

Abstract
To determine whether the EGFR tyrosine kinase inhibitor, erlotinib may cause hypomagnesemia, inflammation and cardiac stress, erlotinib was administered to rats (10 mg/kg/day) for 9 weeks. Plasma magnesium decreased progressively between 3-9 weeks (-9 to -26%). Modest increases in plasma substance P (SP) occurred at 3 (+27%) and 9 (+25%) weeks. Neutrophil superoxide-generating activity increased 3-fold, and plasma 8-isoprostane rose 210%, along with noticeable appearance of cardiac peri-vascular nitrotyrosine. The neurokinin-1 (NK-1) receptor antagonist, aprepitant (2 mg/kg/day), attenuated erlotinib-induced hypomagnesemia up to 42%; reduced circulating SP, suppressed neutrophil superoxide activity and 8-isoprostane elevations; cardiac nitrotyrosine was diminished. Echocardiography revealed mild to moderately decreased left ventricular ejection fraction (-11%) and % fractional shortening (-17%) by 7 weeks of erlotinib treatment, and significant reduction (-17.5%) in mitral valve E/A ratio at week 9, indicative of systolic and early diastolic dysfunction. Mild thinning of the left ventricular posterior wall suggested early dilated cardiomyopathy. Aprepitant completely prevented the erlotinib-induced systolic and diastolic dysfunction, and partially attenuated the anatomical changes. Thus, chronic erlotinib treatment does induce moderate hypomagnesemia, triggering SP-mediated oxidative/inflammation stress, and mild to moderate cardiac dysfunction, which can largely be corrected by administration of the SP receptor blocker.
Keywords: EGFR Tyrosine Kinase Inhibitor erlotinib (Tarceva), Hypomagnesemia, Neutrophil Activation, Total 8-isoprostane, Cardiac dysfunction, Aprepitant (Emend), NK-1 receptor blocker
Introduction
Therapies targeted to inhibit the activity of the epidermal growth factor (EGF) pathway through its receptor (EGFR) have seen increased application in anti-cancer treatment (1). The EGF pathway can be inhibited by injections of monoclonal antibodies targeting an extracellular epitope of the EGFR, or by chemical agents that inhibit the receptor tyrosine kinase on the cytoplasmic side of cancer cells (1, 2). Tarceva® (erlotinib) is approved as a first-line and maintenance treatment, and 2nd- or 3rd-line treatment for advanced-stage non-small cell lung cancer (NSCLC). It is an orally administered reversible tyrosine kinase inhibitor (TKI) targeting the EGF receptor, that is up-regulated in the majority of lung, colorectal, and head and neck cancers (3). However, EGFR activation is also required for active epithelial magnesium (Mg)-absorption that is mediated by the transient receptor potential melastatin 6 (TRPM6) channel in the kidney and colon (4). Treatment of patients with monoclonal antibodies (cetuximab and panitumumab) targeting the EGFR in colorectal cancer was found to cause pronounced hypomagnesemia (4, 5). Noticeable hypomagnesemia was also observed in patient receiving cetuximab plus erlotinib (6). However, preclinical in vitro patch clamp analyses in TRPM6-expressing renal cells showed that a physiological concentration (0.3 μM) of erlotinib did not inhibit EGF-induced changes in TRPM6 current density and tyrosine phosphorylation of EGFR (7). Erlotinib can provoke cellular oxidative stress in cancer cells through NOX-4 up-regulation (8.9). As a class, TKIs are known to display varying degrees of cardiotoxicity generally attributed to off-target kinase inhibition (10, 11); yet the systemic oxidative impact and the long term effects of erlotinib on Mg handling remain unexplored. We previously reported that an experimental TKI, tyrphostin AG 1478, which is chemically similar to erlotinib, displayed substantial cardiac dysfunctional effects that were associated with enhanced neurogenic inflammation (elevated circulating substance P [SP], oxidative stress, and hypomagnesemia. (12) In the current study, we found that chronic treatment of rats with erlotinib also induced significant hypomagnesemia and systemic oxidative stress with associated cardiac dysfunction. Furthermore, we found that these effects can be significantly inhibited by substance P-receptor blockade using aprepitant.
Materials and Methods
Animal Model and Treatment Protocol
Experiments on animals were conducted in accordance with the principles given in the US Department of Health and Human Services Guide for the Care and Use of Laboratory Animals and were approved by The George Washington University Institutional Animal Care and Use Committee. Male Sprague-Dawley rats (125-150 g) were purchased from Hilltop Lab Animals, Inc. (Scottdale, PA). After 1 week of quarantine, all age-matched rats were placed on an ad libitum Mg normal diet (25 mmole magnesium oxide/kg food regarded as 100% recommended daily allowance for rodents) obtained from Harlan-Teklad (Madison, WI) containing extracted casein as the diet base supplemented with essential vitamins and nutrients; or the same diet supplemented with erlotinib (OSI Pharmaceuticals, LLC, Northbrook, IL 60062, USA) to obtain a starting dose of 10 mg/kg/day, Emend ® (as aprepitant, Merck & Co., INC. USA) to obtain a starting dose of 2 mg/kg/day, or both agents at these doses. Animal groups include: control (n=5), erlotinib treated (n=5), erlotinib + aprepitant-treated (n=7) and aprepitant treated (n=5). Individually-housed rats were weighed and food consumption recorded daily to obtain actual drug dosage: time-range average erlotinib dose over 9 weeks was 7.07 mg/kg/day, and time-range average aprepitant dose over 9 weeks was 1.41 mg/kg/day. Rats had free access to distilled-deionized water, and were on a 12 h light/dark cycle for up to 9 weeks.
Blood Sample Collection/Preparation
At periodic intervals, blood was collected (∼0.5 ml) aseptically from the tail tip of anaesthetized rats (2 % isoflurane, EZ Anesthesia Chamber with nose cone) (13, 14) in sterile microtainer plasma separator tubes containing heparin and the protease inhibitor, aprotinin (Sigma Chemicals, St. Louis MO) to yield final blood concentrations of 10.74 units/ml and 0.016 units/ml, respectively. For subsequent samplings, the scab was carefully removed, and the process was repeated. Plasma was obtained after centrifugation (12,000 rpm, 2 min, RT, IDEXX StatSpin VT, Iris International, Inc., Westwood, MA). Tail bleed samples were used for assessment of plasma Mg, and substance P levels. Sacrifice blood samples (∼ 8 ml collected in heparin plus aprotinin containing BD vacutainer SST tubes) were taken from anaesthetized, heparinized (0.3-0.4 ml 358 units/ml heparin in 0.9% NaCl, i.p.) rats from either the vena cava or by cardiac puncture, and were centrifuged (3,500 rpm, 10 min, RT). In addition to the above plasma parameters, sacrifice plasma was also assayed for 8-isoprostane levels, and the whole blood samples were processed for neutrophil isolation and assessment of superoxide anion production.
Plasma Magnesium
Magnesium levels in 1:50 or 1:100 diluted, acidified (nitric acid) plasma samples were determined by atomic absorption flame emission spectroscopy (wavelength = 285.2 nm) using an AA-6200 Shimadzu spectrophotometer (Columbia, MD) as described (13). Values obtained from standard curves were mg/dl, and reported as % of control.
Plasma Substance P Determination
Plasma SP levels (14,15) were assessed using an ELISA kit from R&D Systems (Minneapolis, MN). This is a competitive binding assay in which SP within samples (50 μl of 1:1 diluted plasma) competes with a fixed amount of horseradish peroxidase- labeled SP for sites on a murine monoclonal antibody. Color development was inversely proportional to SP concentration and absorbance was read at 450 nm with background subtraction at 540 nm. Values represent % changes in plasma SP levels compared to time-paired controls (100%) and were means ± SE of 4-6 rats. Overall average (n=9) of control rat plasma SP levels at 3, 5 and 9 weeks was 593.13 ± 15.6 pg/ml.
Plasma total 8-isoprostane
Plasma samples (with 0.005% BHT added) were processed to obtained total 8-isoprostane. Briefly, plasma aliquots (100 ul) were alkalinized in 2N NaOH at 45°C for 2 hrs to release lipid bound and esterified isoprostanes. The alkalinized aliquots were then neutralized in 2N HCl, prior to centrifugation (at room temperature) for 5 min at 12,000 rpm in a microcentrifuge (Marathon Micro A, Fisher Scientific). The final supernatants were used for the determination of total 8-isoprostane content by an EIA kit (Cayman Chemical, MI) (13, 16).
Neutrophil basal and stimulated superoxide production
At sacrifice, whole blood (3 ml) samples were obtained; neutrophils were isolated by a step-gradient centrifugation method (13-16). Superoxide anion production from neutrophils (0.5-0.75×106/ml) with (stimulated) or without (basal) PMA (phorbol myristate acetate, 100 ng/ml) was assayed in a phosphate buffer (pH 7.6) containing 5 mM glucose, 1 mM CaCl2, 1 mM MgCl2, and 75 μM cytochrome c ± 50 μg SOD. Superoxide generation was estimated as SOD-inhibitable reduction of cytochrome c using the extinction coefficient: E550=2.1×104M-1cm-1 .
Cardiac Nitrotyrosine Determination by Histochemical Analysis
Cardiac tissue was rapidly excised, rinsed in saline, embedded in OCT compound, quickly frozen and kept at -80°C until used. Cryosections, 5 um thick, were stained by an indirect, immunohistochemical method (13) using polyclonal rabbit anti-rat Nitrotyrosine antibody (5ug/ml; Millipore, Billerica, MA) and the Vecta-Stain Elite ABC kit immunoperoxidase system (Vector Laboratories, Burlingame, CA). Samples were examined under an Olympus BX60 microscope, and multiple images were taken with a digital camera (Evolution Color MP; Media Cybernetics, Silver Spring, MD). Briefly, fresh frozen sections were fixed in 10% formaldehyde, washed in PBS, incubated with 0.3% hydrogen peroxide in methanol for 30 min to block endogenous peroxidase, washed in PBS and incubated overnight at 4°C with rabbit anti-nitrotyrosine antibody, which was applied on every slide except negative control. ABC kit reagents for horseradish peroxidase and 3-3′diaminebenzidine (DAB) were used to visualize the antigen-antibody reaction product as brown color. The sections were counterstained with hematoxylin, rinsed, dehydrated and cleared in series of ethanol and xylene and cover slipped using mounting medium.
Trans-thoracic Echocardiography
Anesthetized rats were subjected to echocardiography periodically during the treatment regimens using a GE VingMed System Five Echocardiogram System (12, 13, 17, 18). Heart rate and rectal temperature (35.9 - 37.5 °C) were monitored constantly with placement of animals on a warming platform with paws taped down to limit any motion during imaging; a sterile eye lubricant was applied to prevent eye drying during the procedure. Hair over the thorax was removed first with an electric clipper, and then with a depilatory cream (< 2 min exposure). Animals were imaged (10 MHz probe) for about 20 minutes at an image depth of 3 cm. This imaging system allows for both cardiac structural and functional evaluation. Systolic function was measured as % fractional shortening (% FS) using M-mode imaging, and left ventricular ejection fraction (LVEF) was calculated from M-mode measurements as well. Key anatomical parameters (LV posterior wall thickness in diastole or systole = LVPWd or s) were measured to assess the presence of dilated cardiomyopathy and reduced cardiac function. Measurements of aortic and pulmonary artery diameters were used to calculate stroke volumes. Aortic pressure max (AoPmax) was assessed; and spectral Doppler velocities were measured for the pulmonic and aortic outflows to calculate cardiac output (CO), and for the mitral valve inflows to assess ventricular diastolic function (mitral valve E and A wave velocities and the E/A ratio were obtained). After echocardiography, animals were placed in room air until awake, and observed until fully recovered.
Statistics
Data were reported as the mean ± SEM of 4–7 animals per group. Data were checked by F-test for equality of groups' variation, and statistical differences were evaluated by two-tailed Student's t-test. Selected data were analyzed by one-way ANOVA followed by a Tukey's test. Statistical significance was considered at p<0.05.
Results
Lack of effects of erlotinib treatment on food consumption and weight gain
All drug agents were administered orally through commercially prepared food, but neither erlotinib nor aprepitant affected food consumption of the animals compared to controls (data not shown). Erlotinib alone modestly depressed animal weight gain (-13%, N.S.) over 9 weeks of drug treatment (Fig. 1). Erlotinib plus aprepitant slightly improved weight gain (-8%); however, no statistically significant differences were found among any of the treatment groups versus control.
Figure 1.
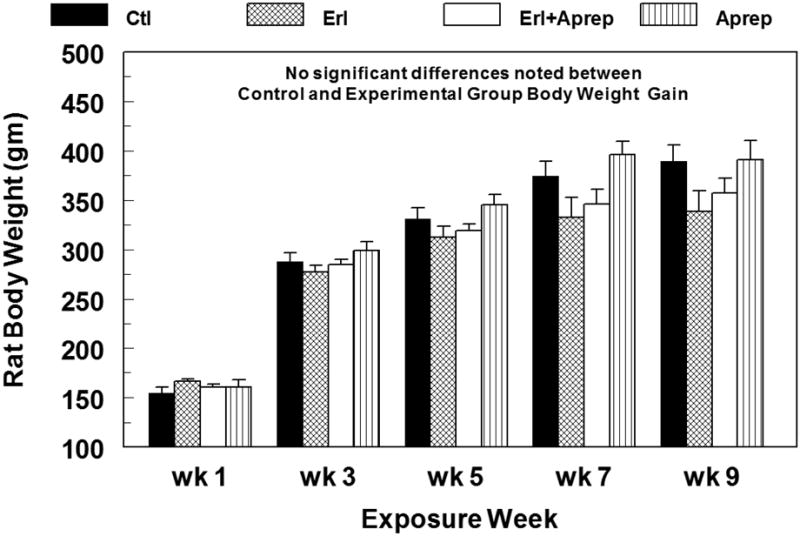
Erlotinib ± aprepitant treatment does not significantly alter the rate of animal weight gain versus control during 9 weeks of exposure. Erlotinib (starting 10 mg/kg/day, oral) and/or aprepitant (starting 2 mg/kg/day, oral) were administered in custom-prepared food. Data are means ± SEM of 5–7 rats per group.
Erlotinib promotes hypomagnesemia and neurogenic inflammation
Erlotinib caused significant hypomagnesemia in rats beginning at week 3 and continuing to week 9 (Fig. 2A): -9 % at week 3, -13% at week 5, -16% at week 7; and a more significant decrease (-26%, p <0.001) at week 9. We also measured circulating SP levels and found modest increases during week 3 (27.3%, p<0.05), week 5 (12.3%, NS), and week 9 (25.4%, p<0.01) (Fig. 2B). Interestingly, these increases were almost completely blocked by co-treatment with aprepitant, a highly specific NK-1 receptor blocker. Fig.2A also indicates that in the presence of SP receptor blockade, the severity of hypomagnesemia due to erlotinib was attenuated by ∼42% at week 9.
Figure 2.
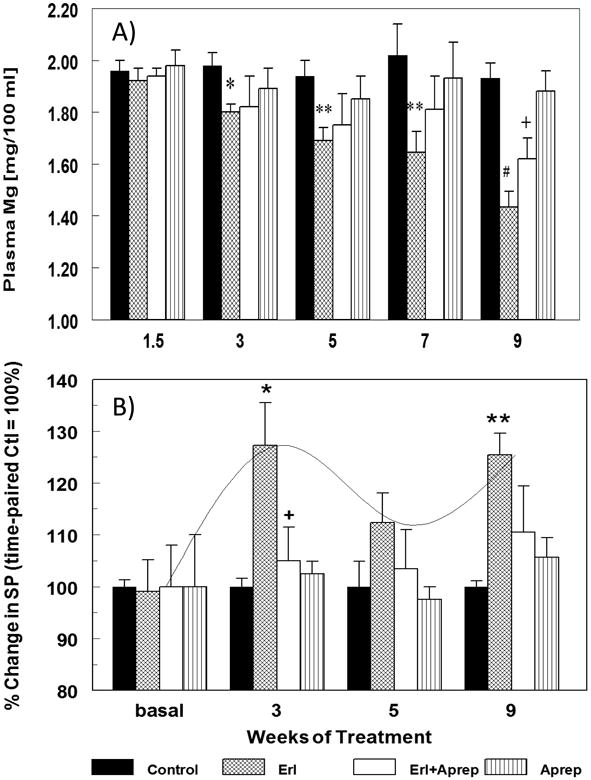
A & B. Temporal effects of erlotinib with or without aprepitant treatment for up to 9 weeks on rat plasma magnesium (A) and substance P (SP) (B) levels. Blood was sampled at specified times from rats treated with erlotinib (starting 10 mg/kg/day, oral) and/or aprepitant (starting 2 mg/kg/day, oral). (A) Plasma was assessed for magnesium by flame emission atomic absorption spectroscopy. Data are the means ± SEM of 5-7 animals per group. * p <0.05,** p <0.01 and # p <0.001 compared with the time-matched control group; + p<0.055 versus erlotinib alone (B) Plasma SP was assessed by a colorimetric ELISA kit. Values for erlotinib ± aprepitant rats were compared to time-matched controls (100%), and are means ± SEM of 4-6 rats per group. * p <0.05 and ** p <0.01 compared with the time-matched control group; + p <0.05 versus erlotinib alone.
Erlotinib and NK-1 blockade on neutrophil activation and oxidative stress
We previously showed that hypomagnesemia alone in rodents can promote neutrophil free radical generating activity (18,19). In this study, we found that neutrophils isolated from the erlotinib-treated rats (9 weeks) displayed a 3-fold higher basal superoxide generating activity (Fig 3A). When stimulated by PMA, the erlotinib-treated samples also exhibited a 2-fold higher activity compared to controls. Importantly, we found that treatment with aprepitant completely prevented the increases in both the basal and stimulated superoxide generating activity caused by erlotinib treatment. Enhanced systemic oxidative stress, as represented by increased total plasma 8-isoprostane content (Fig. 3B), was prominent by 9 weeks (210% increase, p<0.01). Along with the inhibition of neutrophil activity, aprepitant also completely blocked the elevation of total plasma isoprostane. The aprepitant alone group resembled control values and had no effects on neutrophil superoxide activity or isoprostane levels. In data not shown, a modest but significant (p<0.05) level of oxidative stress was observed at 3 weeks when the plasma 8-isoprostane level was elevated 80% above control which was also inhibited by the aprepitant co-treatment.
Figure 3.
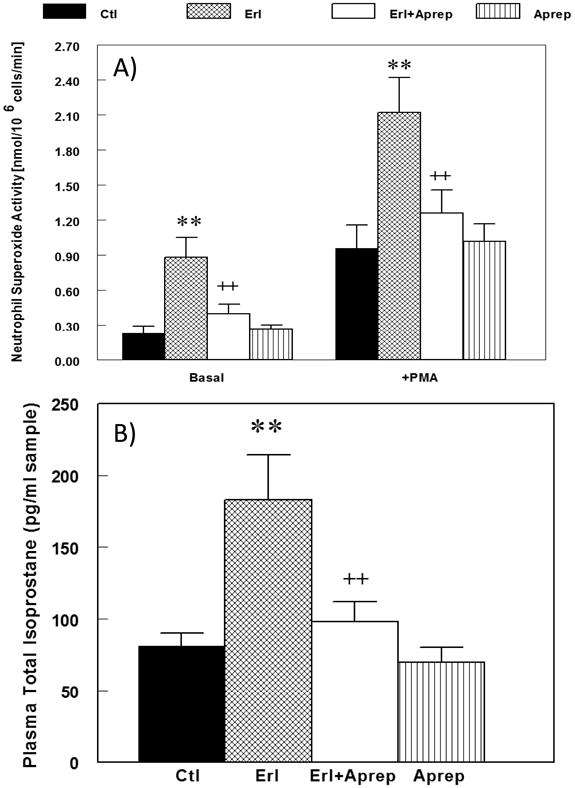
A & B. Effects of erlotinib with or without aprepitant treatment for 9 weeks on rat (A) neutrophil basal superoxide generating activity, and (B) plasma 8-isoprostane content. Blood was sampled from rats treated with erlotinib (starting 10 mg/kg/day, oral) and/or aprepitant (starting 2 mg/kg/day, oral) at 9 weeks of exposure. (A) circulating neutrophil basal superoxide generating activity was determined as SOD-inhibitable reduction of cytochrome c; and (B) plasma 8-isoprostane was assessed by a colorimetric ELISA kit. Values for erlotinib ± aprepitant rats were compared to time-matched controls, and are means ± SEM of 4-6 rats per group. ** p < 0.01 versus control; ++ p < 0.01 versus erlotinib alone.
Nitrosative stress represented by nitrotyrosine expression in cardiac tissue
The polyclonal anti-nitrotyrosine Ab showed slightly diffuse myocardial immunoreactivity throughout the cardiac tissue sections from erlotinib and erlotinib+aprepitant treated animals (9 weeks), but the prominent brown staining was only observed in the vascular wall smooth muscle cells and perivascular connective tissue of tunica adventitia in erlotinib treated animals (Fig. 4). Occasionally endothelial cell staining was seen in the erlotinib treated samples (Fig. 4-Insert). In general, the perivascular nitrotyrosine staining was substantially attenuated in the erlotinib+aprepitant samples. Aprepitant alone did not have any effect on nitrotyrosine formation and the immunohistochemical staining pattern resembled vehicle control samples.
Figure 4.
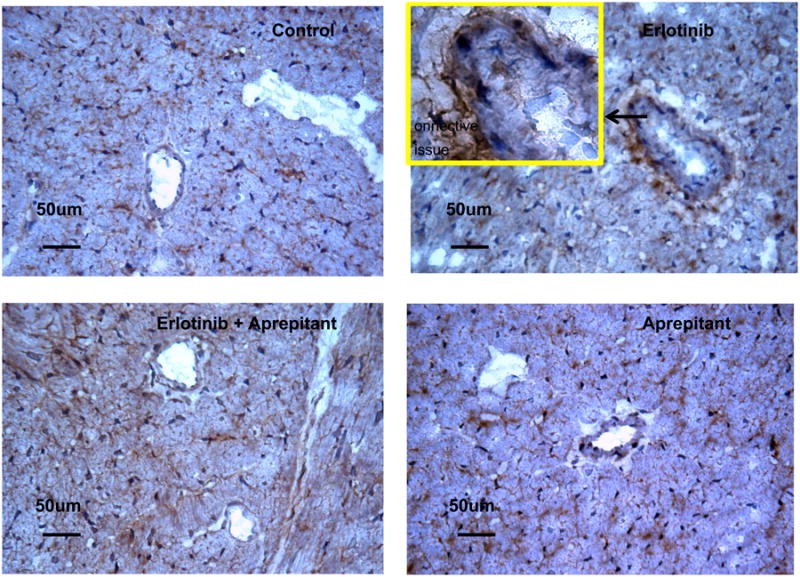
Effects of erlotinib with or without aprepitant treatment of rats for 9 weeks on 3-nitrotyrosine formation in cardiac tissue. After 9 weeks, animals were sacrificed and hearts were quickly excised, trimmed, rinsed, embedded in OCT compound, and frozen at -70°C until used. 3-nitrotyrosine was localized immunohistochemically using polyclonal rabbit anti-rat nitrotyrosine antibody, and the Vecta-Stain Elite ABC kit immunoperoxidase system. Representative samples from control, erlotinib alone, erlotinib + aprepitant, and aprepitant alone treated rat groups are depicted. Diffused brown staining is present in ventricles from erlotinib and erlotinib+aprepitant treated animals; in ventricles from erlotinib treated animals 3-nitrotyrosine is predominantly localized in perivascular connective tissue and occasionally in smooth muscle cells (SMC) of intima tunica as well as in endothelial cells (EC), see the insert. Original magnification 40×, for insert 100×.
Erlotinib and NK-1 blockade on Cardiac Function
No significant changes in cardiac contractile function were detected before 7 weeks in the erlotinib -treated rats; LV ejection fraction (LVEF: left panel, p<0.05) was 10.4 and 11 % lower and LV % fractional shortening (LV %FS: center, p<0.05) was 17.2 and 17.7 % lower than time-matched controls at 7 and 9 weeks, respectively (Fig. 5A & B). Erlotinib also impaired hemodynamic parameters in a similar exposure time- dependent manner: cardiac output was reduced 18.8 (p<0.05) and 20.3 (p<0.02) %, and aortic pressure maximum was 17.1 (NS) and 27.5 % (p<0.05) lower than controls at 7 and 9 weeks, respectively. Mitral valve E/A ratio (Fig. 5C right) was not significantly affected by erlotinib through 7 weeks, but a significant reduction (17.5 %, p<0.05) occurred by week 9, suggesting a more delayed development of LV diastolic dysfunction. We focused on possible changes in LVPWs and LVPWd, which are related to changes in cardiac function and cardiac dilation, respectively. Indeed, both LVPWs and LVPWd were altered by erlotinib: LVPWd fell 15.6 to 20.9 % (all p<0.05) and LVPWs declined 9.3 (NS) to 21.9 (p<0.05) % during 5 to 9 weeks of erlotinib exposure (Fig. 6).
Figure 5.
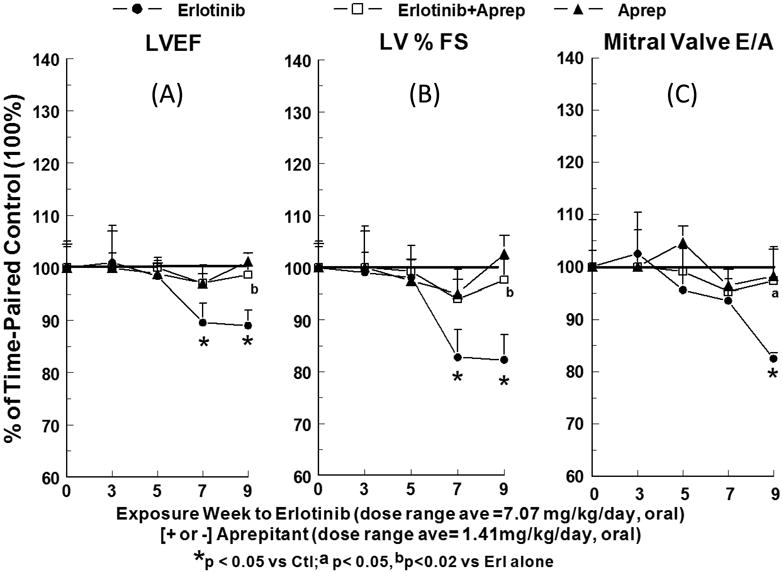
A, B & C. Effects of chronic erlotinib ± aprepitant treatment for 9 weeks on echocardiographic functional parameters in rats. (A) Left ventricular ejection fraction (LVEF); (B) left ventricular percent fractional shortening (LV % FS); and (C) mitral valve E/A ratio. Rats treated with erlotinib (starting 10 mg/kg/day, oral) and/or aprepitant (starting 2 mg/kg/day, oral) received echocardiography at the indicated intervals. Values for erlotinib ± aprepitant rats were compared to time-matched controls (100%), and are means ± SEM of 5–7 rats per group. * p <0.05 compared with the time-matched control group; a p<0.05 and b p<0.02 versus erlotinib alone.
Figure 6.
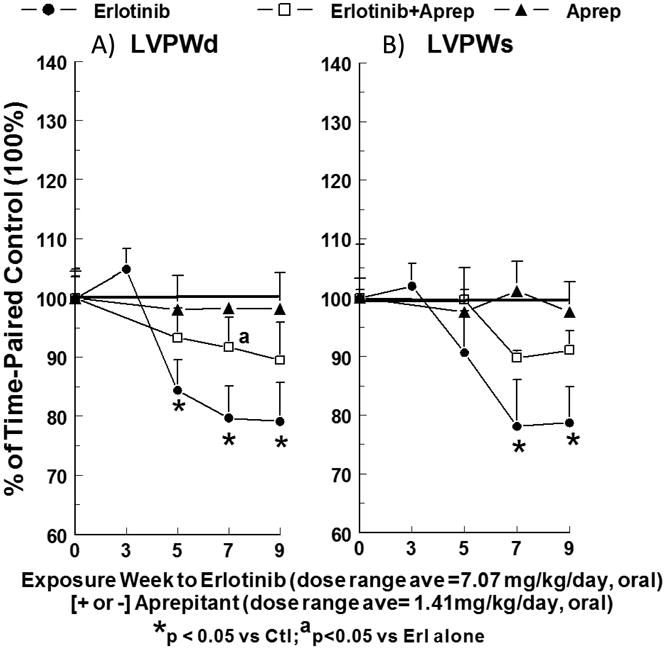
Effects of chronic erlotinib ± aprepitant treatment for 9 weeks on LV posterior wall thickness in diastole (LVPWd) and systole (LVPWs). Rats treated with erlotinib (starting 10 mg/kg/day, oral) and/or aprepitant (starting 2 mg/kg/day, oral) received echocardiography at the indicated intervals. Values (% change) for erlotinib ± aprepitant rats were compared to time-matched controls, and are means ± SEM of 5–7 rats per group. * p <0.05 compared with the time-matched control group; a p<0.05 versus erlotinib alone.
Concurrent treatment of erlotinib-exposed rats with aprepitant attenuated all of the effects of erlotinib on LV systolic, diastolic and anatomical parameters. Compared to erlotinib alone, aprepitant co-treatment improved LVEF 71.3 % and 87.2% at 7 and 9 (p<0.02) weeks (Fig. 5 A); improved LV %FS 65.1 and 86.4% at 7 and 9 (p<0.02) weeks (Fig. 5 B); and improved mitral valve E/A ratio 84.6% at 9 weeks (Fig. 5 C, [p<0.05]). Aprepitant afforded protection against the erlotinib-induced decline in cardiac output by 64.9 and 75.4% at 7 and 9 weeks (p<0.02 vs erlotinib alone), and also improved AoPmax by 86 (NS) and 64 (NS) % at 7 and 9 weeks. The cardiac anatomical parameters also benefited from aprepitant co-treatment of erlotinib-exposed rats (Fig 6): LVPWd improved 59.1 (p<0.05 vs erlotinib alone) and 49.8%; and LVPWs improved 53.4 and 58.2 % at 7 and 9 weeks. Aprepitant alone did not have any significant impact (Figs. 5 & 6) on LV systolic, diastolic, hemodynamic and anatomical parameters compared to time-paired controls.
Discussion
Hypomagnesemia is a major side-effect of some specific mono-clonal antibody EGFR inhibitors (cetuximab, panitumumab)(3), and many commonly-used anti-cancer drugs such as cisplatin, which is directly toxic to the kidney (20,21). However, the TKI, erlotinib, competes reversibly with ATP to bind to the intracellular catalytic site of the EGFR-TK to inhibit EGFR auto-phosphorylation and down-stream signaling (2,3). Therefore small molecules TKIs are inherently less selective and the effects on Mg wasting directly related to EGFR inhibition remained to be confirmed. The short-term effect of erlotinib on Mg-handling in rodents was previously reported to be modest (7). Indeed, our study indicated that erlotinib treatment for 10 days had essentially no effect on plasma Mg levels, and 3 to 5 weeks of erlotinib only produced modest, but significant decreases in plasma Mg (<14 %). In a prior study, Dimke et al (7) reported that a much higher dose of erlotinib (40 mg/kg) for 23 days, caused only a 10% lowering of plasma Mg. Interestingly, when treatment was prolonged to 7- 9 weeks in our study, more pronounced decreases (25-28%) of plasma Mg were revealed, suggesting a time –dependent cumulative toxic effect on the kidney and/or intestine for Mg handling. As discussed earlier, these decreases may be related to the pharmacological inhibition of EGF-EGFR activation, which is a required up-stream event for renal and intestinal magnesium reabsorption by the TRPM6 channel (4, 22). However, the fact that erlotinib caused only minimal effects on the plasma Mg levels during earlier weeks of exposure (< 3 weeks) suggested that EGFR inhibition by erlotinib may not be the only mechanism causing Mg wasting in later weeks. Erlotinib is also known to activate NOX4 (8,9), the NADPH oxidase that is highly enriched in the kidney (23, 24). Thus, chronic erlotinib treatment may lead to activation of kidney NOX4 to generate H2O2; it was previously demonstrated that H2O2 alone can cause dose-dependent inhibition of TRPM6 channel activity and subsequently decreased re-absorption of Mg (25). Although such a mechanism remains to be confirmed, our animal study demonstrated that erlotinib can cause substantial and significant hypomagnesemia during chronic treatment.
Our previous reports documented that hypomagnesemia alone caused systemic neurogenic inflammation (26,27); in a later study (28), the extent of neurogenic inflammation, as signified by plasma SP elevations, depended on the severity of hypomagnesemia. In the current study, the modest (nearly 30% above control), but significant elevations of circulating SP levels were detected in the erlotinib rats. Interestingly, these elevations of plasma SP were almost completely blocked by treatment with aprepitant; this suggests that the enhanced SP production might be through a positive feedback, autocrine- like mechanism (29), mediated by SP/NK-1 receptor activation, associated upregulation of SP messenger RNA expression, and the subsequent de novo SP synthesis in inflammatory cells (29). In the presence of aprepitant, autocrine -promoted new synthesis of SP would be inhibited. We previously observed that neutral endopeptidase (neprilysin or NEP), the main enzyme which degrades SP, was inactivated during hypomagnesemia in rats and that this inactivation was prevented by NK-1R blockade (18). Therefore, it is possible that treatment with aprepitant might preserve NEP enzymatic activity, thereby permitting the degradation of circulating SP.
The transient nature of plasma SP elevation during erlotinib exposure led to the appearance of a biphasic profile (Fig. 2B), which was reminiscent of those observed in rodent models of both diet (28)- and TKI [tyrphostin AG1478(30)-induced hypomagnesemia. This pattern may be the consequence of various events, which either alone or in concert, could modulate circulating SP levels. Neuronal release of this neuropeptide is largely regulated by the voltage-dependent, Mg-gated N-methyl-D-aspartate (NMDA) receptor/channel complex (31); removal of extracellular Mg2+ unblocks the cation channel which is a prerequisite in the process of vesicular SP release. The sensitivity of the NMDA receptor/channel complex to changes in extracellular Mg levels depends on the subunit composition of the heteromeric channel. In the mouse, some channels were found to be more sensitive to Mg blockade than others (32). Similar functional variations were detected when considering the different subunit compositions of the receptor/channel complex in rats (33). These observations imply the existence of fine regulatory control of substance P release in response to small changes (marginal to moderate) in extracellular Mg levels. This could account for the observed transient plasma SP elevations (Fig. 2B), since receptor/channel complexes more resistant to declining Mg levels (< 14 % lower) might release SP during earlier periods of erlotinib treatment (1.5 to 3 weeks), whereas those more sensitive to Mg blockade might release SP at later times (> 7 weeks) in response to further declines in Mg (> 25% lower). Alternatively, other potential reasons for the biphasic profile might reflect: (i) enhanced abilities of non-neuronal (inflammatory) cells, which possess mRNA for SP (de novo synthesis) as well as NK-1 receptors, to release SP upon cell stimulation (29); and/or (ii) the attenuated SP degradative capacity of endogenous NEP, which occurred during prolonged hypomagnesemia (18).
We found that the basal superoxide generating activities of neutrophils from erlotinib treated rats were enhanced 3-fold; since this activity was promoted by PMA (a protein kinase C activator), this suggests that the enhanced superoxide production was from NOX-2, which is the predominant NADPH oxidase system in neutrophils. Previously, we showed that neutrophil NK-1R can be up-regulated by hypomagnesemia (15); the activation of the neutrophils by erlotinib treatment and the suppression by aprepitant suggested that the cascade of inflammatory/oxidative events were governed by enhanced SP/NK-1R interaction (19). In association with increased endogenous neutrophil activation, we also found that plasma 8-isoprostane, an in vivo marker of non-enzymatic lipid peroxidation, was significantly increased in the erlotinib -treated rats, but this was completely attenuated by aprepitant administration, confirming the role of neurogenic inflammation in causing the oxidative stress. Increased peri-vascular nitrotyrosine content in the erlotinib-treated hearts (Fig. 4) suggests elevated nitrosative stress, likely mediated by peroxynitrite; the extent of this accumulation was reduced by treatment with aprepitant.
Rats receiving erlotinib for at least 7 weeks or longer exhibited mild to moderate compromised systolic and diastolic function (Fig. 5). Modest cardiac wall thinning (LVPW) also occurred, which suggests early dilated cardiomyopathy. All of these changes were substantially attenuated by aprepitant, suggesting that erlotinib-induced cardiac dysfunction and structural changes occurred subsequent to SP-mediated neurogenic inflammation. Alternatively, elevated SP may have a direct effect on the heart through its interplay at the cardiac NK-1R; this possibility was recently raised by the demonstration that SP can directly regulate adverse myocardial remodeling via cardiomyocyte NK-1R in a hypertensive rat model (34).
It is generally agreed that the TKI-induced cardiotoxicity is due to off-target effects as opposed to on-target actions such as those produced by Herceptin (35, 36). Earlier clinical trials did not report major cardiac events associated with erlotinib use (37); however, in those studies, LV systolic function was not monitored, nor was it clear whether they were carried out long enough to reveal substantial impairment in cardiac function (37). Our data (decreased LVEF and LV %FS) demonstrated that mild to moderate cardiac dysfunction occurred by 7 weeks of treatment; in human equivalence, we postulate that this would be approximately 2 years of clinical treatment. Although acute in vitro studies indicated that erlotinib caused minimal toxicity in isolated cardiomyocytes (11, 35), the mechanism leading to cardiotoxicity following prolonged exposure may not involve erlotinib- mediated inhibition of cardiac kinases, but might be the consequence of a secondary pathological event. We postulate that much of the cardiac injury/pathology attributed to erlotinib was caused by SP mediated neurogenic inflammation/oxidative stress subsequent to the progressive hypomagnesemia. Major support for this hypothesis was the observation that NK-1 receptor blockade almost completely prevented the development of cardiac systolic and diastolic dysfunction and the associated cardiac wall thinning in erlotinib -treated animals. Co-existing hypomagnesemia is common among cancer patients receiving chemotherapy with agents such as cisplatin. In rodent models, it was recently reported that cisplatin caused injury of the renal distal convsoluted tubule, and down-regulated the renal TRPM6/EGF pathway leading to significant Mg-wasting (21). When combined with cisplatin to enhance the cancer cell cytotoxicity in advanced NSCLC (38), one may anticipate that erlotinib would cause more severe hypomagnesemia, and heightened neurogenic inflammation. Under these circumstances, supplementation with Mg along with aprepitant might attenuate the more severe systemic stress and cardiac dysfunction accompanying combined cisplatin plus erlotinib therapy.
Acknowledgments
This study was supported by United States Public Health Service grants NIH RO1-HL-62282 and 1 R21 HL108311.
References
- Sawyers C. Targeted cancer therapy. Nature. 2004;432:294–297. doi: 10.1038/nature03095. [DOI] [PubMed] [Google Scholar]
- Imai K, Takaoka A. Comparing antibody and small-molecule therapies for cancer. Nat Rev Cancer. 2006;6:714–27. doi: 10.1038/nrc1913. [DOI] [PubMed] [Google Scholar]
- Schettino C, Bareschino MA, Ricci V, Ciardiello F. Erlotinib: An EGF receptor tyrosine kinase inhibitor in non-small-cell lung cancer treatment. Expert Rev Respir Med. 2008;2:167–78. doi: 10.1586/17476348.2.2.167. [DOI] [PubMed] [Google Scholar]
- Tejpar S, Piessevaux H, Claes K, Piront P, Hoenderop JGJ, Verslype C, Van Cutsem E. Magnesium wasting associated with epidermal-growth-factor receptor-targeting antibodies in colorectal cancer: a prospective study. Lancet Oncol. 2007;8:387–394. doi: 10.1016/S1470-2045(07)70108-0. [DOI] [PubMed] [Google Scholar]
- Petrelli F, Borgonovo K, Cabiddu M, Ghilardi M, Barni S. Risk of anti-EGFR monoclonal antibody-related hypomagnesemia: systematic review and pooled analysis of randomized studies. Expert Opinion on Drug Safety. 2012;11:S9–S19. doi: 10.1517/14740338.2011.606213. No. S1. [DOI] [PubMed] [Google Scholar]
- Janjigian YY, Azzoli CG, Krug LM, Pereira LK, Rizvi NA, Pietanza MC, Kris MG, Ginsberg MS, Pao W, Miller VA, Riely GJ. Phase I/II trial of cetuximab and erlotinib in patients with lung adenocarcinoma and acquired resistance to erlotinib. Clin Cancer Res. 2011;17:2521–7. doi: 10.1158/1078-0432.CCR-10-2662. [DOI] [PubMed] [Google Scholar]
- Dimke H, van der Wijst J, Alexander TR, Meijer IM, Mulder GM, van Goor H, Tejpar S, Hoenderop JG, Bindels RJ. Effects of the EGFR Inhibitor Erlotinib on Magnesium Handling. J Am Soc Nephrol. 2010;21:1309–16. doi: 10.1681/ASN.2009111153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher EV, Love-Homan L, Sobhakumari A, Feddersen CR, Koch AT, Goel A, Simons AL. EGFR inhibition induces proinflammatory cytokines via NOX4 in HNSCC. Mol Cancer Res. 2013;11:1574–84. doi: 10.1158/1541-7786.MCR-13-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orcutt KP, Parsons AD, Sibenaller ZA, Scarbrough PM, Zhu Y, Sobhakumari A, Wilke WW, Kalen AL, Goswami P, Miller FJ, Jr, Spitz DR, Simons AL. Erlotinib-mediated inhibition of EGFR signaling induces metabolic oxidative stress through NOX4. Cancer Res. 2011;71:3932–40. doi: 10.1158/0008-5472.CAN-10-3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Papoian T. Tyrosine kinase inhibitor (TKI)-induced cardiotoxicity: approaches to narrow the gaps between preclinical safety evaluation and clinical outcome. J Appl Toxicol. 2012;32:945–51. doi: 10.1002/jat.2813. [DOI] [PubMed] [Google Scholar]
- Doherty KR, Wappel RL, Talbert DR, Trusk PB, Moran DM, Kramer JW, Brown AM, Shell SA, Bacus S. Multi-parameter in vitro toxicity testing of crizotinib, sunitinib, erlotinib, and nilotinib in human cardiomyocytes. Toxicol Appl Pharmacol. 2013;272:245–55. doi: 10.1016/j.taap.2013.04.027. [DOI] [PubMed] [Google Scholar]
- Weglicki WB, Kramer JH, Spurney CF, Chmielinska JJ, Mak IT. The EGFR tyrosine kinase inhibitor tyrphostin AG-1478 causes hypomagnesemia and cardiac dysfunction. Can J Physiol Pharmacol. 2012;90:1145–9. doi: 10.1139/y2012-023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak IT, Kramer JH, Chen X, Chmielinska JJ, Spurney CF, Weglicki WB. Mg-supplementation Attenuates Ritonavir-induced Hyperlipidemia, Oxidative Stress and Cardiac Dysfunction in Rats. Am J Physiol Regul Integr Comp Physiol. 2013;305:R1102–R1111. doi: 10.1152/ajpregu.00268.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weglicki WB, Chmielinska JJ, Kramer JH, Spurney CF, Lu B, Mak IT. Neutral endopeptidase inhibition enhances substance P mediated inflammation due to hypomagnesemia. Mag Res. 2009;22:1–7. [PMC free article] [PubMed] [Google Scholar]
- Weglicki WB, Mak IT, Chmielinska JJ, Tejero-Taldo MI, Komarov A, Kramer JH. The role of Magnesium Deficiency in Cardiovascular and Intestinal Inflammation. Magnes Res. 2012;23:S199–206. doi: 10.1684/mrh.2010.0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak IT, Chmielinska JJ, Kramer JH, Weglicki WB. AZT-Induced cardiovascular toxicity - attenuation by Mg-supplementation. Cardiovascular Toxicol. 2009;9:78–85. doi: 10.1007/s12012-009-9040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer JH, Spurney C, Iantorno M, Tziros C, Mak IT, Tejero-Taldo MI, Chmielinska JJ, Komarov AM, Weglicki WB. Neurogenic inflammation and cardiac dysfunction due to hypomagnesemia. Am J Med Scis. 2009;338:22–27. doi: 10.1097/MAJ.0b013e3181aaee4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak IT, Chmielinska JJ, Kramer JH, Spurney CF, Weglicki WB. Loss of neutral Endopeptidase activity contributes to neutrophil activation and cardiac dysfunction during chronic hypomagnesemia: Protection by substance P receptor blockade. Exp Clin Cardiol. 2011;16:121–124. [PMC free article] [PubMed] [Google Scholar]
- Mak IT, Kramer JH, Weglicki WB. Suppression of neutrophil and endothelial activation by substance P receptor blockade in the Mg-deficient rat. Mag Res. 2003;16:91–97. [PubMed] [Google Scholar]
- Van Angelen AA, Glaudemans B, van der Kemp AW, Hoenderop JG, Bindels RJ. Cisplatin-induced injury of the renal distal convoluted tubule is associated with hypomagnesaemia in mice. Nephrol Dial Transplant. 2013;28:879–89. doi: 10.1093/ndt/gfs499. [DOI] [PubMed] [Google Scholar]
- Ledeganck KJ, Boulet GA, Bogers JJ, Verpooten GA, De Winter BY. The TRPM6/EGF pathway is downregulated in a rat model of cisplatin nephrotoxicity. PLoS One. 2013;8:e57016. doi: 10.1371/journal.pone.0057016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thebault S, Alexander RT, Tiel Groenestege WM, Hoenderop JG, Bindels RJ. EGF increases TRPM6 activity and surface expression. J Am Soc Nephrol. 2009;20:78–85. doi: 10.1681/ASN.2008030327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill PS, Wilcox CS. NADPH oxidases in the kidney. Antioxid Redox Signal. 2006;8:1597–1607. doi: 10.1089/ars.2006.8.1597. [DOI] [PubMed] [Google Scholar]
- Nistala R, Whaley-Connell A, Sowers JR. Redox control of renal function and hypertension. Antioxid Redox Signal. 2008;10:2047–89. doi: 10.1089/ars.2008.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao G, Lee KP, van der Wijst J, de Graaf M, van der Kemp A, Bindels RJ, Hoenderop JG. Methionine sulfoxide reductase B1 (MsrB1) recovers TRPM6 channel activity during oxidative stress. J Biol Chem. 2010;285:26081–7. doi: 10.1074/jbc.M110.103655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weglicki WB, Phillips TM. Pathobiology of magnesium deficiency: A cytokine/neurogenic inflammation hypothesis. Am J Physiol. 1992;263:R734R737. doi: 10.1152/ajpregu.1992.263.3.R734. [DOI] [PubMed] [Google Scholar]
- Weglicki WB, Mak IT, Kramer JH, Dickens BF, Cassidy MM, Stafford RE, Phillips TM. Role of free radicals and substance P in magnesium deficiency. Cardiovasc Res. 1996;31:677–682. [PubMed] [Google Scholar]
- Kramer JH, Mak IT, Phillips TM, Weglicki WB. Dietary Mg-intake influence circulating pro-inflammatory neuropeptide levels and loss of myocardial tolerance to postischemic stress. Exp Biol Med. 2003;228:665–673. doi: 10.1177/153537020322800604. [DOI] [PubMed] [Google Scholar]
- Ho WZ, Lai JP, Zhu XH, Uvaydova M, Douglas SD. Human monocytes and macrophages express substance P and neurokinin-1 receptor. J Immunol. 1997;159:5654–5660. [PubMed] [Google Scholar]
- Weglicki WB, Mak IT, Chmielinska JJ, Spurney CF, Kramer JH. Epidermal growth factor receptor-tyrosine kinase inhibition by erlotinib causes hypomagnesemia, oxidative stress and cardiac dysfunction (abstract) FASEB J. 2013;27:1128.18. [Google Scholar]
- Liu H, Mantyh PW, Basbaum AI. NMDA-receptor regulation of substance P release from primary afferent nociceptors. Nature. 1997;386:721–724. doi: 10.1038/386721a0. [DOI] [PubMed] [Google Scholar]
- Mishina M, Mori H, Araki K, Kushiya E, Meguro H, Kutsuwada T, Kashiwabuchi N, Ikeda K, Nagasawa M, Yamazaki M, Masaki H, Yamakura T, Morita T, Sakimura K. Molecular and functional diversity of the NMDA receptor channel. Ann N Y Acad Sci. 1993;707:136–152. doi: 10.1111/j.1749-6632.1993.tb38049.x. [DOI] [PubMed] [Google Scholar]
- Masu M, Nakajima Y, Moriyoshi K, Ishii T, Akazawa C, Nakanashi S. Molecular characterization of NMDA and metabotropic glutamate receptors. Ann N Y Acad Sci. 1993;707:153–164. doi: 10.1111/j.1749-6632.1993.tb38050.x. [DOI] [PubMed] [Google Scholar]
- Dehlin HM, Manteufel EJ, Monroe AL, Reimer MH, Jr, Levick SP. Substance P acting via the neurokinin-1 receptor regulates adverse myocardial remodeling in a rat model of hypertension. Int J Cardiol. 2013;168:4643–4651. doi: 10.1016/j.ijcard.2013.07.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasinoff BB. The cardiotoxicity and myocyte damage caused by small molecule anticancer tyrosine kinase inhibitors is correlated with lack of target specificity. Toxicol Appl Pharmacol. 2010;244:190–195. doi: 10.1016/j.taap.2009.12.032. [DOI] [PubMed] [Google Scholar]
- Hahn VS, Lenihan DJ, Ky B. Cancer therapy-induced cardiotoxicity: basic mechanisms and potential cardioprotective therapies. J Am Heart Assoc. 2014 Apr 22;3(2):e000665. doi: 10.1161/JAHA.113.000665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Kerkelä R, Force T. Mechanisms of cardiac dysfunction associated with tyrosine kinase inhibitor cancer therapeutics. Circulation. 2008;117:84–95. doi: 10.1161/CIRCULATIONAHA.108.776831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metro G, Finocchiaro G, Toschi L, Bartolini S, Magrini E, Cancellieri A, Trisolini R, Castaldini L, Tallini G, Crino L, Cappuzzo F. Epidermal growth factor receptor (EGFR) targeted therapies in non-small cell lung cancer (NSCLC) Rev Recent Clin Trials. 2006;1:1–13. doi: 10.2174/157488706775246157. [DOI] [PubMed] [Google Scholar]