

Abstract
A facile methodology to prepare water-dispersible nanogels based on pentafluorophenyl acrylate and polyethylene glycol methacrylate random copolymer and diamine cross-linkers has been developed. Cross-linking reaction was characterized by FTIR and 19F NMR. We show that those nanogels : (i) are water-dispersible; (ii) can conveniently encapsulate lipophilic guest molecules; (iii) can be prepared with different nanosizes; (iv) are engineered to allow for surface decoration with additional functional groups.
Keywords: Nanogel synthesis, Activated ester, Amphiphilic polymer assembly, Surface modification
Design and synthesis of water-soluble cross-linked polymer nanoparticles or nanogels that can sequester lipophilic guest molecules within their interiors is of great interest in various applications ranging from delivery vehicles for therapeutics, to diagnostics to theranostics, among others.1-4 However, the classical preparative methods including microemulsion or inverse microemulsion ones do not conveniently allow the nanogels to be water-soluble and encapsulate lipophilic guest molecules simultaneously.5-6 Preparations of polymeric nanoparticles by covalently cross-linking the individual components of a micellar assembly have been reported and have shown great promise for producing stable nano-scopic scaffolds.7-9 Such methods use block-copolymers to form core-shell nanosized assemblies, which are cross-linked with complementary cross-linkers.
Recently, we have introduced an emulsion-free method for the synthesis of cross-linked nanogels in aqueous media that also provides for convenient hydrophobic guest encapsulation.10,11 In this method, we use amphiphilic random copolymers that form a stable self-organized nano-assembly in solutions, which are induced to undergo a self-crosslinking reaction to produce well-controlled nanogels. This self-crosslinking reaction was executed using a dithiopyridyl unit as the reactive moiety, which affords disulfide crosslinks. These disulfide crosslinks are useful for systems that respond to differential redox environments, such as glutathione concentrations that are different in extracellular and intracellular environments by about three orders of magnitude. To further improve the versatility of this approach, it is essential that our method allows for achieving nanogels beyond the disulfide-based crosslinks. The ability to incorporate a variety of functional cross-linkers into nanogels will tremendously facilitate our capacity to orthogonally tailor them with multifunctional cross-linkers. Among reactive functionalities, pentafluorophenyl (PFP) activated polymers with a variety of functional amines have been intensively studied for making well-defined block block copolymers, biofunctionalized polymers, and responsive surface coatings.12-15, 26-27 Although the amidation of PFP is similar to that of other activated esters, such as the N-hydroxy-succinamide (NHS) ester, PFP has relatively higher hydrophobicity, reactive and higher hydrolytic stability providing possibilities of running the reaction in either organic solvent or aqueous solution - all features desired in a functional group for this synthetic method.16,17 In this communication, we introduce a new methodology for crosslinking polymers based on a random copolymer containing PFP moieties and diamino cross-linker molecules. The critical feature that needs to be tested here is whether a set of intermolecular reactions would be as facile and provide as well-defined a nanogel, as the previously reported self-crosslinking reaction. We show here that this indeed can be achieved.
The key feature in our strategy involves the utility of an amphiphilic random copolymer, where the reactive lipophilic functional groups are utilized for crosslinking (Figure 1). In this case, we envisioned the use of pentafluorophenyl moiety as the lipophilic functional group that provides such reactivity. Thus, we prepared random copolymers, represented by P1, containing polyethylene glycol methacrylate (PEGMA) as the hydrophilic unit and the pentafluorophenyl acrylate (PFPA) as the lipophilic unit. We envisaged that addition of a calculated amount of diamine to a solution of the PPFPA-r-PEGMA random copolymer will cause inter- and intra-chain crosslinking amidation reactions to afford the nanogel (Figure 1). In our previous report involving disulfide crosslinks, these crosslinks were simply achieved by adding a reagent that affords a self-crosslinked nanogel product. In the present case, we are executing an intramolecular reaction where the added diamine is integrated to the nanogel product formed. We also envisaged that since amphiphilic assembly can accommodate lipophilic guest molecules, these will be incorporated in the assembly during the crosslinking in aqueous solution. This causes the lipophilic molecules to be encapsulated within the crosslinked interiors of the nanogel. Since we are using only a percentage of the PFP moiety for the crosslinking reaction, we could also potentially use the remaining reactive PFP functionalities for surface functionalization, as illustrated in Figure 1.
Figure 1.
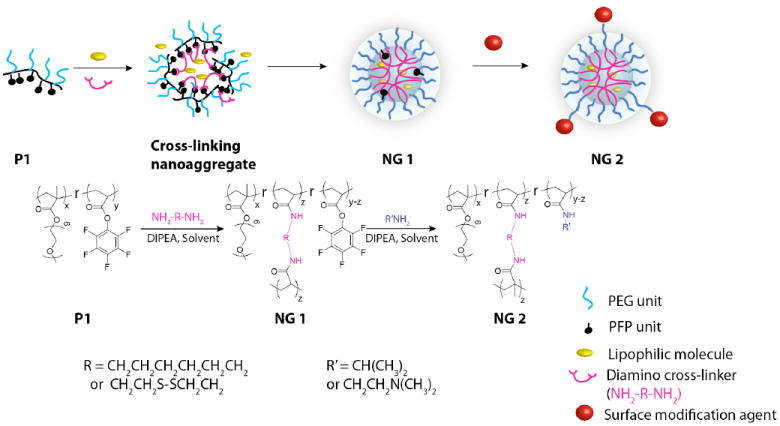
Schematic representation of design and synthesis of the cross-linked polymer nanogels
To test our hypothesis, cross-linked nanogels with 50% and 100% cross-linking density (with respect to the PFP units) were prepared from a PPFPA-r-PEGMA solution (10 mg/mL in THF) by adding a calculated amount of cystamine (CYS) and hexamethylenediamine (HMDA) followed by the addition of water and evaporation of THF. Reaction between the amine the activated ester results in the formation of the amide bond and the concurrent release of the PFP moiety in the form of pentafluorophenol (Figure 2). The cross-linking reaction was thus monitored by following the amidation by FTIR and the release of PFP by 19F NMR. IR spectra of reaction mixtures, listed in Figure 2a, were recorded after heating at 50 °C for 4 hours. We noted that the peak at 1780 cm-1, corresponding to the activated ester C=O stretching, disappeared with the concurrent appearance of the peak at 1640 cm-1 ascribed to the amide C=O stretching. The disappearance of the activated carbonyl group and appearance of the amide carbonyl group designate that the PFP was completely converted to amide when 0.5 eq. of cross-linker with respect to PFP groups was employed. When 0.25 eq. of cross-linker was used, half of the PFP esters were converted to amide. The remaining PFP activated ester C=O signal can still be observed.
Figure 2.
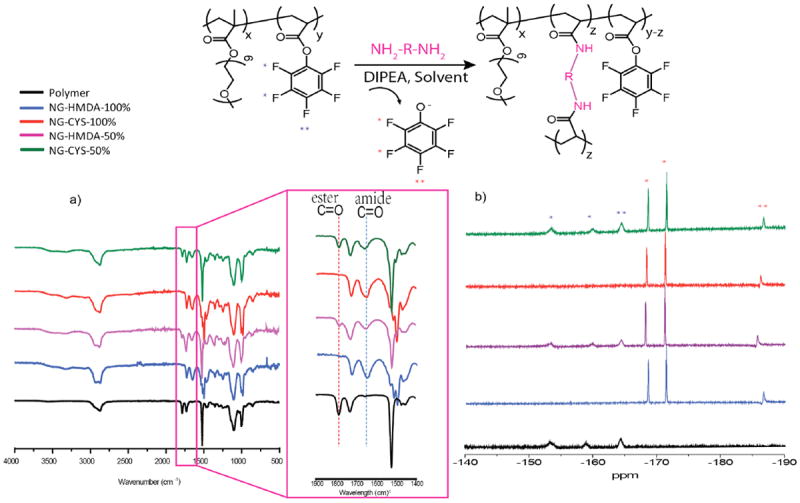
Characterization of cross-linking in the presence of hexamethylenediamine and cystamine: a) amidation monitored by FTIR; b) release of pentafluorophenyl groups tracked by 19F-NMR
To further confirm the cross-linking, the release of PFP was tracked by 19F NMR (shown in Figure 2b). The 19F NMR spectrum of the polymer shows three broad peaks at -153.4, -159.9, and -164.3 ppm. After adding CYS or HMDA cross-linkers, the polymer was cross-linked to affording nanogels, releasing C6F5OH groups, which show sharp signals located at -168.5, -171.4 and - 186.6 ppm. The broad peaks fully disappeared with the addition of 0.5 eq. of cross-linker, suggesting 100% cross-linking density in yielded nanogels. The remaining broad peaks and new sharp peaks are simultaneously observed when 50% of the PFP groups were cross-linked. The dramatic change in chemical shift indicates the conversion from activated ester to amide and hence cross-linking. Additionally, end group analysis by 1H NMR reveals that most of end groups remain intact after cross-linking (Figure S3). The size of the nanogels prepared from a 10 mg/mL polymer solution in THF with CYS were measured by dynamic light scattering (DLS) shown in Figure 3.
Figure 3.
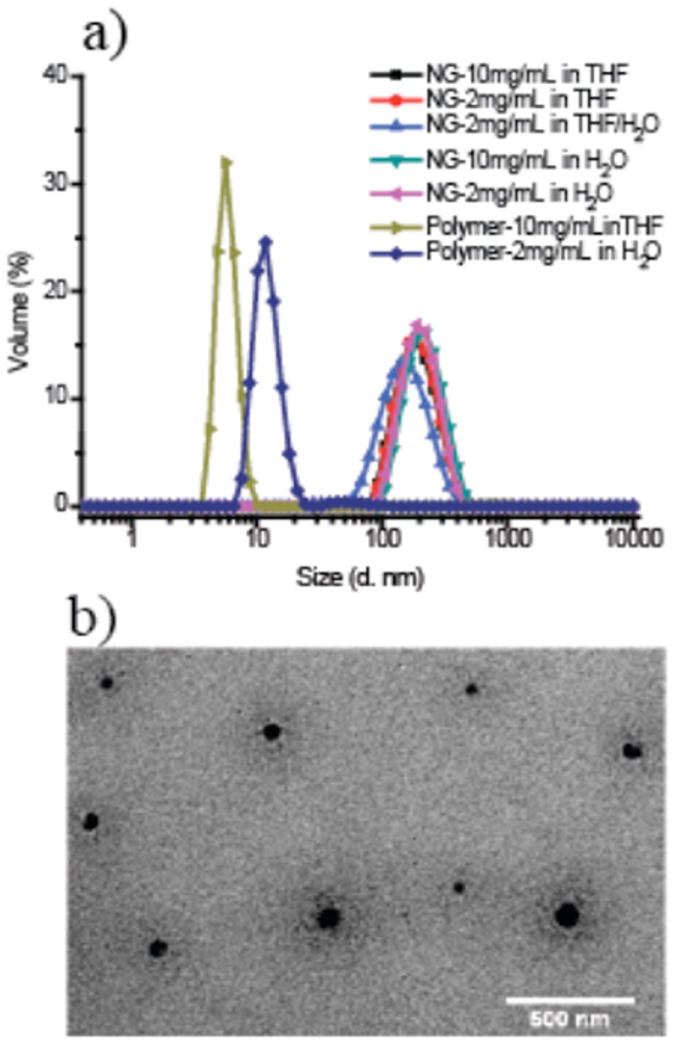
a) Size distributions of nanogel cross-linked by CYS, and then re-dispersed in different solvents with various concentrations; b) TEM image of nanogel; The scale bar is 500 nm
The size of the nanogel dispersion in water was ~180 nm, which is coincident with what was observed from TEM. In order to show that the synthesized nanogels are stable cross-linked networks, rather than the simple aggregation of the polymer, nanogels were re-dispersed in THF and a THF/H2O mixture at various concentrations and were compared. Sizes of the same nanogel dispersed in THF or THF/H2O mixture are similar to that in H2O alone. Also, dilution of the nanogels to 2 mg/mL in THF or H2O does not result in a size change (remains at ~180 nm). However, the same dilution of the polymer aggregates, prior to the crosslinking reaction, reduces the aggregate sizes to ~13 nm. These results suggest that the nanogel is from a covalently crosslinked network and has no concentration and solvent dependency. We observed a similar result from the nanogel cross-linked by HMDA, the details of which are shown in the Supporting Information (Figure S1). Although PPFPA-r-PEGMA dissolved in THF shows peaks at around 10 nm, the poor correlation function is taken to indicate ill-defined aggregation. Since the nanogel sizes are much larger than the aggregate sizes of PPFPA-r-PEGMA in THF, we believe that there is some inter-aggregate crosslinking under these reaction conditions. This is not surprising considering that the crosslinking is based on intermolecular reactions.
A lipophilic guest molecule, 3,3’-dioctadecyl-oxacarbocyanine perchlorate (DiI) was encapsulated in nanogels during cross-linking. During this encapsulation, 1 wt% of DiI with respect to polymer was initially fed. Guest molecule encapsulation was measured by absorbance spectroscopy shown in Figure 4.
Figure 4.
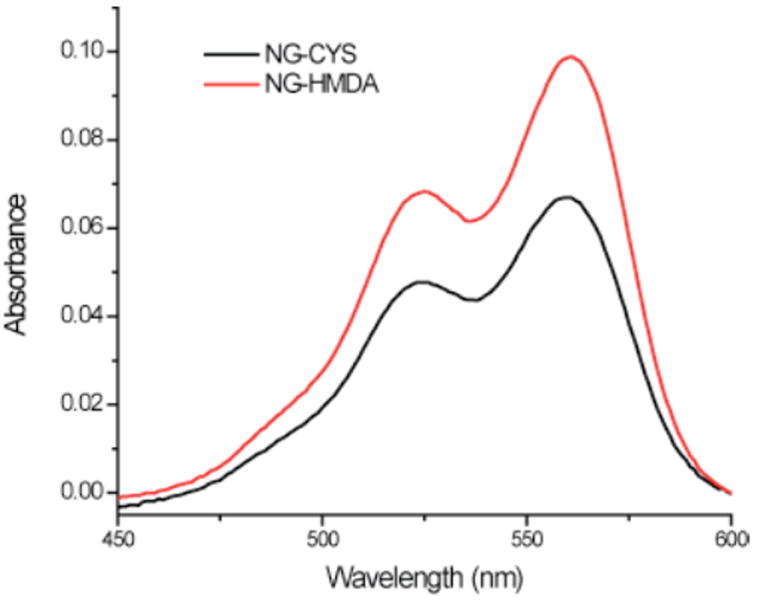
Absorbance of nanogel respectively cross-linked by CYS and HMDA and loaded with DiI. Concentration of both measured nanogel solution is 0.2 mg/mL.
Nanoparticle size is known to play a significant role in several applications. For example, in the case of drug delivery, size has impact on biodistribution, cellular uptake, and permeability in disease sites, thus affecting the therapeutic efficacy.18-20 Based on our previous hypothesis, larger nanogel size is a result of small percentage of inter-aggregate cross-linking in addition to the desired intra-aggregate crosslinking. The inter-aggregate phenomenon is expected to be highly concentration dependent. Hence, the nanogel size would be significantly affected by the original concentration of the PPFPA-r-PEGMA solution. We prepared four nanogels from 2mg/mL, 5mg/mL, 10mg/mL and 20mg/mL PPFPA-PEGMA solution in THF. As shown in Figure 5, the size of the nanogel dispersed in H2O shifts from 100 nm to 200 nm along with the increase in PPFPA-PEGMA concentration.
Figure 5.
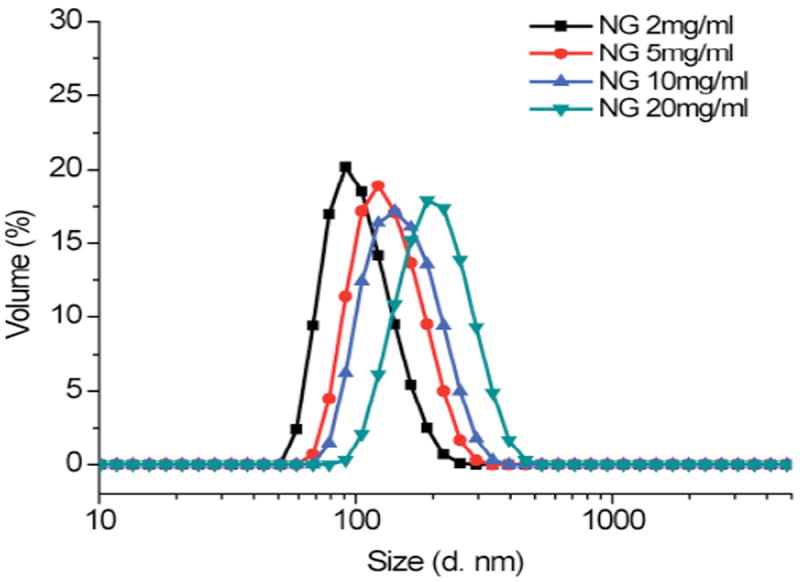
Size of nanogels prepared in a variety of PPFPA-PEGMA concentrations
In addition to this method, we were also interested in preparing the nanogels directly in water. This has several advantages: (i) the organic solvent free nanocarrier preparation method is much preferred in terms of removing the trace amounts of solvent and its environment-friendly nature; (ii) allows for an additional handle to tune the particle size; (iii) allows for efficient incorporation of a broader range of lipophilic guest molecules within the nanogels. Since it has been reported that PFP activated esters are much less susceptible toward hydrolysis, the preparation of nanogels in water should be feasible.21-22 We attempted to make nanogels in aqueous solutions using a PPFPA-r-PEGMA solution at a variety of polymer concentrations. The cross-linking reaction in H2O was also detected by FTIR. The collected FTIR spectrum of the aqueous reaction mixture after 4 hours heating was shown in Figure 6a. No remaining PFP groups were observed after cross-linking, indicated by the complete disappearance of the activated ester C=O absor-bance. Evolution of a peak at 1640 cm-1 further confirms the amidation. Another potential issue is the extent to which the PFP activated ester was converted to amide, compared to the potential hydrolysis of the ester during the cross-linking process. This can be addressed by monitoring the IR spectrum. The absorbance of the poly methacrylic acid, the product of hydrolysis, should be observed if hydrolysis occurred during the cross-linking process. However, we could not find any significant evidence for the hydrolysis peak, which is expected to be at around 1710 cm-1.23-25 This suggests that cross-linking is the predominant reaction. Interestingly, the sizes of the nanogels prepared in H2O were around 10 nm independent of the concentration of PPFPA-PEGMA solutions used. Size measurements from DLS are in good agreement with the TEM results shown in Figure 6b.
Figure 6.
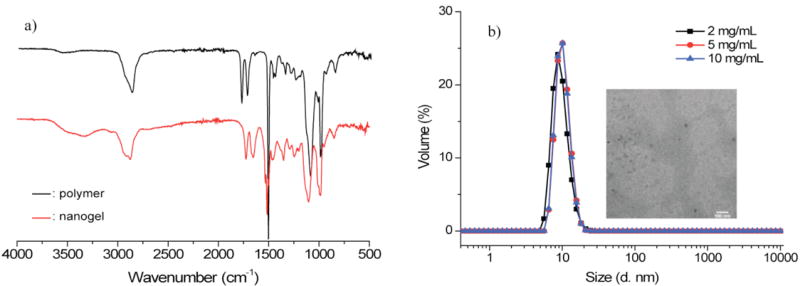
Synthesis of nanogel in H2O: a) Cross-linking followed by FTIR; b) Size of nanogel prepared from 2mg/mL, 5mg/mL, 10mg/mL of PFPA-PEG solution in H2O; Inset is TEM image of nanogel where the scale bar is 100 nm.
We were also interested in demonstrating that the residual PFP moieties can be utilized to additionally functionalize the surface of the nanogels. This provides at least two key advantages: (i) this post-nanogel formation reaction can be used to eliminate the remaining reactive PFP moieties. This is useful in applications such as drug delivery, where the reactivity of the PFP moiety could be a source of toxicity; (ii) this allows for the incorporation of functionalities on the surface of the nanogels. An example of an implication of such a capability includes incorporation of ligands for sensing and targeted delivery. In order to demonstrate the possibility of surface engineering on these nanogels, isopropylamine (IPA) and N,N-dimethylethylenediamine (DMEDA) were incorporated onto the 50% crosslinked nanogel after the nanogel synthesis. This reaction with IPA and DMEDA was monitored by FTIR (Figure S2), which clearly indicates that the amidation step can indeed be carried out in the post nanogel synthesis steps. Moreover, zeta potentials of these nanogels were measured to obtain additional evidence for surface modification (Figure 7). Zeta potentials of the 50% and 100% cystamine crosslinked nanogels were around -30 mV. Similarly, the 50% cross-linked nanogel modified by IPA also has a zeta potential of -30 mV. The observation of negatively charged nanogel surface is attributed to the carboxylate groups in the chain transfer agent. This is supported by the fact that utilization of charge-neutral initiators for our polymers in the disulfide crosslinked nanogels affords charge neutral surfaces. However, when the nanogel was modified by DMEDA, the zeta potential was found to shift to 5 mV. The positively charged surface is attributed to the protonation of DMEDA and therefore taken to designate surface modification. The presence of these functionalities on the nanogel surface can also be presumed, because our prior studies with the disulfide crosslinked nanogels show that cell penetrating peptide ligands are indeed available on the suface after a similar functionalization step.11
Figure 7.
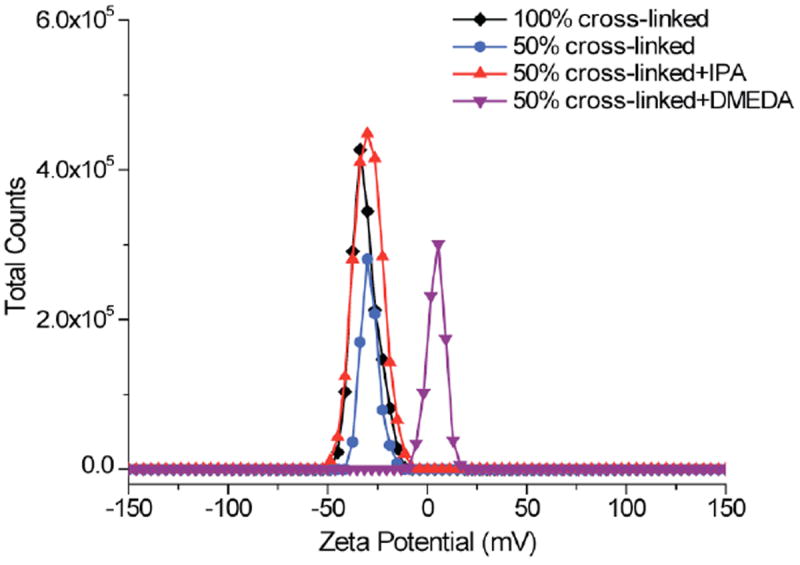
Charged surface of nanogels without and with post-nanogel modification
In summary, we have developed a facile methodology to achieve polymeric nanogels using a simple reaction between the lipophilic activated PFP ester and diamines. This strategy has an advantage that the syntheses of nanogels are not limited to disulfide crosslinked systems or self-crosslinking reactions. The intermolecular nature of the crosslinking reaction allows for incorporating a broader variety of stimuli-sensitive features in the diamine crosslinkers and thus in the nanogel. We have also shown that: (i) there is significant tunability in the size of the nanogels, especially in THF; (ii) these nanogels can encapsulate lipophilic guest molecules during the crosslinking step of the nanogel synthesis; (iii) the nanogels are dispersible in water, irrespective of whether they were prepared in THF or water; (iv) residual PFP moiety can be used in a post-nanogel assembly step to incorporate surface functionalities. We believe that expansion of our ability to achieve new nanogels using PFP moieties will significantly enhance the repertoire of these polymer nanogels in a variety of applications.
Supplementary Material
Acknowledgments
We thank DARPA and the NSF through MRSEC at UMass Amherst for support
Footnotes
Supporting Information. Experiment details, spectroscopic data, DLS. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Farokhzad OC, Langer R. Acs Nano. 2009;3:16. doi: 10.1021/nn900002m. [DOI] [PubMed] [Google Scholar]
- 2.O’Reilly RK, Hawker CJ, Wooley KL. Chem Soc Rev. 2006;35:1068. doi: 10.1039/b514858h. [DOI] [PubMed] [Google Scholar]
- 3.Wooley KL. J Polym Sci Pol Chem. 2000;38:1397. [Google Scholar]
- 4.Allen TM, Cullis PR. Science. 2004;303:1818. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 5.Bachelder EM, Beaudette TT, Broaders KE, Dashe J, Frechet JMJ. J Am Chem Soc. 2008;130:10494. doi: 10.1021/ja803947s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oh JK, Siegwart DJ, Lee HI, Sherwood G, Peteanu L, Hollinger JO, Kataoka K, Matyjaszewski K. J Am Chem Soc. 2007;129:5939. doi: 10.1021/ja069150l. [DOI] [PubMed] [Google Scholar]
- 7.Joralemon MJ, O’Reilly RK, Hawker CJ, Wooley KL. J Am Chem Soc. 2005;127:16892. doi: 10.1021/ja053919x. [DOI] [PubMed] [Google Scholar]
- 8.van der Ende AE, Harrell J, Sathiyakumar V, Meschievitz M, Katz J, Adcock K, Harth E. Macromolecules. 2010;43:5665. [Google Scholar]
- 9.van der Ende AE, Kravitz EJ, Harth E. J Am Chem Soc. 2008;130:8706. doi: 10.1021/ja711417h. [DOI] [PubMed] [Google Scholar]
- 10.Ryu JH, Chacko RT, Jiwpanich S, Bickerton S, Babu RP, Thayumanavan S. J Am Chem Soc. 2010;132:17227. doi: 10.1021/ja1069932. [DOI] [PubMed] [Google Scholar]
- 11.Ryu JH, Jiwpanich S, Chacko R, Bickerton S, Thayumanavan S. J Am Chem Soc. 2010;132:8246. doi: 10.1021/ja102316a. [DOI] [PubMed] [Google Scholar]
- 12.Kessler D, Metz N, Theato P. Macromol Symp. 2007;254:34. [Google Scholar]
- 13.Schattling P, Jochum FD, Theato P. Chem Comm. 2011;47:8859. doi: 10.1039/c1cc12652k. [DOI] [PubMed] [Google Scholar]
- 14.Nilles K, Theato P. J Polym Sci Polym Chem. 2010;48:3683. [Google Scholar]
- 15.Roth PJ, Jochum FD, Zentel R, Theato P. Biomacromolecules. 2010;11:238. doi: 10.1021/bm901095j. [DOI] [PubMed] [Google Scholar]
- 16.Roth PJ, Wiss KT, Zentel R, Theato P. Macromolecules. 2008;41:8513. [Google Scholar]
- 17.Eberhardt M, Mruk R, Zentel R, Theato P. Eur Polym J. 2005;41:1569. [Google Scholar]
- 18.He CB, Hu YP, Yin LC, Tang C, Yin CH. Biomaterials. 2010;31:3657. doi: 10.1016/j.biomaterials.2010.01.065. [DOI] [PubMed] [Google Scholar]
- 19.Balogh L, Nigavekar SS, Nair BM, Lesniak W, Zhang C, Sung LY, Kariapper MST, El-Jawahri A, Llanes M, Bolton B, Mamou F, Tan W, Hutson A, Minc L, Khan MK. Nanomed-Nanotechnol. 2007;3:281. [Google Scholar]
- 20.Wong C, Stylianopoulos T, Cui JA, Martin J, Chauhan VP, Jiang W, Popovic Z, Jain RK, Bawendi MG, Fukumura DP. Natl Acad Sci USA. 2011;108:2426. doi: 10.1073/pnas.1018382108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nilles K, Theato P. Polym Chem. 2011;2:376. [Google Scholar]
- 22.Gibson MI, Frohlich E, Klok HA. J Polym Sci Pol Chem. 2009;47:4332. [Google Scholar]
- 23.Billingham J, Breen C, Yarwood J. Vibrational Spectroscopy. 1997;14:19. [Google Scholar]
- 24.Arndt KF, Richter A, Ludwig S, Zimmermann J, Kressler J, Kuckling D, Adler HJ. Acta Polym. 1999;50:383. [Google Scholar]
- 25.Dubinsky S, Lumelsky Y, Grader GS, Shter GE, Silverstein MS. J Polym Sci Polym Phys. 2005;43:1168. [Google Scholar]
- 26.Singha NK, Gibson MI, Koiry BP, Danial M, Klok HA. Biomacromolecules. 2011;12:2908. doi: 10.1021/bm200469a. [DOI] [PubMed] [Google Scholar]
- 27.Haberkorn N, Nilles K, Schattling P, Theato P. Polym Chem. 2011;2:645. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.