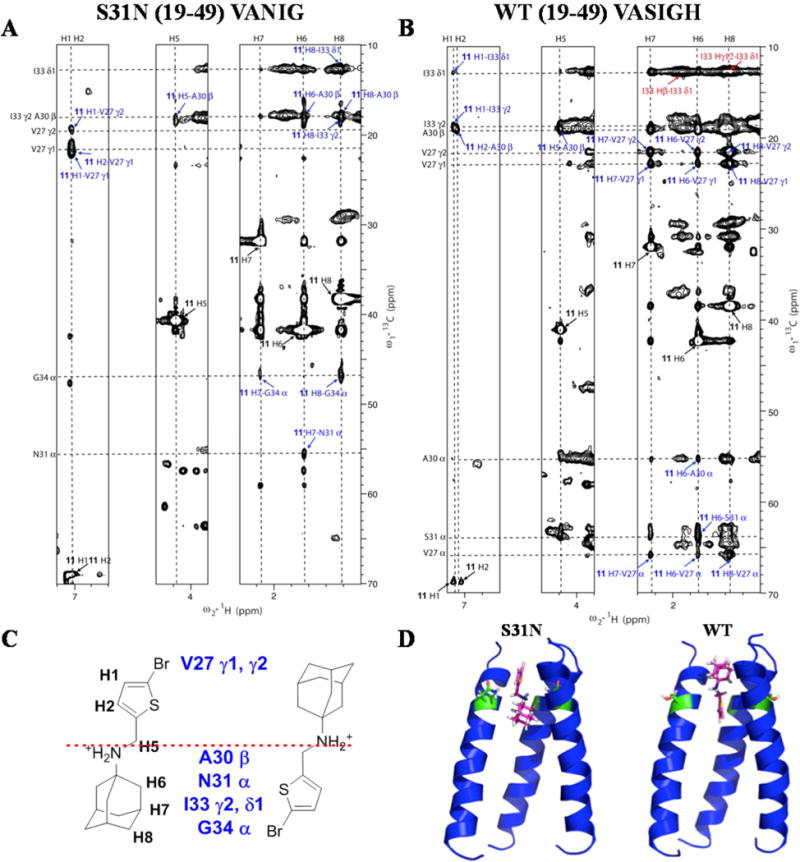
Flip-flop orientation of 11 in M2. (A) 2D 13C–1H NOESY (150 ms) experiment for S31N (19–49) VANIG sample in the presence of 11. (B) 2D 13C–1H NOESY (150 ms) experiment for WT (19–49) VASIGH sample in the presence of 11. Assignments for drug–peptide cross peaks are labeled in blue and listed in Supporting Information Table S3. Assignments for peptide–peptide cross peaks are labeled in red. The diagonal peaks from the drug are labeled in black. (C) Illustration of relative position of 11 in S31N and WT, based on intermolecular peptide–drug NOEs. (D) Models calculated with those peptide–drug NOEs in addition to distance constraints for the peptide obtained for S31N-WJ332 structure. Left is for S31N, and right is for WT in both parts C and D. Positions of the dual inhibitor 11 in S31N and WT are flipped and mirrored along residue 31, providing a direction to design an inhibitor for all M2 mutants. Models generated using the drug-M2 NOEs were deposited in PDB with codes of 2MUW and 2MUV for drug-bound WT-M2 and the S31N mutant, respectively.
The assignment of the complex of S31N (19–49) VANIG in complex with 11 (Figure 2A) is similar to that used previously to solve the corresponding complex with M2WJ332 (2).36 NOE peaks between G34 Cα and the drug protons H7 and H8 were assigned straightforwardly because the Cα chemical shift of Gly is very unique and easily identified. Chemical shifts of drug protons H1 and H2 are overlapped, but the two protons have strong NOEs to the V27 methyl groups that do not overlap with any other peaks, clearly showing that the Val27 methyl groups and the thiophene ring of 11 are very close in space. These NOEs unambiguously demonstrate the drug is up in the S31N channel. Next, the isolated peak of N31 Cα and the drug H6 was assigned. Furthermore, additional NOE assignments for peaks that were overlapped with another were assigned on the basis of the network anchoring approach51–53 with aid of the intermediate models.
Similarly, for the WT (19–49) VASIGH sample, we began with the NOE peaks that could be assigned unambiguously (Figure 2B). A cross-peak between I33 methyl groups and the thiophene ring (11 H1 to I33 γ2 and δ1), together with the lack of NOEs to Val27 methyl groups (as were the case for S31N), place the aromatic group pointing down into the channel. Additionally, NOEs between V27 methyl groups and the adamantane protons H6, H7, and H8 indicate that the adamantane is locating near the N-terminus. At this stage, we generated an intermediate model, which aided the assignment of additional peptide–drug NOEs. From the adamantane protons (H6, H7, and H8), we assigned NOEs to the Cα atoms of V27, A30, and S31. NOEs were also assigned between the thiophene proton H2 and the methylene protons H5 from the drug 11 and the methyl group of Ala30, respectively.
For the WT (19–49) VASIGH sample, we also observed a series of NOEs from the I33δ1 methyl and a number of aliphatic protons, near the chemical shifts of adamantane protons (upper right of Figure 2B). We were unable to assign this region of the spectrum, because it is heavily congested with intraresidue NOEs from I33 as well as NOEs to other regions of the protein (the NOEs seen in a 13C-filtered experiment do not differentiate signals from drug versus other protons in the protein). Given the heavy congestion of the spectrum in this region, we could not rule out the possibility of NOEs from drug 11 to I33, which would have been consistent with the drug binding with thiophene in the up orientation. However, the lack of systematic NOEs between the thiophene protons (H1 and H2) and V27 as were seen in the complex with S31N (19–49) VANIG (Figure 2A) together with the presence of NOEs locating the thiophene ring in the channel near Ala30 indicate that 11 is primarily bound with the heteroaryl group pointing down. Thus, while we are unable to rule out the possibility of other minor conformers, these data clearly confirm that the predominant orientation for WT is with the adamantane up near Val27 and the thiophene directed downward.
These peptide–drug NOEs, along with the distance constraints and backbone dihedral angle constraints for defining the peptide’s tetrameric structure obtained for the S31N-M2WJ332 (2) structure,36 were used to build the models using XPLOR-NIH54 as shown in Figure 2D. There were no distance violations (>0.5 Å) for the assigned NOEs.
MD Simulations of the Dual Inhibitor 11 Binding to WT and S31N
We used MD simulations of M2-TM in a phospholipid bilayer to investigate the dual inhibitory behavior adopted by compound 11 against the M2 proton channels (both WT and S31N mutant) (Figure 3). Given the very slow on and off rates of amantadine derivatives (typically on the hour time scale32,60), the extended structure of the inhibitor, and the restricted geometry of the pore, it was necessary to initiate the simulations with the molecule already inserted in the pore in the predominant orientation inferred from solution NMR. In both simulations, the ammonium gradually converges to stable positions along the pore. In the WT M2-TM, we initialized compound 11 to minimize steric clashes: during the first tens of nanoseconds, the ammonium’s position shifts by about 1 Å toward V27 and aligns itself with the backbone carbonyl of G34, which is the native binding mode for amantadine.33,40 The movement of 11 toward its final pose is accompanied by a conformational change of the M2-TM backbone, in the direction toward the low pH structure.62 After 200 ns, the root-mean-square deviation (RMSD) of the backbone atoms from the starting structure42 is 4 Å, whereas the RMSD from the low pH structure62 is 2.5 Å. For comparison, MD simulations of the stable drug-free or inhibitor-bound conformation yield RMSDs of approximately 1 Å, attributable to thermal fluctuations:33,42 therefore, the final structure is similar to, but distinct from, the low pH structure.62 A similar conformational change was also observed in phospholipid bilayers as an effect of mutations of D44.63 Close interactions are formed between the phosphate groups of the lipids with R45, and water molecules between V27 side chains disappear altogether. Within the pore, the bromine atom of 11 forms transient, nonspecific interactions with water molecules, the backbone carbonyl of G34, and the imidazole of H37: this is in agreement with the ranking of the binding energies with these three moieties in quantum mechanical calculations (see Supporting Information).
Figure 3.
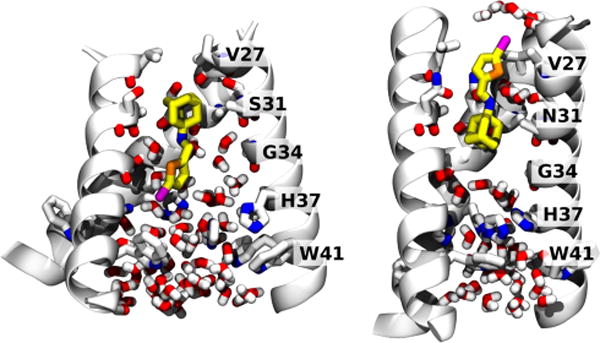
Final MD snapshots of compound 11 bound to the transmembrane segment of WT and S31N M2 in a lipid bilayer. Left: 11 bound within WT M2 after 200 ns. Right: 11 bound within S31N M2 after 100 ns. The protein backbone is shown as ribbons; pore-lining side chains and backbone carbonyls, ligand, and water molecules within the pore are shown as sticks. Water molecules solvating the bromine atom in the outward-facing pose (right) are also shown.
In the S31N mutant, we initialized 11 with the bromo-thiophene moiety facing toward V27, and the ammonium aligned with the side chain of N31, as in the S31N-M2WJ332 (2) structure (PDB: 2LY0).36 This position is maintained throughout the simulation. The V27 side chains are packed tightly around the thiophene group in the starting structure: the Cγ atoms from opposite monomers are separated by 7.8 Å. During the first 100 ns, the N-terminal residues slightly expand to accommodate the thermal fluctuations of 11 (the separation between opposing Cγs increases to 9.2 Å). Water molecules that are initially positioned around 11 leave the region between V27 and G34, with the exception of two water molecules coordinating the ammonium group, also seen in the previous S31N-M2WJ332 (2) simulations.36 In this binding mode, the bromine atom is surrounded by the V27 side chains, and only partially exposed to the outer solvent: the pair correlation function with water oxygens (see Supporting Information) shows 4 or 5 water molecules weakly interacting with the bromine. The first hydration shell is centered at 3.5 Å of distance from the bromine, while the second appears as an even broader peak centered at 6 Å (integrals at 5 and 8 Å, respectively): both features resemble very closely those of a methyl group at an oil–water interface.64 This mode of interaction is in agreement with the SAR of molecules with bromine substituted by other halogens, as well as a methyl (Table 3).
In summary, the drug binding orientation of dual inhibitor 11 as inferred from solution NOEs are supported by MD simulations, showing the relative orientations were indeed stable within the simulation time.
CONCLUSIONS
The heterogeneity of influenza viruses poses a great challenge in the development of anti-influenza drugs. For effective neutralization of influenza viruses, influenza vaccines normally contain two strains of influenza A viruses, A/California/7/2009 (H1N1)-like virus and A/Texas/50/2012 (H3N2) virus, plus at least one strain of influenza B virus, typically B/Massachusetts/2/2012.65 Although influenza vaccines remain the cornerstone in prophylaxis of influenza infection, they are much less active than small molecule antivirals in treating influenza-infected patients.66 Moreover, due to antigenic shift and antigenic drift of influenza viruses, influenza vaccines have to be reformulated each year; plus, there is generally a six-month delay in vaccine production. The limitations of influenza vaccines together with the heterogeneous makeup of influenza viruses call for broad-spectrum small molecule antivirals. We designed a class of such broad-spectrum dual inhibitors with EC50 values against WT and M2 S31N influenza viruses, which is comparable with that of amantadine (1) in inhibiting WT influenza virus. Our design was based on the hypothesis that dual inhibitors bind to WT and M2-S31N channels with opposite orientations: the aromatic headgroup faces down toward C-termini and up toward N-termini in WT and S31N channel, respectively. Starting with our previously discovered lead compounds showing moderate dual inhibition, we identified halide-substituted thiophene compounds having the highest potencies. The antiviral activity of the most potent inhibitor, 11, in inhibiting WT and the S31N mutant is on par with that of amantadine (1) in inhibiting WT virus. NMR investigations were also consistent with the guiding rationale for inhibitor design. All unambiguous NOEs indicated that the inhibitors bind to the WT and S31N channels in opposite inverted orientations, although we cannot rule out the possibility of other minor conformational forms of the drug in the WT protein due to spectral overlap. The dual inhibitors reported herein provide promising lead compounds for further medicinal chemistry optimization with the ultimate goal of developing broad-spectrum anti-influenza drugs.
Supplementary Material
Acknowledgments
This work is supported by NIH grants GM56423 and AI74571 to W.F.D, AI-20201 to R.A.L. and startup funding from the University of Arizona to J.W.
Footnotes
ASSOCIATED CONTENT
Synthesis procedures, compound characterizations, quantum chemical calculations, and molecular dynamics simulation details. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare the following competing financial interest(s): The work was done in collaboration with InfluMedix, and that W.F.D was a member of their Scientific Advisory Board while part of the work was ongoing.
References
- 1.Cox N, Subbarao K. Annu Rev Med. 2000;51:407. doi: 10.1146/annurev.med.51.1.407. [DOI] [PubMed] [Google Scholar]
- 2.Thompson W, Shay D, Weintraub E, Brammer L, Cox N, Anderson L, Fukuda K. JAMA, J Am Med Assoc. 2003;289:179. doi: 10.1001/jama.289.2.179. [DOI] [PubMed] [Google Scholar]
- 3.Thompson W, Shay D, Weintraub E, Brammer I, Bridges C, Cox N, Fukuda K. JAMA, J Am Med Assoc. 2004;292:1333. doi: 10.1001/jama.292.11.1333. [DOI] [PubMed] [Google Scholar]
- 4.Abdel-Ghafar A, Chotpitayasunondh T, Gao Z, Hayden F, Hien N, de Jong M, Naghdaliyev A, Peiris J, Shindo N, Soeroso S, Uyeki T, Consultation, n. W. H. O. Consultation, n. W. H. O. N. Engl J Med. 2008;358:261. doi: 10.1056/NEJMra0707279. [DOI] [PubMed] [Google Scholar]
- 5.Yu H, Cowling BJ, Feng L, Lau EH, Liao Q, Tsang TK, Peng Z, Wu P, Liu F, Fang VJ, Zhang H, Li M, Zeng L, Xu Z, Li Z, Luo H, Li Q, Feng Z, Cao B, Yang W, Wu JT, Wang Y, Leung GM. Lancet. 2013;382:138. doi: 10.1016/S0140-6736(13)61207-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu R, McBride R, Nycholat C, Paulson J, Wilson I. J Virol. 2012;86:982. doi: 10.1128/JVI.06322-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Das K. J Med Chem. 2012;55:6263. doi: 10.1021/jm300455c. [DOI] [PubMed] [Google Scholar]
- 8.Samson M, Pizzorno A, Abed Y, Boivin G. Antiviral Res. 2013;98:174. doi: 10.1016/j.antiviral.2013.03.014. [DOI] [PubMed] [Google Scholar]
- 9.Cheng P, Leung T, Ho E, Leung P, Ng A, Lai M, Lim W. Emerg Infect Dis. 2009;15:966. doi: 10.3201/eid1506.081357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moscona AN. Engl J Med. 2005;353:1363. doi: 10.1056/NEJMra050740. [DOI] [PubMed] [Google Scholar]
- 11.Boltz D, Aldridge J, Webster R, Govorkova E. Drugs. 2010;70:1349. doi: 10.2165/11537960-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.WHO Guidelines for Pharmacological Management of Pandemic Influenza A(H1N1) 2009 and Other Influenza Viruses. World Health Organization; Geneva: Feb, 2010. Available from http://www.ncbi.nlm.nih.gov/books/NBK138515/ [PubMed] [Google Scholar]
- 13.Ison M. Curr Opin Virol. 2011;1:563. doi: 10.1016/j.coviro.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 14.Tsiodras S, Mooney J, Hatzakilis A. BMJ [Br Med J] 2007;334:293. [Google Scholar]
- 15.Webster RG, Govorkova EA. Ann NY Acad Sci. 2014;1323:115. doi: 10.1111/nyas.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheung C, Rayner J, Smith G, Wang P, Naipospos T, Zhang J, Yuen K, Webster R, Peiris J, Guan Y, Chen H. J Infect Dis. 2006;193:1626. doi: 10.1086/504723. [DOI] [PubMed] [Google Scholar]
- 17.Hayden F. Influenza Other Respir Viruses. 2013;7:63. doi: 10.1111/irv.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pinto L, Lamb RJ. Biol Chem. 2006;281:8997. doi: 10.1074/jbc.R500020200. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Qiu JX, Soto C, DeGrado WF. Curr Opin Struct Biol. 2011;21:68. doi: 10.1016/j.sbi.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hay A, Wolstenholme A, Skehel J, Smith M. EMBO J. 1985;4:3021. doi: 10.1002/j.1460-2075.1985.tb04038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown AN, McSharry JJ, Weng Q, Driebe EM, Engelthaler DM, Sheff K, Keim PS, Nguyen J, Drusano GL. Antimicrob Agents Chemother. 2010;54:3442. doi: 10.1128/AAC.01385-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grambas S, Hay AJ. Virology. 1992;190:11. doi: 10.1016/0042-6822(92)91187-y. [DOI] [PubMed] [Google Scholar]
- 23.Shiraishi K, Mitamura K, Sakai-Tagawa Y, Goto H, Sugaya N, Kawaoka Y. J Infect Dis. 2003;188:57. doi: 10.1086/375799. [DOI] [PubMed] [Google Scholar]
- 24.Abed Y, Goyette N, Boivin G. Antimicrob Agents Chemother. 2005;49:556. doi: 10.1128/AAC.49.2.556-559.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Balannik V, Carnevale V, Fiorin G, Levine B, Lamb R, Klein M, DeGrado W, Pinto L. Biochemistry. 2010;49:696. doi: 10.1021/bi901799k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stouffer AL, Ma C, Cristian L, Ohigashi Y, Lamb RA, Lear JD, Pinto LH, DeGrado WF. Structure. 2008;16:1067. doi: 10.1016/j.str.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suzuki H, Saito R, Masuda H, Oshitani H, Sato M, Sato I. J Infect Chemother. 2003;9:195. doi: 10.1007/s10156-003-0262-6. [DOI] [PubMed] [Google Scholar]
- 28.Furuse Y, Suzuki A, Kamigaki T, Oshitani H. Virol J. 2009;6:67. doi: 10.1186/1743-422X-6-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Furuse Y, Suzuki A, Oshitani H. Antimicrob Agents Chemother. 2009;53:4457. doi: 10.1128/AAC.00650-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Das K, Aramini J, Ma L, Krug R, Arnold E. Nat Struct Mol Biol. 2010;17:530. doi: 10.1038/nsmb.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rey-Carrizo M, Torres E, Ma C, Barniol-Xicota M, Wang J, Wu Y, Naesens L, DeGrado WF, Lamb RA, Pinto LH, Vazquez S. J Med Chem. 2013;56:9265. doi: 10.1021/jm401340p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balannik V, Wang J, Ohigashi Y, Jing X, Magavern E, Lamb RA, DeGrado WF, Pinto LH. Biochemistry. 2009;48:11872. doi: 10.1021/bi9014488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang J, Ma C, Fiorin G, Carnevale V, Wang T, Hu F, Lamb RA, Pinto LH, Hong M, Kein ML, DeGrado WFJ. Am Chem Soc. 2011;133:12834. doi: 10.1021/ja204969m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J, Ma C, Wu Y, Lamb RA, Pinto LH, DeGrado WF. J Am Chem Soc. 2011;133:13844. doi: 10.1021/ja2050666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J, Ma C, Wang J, Jo H, Canturk B, Fiorin G, Pinto LH, Lamb RA, Klein ML, DeGrado WF. J Med Chem. 2013;56:2804. doi: 10.1021/jm301538e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang J, Wu Y, Ma C, Fiorin G, Wang J, Pinto LH, Lamb RA, Klein ML, DeGrado WF. Proc Natl Acad Sci USA. 2013;110:1315. doi: 10.1073/pnas.1216526110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharma M, Yi M, Dong H, Qin H, Peterson E, Busath DD, Zhou HX, Cross TA. Science. 2010;330:509. doi: 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miao Y, Qin H, Fu R, Sharma M, Can TV, Hung I, Luca S, Gor’kov PL, Brey WW, Cross TA. Angew Chem, Int Ed. 2012;51:8383. doi: 10.1002/anie.201204666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andreas LB, Eddy MT, Chou JJ, Griffin RG. J Am Chem Soc. 2012;134:7215. doi: 10.1021/ja3003606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cady SD, Schmidt-Rohr K, Wang J, Soto CS, DeGrado WF, Hong M. Nature. 2010;463:689. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Clercq E. Nat Rev Drug Discovery. 2007;6:1001. doi: 10.1038/nrd2424. [DOI] [PubMed] [Google Scholar]
- 42.Acharya R, Carnevale V, Fiorin G, Levine BG, Polishchuk AL, Balannik V, Samish I, Lamb RA, Pinto LH, DeGrado WF, Klein ML. Proc Natl Acad Sci USA. 2010;107:15075. doi: 10.1073/pnas.1007071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Pastor RW. J Phys Chem B. 2010;114(23):7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.MacKerell AD, Brooks B, Brooks CL, Nilsson L, Roux B, Won Y, Karplus M. CHARMM: The Energy Function and Its Parameterization In Encyclopedia of Computational Chemistry. John Wiley & Sons; New York: 2002. [Google Scholar]
- 45.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, MacKerell AD. J Comput Chem. 2010;31:671. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J Chem Phys. 1983;79:926. [Google Scholar]
- 47.Cady SD, Wang J, Wu Y, DeGrado WF, Hong M. J Am Chem Soc. 2011;133:4274. doi: 10.1021/ja102581n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sattler M, Schleucher J, Griesinger C, Smith ME, Eck ERHv. Heteronuclear Multidimensional NMR Experiments for the Structure Determination of Proteins in Solution Employing Pulsed Field Gradients. Elsevier; Amsterdam: 1999. [Google Scholar]
- 49.Cavanagh J, Fairbrother WJ, Palmer AG, III, Rance M, Skelton NJ. Protein NMR Spectroscopy: Principles and Practice. Academic Press; Amsterdam; Boston: 2007. [Google Scholar]
- 50.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax AJ. Biomol NMR. 1995;6:277. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 51.Nilges M, Macias MJ, O’Donoghue SI, Oschkinat H. J Mol Biol. 1997;269:408. doi: 10.1006/jmbi.1997.1044. [DOI] [PubMed] [Google Scholar]
- 52.Huang YJ, Tejero R, Powers R, Montelione GT. Proteins. 2006;62:587. doi: 10.1002/prot.20820. [DOI] [PubMed] [Google Scholar]
- 53.Herrmann T, Guntert P, Wuthrich K. J Mol Biol. 2002;319:209. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- 54.Schwieters CD, Kuszewski JJ, Tjandra N, Marius Clore G. J Magn Reson. 2003;160:65. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 55.Lagoja IM, De Clercq E. Med Res Rev. 2008;28:1. doi: 10.1002/med.20096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wanka L, Iqbal K, Schreiner P. Chem Rev. 2013;113:3516. doi: 10.1021/cr100264t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Duque MD, Ma C, Torres E, Wang J, Naesens L, Juarez-Jimenez J, Camps P, Javier Luque F, DeGrado WF, Lamb RA, Pinto LH, Vazquez S. J Med Chem. 2011;54:2646. doi: 10.1021/jm101334y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Williams JK, Tietze D, Wang J, Wu Y, DeGrado WF, Hong M. J Am Chem Soc. 2013;135:9885. doi: 10.1021/ja4041412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang J, Ma C, Balannik V, Pinto LH, Lamb RA, DeGrado WF. ACS Med Chem Lett. 2011;2:307. doi: 10.1021/ml100297w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang C, Takeuchi K, Pinto LH, Lamb RA. J Virol. 1993;67:5585. doi: 10.1128/jvi.67.9.5585-5594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Torres E, Duque MD, Vanderlinden E, Ma C, Pinto LH, Camps P, Froeyen M, Vazquez S, Naesens L. Antiviral Res. 2013;99:281. doi: 10.1016/j.antiviral.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. Nature. 2008;451:596. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ma C, Fiorin G, Carnevale V, Wang J, Lamb RA, Klein ML, Wu Y, Pinto LH, DeGrado WF. Structure. 2013;21:2033. doi: 10.1016/j.str.2013.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ashbaugh HS, Paulaitis ME. J Am Chem Soc. 2001;123:10721. doi: 10.1021/ja016324k. [DOI] [PubMed] [Google Scholar]
- 65.Grohskopf L, Shay D, Shimabukuro T, Sokolow L, Keitel W, Bresee J, Cox N. MMWR Recomm Rep. 2013;62:1. [Google Scholar]
- 66.Jin H, Chen Z. Curr Opin Virol. 2014;6:34. doi: 10.1016/j.coviro.2014.02.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.