

Abstract
Recent studies have shown that single-stranded (ss) viral RNAs fold into more compact structures than random RNA sequences with similar chemical composition and identical length. Based on this comparison, it has been suggested that wild-type viral RNA may have evolved to be atypically compact so as to aid its encapsidation and assist the viral assembly process. To further explore the compactness selection hypothesis, we systematically compare the predicted sizes of >100 wild-type viral sequences with those of their mutants, which are evolved in silico and subject to a number of known evolutionary constraints. In particular, we enforce mutation synonynimity, preserve the codon-bias, and leave untranslated regions intact. It is found that progressive accumulation of these restricted mutations still suffices to completely erase the characteristic compactness imprint of the viral RNA genomes, making them in this respect physically indistinguishable from randomly shuffled RNAs. This shows that maintaining the physical compactness of the genome is indeed a primary factor among ssRNA viruses’ evolutionary constraints, contributing also to the evidence that synonymous mutations in viral ssRNA genomes are not strictly neutral.
Introduction
Minimalistic organisms, such as single-stranded (ss)RNA viruses, are ideally suited to investigate how the three-dimensional organization of the genome—and not just its sequence composition—is subject to selective evolutionary pressure. We recall, for instance, that several structural features are robustly maintained in the highly-mutating ssRNA viruses. These include RNA structures acting as signals for translation (1), for transcription initiation (2), or as packaging signals to initiate the self-assembly of the virion (3,4). Other conserved structures have also been identified (5–7), including long-range interactions between different genomic regions of RNA (5,8), whose role in the virus life cycle is still unknown.
The preservation of these structural features must act as a powerful constraint on viable RNAs, together with the multiple other, often competing, selection pressures (9–11). The evolutionary mechanisms that maintain the viral protein phenotype clearly impact the genome chemical composition more directly, by largely restricting those mutations which have a deleterious effect on the encoded proteins (12–15). On the other hand, synonymous mutations, i.e., mutations that do not change the amino acid sequences encoded by the genes, are neutral with regard to these mechanisms, but still have an impact on the structural features of RNAs.
It is increasingly becoming recognized that the mechanisms that may constrain synonymous mutations extend beyond the aforementioned conservation of specific genome structures, and are underpinned by general physico-chemical constraints. The latter mostly stem from the polymeric nature of the gene-carrying macromolecules and their steric and electrostatic self-interactions, as well as interactions with the capsid proteins (16–19). These molecular interactions can be long-ranged and depend crucially on the pH of the local aqueous solution environment (20), conferring virions the ability to assemble and disassemble spontaneously at proper bathing solution conditions (21–28), and the ability to recognize and selectively encapsidate only viral RNA even in the absence of packaging signals (19,29–32).
In this study we focus on a general and major structure-related selection constraint, namely the feasibility to efficiently package viral RNA inside the capsid, and address its competition with sequence-based selection mechanisms. The overarching question is whether the viral RNA sequence has evolved not only for encoding a specific protein phenotype but also for promoting an innate fold of the free (unencapsidated) viral RNA itself that is primed for efficient encapsidation.
Major advances toward solving this important conundrum have been recently made by comparing the predicted equilibrium properties of ssRNA folds of several icosahedral viruses with those of random RNA sequences with similar length and nucleotide composition. By using general arguments based on the scaling properties of linear (33) and/or branched polymers (34), the folded wild-type (WT) viral RNA was shown to be significantly more compact than random nucleotide sequences. In addition—and most notably—the average radius of gyration of WT RNA genomes was found to exceed only slightly the inner radius of the fully assembled capsid (35).
In this context, a key and still open problem relates to the extent to which the selective pressure for easily encapsidable RNA genomes directly competes with the other sequence-based mechanisms that are simultaneously at play for selecting biologically viable viral RNA. As a matter of fact, the enhanced compactness of viral RNA has so far been established only by comparison against random sequences that do not retain any specific viral-like characteristics except from the overall nucleotide composition. Because the volume of the sequence phase space that is accessible to viable viral RNA sequences is actually vanishingly small compared to the available combinatorial phase space of random sequences, it is crucial to ascertain the implications of introducing realistic sequence constraints into the picture. Such constraints could even affect the properties of the associated folds to the point of implying genome compactness, which would make the assumption of a distinct selection principle based on RNA compactness superfluous.
To address these issues, we consider the implications of constrained mutations that conserve the encoded protein phenotype and the viral-like nucleotide composition on the compactness of viral RNA genomes. This allows us to examine the concurrence, or possibly the incompatibility, of sequence- and structure-based parallel selection mechanisms, and to ascertain whether the conservation of RNA compactness is among the causes of the sensitivity of ssRNA viruses to synonymous mutations.
Specifically, we consider 128 viral RNA sequences and evolve them synthetically by accumulating exclusively synonymous pointwise mutations, measuring their impact on the properly quantified compactness of the genome. We recall that the constraint of synonymity, i.e., considering only codons that encode for the same amino acids, is particularly severe for viral RNA because of both the high gene density and the frequent presence of overlapping reading frames.
Our study unequivocally shows that, at least for the viruses studied, the accumulation of strictly synonymous mutations—even if they are sparse—is sufficient to cause a systematic drift of the properly quantified compactness of the genome toward values comparable to those of unrestricted random sequences that are systematically much larger than those of the WT genomes. By focusing on the mutational dynamics of four viral genomes, we show that while mutating as few as 5% of a genome is enough to erase its compactness, there is still a nonnegligible portion of the sequence space in the vicinity of the WT sequence in which the genomes are at least as compact as the WT genome, while still coding for the correct proteins.
Furthermore, we show that the typical WT RNA compactness is related neither to the codon usage biases present in viral genomes nor to the particular sequences of the untranslated regions (UTRs) present at the 5′ and 3′ ends of the genomes. These results provide a posteriori evidence that the same viral RNA sequence can encode not only for the expression of the proper protein complement, exposed to canonical selection pressure mechanisms, but can also, on another level, prime the optimal physico-chemical genome-packing organization.
Materials and Methods
Wild-type viral sequences
Viral ssRNA sequences were obtained from the NCBI nucleotide database (36). The dataset we use includes positive-strand ssRNA viruses from the following families: Tymoviridae (from the order Tymovirales); Flaviviridae; Caliciviridae; Picornaviridae; Comovirinae; Bromoviridae; and Tombusviridae (37). All the viruses considered have icosahedral capsids, the majority of them with triangulation number T = 3. Most of the families in the dataset have monopartite genome, with the exception of Comovirinae, which have a bipartite genome, and Bromoviridae, which have a tripartite genome (37). Comovirinae pack the two segments, denoted RNA1 and RNA2, into separate virions; the two largest RNA segments of Bromoviridae genome, denoted RNA1 and RNA2, are also packed into separate virions, and we thus consider only these two segments. All the considered viruses use the eukaryotic genetic code and their genes have no reading gaps. Several sequences among those we consider also have overlapping reading frames, which are known to impose further evolutionary constraints increasing the deleterious effects of mutations (38,39). With these restrictions taken into account, the final dataset of analyzed sequences contains 128 viral genomes (compiled in Table S1 in the Supporting Material).
Synonymous point mutations
Extended models of sequence evolution of overlapping genes can account for the codependency of the nucleotide substitution process in two reading frames (40,41), but are based in computationally very intense simulations and are not always applicable to large sequence datasets. Because in this study we are interested in the statistical properties across various viral families, we adopt a much simpler model that simply conserves the produced amino acids in all reading frames.
Mutated viral ssRNA sequences are obtained using a Monte Carlo (MC) scheme designed to simulate synonymous point substitutions while also conserving dinucleotide frequencies. Starting from a WT sequence, a point substitution is introduced at every step and accepted or rejected using a Metropolis algorithm. Substitutions that change the amino acids encoded by the genes, and are thus nonsynonymous, are rejected. To preserve the dinucleotide frequencies, we additionally introduce a fictitious energy related to the viral dinucleotide odd-ratios (42),
| (1) |
where
| (2) |
Here, N(XY) is the number of XY pairs, N(X), N(Y) are the numbers of X and Y nucleotides in the sequence, and N is the total length of the RNA sequence.
The values of the constants KXY are chosen in such a way that a considerable portion (but not all) of the proposed sequences have dinucleotide odd-ratios lying within 1.5ΔQ, where ΔQ is the interquartile distance evaluated on the OWT(XY) distribution of the corresponding viral family (see the Supporting Material for additional information). We produce an extensive ensemble of point mutations (∼109) to ensure an appropriate sampling of the sequence space. Sequences are sampled every 100N mutations to ensure they are uncorrelated, and filtered a posteriori to have all odd-ratios within 1.5ΔQ. For every WT viral sequence we generate a set of 500–2000 mutated sequences and finally characterize the spatial compactness of the associated fold by computing the thermally averaged maximum ladder distance, 〈MLD〉, described in a later subsection.
As an additional check, we also produce synonymous substitutions using the Fisher-Yates shuffling algorithm (43,44)—in this way, the exact chemical composition of the sequences is conserved, although the dinucleotide odd-ratios are not. While much more complex models for the nucleotide substitutions exist (see, for instance, the review by Anisimova and Kosiol (45) and references therein), we chose these simple ones that conserve the chemical composition of the sequences, because they are sufficient to prove our point, and can most importantly be applied in the same manner to all the genomes we considered.
To investigate the effect of progressively accumulating mutations on viral RNA compactness, quantified by the MLD, we first choose the KXY values in such a way that all produced sequences obey the dinucleotide constraints. The generated MC trajectories are then sampled every N/100 steps. This sampling produces strongly correlated sequences that show the evolution of the genome MLDs toward the values of their random counterparts.
Synonymous mutations preserving codon bias
As an optional additional constraint, we fix the WT codon population by shuffling equivalent codons, as done in Gu et al. (46). The shuffling is performed at the genewise level by first enumerating and pooling the synonymous codons in the WT gene sequence. Each codon in the latter is then replaced by one picked randomly from its synonymous pool. The pools are thus progressively depleted until all reassignments are completed, as in the standard Fisher-Yates shuffling algorithm (43,44). This shuffling procedure, which clearly preserves the WT codon bias at the gene level, is applicable to viral genomes without overlapping genes; these are 86 in our case.
Random RNA sequences
Random ssRNA sequences, used to obtain the scaling law for the MLD of random RNAs, are produced by shuffling RNA sequences with the Fisher-Yates algorithm (43,44). Random numbers, here as well as in the rest of the article, are generated by the SIMD-oriented Fast Mersenne Twister random generator, Ver. 1.4 (47). The SIMD-oriented Fast Mersenne Twister has a period of 2216091 −1, which suffices to produce random permutations of even 10 Knt-long RNA sequences. We use the same viral-like composition for the random sequences as in Yoffe et al. (33), that is, 0.26 A, 0.28 U, 0.24 G, and 0.22 C, to obtain the scaling law for random viral-like RNAs. This average composition is computed excluding Tymoviridae, which differ significantly in their composition. For the Tymoviridae family, we use the averaged composition of the viruses in our sample belonging to this family only (see Table S1 for the list), with the corresponding nucleotide composition: 0.219 A, 0.254 U, 0.163 G, and 0.364 C.
Maximum ladder distance
To investigate the possibility that synonymous substitutions, while being neutral with respect to the encoded protein complement, can affect the secondary structure of viral RNA, we use the (thermally averaged) maximum ladder distance (MLD), a quantitative, albeit coarse-grained indicator of the compactness of RNA folds introduced by Yoffe et al. (33). While the MLD of random RNAs with viral-like nucleotide composition follows a simple scaling law, the MLDs of viral ssRNA genomes are, on the other hand, significantly lower, indicating that their folds are more compact than those of random RNAs.
By modelling DNA as an ideal polymer chain, one can use graph-theoretical arguments to compute its MLD (33,48): For every pair of nucleotides i and j in an RNA sequence we compute the ladder distance, i.e., the number of steps on the ladder separating the two nucleotides on the folded RNA. The maximum value of all the ladder distances in a fold is then its MLD; an example is shown in Fig. 1 a. By treating the MLD contour as the backbone of a linear polymer chain, this provides a measure of compactness/extendedness of the RNA molecule, even though it is not a direct measure of the three-dimensional size of the RNA. This simple measure yields the same scaling relationships as in the case when one treats the RNA as an ideal branched polymer, computing its root-mean-square radius of gyration to determine its extendedness (34).
Figure 1.
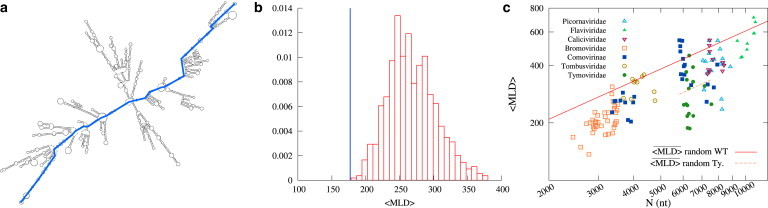
(a) Example of a typical fold of the entire brome mosaic virus (BMV) RNA2 sequence. The maximum ladder distance (MLD) of the folded sequence is highlighted. (b) Thermally averaged MLD, 〈MLD〉, of the WT BMV RNA2 sequence (blue line) and the distribution of 〈MLD〉 values obtained for random RNA sequences of same length and composition as the WT sequence. (c) 〈MLD〉 value of viral ssRNA sequences versus the sequence length N (in nucleotides). Different virus families are represented by different colors and symbols. (Red solid line) Power law of Eq. 3 for the expected values of 〈MLD〉 for random RNA sequences, constrained only by their overall viral-like nucleotide composition. Due to their atypical nucleotide composition, Tymoviridae are not represented by Eq. 3, and the corresponding scaling law for Tymoviridae-like random RNA sequences, , is shown (orange dashed line). See the Supporting Material for further information. (To see this figure in color, go online.)
The secondary structures of viral and random RNA sequences for which we determine their MLDs are obtained by folding the sequences with the RNASUBOPT program available in the VIENNARNA Package, Ver. 2.1 (49). Due to the length of viral RNA, a population of different folds having comparable energy is expected. Therefore, instead of looking for the minimum energy fold, we produce 500 folds at thermal equilibrium for every RNA sequence. This results in a thermal average for the MLD of every sequence, obtained by averaging over this ensemble.
Results and Discussion
Validation: compactness of WT and random RNA sequences
As a starting point for our analysis we considered an extensive set of 128 WT viral sequences listed in Table S1. We characterized their compactness by following the method introduced by Yoffe et al. (33), which entails two steps, detailed in the Materials and Methods section. The first step consists of computing an ensemble of several hundred representative planar RNA folds using the VIENNARNA package (49). Next, one calculates the MLD of each fold. We recall that the ladder distances are obtained by considering in turn all possible pairs of nucleotides and identifying their shortest connecting path, i.e., the one with the minimal number of rungs-on-the-ladder along the duplexed parts of the folds. The number of rungs of the longest minimal path is the MLD, an example of which is shown in Fig. 1 a.
As discussed in Yoffe et al. (33) and Fang et al. (34), the thermal average of the MLD, denoted by 〈MLD〉, is a viable, albeit coarse-grained proxy for the equilibrium spatial compactness of a folded sequence. Because it can be calculated by highly efficient algorithms, it is particularly apt for numerical implementation in extensive enumerative contexts such as this one.
The comparison of the 〈MLD〉 values computed for the 128 viral sequences considered in our study with the 〈MLD〉 values of random sequences with viral-like nucleotide composition (see Materials and Methods) conforms to the earlier conclusion of Yoffe et al. (33) that WT RNA genomes have an enhanced fold compactness compared to arbitrary RNA sequences. This point is illustrated in Fig. 1, b and c. As can be seen in Fig. 1 c, the 〈MLD〉 values of random RNA sequences, additionally averaged over several possible mutations, follow the power law
| (3) |
where the overline indicates the additional averaging over different possible mutations. On the other hand, the 〈MLD〉 values of WT sequences are almost always more compact than the corresponding random values given by Eq. 3. We also note that the parameters of the power law given by Eq. 3 are in good accord with the findings of Yoffe et al. (33).
Compactness of WT and synonymously-mutated RNA sequences
Because the fixation of mutations in viral genomes is subject to a number of evolutionary pressures, the fact that WT RNA sequences of icosahedral viruses tend to be more compact than predicted by Eq. 3 is not enough to conclude that they have been evolutionarily selected for optimal compactness. In fact, the sequence space accessible to random mutations is unrealistically large because it does not account for the several selection constraints that viable RNA sequences have to obey.
Arguably, the most severe of such constraints reflects the necessity for the viruses to preserve their protein phenotype. Accordingly, we explore its implications for genome compactness by considering only sequences that encode for the same proteins as the WT RNA. This amounts to restricting our considerations only to the rather limited combinatorial subspace of synonymous variants of WT viral RNA sequences.
We recall that synonymous mutations originate in the degenerate mapping of the 61 possible codons, which are nucleotide triplets, to the 20 canonical amino acids. Equivalent codons typically differ only at the third nucleotide (50). Accordingly, we shall assume, for simplicity, that the A, U, G, and C nucleotides can appear with equal probability at the third codon position. One can then estimate that two synonymous versions of a gene have a nucleotide sequence identity of ∼75%. Because, in the set of viruses considered in our study, on average (90 ± 7)% of the genome codes for at least one gene, and additionally assuming for simplicity that the four nucleotides have equal probability in the noncoding region that we are not constraining, we can estimate that at least ∼66–73% of the whole genome will be conserved under synonymous mutation flow.
This limited genome composition variability is further thinned down by the imposed conservation of the dinucleotide composition characteristic for the virus family and, in some viruses, by the presence of overlapping reading frames that dramatically reduce the possibility to mutate the third nucleotide in a codon. Due to these two factors, it is found that typical sequence identity between WT sequences and their synonymous mutations are in the ∼66–85% range as shown in Fig. 2 B.
Figure 2.
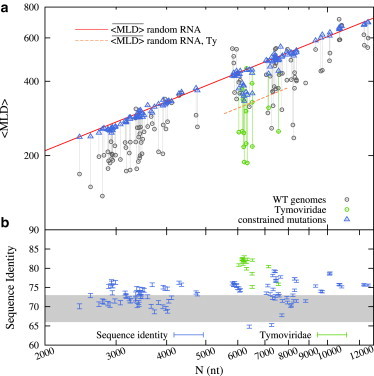
(a) Influence of synonymous point mutations on MLD. (Gray circles) The 〈MLD〉 values of WT viral sequences from Fig. 1b; (blue triangles) values of synonymously mutated sequences. Scaling laws for values of random RNA sequences with viral-like and Tymoviridae-like composition are shown as in Fig. 1. (b) The average degree of sequence identity between the mutated and WT sequences. (Gray-shaded area) Values one would expect if only one in three nucleotides were allowed to mutate in the coding regions of the genomes. Note that Tymoviridae genomes (green) are more conserved than the others. This is due to the presence of overlapping reading frames covering, on average, 30% of their genome. (To see this figure in color, go online.)
The sequence space of synonymous mutations is thus so severely restricted that there is no reason to expect that their progressive accumulation has the same effect on compactness as the unrestricted random shuffling of viral RNA sequences. As a matter of fact, the constrained synonymously-mutated sequences could have, a priori, approximately the same compactness as WT sequences or even improve it! To support the earlier observations that WT RNAs are optimized for their spatial compactness, one must therefore necessarily demonstrate that the accumulation of synonymous mutations, while leaving the encoded protein phenotype and the chemical composition of the sequence unchanged, progressively destroys the spatial compactness that is observed in WT sequences, which is quantified by their respective MLDs.
To address this point, we start from WT viral RNA sequences and generate a mutation flow in the sequence space using a Monte Carlo algorithm that proposes point mutations of the sequence and accepts or rejects them based on the constraints of synonymity and the conservation of the dinucleotide frequencies characteristic for a given virus family (see also Materials and Methods). The typical compactness of the resulting synonymously mutated WT genomes is again characterized by the asymptotic value of 〈MLD〉, averaged additionally over different mutated sequences and denoted by .
The resulting MLDs are shown in Fig. 2 a. It is indeed striking to notice that despite the strongly reduced available sequence space, the value of synonymously mutated sequences falls on the same curve that describes the of random sequences, given by the power law in Eq. 3. This fundamental observation can be condensed in the symbolic statement
| (4) |
where N is the genome length and the arrow is a shorthand for indicating the flow in the synonymous mutations subspace.
This result proves the conjecture that the WT genomes are indeed characterized by a certain optimality of the MLD which, in turn, reflects atypically high degrees of RNA fold compactness. In fact, the results of Fig. 2 b demonstrate that the WT MLD/compactness can be obliterated even within a much restricted subset of mutations that otherwise leave the viral phenotype and sequence composition unchanged.
As an aside, we note that Tymoviridae exhibit an atypical behavior, with the limiting value of under the synonymous mutation flow approaching values that are still below the ones characteristic for random RNAs. The reason for this lies in the fact that Tymoviridae have a different nucleotide composition with respect to other viral families; upon accounting for this different composition, one obtains a different prefactor for the scaling law in Eq. 3, corresponding to smaller values of MLD, which are indicative of higher compactness (as shown in Fig. 1 c; see Fig. S3 for more details).
Synonymous mutation flow and the stability of genome MLD
The previous result leads us to examine the details of the implied synonymous mutation flow (Eq. 4) and the stability of the terminal, asymptotic state of the mutated sequence. In particular, we wish to establish the minimal number of point nucleotide mutations that are needed to bring the MLD of a viral RNA from its WT value to the random reference value. It is especially interesting to ascertain whether this change in compactness happens progressively, indicating that a continuous accumulation of mutations is responsible for disrupting the WT RNA spatial compactness, or that the change is due to sporadic, punctuated events, which would suggest the presence of specific RNA hotspots, where mutations can dramatically affect fold compactness.
To illuminate this point, we considered nine synthetic synonymous mutation flow trajectories for four different viral sequences extracted from three viruses picked at random from three different families: brome mosaic virus (BMV), ononsis yellow mosaic virus (OnYMV), and equine rhinitis B virus 1 (ERBV1). The considered sequences were chosen in order to probe the whole range of genome lengths spanning from N ≃ 2800 nt to N ≃ 8800 nt. The trajectories were generated using the same MC scheme used to generate the equilibrium data presented in Fig. 2 (see also Materials and Methods), but with a much more frequent sampling of the mutated sequences (every N/100 attempted synonymous mutations) so as to leave detectable correlations in the series of generated sequences—in this way mimicking the viral mutation dynamics.
The results are shown in Fig. 3. From the mutation flow trajectories we discern that, at least for the sequences considered, the change in compactness follows the continuous and gradual accumulation of synonymous mutations, and does not take place in a punctuated manner. Nonetheless, not many mutations are needed to make the MLD of these sequences already indistinguishable from that of randomized RNAs. In fact, mutating not more than ∼5% of the full genome suffices to erase the characteristic WT RNA compactness imprint.
Figure 3.
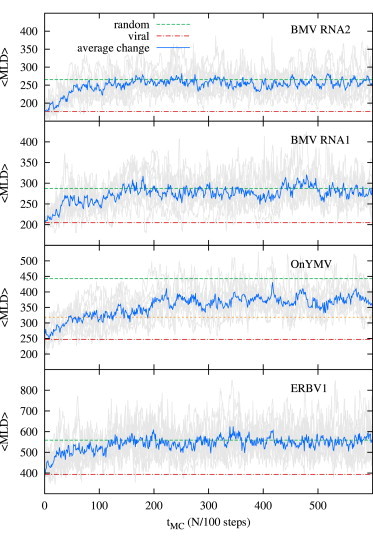
Mutation dynamics trajectories for four viral ssRNA sequences. (Top to bottom) BMV RNA2 and RNA1 segments from the tripartite genome of BMV (Bromoviridae), OnYMV (Tymoviridae), and ERBV1 (Picornaviridae). Each panel shows nine 〈MLD〉 trajectories and their average value (blue) for each sequence in units of MC steps, N/100. (Red dot-dashed lines and green dashed lines) 〈MLD〉 values of WT RNAs and the values of random RNAs (for viral-like composition, Eq. 3), respectively. Note that in the case of OnYMV, a Tymovirus, we must consider the appropriate asymptotic value of for random RNAs with Tymoviridae-like composition (see Fig. 1). This value is shown in the figure (orange short-dashed line). (To see this figure in color, go online.)
A further interesting point clarified by the mutation flow trajectories shown in Fig. 3 is that the genome-fold compactness is not completely optimized even in the case of WT sequences. In fact, for the four sequences considered in Fig. 3, one occasionally observes more compact folded states, particularly during the initial part of the trajectories.
To better explore this interesting observation, we computed the probability density of finding mutated sequences with given 〈MLD〉 as a function of the sequence identity to the WT sequence ratio, and plotted it as a color-coded heatmap. These probability density plots are shown in Fig. 4, and we can observe that, for some of the genomes considered (such as BMV RNA1 and ERBV1), more compact structures are reachable even when nearly all the unconstrained nucleotides have already been mutated. This point is most relevant in this context. In fact, it demonstrates that the sequence-based synonymity constraint and the structure-based one for fold compactness, despite being in competition, can still be compatible.
Figure 4.
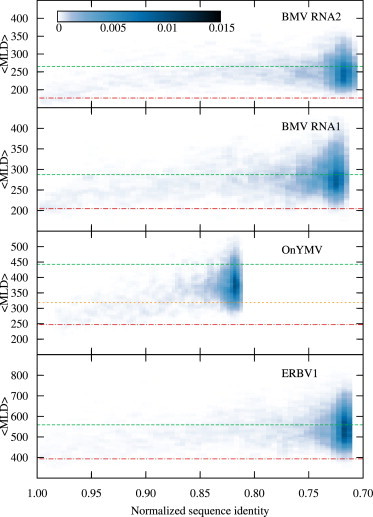
Color-coded heat maps for the probability density of finding mutated sequences with given 〈MLD〉 and sequence identity with the WT sequence. The probability density for each virus is computed and normalized over the whole length of the nine mutation trajectories (1500 MC steps) shown in Fig. 3. (Red dot-dashed lines and green dashed lines) 〈MLD〉 values of WT RNA and the values of random RNAs (with viral-like composition, Eq. 3), respectively. (Orange short-dashed line) In the OnYMV case, the random value for Tymoviridae-like composition is shown. (To see this figure in color, go online.)
This point is made more poignantly by considering the near-native pool of synonymous sequences (e.g., those with sequence identity ≥95%) for the four cases presented in Fig. 4. Across these instances, it is found that 12–21% of the near-native synonymous sequences have a predicted fold compactness equal to or higher than that of the wild-type one. This indicates that the well-optimized viral sequences still have a portion of phase space available for evolving while respecting both sequence- and structure-based stringent constraints. This appreciable residual mutation freedom may be clearly necessary to simultaneously accommodate other concurrent selection constraints.
Taking into account codon usage bias and untranslated regions
Finally, we examine the effect of two additional constraints that are known to be relevant for some viruses, and which may play a role in maintaining viral RNA compactness. The first constraint is given by the presence of functionally important secondary RNA structures in the UTRs at the 3′ and 5′ ends of several viral genomes (51–53). We take into account this constraint by simply limiting the mutation flow to the coding regions of the genomes. Note that with this additional constraint, our theoretical estimate of the overall sequence identity between WT sequences and sequences mutated asymptotically to saturation, moves from the 66–73% range to the 76–83% one.
The second additional constraint is given by the fact that, because viruses adapt to their hosts, not all the codons that translate into the same amino acid are statistically equivalent: some of them are more probable than others. This codon usage bias is known to be an important constraint for several viruses. In fact, changing the codon bias or the codon-pair bias leads to attenuated viruses and has been proposed as a possible vaccination strategy (54,55). To produce mutated sequences with WT codon populations, we shuffled the equivalent codons within every viral gene (see Materials and Methods for details regarding the implementation of codon-bias-preserving synonymous mutations).
The results obtained with both of these constraints are compared in Fig. 5 against those previously obtained using synonymous point mutations. It is important to note that even with these additional constraints, which further thin out the phase space available to mutations, our results remain valid—confirming the presence of an evolutionary pressure to produce compact RNA folds.
Figure 5.
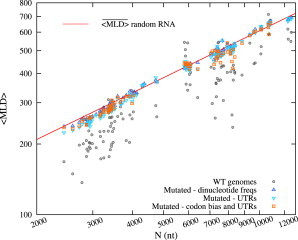
The values for the synonymous constraint only (upward triangles), and for the additional constraints of preserving UTR sequences (downward triangles) and UTR sequences with codon biases (squares). The values for these last two cases are evaluated over a set of 150 mutated sequences for each virus. Data are presented in the same manner as in Fig. 2 (see also Fig. S3 for UTRs preserving synonymous point mutations of Tymoviridae). (To see this figure in color, go online.)
Conclusions
While the fundamental mechanisms by which point mutations affect the fitness of the organisms in their respective environments (via the transcription of the mutated nucleotide sequence into the modified protein products) are well understood (12–14), it is less known what the effects are on the purely physico-chemical properties of their genomes. In order to investigate possible parallel selection mechanisms and eventual embedded levels of coding that control the compactness of viral ssRNA folds, we analyzed a synthetic model for accumulating synonymous mutations in viral RNAs and assessed their impact on the spatial compactness of the genome as quantified by the MLD measure, introduced by Yoffe et al. (33). We have analyzed the effects of synonymous mutations under different constraints on ssRNA genomes for a large number of different viral families with icosahedral capsids, and compared the changes in their compactness to randomly shuffled RNA sequences with the same nucleotide composition, which are in general significantly less compact than those encapsidated by viruses.
By using extensive computational analysis, we have shown that progressive accumulation of synonymous point mutations (although neutral from the functional point of view because they conserve the expressed protein complement) completely erases the typical compactness of viral WT RNA folds. In fact, under the synonymous mutation flow, the MLDs of WT RNAs approach their corresponding random RNA values in a continuous manner even after a relatively small number of mutations. Although, in principle, the emergence of viral RNA fold compactness may still be related to some other evolutionary pressure, our results rule out the principal ones, including codon bias and the preservation of functional UTRs, and thus strongly support the independent evolution of viral RNA fold compactness. Arguably, such a dramatic reduction in RNA fold compactness, which in this respect eventually makes it undistinguishable from a random RNA sequence, has a relevant impact on the virion assembly and therefore on the ability of viruses to replicate and propagate their infection. These results are strengthened by the observation that the typical WT RNA compactness is not related to codon usage bias nor is it dictated by the particular sequence/structure of its noncoding regions. In fact, synonymous mutations that preserve both these properties are still found to destroy the typical WT RNA compactness.
The connection between the viral RNA sequence and its physical properties, such as its compactness, may in future allow control of the physical properties of viral RNAs and specifically their aptitude for efficient packing. This, we believe, may lead to improving and broadening the scope of existing strategies that harness viral mutation rates to achieve virus attenuation.
Acknowledgments
L.T., A.L.B., and R.P. acknowledge support from Slovenian Agency for Research and Development (ARRS) grant Nos. P1-0055, J1-4297, and J1-4134. C.M. acknowledges support from the Italian Ministry of Education, Projects of National Interest grant No. 2010HXAW77.
Supporting Material
References
- 1.Olsthoorn R.C., van Duin J. Evolutionary reconstruction of a hairpin deleted from the genome of an RNA virus. Proc. Natl. Acad. Sci. USA. 1996;93:12256–12261. doi: 10.1073/pnas.93.22.12256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klovins J., Berzins V., van Duin J. A long-range interaction in Qβ RNA that bridges the thousand nucleotides between the M-site and the 3′ end is required for replication. RNA. 1998;4:948–957. doi: 10.1017/s1355838298980177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dykeman E.C., Stockley P.G., Twarock R. Packaging signals in two single-stranded RNA viruses imply a conserved assembly mechanism and geometry of the packaged genome. J. Mol. Biol. 2013;425:3235–3249. doi: 10.1016/j.jmb.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 4.Dykeman E.C., Stockley P.G., Twarock R. Solving a Levinthal’s paradox for virus assembly identifies a unique antiviral strategy. Proc. Natl. Acad. Sci. USA. 2014;111:5361–5366. doi: 10.1073/pnas.1319479111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simmonds P., Tuplin A., Evans D.J. Detection of genome-scale ordered RNA structure (GORS) in genomes of positive-stranded RNA viruses: implications for virus evolution and host persistence. RNA. 2004;10:1337–1351. doi: 10.1261/rna.7640104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanjuán R., Bordería A.V. Interplay between RNA structure and protein evolution in HIV-1. Mol. Biol. Evol. 2011;28:1333–1338. doi: 10.1093/molbev/msq329. [DOI] [PubMed] [Google Scholar]
- 7.Cuevas J.M., Domingo-Calap P., Sanjuán R. The fitness effects of synonymous mutations in DNA and RNA viruses. Mol. Biol. Evol. 2012;29:17–20. doi: 10.1093/molbev/msr179. [DOI] [PubMed] [Google Scholar]
- 8.Davis M., Sagan S.M., Simmonds P. Bioinformatic and physical characterizations of genome-scale ordered RNA structure in mammalian RNA viruses. J. Virol. 2008;82:11824–11836. doi: 10.1128/JVI.01078-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holmes E.C. Oxford University Press; New York: 2009. The Evolution and Emergence of RNA Viruses. [Google Scholar]
- 10.Belshaw R., Gardner A., Pybus O.G. Pacing a small cage: mutation and RNA viruses. Trends Ecol. Evol. (Amst.) 2008;23:188–193. doi: 10.1016/j.tree.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eigen M. Viruses: evolution, propagation, and defense. Nutr. Rev. 2000;58:S5–S16. doi: 10.1111/j.1753-4887.2000.tb07798.x. [DOI] [PubMed] [Google Scholar]
- 12.Wylie C.S., Shakhnovich E.I. A biophysical protein folding model accounts for most mutational fitness effects in viruses. Proc. Natl. Acad. Sci. USA. 2011;108:9916–9921. doi: 10.1073/pnas.1017572108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen P., Shakhnovich E.I. Lethal mutagenesis in viruses and bacteria. Genetics. 2009;183:639–650. doi: 10.1534/genetics.109.106492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duffy S., Shackelton L.A., Holmes E.C. Rates of evolutionary change in viruses: patterns and determinants. Nat. Rev. Genet. 2008;9:267–276. doi: 10.1038/nrg2323. [DOI] [PubMed] [Google Scholar]
- 15.Gong L.I., Suchard M.A., Bloom J.D. Stability-mediated epistasis constrains the evolution of an influenza protein. eLife. 2013;2:e00631. doi: 10.7554/eLife.00631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hyeon C., Dima R.I., Thirumalai D. Size, shape, and flexibility of RNA structures. J. Chem. Phys. 2006;125:194905. doi: 10.1063/1.2364190. [DOI] [PubMed] [Google Scholar]
- 17.Marenduzzo D., Orlandini E., Micheletti C. DNA-DNA interactions in bacteriophage capsids are responsible for the observed DNA knotting. Proc. Natl. Acad. Sci. USA. 2009;106:22269–22274. doi: 10.1073/pnas.0907524106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marenduzzo D., Micheletti C., Sumners W. Topological friction strongly affects viral DNA ejection. Proc. Natl. Acad. Sci. USA. 2013;110:20081–20086. doi: 10.1073/pnas.1306601110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Erdemci-Tandogan G., Wagner J., Zandi R. RNA topology remolds electrostatic stabilization of viruses. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2014;89:032707. doi: 10.1103/PhysRevE.89.032707. [DOI] [PubMed] [Google Scholar]
- 20.Nap R.J., Lošdorfer Božič A., Podgornik R. The role of solution conditions in the bacteriophage PP7 capsid charge regulation. Biophys. J. 2014;107:1970–1979. doi: 10.1016/j.bpj.2014.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caspar D.L.D., Namba K. Switching in the self-assembly of tobacco mosaic virus. Adv. Biophys. 1990;26:157–185. doi: 10.1016/0065-227x(90)90011-h. [DOI] [PubMed] [Google Scholar]
- 22.Bruinsma R.F., Gelbart W.M., Zandi R. Viral self-assembly as a thermodynamic process. Phys. Rev. Lett. 2003;90:248101. doi: 10.1103/PhysRevLett.90.248101. [DOI] [PubMed] [Google Scholar]
- 23.Reguera J., Carreira A., Mateu M.G. Role of interfacial amino acid residues in assembly, stability, and conformation of a spherical virus capsid. Proc. Natl. Acad. Sci. USA. 2004;101:2724–2729. doi: 10.1073/pnas.0307748101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh S., Zlotnick A. Observed hysteresis of virus capsid disassembly is implicit in kinetic models of assembly. J. Biol. Chem. 2003;278:18249–18255. doi: 10.1074/jbc.M211408200. [DOI] [PubMed] [Google Scholar]
- 25.Nguyen H.D., Reddy V.S., Brooks C.L., 3rd Deciphering the kinetic mechanism of spontaneous self-assembly of icosahedral capsids. Nano Lett. 2007;7:338–344. doi: 10.1021/nl062449h. [DOI] [PubMed] [Google Scholar]
- 26.Castellanos M., Pérez R., Mateu M.G. Mechanical elasticity as a physical signature of conformational dynamics in a virus particle. Proc. Natl. Acad. Sci. USA. 2012;109:12028–12033. doi: 10.1073/pnas.1207437109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roos W.H., Gertsman I., Wuite G.J.L. Mechanics of bacteriophage maturation. Proc. Natl. Acad. Sci. USA. 2012;109:2342–2347. doi: 10.1073/pnas.1109590109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polles G., Indelicato G., Micheletti C. Mechanical and assembly units of viral capsids identified via quasi-rigid domain decomposition. PLOS Comput. Biol. 2013;9:e1003331. doi: 10.1371/journal.pcbi.1003331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cadena-Nava R.D., Comas-Garcia M., Gelbart W.M. Self-assembly of viral capsid protein and RNA molecules of different sizes: requirement for a specific high protein/RNA mass ratio. J. Virol. 2012;86:3318–3326. doi: 10.1128/JVI.06566-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Comas-Garcia M., Cadena-Nava R.D., Gelbart W.M. In vitro quantification of the relative packaging efficiencies of single-stranded RNA molecules by viral capsid protein. J. Virol. 2012;86:12271–12282. doi: 10.1128/JVI.01695-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perlmutter J.D., Qiao C., Hagan M.F. Viral genome structures are optimal for capsid assembly. eLife. 2013;2:e00632. doi: 10.7554/eLife.00632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harvey S.C., Zeng Y., Heitsch C.E. The icosahedral RNA virus as a grotto: organizing the genome into stalagmites and stalactites. J. Biol. Phys. 2013;39:163–172. doi: 10.1007/s10867-013-9312-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoffe A.M., Prinsen P., Ben-Shaul A. Predicting the sizes of large RNA molecules. Proc. Natl. Acad. Sci. USA. 2008;105:16153–16158. doi: 10.1073/pnas.0808089105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fang L.T., Gelbart W.M., Ben-Shaul A. The size of RNA as an ideal branched polymer. J. Chem. Phys. 2011;135:155105. doi: 10.1063/1.3652763. [DOI] [PubMed] [Google Scholar]
- 35.Gopal A., Zhou Z.H., Gelbart W.M. Visualizing large RNA molecules in solution. RNA. 2012;18:284–299. doi: 10.1261/rna.027557.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.National Center for Biotechnology Information (NCBI) Nucleotide Database. 2013. http://www.ncbi.nlm.nih.gov/nucleotide. Accessed May 27, 2013.
- 37.Hulo C., de Castro E., Le Mercier P. ViralZone: a knowledge resource to understand virus diversity. Nucleic Acids Res. 2011;39:D576–D582. doi: 10.1093/nar/gkq901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simon-Loriere E., Holmes E.C., Pagán I. The effect of gene overlapping on the rate of RNA virus evolution. Mol. Biol. Evol. 2013;30:1916–1928. doi: 10.1093/molbev/mst094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chirico N., Vianelli A., Belshaw R. Why genes overlap in viruses. Proc. Biol. Sci. 2010;277:3809–3817. doi: 10.1098/rspb.2010.1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pedersen A.-M.K., Jensen J.L. A dependent-rates model and an MCMC-based methodology for the maximum-likelihood analysis of sequences with overlapping reading frames. Mol. Biol. Evol. 2001;18:763–776. doi: 10.1093/oxfordjournals.molbev.a003859. [DOI] [PubMed] [Google Scholar]
- 41.Chung W.-Y., Wadhawan S., Nekrutenko A. A first look at ARFome: dual-coding genes in mammalian genomes. PLOS Comput. Biol. 2007;3:e91. doi: 10.1371/journal.pcbi.0030091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nussinov R. Nearest neighbor nucleotide patterns. Structural and biological implications. J. Biol. Chem. 1981;256:8458–8462. [PubMed] [Google Scholar]
- 43.Durstenfeld R. ALGORITHM 235: random permutation. Commun. ACM. 1964;7:420. [Google Scholar]
- 44.Knuth D.E. 2nd Ed. Vol. 2. Addison-Wesley; Reading, MA: 1981. Seminumerical algorithms. (The Art of Computer Programming). [Google Scholar]
- 45.Anisimova M., Kosiol C. Investigating protein-coding sequence evolution with probabilistic codon substitution models. Mol. Biol. Evol. 2009;26:255–271. doi: 10.1093/molbev/msn232. [DOI] [PubMed] [Google Scholar]
- 46.Gu W., Zhou T., Wilke C.O. A universal trend of reduced mRNA stability near the translation-initiation site in prokaryotes and eukaryotes. PLOS Comput. Biol. 2010;6:e1000664. doi: 10.1371/journal.pcbi.1000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saito M., Matsumoto M. SIMD-oriented fast Mersenne Twister: a 128-bit pseudo-random number generator. In: Keller A., Heinrich S., Niederreiter H., editors. Monte Carlo and Quasi-Monte Carlo Methods 2008. Springer; Berlin, Germany: 2008. pp. 607–622. [Google Scholar]
- 48.Bundschuh R., Hwa T. Statistical mechanics of secondary structures formed by random RNA sequences. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2002;65:031903. doi: 10.1103/PhysRevE.65.031903. [DOI] [PubMed] [Google Scholar]
- 49.Lorenz R., Bernhart S.H., Hofacker I.L. VIENNARNA Package 2.0. Algorithms Mol. Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nelson D.L., Cox M.M. 5th Ed. W. H. Freeman; New York: 2008. Lehninger Principles of Biochemistry. [Google Scholar]
- 51.Marz M., Beerenwinkel N., Töpfer A. Challenges in RNA virus bioinformatics. Bioinformatics. 2014;30:1793–1799. doi: 10.1093/bioinformatics/btu105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alvarez D.E., De Lella Ezcurra A.L., Gamarnik A.V. Role of RNA structures present at the 3′UTR of dengue virus on translation, RNA synthesis, and viral replication. Virology. 2005;339:200–212. doi: 10.1016/j.virol.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 53.Tsukiyama-Kohara K., Iizuka N., Nomoto A. Internal ribosome entry site within hepatitis C virus RNA. J. Virol. 1992;66:1476–1483. doi: 10.1128/jvi.66.3.1476-1483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bull J.J., Molineux I.J., Wilke C.O. Slow fitness recovery in a codon-modified viral genome. Mol. Biol. Evol. 2012;29:2997–3004. doi: 10.1093/molbev/mss119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Coleman J.R., Papamichail D., Mueller S. Virus attenuation by genome-scale changes in codon pair bias. Science. 2008;320:1784–1787. doi: 10.1126/science.1155761. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.