

Abstract
Objective
Mercury is a ubiquitous environmental contaminant with toxic outcomes over a range of exposures. In this study, we investigated the effects of mercury exposure on early immune responses to coxsackievirus B3 (CVB3) infection in a murine model of autoimmune heart disease.
Materials and Methods
Female BALB/c mice, susceptible to CVB3-induced autoimmune myocarditis, were treated with mercuric chloride (200 μg/kg body weight every other day for 2 weeks) prior to infection with CVB3. Six hours post-infection, immune cells were isolated from the spleen and peritoneum for flow cytometry, gene expression, and cytokine profiling. Thirty-five days post-infection, hearts were collected for histological examination of immune cell infiltration.
Results
As for male mice, mercury exposure significantly increased autoimmune myocarditis and immune infiltration into the heart. During the innate response six hours post-infection, mercury increased expression of co-stimulatory molecules and innate immune receptors on peritoneal macrophages. At the same time point, the alternatively-activated macrophage gene, arginase, was increased while the classically-activated macrophage gene, inducible nitric oxide synthase, was unaffected. Expression of activation markers were decreased on peritoneal B cells with mercury exposure while T cells were unaffected. Mercury increased production of pro-inflammatory mediators in the spleen. Macrophage-recruiting chemokines and activating cytokines such as CCL2, CCL4, and IL-6 were increased with mercury following CVB3 infection.
Conclusions
Thus, mercury treatment exacerbates autoimmune myocarditis in female mice and alters early innate signaling on peritoneal macrophages. Mercury also modulates the cytokine profile in the spleen toward a macrophage-activating milieu, and upregulates alternatively-activated macrophage genes, providing evidence that mercury exposure promotes inflammation in the context of infection.
Keywords: mercury, innate immunity, cytokine, coxsackievirus, autoimmunity
Introduction
Mercury is a heavy metal with well-defined neurotoxic properties and increasing evidence for immunotoxic effects. Mercury exposure is of global public health importance and many individuals in the US and worldwide are exposed at doses exceeding the level of concern [1]. Exposure to mercury compounds can occur through occupational exposures, primarily inorganic mercury (iHg) [2], or environmentally through consumption of fish contaminated by methyl mercury (MeHg) [3]. An epidemiological study found an association between clinically diagnosed systemic lupus erythematosus (SLE) and self-reported occupational exposure to mercury primarily in those working with mercury-based dental amalgams [4]. We, and others, have reported that exposure to mercury increases serum biomarkers of autoimmune dysfunction including anti-nuclear (ANA) and anti-nucleolar autoantibodies (ANoA) [5–8], thyroid autoantibodies [9], and other autoantibodies such as anti-glutathione S-transferase autoantibody [10].
Many studies in rats and mice have demonstrated that relatively high dose exposure to mercury influences the immune system resulting in autoimmunity. In certain inbred strains of mice mercury compounds induce a lupus-like autoimmune disease in females [11–14]. In lupus-prone female BXSB mice, low dose mercury can accelerate kidney disease [15] and pretreatment with low dose mercury accelerates disease in the chronic Graft-versus-Host model of lupus in female, but not in male mice [16]. We recently reported that pretreatment with low dose mercury increases the severity of chronic autoimmune myocarditis and the prevalence of dilated cardiomyopathy in the coxsackievirus B3 (CVB3) and experimental autoimmune myocarditis models [17, 18]. However, the precise mechanisms for enhanced disease remain unclear.
In this study, we explore the hypothesis that the immunomodulatory effects of mercury are elicited in the context of genetic predisposition and co-exposure to a triggering event by using CVB3 as a trigger and studying the early innate immune response to mercury and CVB3 co-exposure. Previously, we found that in order for mercury to increase chronic myocarditis and dilated cardiomyopathy, mercury exposure had to occur prior to CVB3 infection [17]. Thus, we hypothesize that mercury modulates the early innate immune response to CVB3 infection and thereby sets in place a cascade of events ending in increased severity of myocarditis and prevalence of dilated cardiomyopathy. We showed that the innate immune response to CVB3 directs the progression to acute myocarditis and dilated cardiomyopathy [19–21], suggesting that alterations in the innate immune response by mercury could have long-term effects. To test this hypothesis, we examined the early innate immune response of peritoneal immune cells and gene expression/cytokine levels in the spleen at 6 hours post-infection. We confirmed that mercury exposure exacerbates chronic autoimmune myocarditis in female BALB/c mice as in our previous report in males. In addition, we have shown that mercury exposure without CVB3 infection does not induce myocarditis in female BALB/c mice [17].
Materials and methods
Treatment protocol
Female BALB/cJ (BALB/c) mice (6–8 weeks old) were purchased from Jackson Laboratory (Bar Harbor, ME) and were pretreated with mercuric chloride (HgCl2, 200μg/kg body weight every other day for 2 weeks, for a total of 8 doses, 100μl per mouse) by subcutaneous (sc) injection before infection with CVB3. This is the dosing regimen most commonly used in studies of mercury-induced immunotoxicity [22]. Female mice were used in this study because most published studies of the immune response to mercury have been conducted in female mice, and female BALB/c mice respond with a similar innate immune response to CVB3 infection in many respects, but at a lower level [19, 20]. Control animals were treated with PBS (vehicle, 100μl per mouse) following the same schedule of dosing prior to infection. Mice were maintained under pathogen-free conditions in the animal facility at Johns Hopkins University School of Medicine or the University of South Carolina School of Medicine, and approval was obtained from the Animal Care and Use Committees of the Johns Hopkins Bloomberg School of Public Health and the University of South Carolina for all procedures.
Infection with CVB3
Five days after the last mercury or vehicle treatment, mice were inoculated by intraperitoneal (ip) injection with 103 plaque-forming units (PFU) of a heart-passaged stock of CVB3 that contained cardiac proteins (Nancy strain; American Type Culture Collection) [23, 24]. We include this brief wash-out period to allow the immune system time to recover and respond to the mercury exposure. Individual experiments were conducted at least three times with 15 mice per treatment group. In two of these experiments, an additional 10 mice per treatment group were kept until day 35 post-infection for examination of chronic autoimmune myocarditis.
Examination of chronic autoimmune myocarditis
Thirty-five days post-infection, a subset of mice were euthanized by cervical dislocation under anesthesia and hearts were cut longitudinally, fixed in 10% phosphate-buffered formalin, and embedded in paraffin. Sections were cut 5 μm thick at various depths in the tissue section and stained with hematoxylin and eosin (H&E) to determine the level of inflammation. Sections were examined by two independent investigators in a blinded manner. Myocarditis was assessed as the percentage of the heart section with inflammation compared with the overall size of the heart section, with the aid of a microscope eyepiece grid, as described previously [25].
Evaluation of cellular phenotypes
At 6 hours post-infection, animals were euthanized by cervical dislocation under anesthesia and peritoneal lavage was performed by standard methods [20]. Lavage samples were pooled from 3 mice from a total of 15 mice per treatment group (resulting in N=5 per treatment) and mast cells and macrophages were separated from peritoneal lavage cells using anti-CD117 or anti-FITC (to cells stained with anti-F4/80 FITC) paramagnetic beads on a magnetic column (Miltenyi Biotec), as previously described [20]. Six hours was chosen because previous studies have found this to be the optimal time for change in the cellular markers of interest in these cells in the peritoneum [26, 19, 20]. Pooled immune cells were stained with the following monoclonal antibodies in various 5-color combinations (BD Pharmingen or eBioscience) diluted in 1% FBS in PBS: F4/80 (clone BM8), CD11b (clone M1/70), CD3 (clone 17A2), CD11c (clone HL3), CD19 (clone 1D3), toll-like receptor (TLR)-4 (clone MTS5510), CD25 (clone 7D4), CD80 (B7-1, clone 16-10A1), CD86 (B7-2, clone GL1), CD117 (clone 2B8), CTLA-4 (CD152, clone UC10-4B9), T cell Ig mucin (Tim)-3 (clone 8B.2C12). For intracellular staining, cells were fixed and permeabilized using BD Cytofix/perm (BD Pharmingen). Cell fluorescence was measured using a LSR II flow cytometer (BD Biosciences) or FACSAria (BD Bioscience) and data analyzed using FlowJo software (Treestar, Inc). The experiments were conducted four times with 15 mice/treatment group.
Cytokine measurement
The spleen was also removed for cytokine analyses. The spleen was cut in half and frozen in dry ice immediately after harvest and stored at −80°C until homogenized (or utilized for RNA isolation, see section 2.5 below). Spleens were weighed, thawed, and homogenized at 10% w/v in 2% fetal bovine serum/minimal essential medium, and supernatants were stored at −80°C until used in cytokine assays. Cytokines were measured in tissue supernatants using bead-based multiplex cytokine kits (Bio-Plex; Bio-Rad), according to manufacturer's instructions. The limits of detection for the cytokines were as follows: interleukin (IL)-1α, 1.3 pg/ml; IL-1β, 3.0 pg/ml; IL-3, 1.3 pg/ml; IL-4, 2.4 pg/ml; IL-5, 1.7 pg/ml; IL-6, 1.2 pg/ml; IL-9, 1.4 pg/ml; IL-10, 1.0 pg/ml; IL-12/23 p40, 1.2 pg/ml; IL-12 p70, 2.5 pg/ml; IL-13, 1.6 pg/ml; IL-17a, 1.4 pg/ml; IFN-γ, 2.0 pg/ml; eotaxin, 1.7 pg/ml; granulocyte colony-stimulating factor (G-CSF), 1.7 pg/ml; granulocyte-macrophage colony-stimulating factor (GM-CSF), 1.6 pg/ml; CCL2, 1.7 pg/ml; CCL3, 1.6 pg/ml; CCL4, 1.2 pg/ml; regulated upon activation normal T-cell expressed and presumably secreted (RANTES), 1.0 pg/ml; tumor necrosis factor (TNF)-α, 1.7 pg/ml. Cytokines were expressed as pg/g of spleen tissue ± SEM.
qPCR gene expression analysis
Spleen tissue samples were placed into RNAlater©-ice (Invitrogen; Carlsbad, CA) according to manufacturer's instructions. Tissues were soaked in at −20°C for 16 hours, removed from RNAlater©-ice, and then stored at −20°C until use. Tissues were homogenized and total RNA was isolated using RNeasy isolation kit (Qiagen; Valencia, CA) according to manufacturer's instructions. RNA quantity and quality was assessed by 260/280 ratio. Primers used were designed through NCBI primer-blast for Rplp0 (house-keeping gene), Arg1, and Nos2 and purchased from Integrated DNA Technologies (Coralville, IA). Primer sequences are presented in Table 1. Gene expression was analyzed by real time qPCR on a CFX Connect (Bio-Rad) using SensiFast SYBR No-ROX One-Step kit (Bioline; Randolph, MA) with the following protocol: 1 cycle at 45°C for 10 minutes, 1 cycle at 95°C for 1 minutes, 35 cycles of 95°C for 5 seconds, 60°C for 10 seconds, and 72°C for 5 seconds, followed by a melt curve. qPCR results were analyzed using Bio-Rad CFX Manager software (Bio-Rad). Data are expressed as relative normalized expression: ΔΔCq for each gene of interest relative to the house-keeping gene.
Table 1.
qPCR primer sequences.
| Primer sequence (5' – 3') | ||
|---|---|---|
| Argl | forward | CTG TGG GGA AAG CCA ATG AA |
| reverse | CAG CAC CAC ACT GAC TCT TC | |
| Nos2 | forward | GAA GTT CAG CAA CAA CCC CA |
| reverse | TTC AAG ATA GGG AGC TGC GA | |
| RplpO (house-keeping) | forward | CGT TGG AGT GAC ATC GTC TTT A |
| reverse | GGA TGA TCT TGA GGA AGT AGT TGG |
Statistical analysis
The data were analyzed by the Student's t-test (significance of observed differences between treatment groups) for normally distributed data. The Mann-Whitney U test was used to evaluate nonparametric data comparing two groups. Differences were considered significant at p<0.05.
Results
Mercury exacerbates chronic autoimmune myocarditis in female mice
Mercury exposure significantly increased chronic CVB3-induced myocarditis at day 35 post-infection (Figure 1). Treatment of mice with mercury or PBS (vehicle) alone did not result in the development of myocarditis or immune infiltration into the heart. We previously reported similar findings in male mice, which are susceptible to more severe myocarditis in response to CVB3 [17].
Figure 1. Mercury exposure exacerbates chronic autoimmune myocarditis.
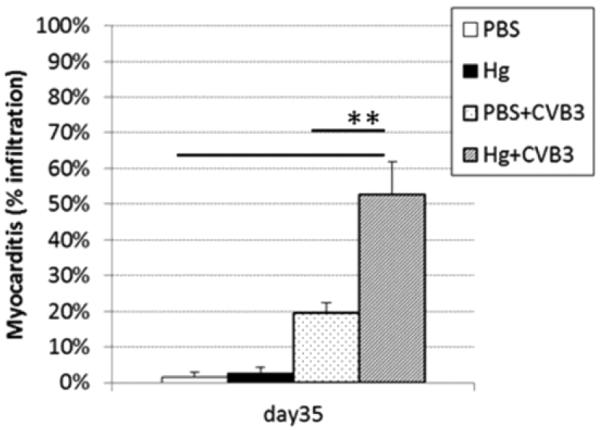
PBS-treated (vehicle control) and mercury-treated mice were compared for the percent inflammation in the heart at day 35 post-infection. Mice received 200 μl HgCl2 at 200 μg/kg in PBS or vehicle alone every other day for 2 weeks. Data shown are the mean ± SEM of 8–10 mice per treatment group. Data are from one representative experiment of two. *indicates p<0.05 between groups.
Mercury increases co-stimulatory and innate signaling molecule expression on macrophages
Activation of the innate immune system is necessary for protection from infection and the development of an adaptive immune response. Due to the small number of macrophages and mast cells present in the peritoneal lavage at 6 hours post-infection, we pooled subsets of the samples (cells isolated from 3 mice) by treatment group, resulting in N=5 per treatment group for flow cytometric analyses. Further, we sorted cells for macrophage positivity using paramagnetic beads conjugated to F4/80 antibody. To verify sorting, we stained cells with CD11b and demonstrate a high percent of macrophage cells (Figure 2E). Similar enrichment for mast cells was used on the F4/80negative fraction (data not shown).
Figure 2. Mercury exposure modulates co-stimulatory and signaling molecules on immune cells.

Peritoneal (A) macrophages, (B) mast cells, (C) B cells, and (D) T cells were isolated from PBS (vehicle) and mercury-treated mice in the absence of or six-hours after CVB3 infection. Cells were analyzed by flow cytometry for expression of the molecules shown. Results for pooled (3 mice each) samples from 15 mice per treatment group are shown as percent of total sorted and gated cells (gated cells are identified on the y-axis). Representative histograms are shown for peritoneal macrophages paramagnetically sorted for F4/80 positivity and stained for CD11b (E). Within the CD11b gate shown in (E), cells were determined to be positive for (F) CD80, (G) CD86, or (H) TLR4. Similar sorting and gating parameters were utilized for the other cell subsets (B-D). Data are from one representative experiment of four. Bars represent the means of each group ± SEM. *indicates p<0.05 between groups.
Exposure to mercury alone has no effect on the total number of macrophages in the peritoneum (Table 2) nor does expression of the macrophage markers we examined (PBS and Hg groups in Figure 2A) change. However, mercury exposure combined with viral infection increases the percentage of macrophages expressing the co-stimulatory molecule CD86 (Figure 2A, G) or the innate receptor TLR4 (PBS+CVB3 and Hg+CVB3 groups in Figure 2A, H). While the total number of CD86+ macrophages is not different between these two groups, the number of TLR4+ macrophages is significantly reduced (Table 3).
Table 2.
Mercury exposure changes total cell numbers in the absence of CVB3 infection.
| PBS (average total cell # ±SD) | Mercury (average total cell # ±SD) | ||
|---|---|---|---|
| Macrophages (CD11b+) | CD80+ | 4027 ±355 | 3312 ±642 |
| CD86+ | 4568 ±327 | 4622 ±821 | |
| TLR4+ | 1797 ±261 | 1628 ±186 | |
| Mast cells (CD117+) | CD80+ | 935 ±215 | 1048 ±156 |
| CD86+ | 51 ±11 | 130 ±25 * | |
| TLR4+ | 161 ±45 | 358 ±82 | |
| B cells (CD19+) | CD80+ | 6685 ±798 | 1882 ±256 * |
| CD86+ | 760 ±239 | 197 ±82 | |
| TLR4+ | 3554 ±1039 | 798 ±177 * | |
| T cells (CD4+) | TLR4+ | 28 ±7 | 16 ±4 |
| CD25+ | 134 ±10 | 144 ±30 | |
| CTLA4+ | 144 ±38 | 323 ±140 |
indicates significant differences between groups
Table 3.
Mercury changes total cell numbers with CVB3 infection.
| PBS (average total cell # ±SD) | Mercury (average total cell # ±SD) | ||
|---|---|---|---|
| Macrophages (CD11b+) | CD80+ | 1153 ±266 | 1592 ±286 |
| CD86+ | 568 ±150 | 994 ±177 | |
| TLR4+ | 196 ±45 | 563 ±102 * | |
| Mast cells (CD117+) | CD80+ | 2149 ±196 | 323 ±49 * |
| CD86+ | 5 ±1 | 4 ±1 | |
| TLR4+ | 157 ±10 | 489 ±124 * | |
| B cells (CD19+) | CD80+ | 1617 ±103 | 19 ±8 * |
| CD86+ | 213 ±16 | 30 ±7 * | |
| TLR4+ | 68 ±15 | 2 ±1 * | |
| T cells (CD4+) | TLR4+ | 0 ±0 | 0 ±0 |
| CD25+ | 4 ±1 | 3 ±1 | |
| CTLA4+ | 10 ±1 | 12 ±3 |
indicates significant differences between groups
Mercury decreases regulatory co-stimulatory but increases innate signaling molecules on mast cells
We found that mercury exposure decreases the regulatory co-stimulatory molecule CD80 on mast cells (Figure 2B) both in the presence and absence of viral infection. The total number of CD80+ mast cells is not changed between treatment groups (Table 2). These data are consistent with our previous findings that reduction in CD80 expression is associated with reduced CTLA-4 levels in CD4+ T cells and thus, decreased regulation [20].
Conversely, TLR4 and Tim-3 molecule expression increases with mercury in the presence of viral infection (PBS+CVB3 and Hg+CVB3 groups in Figure 2B). At the same time, the total number of TLR4+ mast cells also increases with mercury and viral infection (Table 3), but not in the absence of infection. These results are consistent with previous studies which determined that increased TLR4 on innate immune cells was associated with increased myocarditis in male mice [25, 19]. The ability of mercury to increase TLR4 expression on mast cells and macrophages, which promote cardiac remodeling and fibrosis, provides a mechanism for mercury pretreatment exacerbation of chronic inflammatory dilated cardiomyopathy [17].
Mercury exposure decreases co-stimulatory and innate signaling molecule expression on B cells but does not affect T cells
Regardless of the presence or absence of CVB3 infection, we found that mercury decreases CD80, CD86, and TLR4 expression on B cells (Figure 2C). The total number of B cells decreases with mercury exposure with and without the presence of virus (Tables 2 and 3). T cells were unaffected by mercury exposure (Figure 2D).
Mercury modulates cytokines in the spleen
Because mercury has previously been shown to increase by Th1 and Th2 cytokine levels in animal models of mercury-induced autoimmunity [27], we examined a wide panel of cytokines in the spleen using multiplex kits. Mercury exposure on its own induces elevated CCL4, and significantly decreases TNF-α, IL-1β, and RANTES (PBS and Hg groups in Figure 3).
Figure 3. Mercury exposure modulates cytokine levels in spleen.

PBS (vehicle) and mercury-treated mice in the absence of or six-hours after CVB3 infection were compared for the level of cytokines. Spleens were collected, homogenized, and homogenates analyzed for cytokine levels by bead-based multiplex assay as previously described. Data are expressed as mean cytokine level (pg)/g spleen tissue ± SEM of 15 mice per treatment group. Data are shown from one representative experiment of four. *indicates p<0.05 between groups.
Six hours post-infection with CVB3, CCL4 remains elevated and RANTES remains decreased with mercury exposure; however TNF-α and IL-1β are not different between treatment groups (PBS+CVB3 and Hg+CVB3 groups in Figure 3). The pro-inflammatory cytokines IL-6 and CCL2 are significantly increased with mercury in the context of viral infection. Mercury exposure also significantly decreases cytokines associated with Th2 responses such as IL-9 and IL-13, but only in the presence of virus (PBS+CVB3 and Hg+CVB3 groups in Figure 3).
Mercury pretreatment increases alternatively-activated macrophage gene expression
We previously demonstrated that mercury pretreatment increases the number of macrophages in the heart at 12 days post-infection [17]. Since flow cytometric analysis demonstrated an increase in the number and activity of macrophages in the peritoneum and macrophage-activated/associated cytokine and chemokine levels were also increased in the spleen, we examined gene expression of two genes specific to classically- and alternatively-activated macrophages. Specifically, we examined inducible nitric oxide synthase (iNOS) and arginase (Arg), as markers of classically- and alternatively-activated macrophages, respectively. As expected, CVB3 infection increases iNOS expression, but this effect does not change with mercury exposure (Figure 4A). Alternatively, Arg expression was significantly increased with mercury exposure and CVB3 infection (Figure 4B).
Figure 4. Mercury exposure modulates alternatively-activated macrophage gene expression in the spleen.
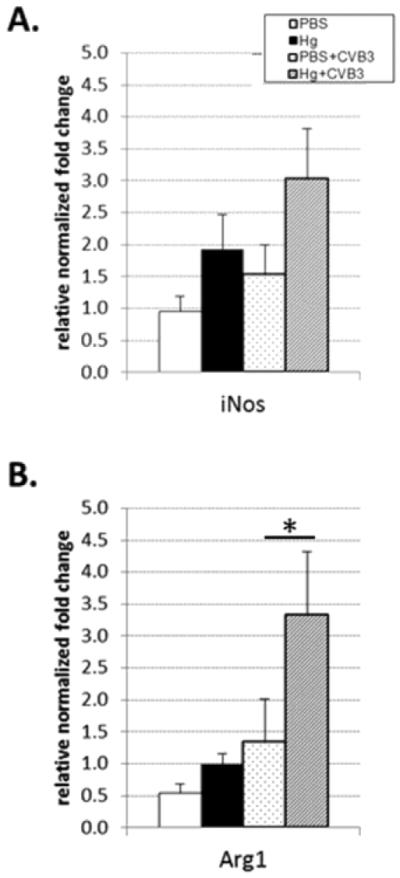
Spleens were isolated from PBS (vehicle) and mercury-treated mice in the absence of or six-hours after CVB3 infection. RNA was isolated and expression of genes analyzed by qPCR. Data are expressed as relative normalized fold change for gene of interest compared to house-keeping gene for 15 mice per treatment group. Data are shown from one representative experiment of three. *indicates p<0.05 between groups.
Discussion
Mercury has been shown to exacerbate autoimmunity in several models including lupus-prone mouse strains, in mice where autoimmune disease is induced by treatment with allografts [16], antigenic stimuli [18], or by infection [28]. These effects are clearly sex-dependent since the disease severity varies with the sex of the mouse in these models. The mechanism(s) by which mercury interacts with the immune system is not fully understood, although it has been suggested that both T and B cells play important roles in driving mercury-induced effects [29, 30, 13, 31, 32]. In the lupus-like mouse model of mercury-induced autoimmune disease, activation of the innate immune response, specifically TLR4 signaling through LPS stimulation, exacerbates disease [33]. In the collagen-induced arthritis model, mercury exposure during the induction phase, or innate immune response, also exacerbates the disease [34]. These studies indicate that mercury alters the innate immune response to infection or adjuvant treatment resulting in increased disease.
In this study, we examined the effects of low dose mercury exposure on the early innate immune response to CVB3 infection. Studies have demonstrated the importance of the innate immune response to CVB3 infection in the progression of inflammatory heart disease [26, 19, 35, 21, 36]. Specifically, the production of innate cytokines, degranulation of mast cells, and induction of pro-fibrotic alternatively-activated macrophages have been shown to be critical for exacerbation of disease. We found that in the presence of an active viral infection, mercury up-regulated the expression of TLR4 on both macrophages and mast cells. These results are consistent with those of Abedi-Valugerdi et al. [33] in that the effects of mercury and either virus (in our studies) or LPS (in their studies) are acting on similar pathways downstream of LPS signaling (i.e. TLR4) [37]. Furthermore, myocardial inflammation is significantly decreased in TLR4 signaling defective mice indicating the importance of this pathway in promoting inflammation following infection [25].
We found up-regulated expression of CD86 on macrophages but down-regulated expression of CD80 and CD80/CD86 expression on mast cells and B cells, respectively, in the presence of viral infection and mercury exposure. We have previously observed increased numbers of macrophages in the heart with mercury pretreatment [17] during acute disease and others have found that CD80/CD86 co-stimulatory molecules are required for mercury-induced autoimmune disease [38]. Importantly, we found that mercury exposure increased the expression of arginase, a marker of alternatively-activated macrophages, in the spleen following CVB3 infection. This finding supports our hypothesis that the mercury-induced exacerbation of CVB3-induced cardiac fibrosis and dilated cardiomyopathy is at least partially driven by upregulation of alternatively-activated macrophages.
Mercury did not change the expression of regulatory or activation molecules on T cells. However, while the peritoneal T cell phenotypes did not change with mercury exposure, T cell-mediated cytokines such as IL-6 and IL-13 did change in the spleen.
Previous studies have shown that cytokines play an important role in the development and severity of autoimmune myocarditis. Elevations in the level of IL-1β and IL-18 in the heart predict a more severe acute disease [25] and IL-6 [39] and IL-12 [40] up-regulate autoimmune myocarditis. IL-17A is required for progression to dilated cardiomyopathy in the experimental autoimmune myocarditis model, but not necessary for acute disease [41]. CCL2, CCL3 [42] and IL-4 [43] are important in the induction of myocarditis. Whereas, IL-10 [44], IL-13 [35], and IFN-γ [45, 40, 46] are protective in autoimmune myocarditis.
The increases in IL-6 that we observed in mercury exposed CVB3 infected mice are consistent with previous findings on macrophage-lineage microglia in vitro in which mercury exposure increased IL-6 levels in primary neuronal cultures [47]. Increased levels of pathogenic IL-6 and CCL2 combined with the loss of regulation provided by the decreased IL-13 levels provide some clues to the mechanism(s) by which mercury exposure exacerbates autoimmune myocarditis [17].
In this study we focused on the effects of mercury on the early immune response to CVB3 infection. Our study differs from other studies of mercury in that we utilize a lower dose of mercury and a shorter dosing time. In addition, we used a non-lethal myocarditic CVB3 that had been passaged through the heart and contained cardiac proteins, making the model more similar to adjuvant-induced myocarditis [48, 23]. There is limited information from the literature on potential mechanisms of interaction between mercury and CVB3. Mercury treatment itself does not induce myocarditis or dilated cardiomyopathy [18, 17]. Furthermore, mercury treatment does not reactivate CVB3 in the heart during chronic myocarditis since infectious virus cannot be detected at day 35 pi [17]. In studies on human peripheral blood mononuclear cells in vitro we found that cytokine responses to mercury required cellular activation [49]. The findings from this study are consistent with our hypothesis that mercury induces changes in the immune response that require a trigger event in order to be elucidated; and that the effect of co-exposure with mercury and trigger (i.e. CVB3) are influenced by host factors. Overall, our findings show that the effects of mercury pretreatment on the early innate immune response to viral infection are complex, with some cells such as macrophages activated while others such as mast cells and B cells are suppressed. These findings emphasize the importance of understanding the particular mechanisms of disease induction for different autoimmune diseases (i.e. TLR4/CD86 for myocarditis and Th2 response/B cells for lupus) in order to understand the effect of mercury exposure on disease outcome.
Acknowledgements
This work was supported by National Institutes of Health [T32 ES07141 and K99/R00 ES015426 to J.F.N.; R01 HL087033 and R01 HL111938 to D.F.] and the Heinz Family Foundation. The authors would like to thank Dr. Sylvia Frisancho-Kiss for her advice on innate flow cytometry.
References
- 1.Mahaffey KR, Clickner RP, Bodurow CC. Blood organic mercury and dietary mercury intake: national health and nutrition examination survey, 1999 and 2000. Environ Health Perspect. 2004;112(5):562–70. doi: 10.1289/ehp.6587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gochfeld M. Cases of mercury exposure, bioavailability, and absorption. Ecotoxicol Environ Saf. 2003;56(1):174–9. doi: 10.1016/s0147-6513(03)00060-5. [DOI] [PubMed] [Google Scholar]
- 3.NRC . Toxicological effects of methyl mercury. National Academy Press; Washington, DC: 2000. [Google Scholar]
- 4.Cooper GS, Parks CG, Treadwell EL, St Clair EW, Gilkeson GS, Dooley MA. Occupational risk factors for the development of systemic lupus erythematosus. J Rheumatol. 2004;31(10):1928–33. [PubMed] [Google Scholar]
- 5.Silva IA, Nyland JF, Gorman A, Perisse A, Ventura AM, Santos EC, et al. Mercury exposure, malaria, and serum antinuclear/antinucleolar antibodies in amazon populations in Brazil: a cross-sectional study. Environ Health. 2004;3(1):11–22. doi: 10.1186/1476-069X-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alves MF, Fraiji NA, Barbosa AC, De Lima DS, Souza JR, Dorea JG, et al. Fish consumption, mercury exposure and serum antinuclear antibody in Amazonians. Int J Environ Health Res. 2006;16(4):255–62. doi: 10.1080/09603120600734147. [DOI] [PubMed] [Google Scholar]
- 7.Nyland JF, Fillion M, Barbosa F, Jr, Shirley DL, Chine C, Lemire M, et al. Biomarkers of methyl mercury exposure immunotoxicity among fish consumers in Amazonian Brazil. Environ Health Perspect. 2011;119:1733–8. doi: 10.1289/ehp.1103741. doi:10.1289/ehp.1103741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gardner RM, Nyland JF, Silva IA, Ventura AM, de Souza JM, Silbergeld EK. Mercury exposure, serum antinuclear/antinucleolar antibodies, and serum cytokine levels in mining populations in Amazonian Brazil: a cross-sectional study. Environ Res. 2010;110(4):345–54. doi: 10.1016/j.envres.2010.02.001. doi:10.1016/j.envres.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gallagher CM, Meliker JR. Mercury and thyroid autoantibodies in U.S. women, NHANES 2007–2008. Environ Int. 2012;40:39–43. doi: 10.1016/j.envint.2011.11.014. doi:S0160-4120(11)00271-6 [pii] 10.1016/j.envint.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 10.Motts JA, Shirley DL, Silbergeld EK, Nyland JF. Novel biomarkers of mercury-induced autoimmune dysfunction: A cross-sectional study in Amazonian Brazil. Environ Res. 2014;132:12–8. doi: 10.1016/j.envres.2014.03.024. doi:10.1016/j.envres.2014.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hultman P, Nielsen JB. The effect of dose, gender, and non-H-2 genes in murine mercury-induced autoimmunity. J Autoimmun. 2001;17(1):27–37. doi: 10.1006/jaut.2001.0521. [DOI] [PubMed] [Google Scholar]
- 12.Monestier M, Losman MJ, Novick KE, Aris JP. Molecular analysis of mercury-induced antinucleolar antibodies in H-2S mice. J Immunol. 1994;152(2):667–75. [PubMed] [Google Scholar]
- 13.Nielsen JB, Hultman P. Mercury-induced autoimmunity in mice. Environ Health Perspect. 2002;110(Suppl 5):877–81. doi: 10.1289/ehp.02110s5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson CJ, White HJ, Rose NR. Murine strain differences in response to mercuric chloride: antinucleolar antibodies production does not correlate with renal immune complex deposition. Clin Immunol Immunopathol. 1997;83(2):127–38. doi: 10.1006/clin.1997.4336. [DOI] [PubMed] [Google Scholar]
- 15.Pollard KM, Pearson DL, Hultman P, Deane TN, Lindh U, Kono DH. Xenobiotic acceleration of idiopathic systemic autoimmunity in lupus- prone bxsb mice. Environ Health Perspect. 2001;109(1):27–33. doi: 10.1289/ehp.0110927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Via CS, Nguyen P, Niculescu F, Papadimitriou J, Hoover D, Silbergeld EK. Low-dose exposure to inorganic mercury accelerates disease and mortality in acquired murine lupus. Environ Health Perspect. 2003;111(10):1273–7. doi: 10.1289/ehp.6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nyland JF, Fairweather D, Shirley DL, Davis SE, Rose NR, Silbergeld EK. Low-dose inorganic mercury increases severity and frequency of chronic coxsackievirus-induced autoimmune myocarditis in mice. Toxicol Sci. 2012;125(1):134–43. doi: 10.1093/toxsci/kfr264. doi:10.1093/toxsci/kfr264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Silbergeld EK, Silva IA, Nyland JF. Mercury and autoimmunity: implications for occupational and environmental health. Toxicol Appl Pharmacol. 2005;207(2 Suppl):282–92. doi: 10.1016/j.taap.2004.11.035. [DOI] [PubMed] [Google Scholar]
- 19.Frisancho-Kiss S, Davis SE, Nyland JF, Frisancho JA, Cihakova D, Barrett MA, et al. Cutting edge: cross-regulation by TLR4 and T cell Ig mucin-3 determines sex differences in inflammatory heart disease. J Immunol. 2007;178(11):6710–4. doi: 10.4049/jimmunol.178.11.6710. [DOI] [PubMed] [Google Scholar]
- 20.Frisancho-Kiss S, Nyland JF, Davis SE, Barrett MA, Gatewood SJ, Njoku DB, et al. Cutting edge: T cell Ig mucin-3 reduces inflammatory heart disease by increasing CTLA-4 during innate immunity. J Immunol. 2006;176(11):6411–5. doi: 10.4049/jimmunol.176.11.6411. [DOI] [PubMed] [Google Scholar]
- 21.Onyimba JA, Coronado MJ, Garton AE, Kim JB, Bucek A, Bedja D, et al. The innate immune response to coxsackievirus B3 predicts progression to cardiovascular disease and heart failure in male mice. Biol Sex Differ. 2011;2:2. doi: 10.1186/2042-6410-2-2. doi:2042-6410-2-2 [pii] 10.1186/2042-6410-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lawrence DA, McCabe MJ., Jr Immunomodulation by metals. Int Immunopharmacol. 2002;2(2–3):293–302. doi: 10.1016/s1567-5769(01)00180-1. [DOI] [PubMed] [Google Scholar]
- 23.Fairweather D, Rose NR. Coxsackievirus-induced myocarditis in mice: a model of autoimmune disease for studying immunotoxicity. Methods. 2007;41(1):118–22. doi: 10.1016/j.ymeth.2006.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fairweather D, Stafford KA, Sung YK. Update on coxsackievirus B3 myocarditis. Curr Opin Rheumatol. 2012;24(4):401–7. doi: 10.1097/BOR.0b013e328353372d. doi:10.1097/BOR.0b013e328353372d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fairweather D, Yusung S, Frisancho S, Barrett M, Gatewood S, Steele R, et al. IL-12 Receptor beta1 and Toll-Like Receptor 4 Increase IL-1beta- and IL-18-Associated Myocarditis and Coxsackievirus Replication. J Immunol. 2003;170(9):4731–7. doi: 10.4049/jimmunol.170.9.4731. [DOI] [PubMed] [Google Scholar]
- 26.Fairweather D, Frisancho S, Gatewood S, Njoku D, Steele R, Barrett M, et al. Mast cells and innate cytokines are associated with susceptibility to autoimmune heart disease following Coxsackievirus B3 infection. Autoimmunity. 2004;37:131–45. doi: 10.1080/0891693042000196200. [DOI] [PubMed] [Google Scholar]
- 27.Schiraldi M, Monestier M. How can a chemical element elicit complex immunopathology? Lessons from mercury-induced autoimmunity. Trends in Immunology. 2009;30(10):502–9. doi: 10.1016/j.it.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 28.Ilback NG, Wesslen L, Fohlman J, Friman G. Effects of methyl mercury on cytokines, inflammation and virus clearance in a common infection (coxsackie B3 myocarditis) Toxicol Lett. 1996;89(1):19–28. doi: 10.1016/s0378-4274(96)03777-0. [DOI] [PubMed] [Google Scholar]
- 29.Johansson U, Sander B, Hultman P. Effects of the murine genotype on T cell activation and cytokine production in murine mercury-induced autoimmunity. J Autoimmun. 1997;10(4):347–55. doi: 10.1006/jaut.1997.0149. [DOI] [PubMed] [Google Scholar]
- 30.Kono DH, Balomenos D, Pearson DL, Park MS, Hildebrandt B, Hultman P, et al. The prototypic Th2 autoimmunity induced by mercury is dependent on IFN- gamma and not Th1/Th2 imbalance. J Immunol. 1998;161(1):234–40. [PubMed] [Google Scholar]
- 31.Pollard KM, Hultman P. Effects of mercury on the immune system. Met Ions Biol Syst. 1997;34:421–40. [PubMed] [Google Scholar]
- 32.Hultman P, Bell LJ, Enestrom S, Pollard KM. Murine susceptibility to mercury. I. Autoantibody profiles and systemic immune deposits in inbred, congenic, and intra-H-2 recombinant strains. Clin Immunol Immunopathol. 1992;65(2):98–109. doi: 10.1016/0090-1229(92)90212-7. [DOI] [PubMed] [Google Scholar]
- 33.Abedi-Valugerdi M, Nilsson C, Zargari A, Gharibdoost F, DePierre JW, Hassan M. Bacterial lipopolysaccharide both renders resistant mice susceptible to mercury-induced autoimmunity and exacerbates such autoimmunity in susceptible mice. Clin Exp Immunol. 2005;141(2):238–47. doi: 10.1111/j.1365-2249.2005.02849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hansson M, Djerbi M, Rabbani H, Mellstedt H, Gharibdoost F, Hassan M, et al. Exposure to mercuric chloride during the induction phase and after the onset of collagen-induced arthritis enhances immune/autoimmune responses and exacerbates the disease in DBA/1 mice. Immunology. 2005;114(3):428–37. doi: 10.1111/j.1365-2567.2005.02105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cihakova D, Barin JG, Afanasyeva M, Kimura M, Fairweather D, Berg M, et al. Interleukin-13 protects against experimental autoimmune myocarditis by regulating macrophage differentiation. Am J Pathol. 2008;172(5):1195–208. doi: 10.2353/ajpath.2008.070207. doi:S0002-9440(10)61880-9 [pii] 10.2353/ajpath.2008.070207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaya Z, Afanasyeva M, Wang Y, Dohmen KM, Schlichting J, Tretter T, et al. Contribution of the innate immune system to autoimmune myocarditis: a role for complement. Nat Immunol. 2001;2(8):739–45. doi: 10.1038/90686. [DOI] [PubMed] [Google Scholar]
- 37.Fairweather D, Frisancho-Kiss S, Rose NR. Viruses as adjuvants for autoimmunity: evidence from Coxsackievirus-induced myocarditis. Rev Med Virol. 2005;15(1):17–27. doi: 10.1002/rmv.445. [DOI] [PubMed] [Google Scholar]
- 38.Bagenstose LM, Class R, Salgame P, Monestier M. B7-1 and B7-2 co-stimulatory molecules are required for mercury-induced autoimmunity. Clin Exp Immunol. 2002;127(1):12–9. doi: 10.1046/j.1365-2249.2002.01700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eriksson U, Kurrer MO, Schmitz N, Marsch SC, Fontana A, Eugster HP, et al. Interleukin-6-deficient mice resist development of autoimmune myocarditis associated with impaired upregulation of complement C3. Circulation. 2003;107(2):320–5. doi: 10.1161/01.cir.0000043802.38699.66. [DOI] [PubMed] [Google Scholar]
- 40.Fairweather D, Frisancho-Kiss S, Yusung SA, Barrett MA, Davis SE, Steele RA, et al. IL-12 protects against coxsackievirus B3-induced myocarditis by increasing IFN-gamma and macrophage and neutrophil populations in the heart. J Immunol. 2005;174(1):261–9. doi: 10.4049/jimmunol.174.1.261. [DOI] [PubMed] [Google Scholar]
- 41.Baldeviano GC, Barin JG, Talor MV, Srinivasan S, Bedja D, Zheng D, et al. Interleukin-17A is dispensable for myocarditis but essential for the progression to dilated cardiomyopathy. Circ Res. 2010;106(10):1646–55. doi: 10.1161/CIRCRESAHA.109.213157. [DOI] [PubMed] [Google Scholar]
- 42.Goser S, Ottl R, Brodner A, Dengler TJ, Torzewski J, Egashira K, et al. Critical role for monocyte chemoattractant protein-1 and macrophage inflammatory protein-1alpha in induction of experimental autoimmune myocarditis and effective anti-monocyte chemoattractant protein-1 gene therapy. Circulation. 2005;112(22):3400–7. doi: 10.1161/CIRCULATIONAHA.105.572396. doi:112/22/3400 [pii] 10.1161/CIRCULATIONAHA.105.572396. [DOI] [PubMed] [Google Scholar]
- 43.Afanasyeva M, Wang Y, Kaya Z, Park S, Zilliox MJ, Schofield BH, et al. Experimental Autoimmune Myocarditis in A/J mice Is an Interleukin-4-Dependent Disease with a Th2 Phenotype. Am J Pathol. 2001;159(1):193–203. doi: 10.1016/S0002-9440(10)61685-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaya Z, Dohmen KM, Wang Y, Schlichting J, Afanasyeva M, Leuschner F, et al. Cutting edge: a critical role for IL-10 in induction of nasal tolerance in experimental autoimmune myocarditis. J Immunol. 2002;168(4):1552–6. doi: 10.4049/jimmunol.168.4.1552. [DOI] [PubMed] [Google Scholar]
- 45.Fairweather D, Frisancho-Kiss S, Yusung SA, Barrett MA, Davis SE, Gatewood SJ, et al. Interferon-gamma protects against chronic viral myocarditis by reducing mast cell degranulation, fibrosis, and the profibrotic cytokines transforming growth factor-beta 1, interleukin-1 beta, and interleukin-4 in the heart. Am J Pathol. 2004;165(6):1883–94. doi: 10.1016/s0002-9440(10)63241-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barin JG, Talor MV, Baldeviano GC, Kimura M, Rose NR, Cihakova D. Mechanisms of IFNgamma regulation of autoimmune myocarditis. Exp Mol Pathol. 2010;89(2):83–91. doi: 10.1016/j.yexmp.2010.06.005. doi:S0014-4800(10)00089-4 [pii] 10.1016/j.yexmp.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sass JB, Haselow DT, Silbergeld EK. Methylmercury-induced decrement in neuronal migration may involve cytokine-dependent mechanisms: a novel method to assess neuronal movement in vitro. Toxicol Sci. 2001;63(1):74–81. doi: 10.1093/toxsci/63.1.74. [DOI] [PubMed] [Google Scholar]
- 48.Fairweather D, Rose NR. Models of coxsackievirus-B3-induced myocarditis: recent advances. Drug Discovery Today: Disease Models. 2004;1(4):381–6. [Google Scholar]
- 49.Gardner RM, Nyland JF, Evans SL, Wang SB, Doyle KM, Crainiceanu CM, et al. Mercury induces an unopposed inflammatory response in human peripheral blood mononuclear cells in vitro. Environ Health Perspect. 2009;117(12):1932–8. doi: 10.1289/ehp.0900855. [DOI] [PMC free article] [PubMed] [Google Scholar]