

Abstract
Background
Diabetes is a pandemic disease with a higher occurrence in minority populations. The molecular molecular mechanism to initiate diabetes-associated retinal angiogenesis remains largely unknown. We propose an inflammatory pathway of diabetic retinopathy in which macrophages in the diabetic eye provides TGFβ to retinal endothelial cells (REC) in the retinal microvasculature. In response to TGFβ, REC synthesize and secrete a pro-apoptotic BIGH3 (TGFβ-Induced Gene Human Clone 3) protein, which acts in an autocrine loop to induce REC apoptosis.
Methods
Rhesus monkey retinal endothelial cells (RhREC) were treated with dMCM (cell media of macrophages treated with high glucose and LDL) for apoptosis (TUNEL assays) and BIGH3 mRNA (qPCR) and protein (Western blots) expressions. Cells were also treated with TGFβ1 and 2 for BIGH3 mRNA and protein expression. Inhibition assays were carried out using antibodies for TGFβ1 and for BIGH3 to block apoptosis and mRNA expression. BIGH3 in cultured RhREC cells were identified by immunohistochemistry (IHC). Distribution of BIGH3 and macrophages in the diabetic mouse retina was examined with IHC.
Results
RhRECs treated with dMCM or TGFβ showed a significant increase in apoptosis and BIGH3 protein expression. Recombinant BIGH3 added to RhREC culture medium led to a dose-dependent increase in apoptosis. Antibodies (Ab) directed against BIGH3 and TGFβ, as well as TGFβ receptor blocker resulted in a significant reduction in apoptosis induced by either dMCM, TGFβ or BIGH3. IHC showed that cultured RhREC constitutively expressed BIGH3. Macrophage and BIGH3 protein were co-localized to the inner retina of the diabetic mouse eye.
Conclusion
Our results support a novel inflammatory pathway for diabetic retinopathy. This pathway is initiated by TGFβ released from macrophages, which promotes synthesis and release of BIGH3 protein by REC and REC apoptosis.
Keywords: BIGH3, macrophage, retinal endothelial cells, apoptosis, diabetic retinopathy, TGFβ
Introduction
Apoptosis of retinal pericyte and retinal endothelial cell has been a major focus of investigation on the cellular pathway of diabetic retinopathy. In an earlier report, diabetes and high glucose were shown to activate nuclear factor-κB in human retinal pericyte (but not in endothelial cell) leading to pericyte apoptosis (1–4). In a separate pathway independent of NF-κB, hyperglycemia causes mouse retinal pericyte apoptosis by the activation of PKC-δ and MARK to increase the expression of a protein tyrosine phosphatase (SHP-1) which results in PGDF receptor-β dephosphorylation (2). High glucose, transforming growth factor beta-1 (TGFβ1) and TGFβ–Induced Protein (BIGH3), also known as TGF-Beta-Induced Gene Human Clone 3, beta-ig, and keratoepithelin, produced apoptosis in cultured bovine retinal pericytes (3). In comparison to reports on cellular pathway of diabetic retinopathy associated with retinal pericyte apoptosis, Busik et al. proposed that retinal endothelial cells apoptosis may also be involved in the development of retinal angiogenesis and diabetic retinopathy. In this pathway, hyperglycemia causes other retinal cells to release inflammatory cytokines – IL1β and INFα which induces reactive oxygen species toxicity and apoptosis in retinal endothelial cells (4).
Diabetic retinopathy and nephropathy are known to be associated with chronic inflammation and macrophage activation.(5–7) Tissue macrophages constitutively secrete TGFβ1 (6), which is further increased in response to inflammatory stimuli(7) and following phagocytosis of apoptotic cells (5). TGFβ induces the synthesis and secretion of the BIGH3 protein from mesenchymal and other target cells(8, 9) leading to apoptosis. However, a direct link between metabolic stress, macrophage-derived TGFβ1 secretion and BIGH3 deposition in inflamed, diabetic ocular tissues has not been explored for a possible pathway on retinal vascular cells apoptosis in diabetic retinopathy.
The human BIGH3 gene is localized on chromosome 5q31. Human BIGH3 protein comprises 683 amino acid residues. In its secreted form BIGH3 has a reported molecular weight of 69 kDa (10). BIGH3 is a cell-adhesion class protein that has four fasciclin-1 like domains (Fas 1-4) homologous to fasciclin-1, a Drosophila nerve growth cone guidance molecule (8). There are several different sequences that in vitro are recognized as ligands for integrins, including integrins α3β1, αvβ3 and αvβ5 (11–14). Endothelial cells use αvβ5 in cytoplasmic signaling to mediate cell adhesion and migration,(15) suggesting that BIGH3 may provide a site for macrophage adhesion and retention. BIGH3 is expressed by a wide range of cell types: human corneal epithelial cells (13), human umbilical vein endothelial cells (16), osteoblasts(11), and vascular smooth muscle cells(17). It also functions as a substratum ligand for a number of different integrins on different cell types. In two separate reports, Han et al showed that the gene for BIGH3 protein is also a diabetes-risk gene affecting pancreatic β-islet cell proliferation based on results from a mouse (in-vitro and in-vivo KO) model and on human genetic analysis(18, 19).
Recently, we found that macrophage-conditioned medium is a potent stimulus of BIGH3 synthesis in cultured renal cells (LeBaron et al., unpublished data). In a preliminary study, we have also collected experimental evidence to show that these conditioned media, as well as TGFβ, induced overproduction of BIGH3 in retinal endothelial cells and apoptosis (Mondragon et al ARVO 2012). Subsequently, we performed detailed in-vitro analyses on the response of retinal capillary endothelial cells (RhREC) to macrophage-derived TGFβ and to the BIGH3 protein. Our results indicate that macrophage TGFβ increased BIGH3 mRNA and BIGH3 protein synthesis, which led to a dose-dependent increase of RhREC apoptosis. Using an in vivo model, we further confirm the co-localization of macrophages and the BIGH3 protein in the inner retina of the diabetic mice. Thus we propose that macrophage-associated increase in BIGH3 expression induces retinal endothelial cell apoptosis to weaken retinal capillaries leading to angiogenesis and diabetic retinopathy.
Methods
Cell Culture
Rhesus Retinal Endothelial Cells (RhREC) were purchased from ATCC (Cat No: CRL-1780, RF/6A). Cells were transformed at an early passage and were maintained in minimum essential media (MEM) as described previously(20).
Macrophage Conditioned Medium
Mononuclear cells were isolated from blood obtained from healthy human donors (South Texas Blood and Tissue Center), and mature human monocyte-derived macrophages (HMDM) were prepared as described previously.(21) To generate conditioned media, HMDM were pretreated for 24 hours with RPMI medium supplemented with 10% human AB serum (“healthy” condition) or RPMI medium supplemented with 10% human AB serum and with 25 mM D-glucose plus 100 mg/mL freshly isolated human low-density lipoprotein (“diabetic” condition). After the pre-incubation period, the HMDMs were washed and incubated for 24 hours in serum-free RPMI medium. Conditioned media from “healthy” macrophages (MCM), and macrophages cultured in “diabetic condition” (dMCM) were collected and centrifuged to remove any floating cells. Such conditioned media are hereafter referred to as either “MCM” or “dMCM” based on the medium in which HMDM were cultured. Aliquots of MCMs were kept at −20°C until use.
Transforming Growth Factor β
TGFβ1 and TGFβ2 and small chemical TGFβ1 receptor blocker were purchased from Sigma Aldrich (Cat No: T7039, T2815, SB-431542).
Animals
Female LDL-R−/− mice (B6.129S7-LdlrtmIHer/J, stock no. 002207) on a C57BL/6J background and C57BL/6J donor mice were obtained from The Jackson Laboratories (Bar Harbor, ME). After two weeks on a maintenance diet (MD, AIN-93G, BioServ), mice were rendered diabetic by intraperitoneal injection of STZ (50 mg × kg−1 × day−1) dissolved in citrate buffer (50 mM; pH=4.5) for five consecutive days and after two day’s rest, again for two consecutive days (22). Non-diabetic mice received a comparable volume of citrate buffer. To induce hypercholesterolemia, mice were fed a high fat diet (HFD) supplemented with fat (21% wt/wt) and cholesterol (0.15% wt/wt; AIN-76A, BioServ) beginning 3 weeks after the first STZ injection and continued for a total of 12 weeks. All experiments in the present study conformed to the ARVO (Association for Research in Vision and Ophthalmology) resolution on the humane use of animals in ophthalmic and vision research(23) and were approved by the UTHSCSA-IACUC (Institutional Animal Care and Use Committee).
Antibodies
BIGH3 bacterial fusion protein was used to immunize New Zealand white rabbits. The resultant anti-BIGH3 antiserum has been previously characterized (24). Pre-immune serum from the immunized animal was used as negative control where indicated in the individual experiments. Rat anti-CD68 antibody was obtained from ABD seroTec (Cat No. MCA1957T). Mouse anti-TGFβ1 antibody (clone 9016) was from R&D Systems.
ELISA
Quantitative enzyme linked immunosorbent assay (ELISA) was performed according to manufacturer’s instructions (R&D Systems Cat No: DB100B and DB-250) on different batches of MCM and dMCM to quantify the amount of TGFβ1 and TGFβ2.
Immunohistochemistry
Eyes from adult MD, HFD and diabetic mice were dissected after mice were euthanized by CO2 inhalation and cervical luxation (7). Eyes were embedded in paraffin and frozen at −80°C until use. Eyes were mounted in freezing medium and sections 10-μm thick were mounted on glass microscope slides and fixed for 30 minutes in 10% fresh paraformaldehyde in PBS. Fixed sections were rinsed with 20 mM glycine in PBS (Buffer B), blocked for 1 hour in 3% BSA in PBS, and incubated 12 hours with anti-BIGH3 (1:200) and anti-CD68 (1:300) in Buffer B. After rinsing, sections were incubated for 1 hour with anti-rabbit or anti-rat antibodies conjugated to Alexa Fluor 488 and Alexa Fluor 594 fluorescent dyes, respectively. Sections were again rinsed with PBS, and sealed with Vectashield and a glass coverslip. Unless indicated otherwise, all incubations were at ambient temperature, and all rinses were 3 × 15 minutes in Buffer B.
TUNEL Staining
Wells of a Lab-Tek 16-well chamber slide from Fisher Scientific (Cat No: 12-565-110N) were each seeded with 5 × 103 RhREC cells in 300 μL of MEM. After 24-hour incubation, the MEM was aspirated and replaced with either BIGH3 in MEM growth media, 200 μL of dMCM or MCM (1:3 in RPMI). After 24-hour incubation, MCM was aspirated and the cells were fixed for one hour in 4% paraformaldehyde in PBS. After washing with PBS, cells were treated for two minutes with 250 μL sodium citrate buffer containing 0.1% Triton X-100 at 4°C. TUNEL assay (Roche, Cat No: 11684795910) and 4′,6-diamidino-2-phenylindole (DAPI) staining (Vector Labs, Cat No: H-1500) were performed according to the manufacturer’s recommendations. After washing with PBS, cells were sealed with Vectashield and a glass coverslip. The numbers of stained cells in ten different random high power fields of view (20× magnifications) were counted using a ZEISS Axioplan II fluorescent microscope. A positive control well comprised cells that were pre-treated with DNase1.
Western Blotting
Proteins in equal volumes of conditioned media were resolved on reducing 4–10% SDS polyacrylamide gels and transferred to Immobilon-P membrane. Anti-BIGH3 antibody was incubated (1:5000) with transferred proteins, washed and then incubated with goat anti-rabbit horseradish peroxidase labeled IgG (1:3000). The complex was detected using the substrate diaminobenzidine (DAB). Densitometry was performed using Image J software developed by NIH.
qPCR
Total RNA isolated using Qiagen RNeasy mini kit (Cat No: 74104) was reverse transcribed into cDNA using Taqman Reverse Transcription reagents (Life Technologies, Cat No: 4304134). Quantitative PCR measured BIGH3 mRNA and 18s-rRNA using Syber Green PCR Mix (Life Technologies, Cat No: 4309155). BIGH3 primers were: Forward 5′-TGGACAGACCCTGGAAACTC-3′ and Reverse 5′-GTCTCCCTTCAGGACATCCA-3′. 18s-rRNA primers were Forward 5′-AGACCTGGAGCGACTGAAGA-3′ and Reverse 5′AGAAGTGACGCAGCCCTCTA-3′. The comparative Ct (ΔΔCt) method was used to obtain quantitative data of relative gene expression(25); values were normalized to expression levels of 18s-rRNA.
Results
Cultured RhRECs Synthesized and Secreted BIGH3
To test whether the retinal vasculature contributes to the synthesis and secretion of BIGH3 protein, an in vitro study was conducted by culturing RhREC in MEMα supplemented with 10% FBS. IHC analyses revealed BIGH3 is synthesized by RhREC in culture. The cells secrete BIGH3 that is found in depositions (Fig. 1A) between cells that have a well-organized actin cytoskeleton (Fig. 1B). BIGH3 is localized in both the cytosol of the cells and secreted (see also Supplementary Figure S1), to be incorporated into the substratum and a component of the cells’ conditioned medium, as expected for soluble extracellular matrix molecules. We therefore proceeded to assess the expression and secretion of the BIGH3 protein by RhREC.
Figure 1. BIGH3 is Localized to the Extracellular Space.

RhRECs were cultured in MEMα supplemented with 10% FBS. Cells were fixed in 4% paraformaldehyde and permeabilized with Triton X-100 and stained with BIGH3 antibody (red, A), phalloidin for actin, (green, B) and DAPI for nuclei (blue, C). BIGH3 staining is evident for a secretory network protein and localized to the extracellular space in the cells’ synthesized substratum. Images are shown at 40× magnification. For co-localization of BIGH3 with Fibronectin, a known secretory protein, see Fig. S1.
dMCM, TFGβ and BIGH3 Induced RhREC Apoptosis
In a previous study, we reported that macrophages cultured in growth medium with high glucose and high LDL provided an elevated level of TGFβ in the conditioned medium. Thus, we hypothesized that macrophage-derived TGFβ induces an elevated expression of BIGH3 protein in RhREC’s, which when in sufficient amounts will induce apoptosis. RhRECs were cultured in 100% MEMα, 100% RPMI or 75% RPMI + 25% dMCM for 24 hours and then apoptosis was quantified (See Supplementary Figure S2 for TUNEL staining images). RhRECs in 25% dMCM had a 35% increase (P value <0.01) in apoptosis in comparison to the cells cultured in MEMα and RPMI (Fig. 2A). Fig. 2B shows that TGFβ1, when added to cell media, induced apoptosis in 4% of RhREC in culture while TGFβ2 induced 12% apoptosis. The addition of BIGH3 antibody (Ab) into the cell media reduced the level of RhREC apoptosis induced by TGFβ1 and significantly reduced the level of apoptosis by TGFβ2 (Fig. 2B). In a subsequent experiment, RhREC were cultured in 100% RPMI, 25% dMCM + 75% RPMI, and also co-treated with anti-TGFβ1 antibody (TGFβ Ab), anti-BIGH3 antiserum (BIGH3 Ab), or TGFβ1 receptor blocker (RI) as well as their controls (mouse IgG, rabbit serum, and DMSO). The latter did not block RhREC apoptosis (Fig 2C). However, apoptosis induced by dMCM (27%) was significantly reduced to 8% by co-treatment with TGFβ1 Ab, to 15% by BIGH3 Ab, and to 10% by TGFβ1 receptor inhibitor, RI (see Fig. 2C). Furthermore, the addition of recombinant, full length BIGH3 protein to cell media led to a dose-dependent increase in RhREC apoptosis after a 24 hrs incubation period (Fig. 2D). These results strongly indicate that dMCM-induced RhREC apoptosis is mediated by TGFβ and the BIGH3 protein.
Figure 2. dMCM, TGFβ and BIGH3 Induced RhREC Apoptosis.

TUNEL assays were used to quantify apoptosis (N=3). (A) Increased apoptosis was seen in RhRECs treated with 75% RPMI + 25% dMCM in comparison to cells treated with MEMα only and RPMI only. (B) RhRECs were treated with TGFβ1 or TGFβ2, with and without rabbit polyclonal anti-BIGH3 serum (BIGH3 Ab). BIGH3 Ab significantly reduced TGFβ2-induced apoptosis (Student’s T-test, *P<0.05). (C) RhRECs were cultured in 100% RPMI, 75% RPMI + 25% dMCM and also co-treated with anti-TGFβ1 antibody (TGFβ Ab), anti-BIGH3 antiserum (BIGH3 Ab), or TGFβ1 receptor blocker (RI) as well as their controls (mouse IgG, rabbit serum, and DMSO). (D) Cultured RhRECs were treated with increasing amounts of recombinant BIGH3 protein. After a 24-hour incubation, TUNEL analysis revealed a concentration-dependent increase between BIGH3 amount and percent RhREC apoptosis. Results from one way ANOVA was significant [F(8,62) = 255.1, P<0.0001]. A Dunnett’s multiple comparison test was performed using 0 μg/mL as the control **P<0.01. For representative TUNEL images, see Fig. S2.
dMCM and TGFβ Induced BIGH3 mRNA Expression in RhREC
RhREC treated with dMCM increased the quantity of BIGH3 mRNA (Fig. 3). dMCM treatment increased RhREC mRNA expression of BIGH3 from the two control groups of DMSO and mouse IgG (Fig. 3). The addition of TGFβ 1 receptor inhibitor (RI) and TGFβ1 Ab completely blocked dMCM-induced BIGH3 mRNA expression (Fig. 3A). dMCM treatment resulted in significantly higher BIGH3 mRNA levels in each of the four time points of 3, 6, 24, and 48 hrs (Fig. 3B). A time-dependent increase in BIGH3 expression (from 1 to 3 fold changes) was observed during the first 24 hrs of dMCM treatment (Fig. 3B). Both TGFβ1 and TGFβ2 had a concentration-dependent effect on BIGH3 expression. An increase in TGFβ1 concentration from 0 to10 ng/ml resulted in a 5–6 fold increase in BIGH3 mRNA expression (Fig. 3C). Similarly, concentrations of 0 to 10 ng/ml TGFβ2 increased BIGH3 expression by 3.5 fold (Fig. 3D).
Figure 3. dMCM and TGFβ Induced BIGH3 mRNA Expression in RhREC.

(A) RhRECs were treated with 100% RPMI or 75% RPMI + 25% dMCM. Additionally each condition was co-treated with TGFβ1 receptor blocker (RI), DMSO (vehicle of RI), mouse anti-TGFβ1 antibody (TGFβ Ab), or mouse IgG. BIGH3 mRNA expression was measured by qPCR and was significantly decreased by RI (T-test, *P<0.05) and TGFβ1 Ab (T-test, **P<0.01) (N=3). (B) RhRECs were cultured with 100% RPMI, 75% RPMI + 25% MCM, or 75% RPMI + 25% dMCM. BIGH3 mRNA expression was measured at 3, 6, 24, and 48 hrs after treatments (N=3). A Dunnett’s multiple comparison test was performed and significant difference (**P<0.01) was observed between BIGH3 expression in RhRECs treated with RPMI control and dMCM at all four time points. Two Way ANOVA was significant for dMCM treatment effect [F(2,12) = 17.40, P<0.001]. RhRECs were cultured with TGFβ1 (C) or TGFβ2 (D) added to cell media and BIGH3 expression was measured 24 hrs after initial TGFβ treatment. Dunnett’s multiple comparison tests were performed and significant differences (*P<0.05, **P<0.01) were observed between BIGH3 expression in RhRECs in the control group (0 ng/mL) and in all treatment groups. One way ANOVA also indicated significant treatment effects by TGFβ 1 and 2 [F(4,14) = 25.63, P<0.0001], and [F(4,14) = 12.18, P<0.0001].
TGFβ 1 and 2 Induced BIGH3 Protein Secretion by RhREC
Results from Western blot analyses on cell media of RhRECs treated with TGFβ show an increase in BIGH3 protein synthesis and secretion (Fig. 4A and C). Comparable concentrations of TGFβ1 and TGFβ2 induced a similar amount of BIGH3 protein in the cell media (Fig. 4B and D). Together with results on TGFβ-induced BIGH3 mRNA expressions (see Fig. 3), it is apparent that TGF β1 induced about 5–6 fold increases in BIGH3 mRNA but only 2-fold increases in BIGH3 protein secreted into cell media. In contrast, TGFβ2 induced a 3.5-fold increase in BIGH3 mRNA and also a two-fold increase of BIGH3 protein in culture media. Irrespective of the difference in transcription and translation/secretion of the BIGH3 protein, these results clearly show that the cytokine TGFβ induced RhREC to significantly increase in BIGH3 protein synthesis and secretion.
Figure 4. TGFβ 1 and 2 Induced BIGH3 Protein Secretion by RhREC.

RhRECs were cultured with either TGFβ1 (A) or TGFβ2 (B) at 1, 2, 5 and 10 ng/mL for 24 hrs and refreshed media were collected after a 24 hrs period and probed for the BIGH3 protein by Western blot. Lane 1, 0.1 μg recombinant BIGH3 protein; lane 2–6, BIGH3 protein in cell media of cells treated with TGFβ at 0 ng/mL, 1 ng/mL, 2 ng/mL, 5 ng/mL, 10 ng/mL. Densitometry results from A and C respectively are presented in (B) and (D). One way ANOVA showed no significant dose effect for TGFβ1 but a significant dose effect for TGFβ2 [F(4,14)= 4.081; P<0.05]. Dunnett’s test (D) was performed using 0 ng/mL as control (*P<0.05).
Diabetes Increased Macrophages and BIGH3 in the Inner Retina
IHC analyses using anti-CD68 and anti-BIGH3 antibodies revealed increased macrophages and BIGH3 protein in the inner retina of the diabetic mice (Fig. 5E and F vs. Fig. 5A and B). Macrophage staining was exacerbated throughout the entire retina when LDL-R−/− mice were treated with STZ to induce diabetes (Fig. 5E). In the diabetic mouse retina, staining showed an increase of BIGH3 and macrophages in the ganglion cell layer, inner nuclear layer, outer nuclear layer, and layers where photoreceptors and pigment epithelium are present (Fig. 5H). In contrast, the retina of non-diabetic control mice showed little if any macrophage staining (Fig. 5A) and reduced BIGH3 proteins, particularly in the inner retina (Fig. 5B). These results suggest a potential link between macrophages and BIGH3 protein accumulation, and their co-localization in the diabetic mouse eye.
Figure 5. Diabetes Increased Macrophages and BIGH3 in the Inner Retina.
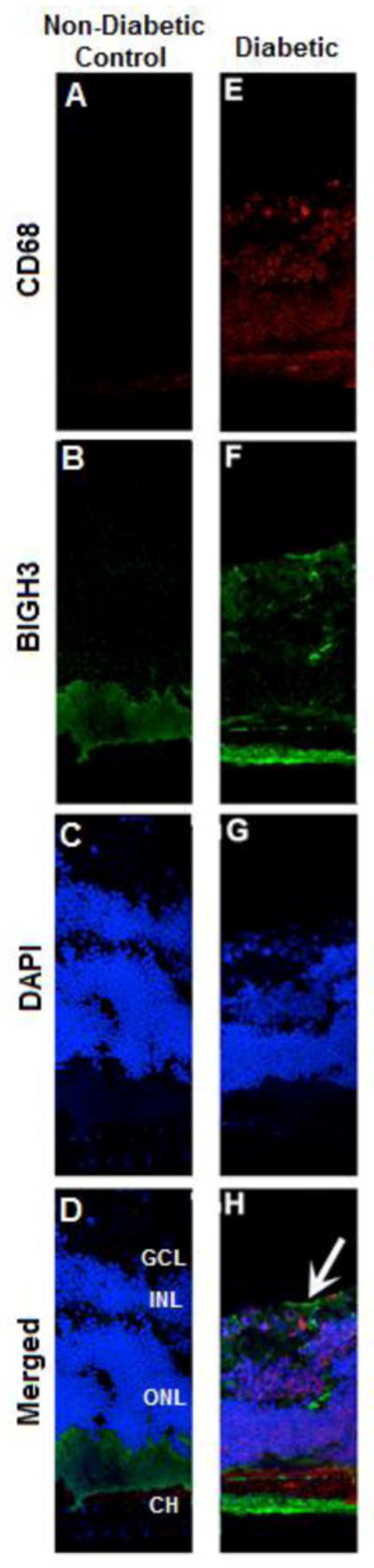
Images are shown from eye sections (A–H) obtained from mice exposed to a normal diet (non-diabetic control; A, B, C, D) and STZ-induced diabetic mice ((LDL-R−/− ; E, F, G, H). Nuclei (blue), macrophages (stained with antibodies directed against CD68, red), and BIGH3 (green) are shown. White arrow (in H) shows the outer edge of an area of the diabetic retina with macrophage and BIGH3 at or near one another. Abbreviations in D depict layers of the retina: Ganglion Cell Layer (GCL), Inner Nuclear Layer (INL), Outer Nuclear Layer (ONL), and Choroid (CH). Images are shown at 20× magnification.
Discussion
In the present study, we tested the hypothesis that RhREC overproduction of BIGH3 mediated by macrophage-derived TGFβ elicits cell apoptosis which promotes tissue damage in the retinal microvasculature. Our results confirm that macrophage-derived TGFβ and recombinant BIGH3 induced apoptosis in RhRECs (Fig. 2). RhREC apoptosis was blocked when macrophage-derived TGFβ and BIGH3 proteins were co-treated with TGFβ antibody, BIGH3 antibody, or TGFβ RI (Fig. 2). Taken together, our results support a pathway of BIGH3 involvement in retinal endothelial cell apoptosis mediated by macrophage-derived TGFβ. This is the first report on a macrophage-mediated inflammatory pathway and RhREC apoptosis on the development of diabetic retinopathy.
It is well known that different cell types express BIGH3 (11, 13, 16, 17, 26), however there is little known about the expression of BIGH3 in normal or diabetic retina and even less so regarding the underlying mechanisms that increase retinal BIGH3 protein and BIGH3-mediated apoptosis. Cultured RhRECs synthesized and secreted BIGH3 protein (Fig. 1) and treatment of RhRECs by dMCM (conditioned media from macrophages cultured in “diabetic condition”) resulted in a significant increase in BIGH3 mRNA (Fig. 3). Treatment of RhRECs by TGFβ also resulted in significant increases in BIGH3 transcript and protein expression (Fig. 3 and 4), suggesting that the macrophage-derived TGFβ may be the bridging factor for the increase in BIGH3 synthesis in RhRECs. It is well known that macrophages are a major source of TGFβ1 (6), and we recently determined that active TGFβ1 levels were 41–53 pg/mL in MCM and 60–76 pg/mL in dMCM, whereas no detectable amounts were ascertained in conditioned medium of macrophages not treated with high glucose and LDL (LeBaron et al. 2014, unpublished data).
The accumulation of microglia/macrophages in the retina has been linked to hyperglycemia in a rat model.(27) In this study we show that LDL receptor knockout mice (rendered diabetic) had an increase in macrophages in the retina opposed to non-diabetic mice (Fig. 5E and A, respectively). Consequently, BIGH3 protein expression increased throughout the retina of diabetic mice (Fig. 5F). BIGH3 noted at the sclera (Fig. 5F) is consistent with in situ hybridization showing BIGH3 mRNA abundance (28). These in vivo data provide initial evidence on macrophages and its co-localization with the BIGH3 protein in the diabetic eye.
In the present study, we have demonstrated that in diabetic conditions, human macrophages synthesize TGFβ1 to an extent that is sufficient to stimulate BIGH3 synthesis and secretion by RhRECs. Moreover, using a paracrine or autocrine mechanism, the newly synthesized BIGH3 induces a significant increase in the number of apoptotic RhRECs. We have included both TGFβ1 and TGFβ2 because the latter was found to be significantly higher in the aqueous and vitreous humor from patients with diabetic retinopathy (29, 30). In the present study, we found that both isomers are potent effectors for the induction of BIGH3 expression and retinal endothelial cell apoptosis (Fig. 2, 3 and 4). Macrophages secrete TGFβ1 even after phagocytosis of apoptotic cells(6), and in response to inflammatory stimuli (7) and are a source of TGFβ1 in this study. However, the origin of TGFβ2 in diabetic conditions in the retina is not readily apparent. It is possible that TGFβ2 is derived from activated retinal microglia (31) or monocyte derived macrophages (dMCM contained a similar level of total and mature TGFβ1 and TGFβ2, unpublished results). Therefore, further study on the role of both isoforms of TGFβ is essential to fully explore its role in this novel pathway of retinopathy development. Moreover, transgenic mice lacking BIGH3 may provide insight on its significance in the development of diabetic retinopathy.
Supplementary Material
Figure S1. Immunohistochemical labelling of RhREC extracellular matrix proteins. RhRECs were fixed in 4% paraformaldehyde and either permeabilized with Triton X-100 (A, B, C) or left with an intact lipid membrane (D, E, F). RhRECs were incubated for 24 hours at room temperature with polyclonal rabbit anti-human BIGH3, rabbit anti-human fibronectin, or normal rabbit IgG. RhRECs were then washed and incubated for one hour in goat anti-rabbit Alexafluor 488. Cell were also labelled with rhodamine Phalloidin and DAPI. Images were captured using a Zeiss Axioplan 2 fluorescent microscope (40× magnification). Comparison of non-permeabilized and permeabilized cells shows immunohistochemical labelling of BIGH3 and fibronectin to be specific to the extracellular matrix (in non-permeabilized cells (arrows in A and B) and, in permeabilized cells, within the secretory system (arrows in D and E). RhRECs incubated with rabbit IgG show no nonspecific labelling of the secondary antibody (C and F).
Figure S2. BIGH3 induces apoptosis in RhREC. Representative TUNEL staining to quantify RhREC apoptosis when treated with PBS vehicle (A) and 5μg/ml BIGH3 (B). Results show minimal TUNEL staining in PBS. BIGH3 protein induced significant TUNEL staining for apoptosis.
Acknowledgments
The authors thank the National Institute on Minority Health and Health Disparities (G12MD007591), the National Heart Lung and Blood Institute (R01HL70963) of the National Institutes of Health, and The San Antonio Life Sciences Institute Grant (SALSI) from Texas Higher Education Coordinating Board for their support. Authors thank Andrew S. Mendiola for technical and editorial assistance and Dr. Jeff Grigsby for insightful comments/suggestions.
Footnotes
Disclosures: None
Conflict of Interest Statement:
The authors declare that they have no conflict of interest.
References
- 1.Romeo G, Liu WH, Asnaghi V, Kern TS, Lorenzi M. Activation of nuclear factor-kappaB induced by diabetes and high glucose regulates a proapoptotic program in retinal pericytes. Diabetes. 2002;51:2241–2248. doi: 10.2337/diabetes.51.7.2241. [DOI] [PubMed] [Google Scholar]
- 2.Geraldes P, Hiraoka-Yamamoto J, Matsumoto M, et al. Activation of PKC-delta and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nature medicine. 2009;15:1298–1306. doi: 10.1038/nm.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Han JH, Ha SW, Lee IK, Kim BW, Kim JG. High glucose-induced apoptosis in bovine retinal pericytes is associated with transforming growth factor beta and betaIG-H3: betaIG-H3 induces apoptosis in retinal pericytes by releasing Arg-Gly-Asp peptides. Clin Exp Ophthalmol. 2010;38:620–628. doi: 10.1111/j.1442-9071.2010.02276.x. [DOI] [PubMed] [Google Scholar]
- 4.Busik JV, Mohr S, Grant MB. Hyperglycemia-induced reactive oxygen species toxicity to endothelial cells is dependent on paracrine mediators. Diabetes. 2008;57:1952–1965. doi: 10.2337/db07-1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fadok VA, Bratton D, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. The Journal of clinical investigation. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McDonald P, Fadock V, Bratton D, Henson PM. Transcriptional and Translational Regulation of Inflammatory Mediator Production by Endogenous TGF-B in Macrophages That Have Ingested Apoptotic Cells. The Journal of Immunogy. 1999;163:6164–6172. [PubMed] [Google Scholar]
- 7.Asmis R, Qiao M, Rossi RR, Cholewa J, Xu L, Asmis LM. Adriamycin promotes macrophage dysfunction in mice. Free Radic Biol Med. 2006;41:165–174. doi: 10.1016/j.freeradbiomed.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 8.Skonier J, Bennett K, Rothwell V, Kosowski S, Plowman G, Wallace P, Edelhoff S, Disteche C, Neubauer M, Marquardt H, et al. beta ig-h3: a transforming growth factor-beta-responsive gene encoding a secreted protein that inhibits cell attachment in vitro and suppresses the growth of CHO cells in nude mice. DNA Cell Biol. 1994;13:571–584. doi: 10.1089/dna.1994.13.571. [DOI] [PubMed] [Google Scholar]
- 9.Porreca E, DiFebbo C, Mincione G, Reale M, Baccante G, Guglielmi MD, Cuccurullo F, Colletta G. Increased transforming growth factor-beta production and gene expression by peripheral blood monocytes of hypertensive patients. Hypertension. 1997;30:134–139. doi: 10.1161/01.hyp.30.1.134. [DOI] [PubMed] [Google Scholar]
- 10.Zamilpa R, Rupaimoole R, Phelix CF, et al. C-terminal fragment of transforming growth factor beta-induced protein (TGFBIp) is required for apoptosis in human osteosarcoma cells. Matrix Biol. 2009;28:347–353. doi: 10.1016/j.matbio.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thapa N, Kang KB, Kim IS. Beta ig-h3 mediates osteoblast adhesion and inhibits differentiation. Bone. 2005;36:232–242. doi: 10.1016/j.bone.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 12.Lee BHBJ, Park RW, Kim JE, Park JY, Kim IS. betaig-h3 triggers signaling pathways mediating adhesion and migration of vascular smooth muscle cells through alphavbeta5 integrin. Exp Mol Med. 2006;38:153–161. doi: 10.1038/emm.2006.19. [DOI] [PubMed] [Google Scholar]
- 13.Kim JE, Kim SJ, Lee BH, Park RW, Kim KS, Kim IS. Identification of motifs for cell adhesion within the repeated domains of transforming growth factor-beta-induced gene, betaig-h3. J Biol Chem. 2000;40 doi: 10.1074/jbc.M002752200. [DOI] [PubMed] [Google Scholar]
- 14.Kim JE, Jeong HW, Nam JO, Lee BH, Choi JY, Park RW, Park JY, Kim IS. Identification of motifs in the fasciclin domains of the transforming growth factor-beta-induced matrix protein betaig-h3 that interact with the alphavbeta5 integrin. J Biol Chem. 2002;277:46159–46165. doi: 10.1074/jbc.M207055200. [DOI] [PubMed] [Google Scholar]
- 15.Liu W, Ahmad SA, Reinmuth N, Shaheen RM, Jung YD, Fan F, Ellis LM. Endothelial cell survival and apoptosis in the tumor vasculature. Apoptosis. 2000;5:323–328. doi: 10.1023/a:1009679307513. [DOI] [PubMed] [Google Scholar]
- 16.Nam JO, Kim JE, Jeong HW, Lee SJ, Lee BH, Choi JY, Park RW, Kim IS. Identification of the alphavbeta3 integrin-interacting motif of betaig-h3 and its anti-angiogenic effect. J Biol Chem. 2003;278:25902–25909. doi: 10.1074/jbc.M300358200. [DOI] [PubMed] [Google Scholar]
- 17.Thapa N, Lee BH, Kim IS. TGFBIp/betaig-h3 protein: a versatile matrix molecule induced by TGF-beta. Int J Biochem Cell Biol. 2007;39:2183–2194. doi: 10.1016/j.biocel.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 18.Han B, Qi S, Hu B, Luo H, Wu J. TGF-beta i promotes islet beta-cell function and regeneration. J Immunol. 2011;186:5833–5844. doi: 10.4049/jimmunol.1002303. [DOI] [PubMed] [Google Scholar]
- 19.Han B, Luo H, Raelson J, Huang J, Li Y, Temblay J, Hu B, Qi S, Wu J. TGFBI (betaIG-H3) is a diabetes risk gene based on mouse and human genetic studies. Hum Mol Genet. 2014;23:4597–4611. doi: 10.1093/hmg/ddu173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grigsby JG, Parvathaneni K, Almanza MA, Botello AM, Mondragon AA, Allen DM, Tsin AT. Effects of tamoxifen versus raloxifene on retinal capillary endothelial cell proliferation. J Ocul Pharmacol Ther. 2011;27:225–233. doi: 10.1089/jop.2010.0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wintergerst ES, Jelk J, Asmis R. Differential expression of CD14, CD36 and the LDL receptor on human monocyte-derived macrophages. A novel cell culture system to study macrophage differentiation and heterogeneity. Histochem Cell Biol. 1998;110:231–241. doi: 10.1007/s004180050285. [DOI] [PubMed] [Google Scholar]
- 22.Qiao M, Zhao Q, Lee CF, Tannock LR, Smart EJ, LeBaron RG, Phelix CF, Rangel Y, Asmis R. Thiol oxidative stress induced by metabolic disorders amplifies macrophage chemotactic responses and accelerates atherogenesis and kidney injury in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2009;29:1779–1786. doi: 10.1161/ATVBAHA.109.191759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Villanza-Espinoza E, Hatch A, Tsin ATC. Effect of light exposure on accumulation depletion of retinyl ester in the chicken retina. Experimental Eye Reseach. 2006;83:871–876. doi: 10.1016/j.exer.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 24.Ferguson JW, Thoma BS, Mikesh MF, Kramer RH, Bennett KL, Purchio A, Bellard BJ, LeBaron RG. The extracellular matrix protein betaIG-H3 is expressed at myotendinous junctions and supports muscle cell adhesion. Cell Tissue Res. 2003;313:93–105. doi: 10.1007/s00441-003-0743-z. [DOI] [PubMed] [Google Scholar]
- 25.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26.LeBaron RG, Bezverkov KI, Zimber MP, Pavelec R, Skonier J, Purchio AF. BIG-H3, a Novel Secretory Protein Inducible by Transforming Growth Factor-B, Is Present in Normal Skin and Promotes the Adhesion and Spreading of Dermal Fibroblasts In Vitro. J Invest Dermatol. 1995;104:844–849. doi: 10.1111/1523-1747.ep12607024. [DOI] [PubMed] [Google Scholar]
- 27.Omri S, Behar-Cohen F, de Kozak Y, Sennlaub F, Verissimo LM, Jonet L, Savoldelli M, Omri B, Crisanti P. Microglia/macrophages migrate through retinal epithelium barrier by a transcellular route in diabetic retinopathy: role of PKCζ in the Goto Kakizaki rat model. Am J Pathol. 2011;179:942–953. doi: 10.1016/j.ajpath.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferguson JW, Mikesh MF, Wheeler EF, LeBaron RG. Developmental expression patterns of Beta-ig (βIG-H3) and its function as a cell adhesion protein. Mechanisms of Development. 2003;120:851–864. doi: 10.1016/s0925-4773(03)00165-5. [DOI] [PubMed] [Google Scholar]
- 29.Ochiai Y, Ochiai H. Higher concentration of transforming growth factor-beta in aqueous humor of glaucomatous eyes and diabetic eyes. Jpn J Ophthalmol. 2002;46:249–253. doi: 10.1016/s0021-5155(01)00523-8. [DOI] [PubMed] [Google Scholar]
- 30.Hirase K, Ikeda T, Sotozono C, Nishida K, Sawa H, Kinoshita S. Transforming growth factor beta2 in the vitreous in proliferative diabetic retinopathy. Arch Ophthalmol. 1998;116:738–741. doi: 10.1001/archopht.116.6.738. [DOI] [PubMed] [Google Scholar]
- 31.Constam DB, Philipp J, Malipiero UV, ten Dijke P, Schachner M, Fontana A. Differential expression of transforming growth factor-beta 1, -beta 2, and -beta 3 by glioblastoma cells, astrocytes, and microglia. J Immunol. 1992;148:1404–1410. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Immunohistochemical labelling of RhREC extracellular matrix proteins. RhRECs were fixed in 4% paraformaldehyde and either permeabilized with Triton X-100 (A, B, C) or left with an intact lipid membrane (D, E, F). RhRECs were incubated for 24 hours at room temperature with polyclonal rabbit anti-human BIGH3, rabbit anti-human fibronectin, or normal rabbit IgG. RhRECs were then washed and incubated for one hour in goat anti-rabbit Alexafluor 488. Cell were also labelled with rhodamine Phalloidin and DAPI. Images were captured using a Zeiss Axioplan 2 fluorescent microscope (40× magnification). Comparison of non-permeabilized and permeabilized cells shows immunohistochemical labelling of BIGH3 and fibronectin to be specific to the extracellular matrix (in non-permeabilized cells (arrows in A and B) and, in permeabilized cells, within the secretory system (arrows in D and E). RhRECs incubated with rabbit IgG show no nonspecific labelling of the secondary antibody (C and F).
Figure S2. BIGH3 induces apoptosis in RhREC. Representative TUNEL staining to quantify RhREC apoptosis when treated with PBS vehicle (A) and 5μg/ml BIGH3 (B). Results show minimal TUNEL staining in PBS. BIGH3 protein induced significant TUNEL staining for apoptosis.