

Abstract
Although the structure, function, conformational dynamics, and controlled thermodynamics of proteins are manifested by their corresponding amino acid sequences, the natural rules for molecular design and their corresponding interplay remain obscure. In this study, we focused on the role of internal cavities of proteins in conformational dynamics. We investigated the pressure-induced responses from the cavity-enlarged L99A mutant of T4 lysozyme, using high-pressure NMR spectroscopy. The signal intensities of the methyl groups in the 1H/13C heteronuclear single quantum correlation spectra, particularly those around the enlarged cavity, decreased with the increasing pressure, and disappeared at 200 MPa, without the appearance of new resonances, thus indicating the presence of heterogeneous conformations around the cavity within the ground state ensemble. Above 200 MPa, the signal intensities of >20 methyl groups gradually decreased with the increasing pressure, without the appearance of new resonances. Interestingly, these residues closely matched those sensing a large conformational change between the ground- and high-energy states, at atmospheric pressure. 13C and 1H NMR line-shape simulations showed that the pressure-induced loss in the peak intensity could be explained by the increase in the high-energy state population. In this high-energy state, the aromatic side chain of F114 gets flipped into the enlarged cavity. The accommodation of the phenylalanine ring into the efficiently packed cavity may decrease the partial molar volume of the high-energy state, relative to the ground state. We suggest that the enlarged cavity is involved in the conformational transition to high-energy states and in the volume fluctuation of the ground state.
Introduction
During the state of equilibrium, proteins in solution fluctuate over a broad spectrum of structural states, ranging from folded to unfolded conformations (1–3). Structural adaptability and the ability to facilitate transitions from the basic folded state (i.e., the ground state) to higher Gibbs free energy states are both necessary for various protein functions, including signal transduction (4) and enzymatic reactions (5). Multiple NMR techniques, such as R2 dispersion (5,6) and 1H/2H exchange NMR spectroscopy (7,8) have been widely used to study the intrinsic conformational fluctuation in proteins. Our group and several other groups have previously shown that multidimensional NMR spectroscopy combined with a pressure perturbation (i.e., high-pressure NMR spectroscopy) is a powerful tool that allows us to study the conformational fluctuations of a protein over a wide spatial and temporal range (9–19). High-pressure NMR spectroscopy has demonstrated that pressure-stabilized states are similar in structure and dynamics to the intrinsically populated states at atmospheric pressure (20,21), and that high-energy states are evolutionarily conserved among homologous proteins (22,23). Although the three-dimensional (3D) structure, conformational dynamics, controlled thermodynamics, and functions of proteins should be manifested by their particular amino acid sequences, the natural rules for molecular design remain largely unknown.
Internal cavities are important structural elements that produce conformational fluctuations in proteins. We have recently shown that cavities are conserved at similar positions in lysozymes derived from a variety of biological species, and that they are responsible for producing certain types of selective disorder in proteins (24). Moreover, from a thermodynamic viewpoint, it was demonstrated that cavity volume in a folded protein represents a major contribution on the volume changes upon protein unfolding (25). To gain further insights into the role of internal cavities of proteins, we investigated the conformational fluctuations in the cavity mutant L99A of T4 lysozyme, using high-pressure NMR spectroscopy. In particular, we focused our attention on the NMR resonances from the methyl groups that line the hydrophobic cavities of this protein.
Wild-type T4 lysozyme is a small hydrolytic enzyme, comprising two domains (N-terminal domain and C-terminal domain), connected by a long central helix, designated as the C-helix (Fig. S1 a in the Supporting Material). The protein contains hydrophobic and hydrophilic cavities. An L99A (Leu-99 to Ala-99) mutation increases the size of the preexisting hydrophobic cavity to ∼150 Å3 in the core of the C-terminal region (26) (Fig. S1 b). The L99A mutant has been used as a model system for understanding protein dynamics in the ligand binding process, as the enlarged cavity allows the ligand binding of substituted benzenes (27) as well as xenon (28). Many physicochemical studies and structure determinations have been performed using x-ray crystallography and NMR spectroscopy (6,27–32). X-ray crystallography showed that the designed hydrophobic cavity in the L99A mutant is sterically inaccessible to the incoming ligands, yet the protein is able to rapidly bind to benzene and to other similar ligands in solution (29). In contrast, the R2 dispersion-based NMR analysis of L99A identified a slow timescale motion around the cavity, which is a conformational transition between the ground state (native state) and an excited state (i.e., high free-energy state) (6,30,31). Structural modeling based on chemical shifts (CS-Rosetta) identified a conformation for the transiently formed state of the protein, in which the aromatic side chain of phenylalanine 114 (F114) was flipped into the enlarged cavity with a simultaneous reorientation of the F-helix (32) (Fig. S1 c). Similarly, in the case of the L121A/L133A cavity mutant of T4 lysozyme, enlargement of the cavity led to a more prominent population of an alternatively packed conformation, in which the cavity was filled, or partially filled, with an engineered side chain (i.e., nitroxide) (33). In this work we show that the application of high pressure to T4 lysozyme L99A leads to a redistribution of enzymes in the stable conformation, resulting in a higher occupancy of enzymes in the excited state.
Materials and Methods
NMR sample preparations
Recombinant cysteine-free T4 lysozyme L99A as a cavity mutant of pseudo wild-type (C54T/C97A; WT∗) was expressed and purified by the methods described previously (34). Uniformly labeled 15N- and 13C-T4 lysozyme was expressed in the M9 media with 15NH4Cl and 13C-glucose as the sole sources of nitrogen and carbon, respectively. The protein sample for the NMR measurements was dissolved in 50 mM phosphate buffer (pH 6.0, 10% 2H2O), containing 25 mM sodium chloride at a concentration of 1 mM. DSS (2, 2-dimethyl-2-silapentane-5-sulfonate) was used as an internal reference for the 1H chemical shifts.
High-pressure NMR measurements and simulation of the NMR spectra
High-pressure NMR experiments were performed using a home-made pressure-resistive quartz cell with an outer diameter of ≈3.5 mm and an inner diameter of ≈1 mm in the pressure range 3–300 MPa, on a DRX 800 spectrometer (Bruker Biospin) (9). To avoid bubbling, we maintained the lowest pressure at 3 MPa, instead of at 0.1 MPa. The one-dimensional (1D) NMR experiments were performed at a 1H frequency of 800.16 MHz, using a 3-9-19 pulsed field gradient for water suppression. In addition, the 1H/13C heteronuclear single quantum correlation (HSQC) measurements were recorded at a proton frequency of 800.16 MHz and a 13C frequency of 201.20 MHz, with 128 increments. At all pressures, the 1H chemical shifts were referenced to the methyl signal of DSS, and the 13C chemical shift was indirectly referenced to DSS (0 ppm for 1H). The data points were extended to 2048 × 256, and 90° shifted sine-bell window functions were applied to each dimension. All the data were processed using the programs NMRPipe (35) and NMRview (36). Crosspeak intensities (volumes) and chemical shifts were obtained by NMRview. When crosspeaks overlap seriously, the intensity data are not analyzed.
The simulations of the 13C NMR spectra were performed using WINDNMR-Pro (37). The NMR spectra were simulated as a function of the population of the high-energy state, assuming a two-state exchange model. The exchange rate constant, kex, and the chemical shift difference (Δω) between the ground- and high-energy states of T4 lysozyme L99A were used in the simulation (31).
3D-RISM calculations
The 3D reference interaction site model (3D-RISM) is a computational solvent representation that is based on the statistical liquid theory (38,39) (see the Supporting Material). 3D-RISM and the prerequisite 1D-RISM solvent susceptibility calculations were performed using AmberTools 12 (40). The solvent was prepared as pure water using the SPC/e water model (with modified hydrogen radii), using 1D-RISM within the rism1d program. The solvent density was modified to meet the experimental water densities at each pressure. Three solutes (the ambient pressure x-ray, high pressure x-ray, as well as the excited state NMR modeled structures of the T4 lysozyme L99A) are 2B6Y, 2B6X, and 2LCB, respectively, in the Protein Data Bank (PDB). The solvent was completely removed, and the protonation states were set at a pH of 7.0, using PROPKA (41–43) in the PDB2PQR program (44,45). The NMR modeled structure included two more C-terminal residues (Asn and Leu), which were removed to allow a fair comparison. An optimized multiple-minimization approach was used to find the lowest local free energy structure (generalized born solvent with conjugate gradient minimization, generalized born solvent with L-BFGS minimization, and 3D-RISM with L-BFGS minimization) using the nab program in AmberTools 12. All the reported data were obtained from the final 3D-RISM distribution functions for the free energy minimized structures.
Cavity occupancies were calculated at the appropriate pressure by integrating the population function, P, within a sphere encapsulating the cavity,
| (1) |
where ρ is the average density of solvent species, gO is the 3D-RISM distribution function of water oxygen (see the Supporting Material). Excess translational entropies were taken into account by simply integrating the expression for the solvent-water excess translational entropy, S, over the appropriate volume (46),
| (2) |
Results and Discussion
Pressure-induced chemical shift changes
Recombinant cysteine-free T4 lysozyme (C54T/C97A) (pseudo wild-type; WT∗) and its cavity mutant L99A (C54T/C97A/L99A) were created previously to study the role of the hydrophobic cavity of the protein (26). The basic folded conformation (the ground state) of L99A obtained by x-ray crystallography was closely similar to that of the WT∗, as shown in Fig. S1.
1D 1H NMR and 2D 1H/13C HSQC spectra of uniformly double-labeled (13C/15N) L99A were collected at different pressures up to 300 MPa. 1D 1H NMR spectra at various pressures from 3 to 300 MPa indicated that the protein maintains its folded conformation even at 300 MPa (Fig. S2). Fig. 1 a shows a superposition of the 1H/13C HSQC spectra at different pressures, up to 300 MPa. The chemical shift changes and the signal intensities are observed as a function of increasing pressure, and the spectral changes are fully reversible. The 1H and 13C chemical shifts varied linearly with pressure, up to a pressure of 100 MPa. Fig. 1 b shows the chemical shift changes of the side-chain methyl groups at a pressure of 100 MPa (1HC, 13CH). The averages and the root mean-square deviations of the pressure-induced shifts in 1HC and 13CH were −0.029 ± 0.017 ppm and 0.19 ± 0.14 ppm, respectively. These linear pressure-induced shifts generally occur because of a small linear compression of the tertiary protein structure, in the ground state ensemble of the protein (47). However, pressure-induced chemical shifts were largely different at different sites (Fig. 1 b). Below 100 MPa, the methyl groups showing large deviations from the average behavior were located around the enlarged cavity for L99A (e.g., I78γ2, L79δ2, L84δ2, V94γ1, V94γ2, A98β, I100γ, M102ε, V103γ1 V103γ2, M106ε on the C- to E-helices, L121δ2 on the G-helix, L133δ2, and A134β on the H-helix, and I150δ1 as well as I150γ2 on the J-helix), as indicated by the green spheres in Fig. 1 c. These results indicate that mechanical compression is much more significant around the cavities than in the rest of the protein.
Figure 1.
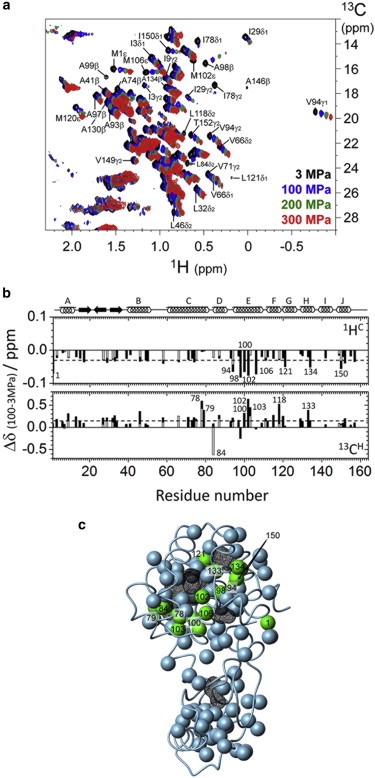
Effects of pressure on L99A T4 lysozyme. (a) 1H/13C HSQC spectra at different pressures: 3 MPa (black), 100 MPa (blue), 200 MPa (green), and 300 MPa (red) for a 1 mM uniformly 13C- and 15N-labeled protein at 25°C in 50 mM phosphate buffer (pH 6.0, 10% 2H2O/90% 1H2O mixture (v/v)) containing 25 mM NaCl. (b) Pressure-induced chemical shift changes in the low-pressure region (Δδ = δ100MPa – δ3MPa). Chemical shift changes of the methyl proton 1HC and the methyl carbon 13CH are plotted along with the residue number. The average values of the pressure shift are −0.03 for 1HC and 0.19 for 13CH, as shown by the dotted lines. Residues showing large pressure-induced chemical shift changes are represented by the residue numbers. The α-helix and β-strand regions are indicated by rings and arrows, respectively, at the top of the panel. (c) Methyl groups showing large pressure-induced shifts (Δδ > |0.05| ppm for 1HC and/or Δδ > |0.3| ppm for 13CH) are depicted by green spheres on the tertiary structure of the L99A mutant (PDB ID: 1L90), whereas the less pressure-sensitive methyl groups are depicted by gray spheres. The internal cavities are drawn with a black wire frame cage calculated using a 1.4 Å probe and the program MOLMOL (54). To see this figure in color, go online.
Pressure-induced changes in the signal intensity
Fig. 2 a shows changes in peak intensities (i.e., maximum peak height) of the 1H/13C HSQC spectra of L99A, as a function of pressure. Errors in intensity estimation are shown in Fig. S3. Several crosspeaks of the side-chain methyl groups displayed a reduced intensity with increasing pressure, up to 200 MPa. Above 200 MPa, several of the other crosspeaks also decreased in intensity, however, new signals corresponding to a disordered polypeptide chain were not observed in the HSQC spectra (Fig. 1 a). The change in peak intensity, namely a broadening or absence of crosspeaks, presumably indicates a heterogeneity of conformations and/or fluctuation among the conformations within the NMR chemical shift timescale (≈ms). The loss of total crosspeak volumes (i.e., integrated peak intensity) in the HSQC at 300 MPa equated to a 26% reduction of the original volumes (the sum of the integrated crosspeak intensities in Fig. 2 a). In the case of conventional 1H/13C HSQC, increases in 13C spin-spin relaxation in the t1 evolution period (i.e., 0–9.6 ms), as well as 1H spin-spin relaxation in t2 period (i.e., 98.4 ms) are likely responsible for the observed crosspeak broadening and loss of crosspeak intensities. In addition, transfer amplitudes of the two INEPT steps, in total 7.5 ms, may decrease with pressure if 1H spin-spin relaxation increases.
Figure 2.
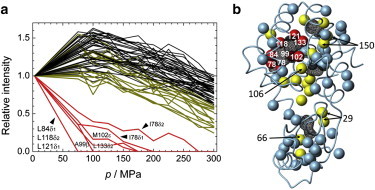
(a) Plots of maximum height (normalized at 3 MPa) of the crosspeaks in 1H/13C HSQC for L99A, obtained at various pressures, from 3 to 300 MPa. Changes in the protein concentration as a function of pressure are corrected, based on the pressure-induced compaction of the solvent water (by ≈9% at 300 MPa) (63). The crosspeaks of the side-chain methyl groups can be classified mainly into three groups: 1) the rapidly decaying group (red lines); 2) the intermediate decaying group (yellow lines); and 3) the slow decaying group (black lines). The rapid and the intermediate decaying groups comprise 1), I78δ1, I78γ2, L84δ1, A99β, M102ε, L118δ2, L121δ1, and L133δ2 and 2), I3δ1, I3γ2, M6ε, L7δ1, I29δ1, I29γ2, L46δ1, L66δ1, V71γ2, V75γ2, L84δ2, V87γ2, A98β, I100γ2, M106ε, A129β, A130β, I150δ1, and I150γ2, respectively. Errors in height estimation are presented in Fig. S3. (b) The observable side-chain methyl groups in the HSQC spectrum at 3 MPa (Fig. 1A) are depicted with spheres. Those belonging to the rapid and intermediate decaying groups are represented by red and yellow spheres, respectively, on the tertiary structure, and residues located around the internal cavities are designated by their residue number. The picture was prepared using MOLMOL (54). To see this figure in color, go online.
The crosspeaks of the methyl groups can be classified into three groups: 1), a rapidly decaying group (red lines); 2), an intermediate decaying group (yellow lines); and 3), a slowly decaying or unaffected group (black lines). The methyl groups belonging to classes (1) and (2) are shown as red and yellow spheres, respectively, in the structure of L99A (Fig. 2 b). All of the rapidly decaying signals from group (1) (I78δ1, I78γ2, L84δ1, A99β, M102ε, L118δ2, L121δ1, and L133δ2) involved the methyl groups lining the enlarged hydrophobic cavity. Members of the intermediate group (2) (I3δ1, I3γ2, M6ε, L7δ1, I29δ1, I29γ2, L46δ1, L66δ1, V71γ2, V75γ2, L84δ2, V87γ2, A98β, I100γ2, M106ε, A129β, A130β, I150δ1, and I150γ2) were located around the enlarged cavity, as well as other cavities of the protein (Fig. 2 b).
Conformational fluctuations within the ground state ensemble
The partial molar volume of a protein fluctuates with thermal agitation in two different ways: A fluctuation either occurs within the same subensemble of conformers, or occurs because of a transition into a different subensemble of conformers possessing a higher Gibbs free energy (referred to as an excited state). Thus, a protein biomolecule adopts a smaller partial molar volume using either or both of the aforementioned adaptations.
Fluctuations occurring within the same subensemble of conformers can be studied by observing the pressure-induced changes in the chemical shifts and peak intensities. The 13C chemical shifts reflect site-specific structural changes at the side chains of the protein, whereas the amide 1H and 15N chemical shifts generally indicate the response of the hydrogen-bond distance and torsion angles to pressure (47). Furthermore, the methyl 13C chemical shifts are more sensitive to atomic packing. Regardless of the underlying dynamics, any change in the measured chemical shifts corresponds to the average of all the structural changes occurring rapidly on the NMR chemical shift time scale (≈ms). We interpret the linear chemical-shift change observed in the lower pressure region (up to ≈100 MPa) to be the result of a linear change in the average internuclear distances and torsion angles (11), the combination of which determines the compressibility coefficient (βT) of the protein.
Larger deviations from the average pressure-induced shifts are observed for the 13C nuclei of methyl groups lining internal cavities, particularly those at the enlarged cavity of L99A. These variations indicated that larger structural changes, including the compression of interatomic distances, take place around the cavities, rather than elsewhere inside the protein structure. As the macroscopic compressibility (which is the net sum of the microscopic compressibilities) is statistically correlated with the volume fluctuation of the system consisting of the solute and the solvent (48), the larger volume fluctuations occurring rapidly on the chemical shift timescale could be attributed to the enlarged cavity of the protein. Similar large pressure-induced changes in the chemical shifts were also observed around the internal cavities of BPTI (49) and hen egg white lysozyme (50). Our past and present results both indicate that volume fluctuations are greater at the residues closer to the internal cavities (at atmospheric pressure), as these regions presumably have a larger structural adaptability to pressure.
In the same pressure range as above (up to 100 MPa), the crosspeaks of the side-chain methyl moieties involved in the rapidly decaying group (1) (e.g., I78δ1, I78γ2, L84δ1, A99β, M102ε, L118δ2, L121δ1, and L133δ2) significantly lose their intensities with increasing pressure. The losses in peak intensities in 2D HSQC spectra at varying external parameters, such as pressure and temperature, are generally attributed to an increase in peak widths (i.e., spin-spin relaxation rates, R2, of the nuclei) in both dimensions, which might result from heterogeneous conformations within the basic folded ensemble, or from conformational exchange among different stable conformational states of the protein on the microsecond to millisecond timescale. As discussed below, the significant decrease in the peak intensities for some of the methyl groups cannot be explained only with the conformational exchange between the ground state and the particular high-energy state of the protein. It is presumably caused by heterogeneous configurations of the corresponding methyl groups resulting from the compression and modified hydration of the enlarged cavity.
In contrast, we observed increases in the crosspeak intensities for many of the intermediate and slow decaying methyl groups, up to 100 MPa. The same was observed for signals of the backbone in the 1H/15N HSQC data (not shown) for L99A. Of importance, we did not observe this effect for WT∗. At the moment we have no explanation for this effect, other than the possibility that it results from the formation of the cavity in the C-terminal domain.
The effect of increasing pressure on the high-energy excited state of L99A
Recently, the structure of the transiently formed high-energy state of T4 lysozyme L99A was characterized by employing a combined strategy comprising R2 dispersion NMR and CS-Rosetta model building (32). Clear structural differences between the ground- and high-energy states were observed, particularly in the vicinity of the enlarged cavity of the protein. In the high-energy state, the side chain of F114 was accommodated inside the cavity, together with a simultaneous reorientation of the F-helix, which required fluctuations outside the ground state ensemble of the protein. 15N-R2 dispersion NMR experiments showed that the high-energy state was populated to ∼3% at 25°C and atmospheric pressure, and was in equilibrium with the ground state (97% populated). The corresponding interconversion occurred on the millisecond timescale, leading to pervasive resonance broadening.
In this high-pressure NMR study, we observed severe line-broadening and missing HSQC crosspeaks, particularly for the residues around the enlarged cavity, at pressures below 200 MPa. Fig. 3 shows a representation of the methyl groups exhibiting a relatively large chemical shift difference (Δω) for the 13C nuclei (Fig. 3 a) between the ground and high-energy states, based on the 13C-R2 dispersion measurements (31), along with the resonance showing severe line-broadening with increasing pressure (Fig. 3 b). Interestingly, the latter group of residues matches closely with the group showing larger chemical shift changes upon conformational transition to the transiently formed high-energy state at atmospheric pressure.
Figure 3.
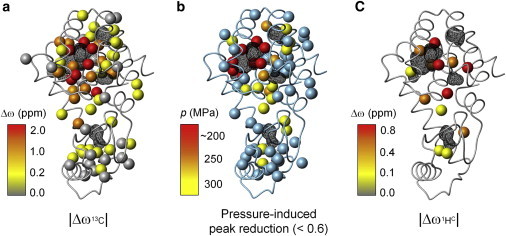
Distribution of residues based on (a) 13C chemical shift differences (|Δω|) for side-chain methyl groups between exchanging sites, as estimated by the R2 dispersion NMR experiments for L99A (31). The amplitude of |Δω| is represented by a color scale from yellow to red. (b) Side-chain methyl groups showing significant loss of their crosspeak intensities at a high pressure. The pressure at which the relative peak intensity is reduced to <0.6 from the original is represented by colors: <200 MPa; red, 250 MPa; orange, and 300 MPa; yellow. (c) Distribution of residues based on the 1H chemical shift differences (|Δω|) for side-chain methyl groups between exchanging sites, as estimated by the ZZ-exchange NMR experiments for L99A/G113A (32). The picture was prepared using MOLMOL (54). To see this figure in color, go online.
Unfortunately, similar data for the methyl protons of T4 lysozyme L99A are not available. However, the closely similar mutant L99A/G113A was studied previously, and was shown to undergo a very similar exchange between ground and excited states, albeit at a slower rate. We were thus able to use the 1H Δω values of L99A/G113A, which were measured by ZZ-exchange NMR at 1°C (32). Exchange crosspeaks in the ZZ-exchange NMR spectrum were observed for at least 13 methyl groups (including I3Hγ2, I27Hγ2, I29Hδ1, L66Hδ1, V71Hγ2, A74Hβ, I78Hγ2, A98Hβ, A99Hβ, M102Hε, I118Hδ2, L121Hδ1, and A146Hβ). Fig. 3 c shows the distribution of these methyl groups based on the chemical shift difference (Δω) for the 1H nuclei. The group showing severe line-broadening with increasing pressure again matches closely with the group showing larger Δω for the 1H nuclei.
These imply that the broadening of the HSQC crosspeaks with increased pressure, and the corresponding increase in the exchange contribution to the 13C and 1H transverse spin relaxation rates, can likely be explained by an increase in the high-energy state population and/or a decrease in the exchange rate constant kex, at high pressure.
Line-shape simulation
To test the hypothesis that the broadening of the HSQC crosspeak intensities with increased pressure can be explained by an increase in the high-energy state population and/or a decrease in the exchange rate constant kex, we simulated the changes in the 13C and 1H spectra using the WINDNMR-Pro program (37). The kex and 13C chemical shift difference (Δω) between the ground- and the transiently formed high-energy states of L99A are available in the literature (31). Because Δω values for the methyl protons of L99A have not been reported, we used the Δω values for the L99A/G113A mutant of T4 lysozyme (32), which were measured by ZZ-exchange NMR at 1°C.
Although, in reality, both the population of the high-energy state and the exchange rate constant kex could change simultaneously with increasing pressure, we first simulated 13C spectral changes as a function of the population of the transiently formed high-energy state. Fig. 4 a shows the simulated 13C spectra for L121Cδ1, which is a representative carbon nucleus showing a rapid decrease in the HSQC peak intensity as a function of pressure (group (1)). When the global exchange rate is 1449 s−1, Δω is 1.88 ppm (1.88 × 201.2 × 2π rad s−1) (31), which corresponds to a slow-to-intermediate exchange condition (kex/Δω = 0.61). Fig. 4 b shows a simulation for L84Cδ2, which belongs to the intermediate decaying group (2). Its Δω value is 0.37 ppm (0.37 × 201.2 × 2π rad s−1) (31), which correlates with an intermediate-to-fast exchange condition (kex/Δω = 3.1). The line-shape simulations clearly show that for both of the residues mentioned previously, the peak width of the ground state rapidly increases with increasing high-energy state population, and that the peak almost disappears when the populations become similar. A new peak, corresponding to the high-energy state, then starts to appear. Other examples of groups that decay at a rapid or intermediate pace are presented in Fig. S4.
Figure 4.
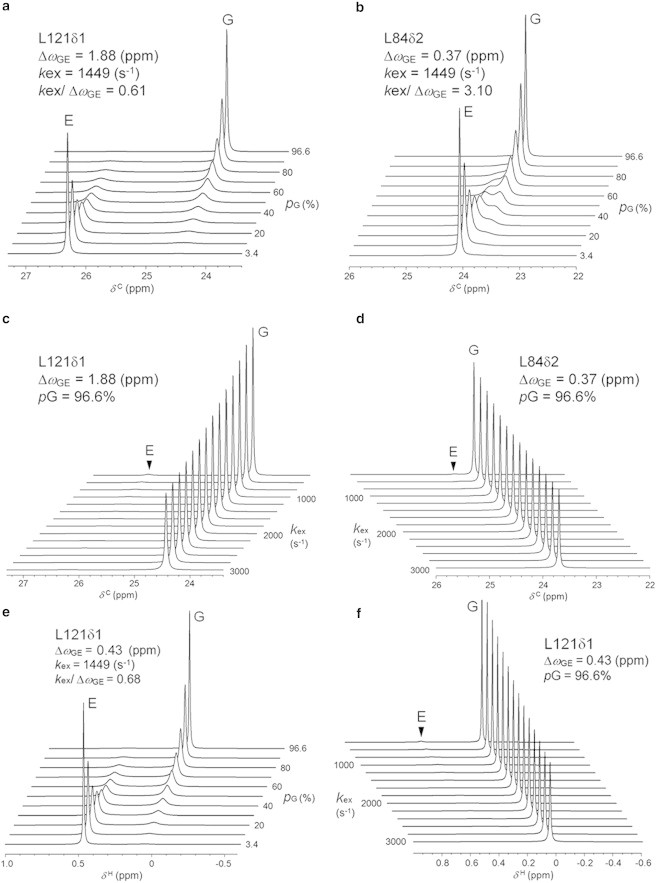
Line-shape simulation for the methyl carbon and proton peaks of L99A, in accordance with the two-state exchange model. (a and b) 13C line-shape spectral simulation for the L121Cδ1 carbon (A) and for the L84Cδ2 carbon (b), as a function of the excited state population. (c and d) 13C line-shape spectral simulation for the L121Cδ1 carbon (c) and the L84Cδ2 carbon (d), as a function of kex. The 13C chemical shift difference ΔωGE between the ground (G) and the transiently populated high-energy state (E), the rate constant kex (1449 s−1) for a chemical exchange between the two states, and a population of the high-energy state pE (3.4%) were all obtained from the literature (31). (e and f) 1H line shape spectral simulation for L121Hδ1 proton, as a function of the high-energy state population (e), and as a function of kex (f). The 1H chemical shift values of the ground and high-energy states of L99A/G113A, measured by ZZ-exchange NMR experiments at 1°C (32), are used in the simulations. The program WINDNMR-Pro was used for the simulations (37).
Second, we simulated the 13C spectra at variable kex values, assuming the population of the high-energy state to be constant (i.e., 3.4%) (Fig. 4, c and d). Changes in peak widths corresponding to the ground and high-energy states are not substantial as long as the high-energy state population is low. However, the peak widths may show larger changes with increased high-energy state populations. The kex usually decrease under high pressure because the positive activation volume for folding is larger than negative activation volume for unfolding (51–53). Thus, in the case of both slow and intermediate exchange conditions, peak widths would be expected to be sharper under high pressure.
Third, we simulated 1H spectra as a function of the high-energy state population and varying kex values (Fig. 4, e and f). In the case of L121 Hδ1, the Δω was 0.43 ppm (0.43 × 800.13 × 2π rad s−1), which corresponds to a slow-to-intermediate exchange condition (kex/Δω = 0.68). When the population of the high-energy state rises, the 1H peak width rapidly increases in a manner similar to the 13C peak width (Fig. 4 a). (see Fig. S5 for other examples of methyl protons involved in the rapid and intermediately decaying groups). Therefore, peak broadening in 1H/13C HSQC spectra would occur due to both 1H and 13C spin relaxation, dependent on the chemical shift difference (Δω). These 13C and 1H spectral simulations strongly suggest that an increase in the high-energy state population is responsible for the selective peak broadening observed with increasing pressure. In the case of the conventional HSQC, peak broadening would occur during the t1-evolution period (i.e., 0–9.6 ms) due to 13C spin-spin relaxation, and during the t2-ditection period (i.e., 98.4 ms) due to 1H spin-spin relaxation. In addition, transfer amplitudes of the two INEPT steps (i.e., 7.5 ms) may decrease with pressure if 1H spin-spin relaxation increases. Because dipolar relaxation on spin-spin relaxation rates (R2) may not alter much with pressure, exchange contribution on R2 of both nuclei could be responsible for the broadening and missing of the signals.
We noticed that signals corresponding to the high-energy state are not observed in the spectra under high pressure. Two explanations for this could be that, in general, a population of the minor state is low and NMR peaks of the minor state have larger peak widths than those of the major state (see Fig. 4), and thus, they exhibit a relatively lower signal/noise ratio. It is also possible that the high-energy state corresponds to much larger peak widths than the ground state because of conformational heterogeneity; note that intrinsic peak width for the high-energy state was assumed to be same as that for the ground state in the spectral simulation.
For some of the signals in the intermediate decaying groups, the peaks showed sigmoidal chemical shift changes, as well as intensity losses, as a function of the high-energy state population (M6ε, I29γ2, and A130β, Fig. S4 b). It is not surprising that, as a function of the high-energy state population, the chemical shifts and the line broadening, depend strongly on the magnitudes of kex and Δω. Our simulations show that the line broadening caused by pressure changes could be explained by an increase in the high-energy state population. A substantial correlation between the line broadening (large R2) at ambient pressure and the peak-intensity reductions (for the same probes) at higher pressures strongly suggests that both methods are probing the same conformational equilibrium, albeit with different populations of the associated states.
Next, we consider the differences in the peak intensity reductions between the intermediately and rapidly decaying groups. When an increase in line width (i.e., R2) occurs in both 1H and 13C dimensions, peak intensities may decrease rapidly with an increase in the high-energy state population. This applies to L121δ1 (the rapidly decaying group), but not to I3γ2 (intermediate decaying group), although each of these has relatively large Δω values for both the 13C and the 1H chemical shifts. We did not find clear differences in the peak intensity reductions between the intermediately and rapidly decaying groups. In other words, the significant loss in the peak intensities for the rapidly decaying methyl groups could not be explained solely based on the conformational exchange between the ground state and the particular high-energy state of the protein.
As a comparison, we studied pressure effects on the WT∗ protein, and found that rapid signal losses were observed for the methyl groups of M102ε, L121δ1, and L133δ2 (data not shown), all of which are located around the same (but smaller) cavity in the C-terminal region. The rapid signal loss in the WT∗ protein cannot be attributed to the transition into the high-energy state, because the volume of the cavity in the WT∗ protein is too small (39 Å3) to bind the F114 side chain. Indeed, 15N spin-spin relaxation NMR analysis did not detect a high-energy state for the WT∗ protein (30). Therefore, we conclude that the rapid loss of the peak intensities at lower pressure observed for L99A should be attributed to changes in the native state subensemble, namely heterogeneous configurations of the corresponding methyl groups.
Origins of chemical shift changes between the ground and high-energy states
Next, we tried to delineate the origins of chemical shift changes by comparing the structures of the ground state (Fig. 5 a, left panel) and the high-energy state (Fig. 5 a, right panel) of the protein. According to the high-energy state structure of L99A (modeled using CS-Rosetta), the F114 aromatic ring is accommodated in the enlarged cavity. Therefore, due to the transition, changes in the ring current shifts are expected. Fig. 5 b, shows the differences in the ring current shifts between the ground- and high-energy states of the 1H and 13C nuclei, respectively, as calculated by the MOLMOL program, which uses the Johnson-Bovey model (54). The methyl groups showing large changes due to ring current shifts for at least one of the two nuclei are I78γ2, L84δ2, L91δ1, V94γ1, A98β, A99β, M102ε, M106ε, T109γ, V111γ2, L118δ2, and L121δ1. All of them are located adjacent to F114, W138, F153, and W158 (Fig. 5 a). It is obvious that larger differences in the ring current shifts between the ground and high-energy states are observed for the groups located in close proximity to the aromatic rings than those located elsewhere in the protein structure. Moreover, the chemical shifts of the methyl protons and the carbons could be used as sensitive probes for detecting the changes in the orientation of the aromatic rings. Interestingly, a substantial loss in the signal was observed, due to an increased pressure, for the methyl groups lying at the cavity (e.g., I78δ1, I78γ2, L84δ1, A99β, M102ε, L118δ2, and L121δ1 indicated with asterisks in Fig. 5 b, respectively), where the F114 aromatic ring is expected to flip-in. The corresponding data for all observed residues is shown in Fig. S6. These results strongly support the idea that the high-energy state, in which the F114 aromatic ring flips into the cavity, is indeed stabilized by higher pressures. It is clear that an increase in the population of the high-energy state would result in the increase in exchange contribution on R2 under high pressure, as the high-energy state exhibits different 13C and/or 1H chemical shifts around the enlarged cavity. The accommodation of the F114 side chain in the cavity would confer the high-energy state a smaller partial molar volume than the ground state, because of improved packing, and because of an alteration in the hydration structure on the molecular surface.
Figure 5.
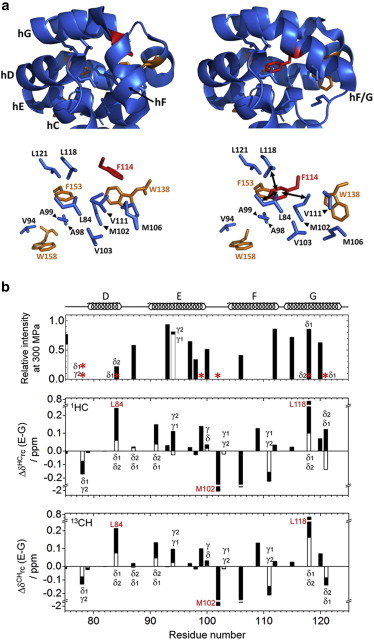
(a, top) A part of the tertiary structure of the ground state (left) and the transiently populated high-energy state (right), as modeled by CS-ROSETTA (32). (A, bottom) The hydrophobic side chains around the enlarged cavity are represented by sticks. F114 is colored in red, whereas the other aromatic side chains near the cavity are depicted in yellow. The hydrophobic side chains facing the aromatic rings are shown in blue. (b, top) The relative intensities of the 1H/13C HSQC crosspeaks at 300 MPa (normalized by the values at 3 MPa) for the methyl groups in the vicinity of the enlarged cavity. Methyl groups showing no intensity at 300 MPa are marked with an asterisk. (b, middle and bottom) The differences in ring current shifts for the methyl groups between the ground- and high-energy states (Δδrc) for the 1H (b, middle) and 13C (b, bottom), as estimated by the program MOLMOL (54). The residues facing the F114 side-chain ring in the high-energy state are denoted by red numbers. The attributes for the side-chain methyl groups are denoted with δ and γ, respectively. When the residue has two methyl groups, the group indicated at the bottom shows a larger absolute value than that of the group on the top. To see this figure in color, go online.
Conformational transitions could also be monitored by nonlinear responses in the pressure-induced chemical shifts, when Δω is much smaller than kex (Δω << kex) (11). Indeed, we observed nonlinear responses for the pressure-induced chemical shifts of several methyl groups for L99A above 100 MPa (e.g., for I9, I17, I29, and L79, shown in Fig. S7). These pressure-induced nonlinear changes may be a part of the sigmoidal changes seen when the high-energy state population is increased. However, we limited our analysis to the pressure-induced line broadening, as it can be easily monitored to delineate the structural motions in the microsecond to millisecond timescale.
Pressure-induced denaturation
Above 200 MPa, the intensities of all crosspeaks decreased in a concerted manner (Fig. 2 a). As predicted by spectral simulation, the peak width for the ground state increased substantially with the increasing population of the high-energy conformation, even in the slow exchange condition. We therefore suggest that the concerted decrease in the crosspeak intensities above 200 MPa marks the onset of an unfolded conformer U, which will not give sharp NMR signals. Because the peaks of the slow decaying group still retain ∼80% of their original integrated intensities at 300 MPa, the population of U should account for <20% of the conformational ensemble. Keep in mind that NMR peaks of the minor state may not give enough signal/noise ratio to be detected (see Fig. 4). An increase in the unfolded state population is expected under high pressure, because L99A is only marginally stable and the unfolded conformation has smaller partial molar volume than the folded one (9,25,55). According to high-pressure fluorescence studies, the unfolded state of L99A should make up ∼82% of the population at 300 MPa and 24°C (e.g., ΔG0=13.3 kJ/mol, ΔV0= −56 mL/mol at pH 7.0 and 20 mM NaCl condition) (55). Although this is inconsistent with the present high-pressure NMR data, it is not surprising because tryptophan fluorescence is sensitive to the polarity of its local environment, and the thermodynamic quantities were calculated from a two-state unfolding model. Moreover, because all three tryptophans (W126, W138, and W158) in the protein are located in the C-terminal domain, the pressure-induced changes in fluorescence quenching may report both on transitions into the excited folded and unfolded states.
Water occupancy in the hydrophobic cavity predicted from 3D-RISM
Although x-ray crystallography suggested that the enlarged hydrophobic cavity in L99A mutant was sterically inaccessible to incoming ligands, spin relaxation NMR studies indicated the presence of conformational fluctuations around the cavity. This fluctuation was much more amplified in the L99A mutant than in WT∗, in solution (6,27–29). It is challenging to understand how the hydration of the cavity correlates with the conformational fluctuation of the protein, and how the conformational fluctuations correlate with the ligand-cavity interaction.
Collins et al. (56) demonstrated an increase in the electron density in the enlarged cavity in the L99A mutant with increasing pressure, using high-pressure x-ray crystallography and molecular dynamics simulations. The number of water molecules existing in the cavity was predicted to be 0 at 0.1 MPa and was predicted to increase to about two at 200 MPa, whereas the protein structure remained intact. However, a recent high-resolution x-ray crystallographic analysis found that the diffuse electron density equivalent to ∼1.5 water molecules was present in the cavity of the crystallized protein, even at a pressure of 0.1 MPa (57).
Clearly, it is desirable to have a general and comprehensive method for predicting the water occupancy in protein cavities. Hence, we employed the 3D reference interaction site model (3D-RISM), which describes molecular solvation based on a rigorous statistical liquid theory (38). 3D-RISM is suited for analyzing the water occupancy at molecular surfaces, including the internal cavities in proteins (see Materials and Methods) (58–60). 3D-RISM analysis was carried out for three different structures; namely, the x-ray structure at 0.1 MPa (blue) (2B6Y), the x-ray structure at 200 MPa (red) (2B6X), and the NMR data-based ROSETTA model of the transiently populated high-energy conformer at 0.1 MPa (green) (2LCB). Free-energy minimized structures were obtained (for the three structures), using 3D-RISM at pressures, 0.1 MPa, 100 MPa, 200 MPa, and 300 MPa, respectively. The partial molar volumes were calculated for the two x-ray structures (2B6Y and 2B6X) at different pressures. The compressed volumes reached ∼1.3% for both the proteins, at 200 MPa (isothermal compressibility, βT ≈ 7 Mbar−1), as shown in Table 1. This is consistent with the observation of the macroscopic compressibility of proteins (4–15 Mbar−1), obtained using sound velocity measurements (61). Fig. 6, a–c, show the enlarged cavities for the three structures (a: 2B6Y, b: 2B6X, c: 2LCB) at each experimental pressure, as estimated by the conventional sphere rotation method. Fig. 6 d shows the water occupancy in the enlarged cavity for the x-ray structures at 0.1 MPa (blue; 2B6Y), 200 MPa (red; 2B6X), and for the NMR model of the transiently populated high-energy conformer, at 0.1 MPa (green; 2LCB). The water occupancy was increased from 3.37 at 0.1 MPa (2B6Y) to 3.71 (2B6X) at 200 MPa, as shown in Table 1. These data indicate that water occupancy does not increase substantially with increasing pressure, when the basic folded conformation is maintained at the elevated pressure. These 3D-RISM analyses qualitatively support the recent high-resolution x-ray crystallographic analysis, showing the presence of diffuse water in the enlarged cavity of the L99A protein, even at a pressure of 0.1 MPa. However, the number of water molecules theoretically present in the enlarged cavity was two times larger than that predicted by high-resolution x-ray crystallography (57). The diffuse electron density detected using high-resolution x-ray crystallography may correspond to the lower limit of detection, which in turn depends on the data quality and refinement method.
Table 1.
3D-RISM analysis
| Pressure | Partial molar volume |
Water occupancy |
Excess translational entropy |
|||
|---|---|---|---|---|---|---|
|
2B6Y |
2B6X |
2B6Y |
2B6X |
2B6Y |
2B6X |
|
| Å3 | Å3 | a | a | J/mol K | J/mol K | |
| 0.1 MPa | 21310 | 21272 | 3.37 | 3.41 | −42.8 | −42.9 |
| 100 MPa | 21141 | 21141 | 3.60 | 3.58 | −46.0 | −46.2 |
| 200 MPa | 21062 | 20988 | 3.84 | 3.71 | −49.5 | −48.4 |
| 300 MPa | 20865 | 20824 | 3.79 | 3.82 | −50.1 | −51.1 |
Number of water molecules equivalent to the water occupancy in the hydrophobic cavity.
Figure 6.
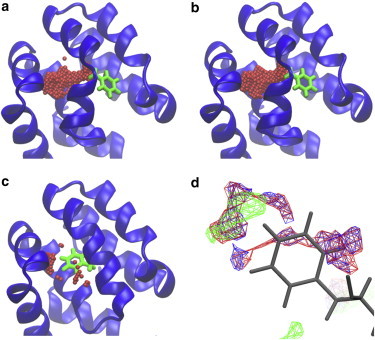
(a–c) A part of the backbone structure of L99A with the enlarged cavities estimated by the conventional sphere rotation method: (a) the x-ray structure at 0.1 MPa (2B6Y), (b) the x-ray structure at 200 MPa (2B6X), and (c) the NMR model of the transiently populated high-energy conformer at 0.1 MPa (2LCB). (d) The water occupancy in the enlarged cavity of L99A for these three structures: blue for x-ray structure at 0.1 MPa, red for x-ray structure at 200 MPa, and green for NMR model at 0.1 MPa. The F114 side chain is shown in stick representation in all panels. To see this figure in color, go online.
Interestingly, the excess translational entropy of the water molecules inside the cavity, as calculated by 3D-RISM, decreased at higher pressures (Table 1, see Materials and Methods). The excess entropy values are negative because they are relative to bulk (bulk excess entropy being zero). Because the values get more negative with increasing pressure suggests that, at higher pressures, the molecular positions are more confined, and therefore would be more easily resolved by x-ray crystallography.
As the 3D-RISM analyses described previously were carried out only on the energy-minimized structure of the protein at each pressure, they did not take into account the conformational heterogeneity of the protein as seen in solution. As compared to the crystallized protein, the protein in solution may exist as a more varied ensemble of structures, with different levels of hydration. Compression, expansion, and hinge-type motions in proteins at different pressures could bring about changes in the shape and hydration of the cavities in the protein structure. Thus, rotational and diffusive motions of water molecules in the cavities could be altered under high pressure. These dynamic motions of the water molecules and the heterogeneous conformations of the cavities would cause the chemical shifts to disperse. Such an effect could be partly involved in the pressure-induced line broadening of the NMR resonances, observed in the low-pressure region (≈100 MPa), as well as in an increase in the population of the high-energy states.
In the case of the transiently populated high-energy state, the aromatic side chain of F114 is flipped into the enlarged cavity, resulting in the displacement of water molecules (Fig. 6 c). The bulky aromatic ring causes an increase in the atomic occupancy of the enlarged cavity, which may be partly responsible for the small partial molar volume of the high-energy state. Note that the partial molar volume analysis performed using 3D-RISM was not performed on the structural models constructed by CS-Rosetta, as these models have a relatively large uncertainty for the side-chain orientations.
Cavity as a source of conformational fluctuations
The present high-pressure NMR experiments and RISM analysis for the T4 lysozyme L99A indicate that the water-containing cavity can serve as a source of conformational fluctuations. Fig. 7 shows a schematic representation of the pressure-induced conformational fluctuations of the protein along with the free-energy landscape. The ground state of the protein has larger volume fluctuations around the cavities as compared to the rest of the protein, as is evident from the chemical shift changes. The ground state of L99A seems to consist of an ensemble of conformations in which the orientation of the methyl groups and the accommodation of water molecules in the cavity are both different. Elevated pressure stabilized the lower volume conformers in the ground state ensemble, resulting in the formation of a heterogeneous ensemble of differently solvated conformers. As the pressure-induced changes cause a chemical shift, as well as a chemical shift-dependent line broadening, the volume fluctuations within the ground state ensemble would occur with rate constants similar to or faster than that of the NMR time scale (τ << ms).
Figure 7.
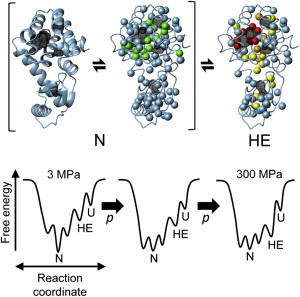
Schematic representation of the conformational equilibria in the T4 lysozyme L99A mutant along with the free-energy landscape. N, HE, and U represent the ground state ensemble, the high Gibbs free-energy state, and the unfolded state, respectively. The methyl groups expected to display a large volume fluctuation within the ground state ensemble are represented by green spheres (see Fig. 1c). Side-chain methyl groups classified into the rapid and intermediate decaying groups are represented by red and yellow spheres, respectively (see Fig. 2b). To see this figure in color, go online.
In addition, the ground state ensemble is in equilibrium with a high-energy state (Fig. 7). We found that the structural characteristics of the pressure-stabilized state of L99A closely resemble those of the transiently formed high-energy state seen at atmospheric pressure, in which the F114 aromatic ring is accommodated in the enlarged hydrophobic cavity. This coincidence validates the argument that an elevated pressure stabilizes the intrinsically existing high-energy conformations of proteins at atmospheric pressure. Under high pressure conditions, the protein uses cavities to adopt a smaller partial molar volume, using either or both of the fluctuations.
A similar loss of the HSQC crosspeaks was also observed around the water-containing cavities in hen egg white lysozyme, when the temperature decreases at high pressure. The disappearance of NMR signals suggests the formation of a heterogeneous ensemble of partially solvated conformations, facilitated by the penetration of more water molecules into cavities of the protein (24). As the water-containing cavity is conserved among lysozymes across a variety of biological species, it seems that water-containing cavities play an important role in biological functions (e.g., allocating a certain degree of mobility to the active site and providing water molecules for hydrolysis reactions). After the atomic occupancy of the cavities becomes substantial because of the hydration or ligand binding under high pressure, proteins usually collapse their cavities and unfold partially or completely, as seen in the case of ubiquitin (21), β-lactoglobulin (62), RalGDS-RBD (15), staphylococcal nuclease (25), and OspA (20). As structural changes are necessary for diverse functions such as ligand binding and signal transduction, the internal cavities of proteins could serve as an important structural element for a variety of biological events.
Acknowledgments
We thank L. E. Kay and G. Bouvignies for providing us with the ZZ-exchange spectra of the L99A/G113A mutant of T4 lysozyme.
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas from the MEXT of Japan to R.K. (23107729).
Supporting Material
Supporting Citation
Reference (64) appears in the Supporting Material.
References
- 1.Eisenmesser E.Z., Millet O., Kern D. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438:117–121. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- 2.Korzhnev D.M., Religa T.L., Kay L.E. A transient and low-populated protein-folding intermediate at atomic resolution. Science. 2010;329:1312–1316. doi: 10.1126/science.1191723. [DOI] [PubMed] [Google Scholar]
- 3.Redfield C. Using nuclear magnetic resonance spectroscopy to study molten globule states of proteins. Methods. 2004;34:121–132. doi: 10.1016/j.ymeth.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 4.Ma B., Nussinov R. Enzyme dynamics point to stepwise conformational selection in catalysis. Curr. Opin. Chem. Biol. 2010;14:652–659. doi: 10.1016/j.cbpa.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boehr D.D., McElheny D., Wright P.E. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313:1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- 6.Mulder F.A.A., Mittermaier A., Kay L.E. Studying excited states of proteins by NMR spectroscopy. Nat. Struct. Biol. 2001;8:932–935. doi: 10.1038/nsb1101-932. [DOI] [PubMed] [Google Scholar]
- 7.Li R., Woodward C. The hydrogen exchange core and protein folding. Protein Sci. 1999;8:1571–1590. doi: 10.1110/ps.8.8.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Englander S.W. Protein folding intermediates and pathways studied by hydrogen exchange. Annu. Rev. Biophys. Biomol. Struct. 2000;29:213–238. doi: 10.1146/annurev.biophys.29.1.213. [DOI] [PubMed] [Google Scholar]
- 9.Akasaka K., Yamada H. On-line cell high-pressure nuclear magnetic resonance technique: application to protein studies. Methods Enzymol. 2001;338:134–158. doi: 10.1016/s0076-6879(02)38218-1. [DOI] [PubMed] [Google Scholar]
- 10.Akasaka K. Probing conformational fluctuation of proteins by pressure perturbation. Chem. Rev. 2006;106:1814–1835. doi: 10.1021/cr040440z. [DOI] [PubMed] [Google Scholar]
- 11.Kitahara R., Hata K., Akasaka K. Pressure-induced chemical shifts as probes for conformational fluctuations in proteins. Prog. Nucl. Magn. Reson. Spectrosc. 2013;71:35–58. doi: 10.1016/j.pnmrs.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Kremer W., Kachel N., Kalbitzer H.R. Species-specific differences in the intermediate states of human and Syrian hamster prion protein detected by high pressure NMR spectroscopy. J. Biol. Chem. 2007;282:22689–22698. doi: 10.1074/jbc.M701884200. [DOI] [PubMed] [Google Scholar]
- 13.Fuentes E.J., Wand A.J. Local stability and dynamics of apocytochrome b562 examined by the dependence of hydrogen exchange on hydrostatic pressure. Biochemistry. 1998;37:9877–9883. doi: 10.1021/bi980894o. [DOI] [PubMed] [Google Scholar]
- 14.Kitazawa S., Kameda T., Kitahara R. Close identity between alternatively folded state N2 of ubiquitin and the conformation of the protein bound to the ubiquitin-activating enzyme. Biochemistry. 2014;53:447–449. doi: 10.1021/bi401617n. [DOI] [PubMed] [Google Scholar]
- 15.Inoue K., Yamada H., Kalbitzer H.R. Pressure-induced local unfolding of the Ras binding domain of RalGDS. Nat. Struct. Biol. 2000;7:547–550. doi: 10.1038/76764. [DOI] [PubMed] [Google Scholar]
- 16.Nisius L., Grzesiek S. Key stabilizing elements of protein structure identified through pressure and temperature perturbation of its hydrogen bond network. Nat. Chem. 2012;4:711–717. doi: 10.1038/nchem.1396. [DOI] [PubMed] [Google Scholar]
- 17.Fu Y., Kasinath V., Wand A.J. Coupled motion in proteins revealed by pressure perturbation. J. Am. Chem. Soc. 2012;134:8543–8550. doi: 10.1021/ja3004655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nielsen G., Jonker H.R.A., Schwalbe H. Kinase in motion: insights into the dynamic nature of p38α by high-pressure NMR spectroscopic studies. ChemBioChem. 2013;14:1799–1806. doi: 10.1002/cbic.201300170. [DOI] [PubMed] [Google Scholar]
- 19.Roche J., Ying J., Bax A. Impact of hydrostatic pressure on an intrinsically disordered protein: a high-pressure NMR study of α-synuclein. ChemBioChem. 2013;14:1754–1761. doi: 10.1002/cbic.201300244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kitahara R., Simorellis A.K., Akasaka K. A delicate interplay of structure, dynamics, and thermodynamics for function: a high pressure NMR study of outer surface protein A. Biophys. J. 2012;102:916–926. doi: 10.1016/j.bpj.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitahara R., Akasaka K. Close identity of a pressure-stabilized intermediate with a kinetic intermediate in protein folding. Proc. Natl. Acad. Sci. USA. 2003;100:3167–3172. doi: 10.1073/pnas.0630309100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kitahara R., Yamaguchi Y., Akasaka K. Evolutionally conserved intermediates between ubiquitin and NEDD8. J. Mol. Biol. 2006;363:395–404. doi: 10.1016/j.jmb.2006.07.074. [DOI] [PubMed] [Google Scholar]
- 23.Kitahara R., Zhao C., Akasaka K. Basic folded and low-populated locally disordered conformers of SUMO-2 characterized by NMR spectroscopy at varying pressures. Biochemistry. 2008;47:30–39. doi: 10.1021/bi7014458. [DOI] [PubMed] [Google Scholar]
- 24.Kamatari Y.O., Smith L.J., Akasaka K. Cavity hydration as a gateway to unfolding: an NMR study of hen lysozyme at high pressure and low temperature. Biophys. Chem. 2011;156:24–30. doi: 10.1016/j.bpc.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 25.Roche J., Caro J.A., Royer C.A. Cavities determine the pressure unfolding of proteins. Proc. Natl. Acad. Sci. USA. 2012;109:6945–6950. doi: 10.1073/pnas.1200915109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eriksson A.E., Baase W.A., Matthews B.W. Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science. 1992;255:178–183. doi: 10.1126/science.1553543. [DOI] [PubMed] [Google Scholar]
- 27.Morton A., Matthews B.W. Specificity of ligand binding in a buried nonpolar cavity of T4 lysozyme: linkage of dynamics and structural plasticity. Biochemistry. 1995;34:8576–8588. doi: 10.1021/bi00027a007. [DOI] [PubMed] [Google Scholar]
- 28.Quillin M.L., Breyer W.A., Matthews B.W. Size versus polarizability in protein-ligand interactions: binding of noble gases within engineered cavities in phage T4 lysozyme. J. Mol. Biol. 2000;302:955–977. doi: 10.1006/jmbi.2000.4063. [DOI] [PubMed] [Google Scholar]
- 29.Feher V.A., Baldwin E.P., Dahlquist F.W. Access of ligands to cavities within the core of a protein is rapid. Nat. Struct. Biol. 1996;3:516–521. doi: 10.1038/nsb0696-516. [DOI] [PubMed] [Google Scholar]
- 30.Mulder F.A.A., Hon B., Kay L.E. Flexibility and ligand exchange in a buried cavity mutant of T4 lysozyme studied by multinuclear NMR. Biochemistry. 2000;39:12614–12622. doi: 10.1021/bi001351t. [DOI] [PubMed] [Google Scholar]
- 31.Mulder F.A.A., Hon B., Kay L.E. Slow internal dynamics in proteins: application of NMR relaxation dispersion spectroscopy to methyl groups in a cavity mutant of T4 lysozyme. J. Am. Chem. Soc. 2002;124:1443–1451. doi: 10.1021/ja0119806. [DOI] [PubMed] [Google Scholar]
- 32.Bouvignies G., Vallurupalli P., Kay L.E. Solution structure of a minor and transiently formed state of a T4 lysozyme mutant. Nature. 2011;477:111–114. doi: 10.1038/nature10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.López C.J., Yang Z., Hubbell W.L. Conformational selection and adaptation to ligand binding in T4 lysozyme cavity mutants. Proc. Natl. Acad. Sci. USA. 2013;110:E4306–E4315. doi: 10.1073/pnas.1318754110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsumura M., Becktel W.J., Matthews B.W. Hydrophobic stabilization in T4 lysozyme determined directly by multiple substitutions of Ile 3. Nature. 1988;334:406–410. doi: 10.1038/334406a0. [DOI] [PubMed] [Google Scholar]
- 35.Delaglio F., Grzesiek S., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 36.Johnson B.A., Blevins R.A. NMR View: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 37.Reich H.J. WinDNMR: Dynamic NMR spectra for Windows. J. Chem. Educ. 1995;72:1086. [Google Scholar]
- 38.Beglov D., Roux B. An integral equation to describe the solvation of polar molecules in liquid water. J. Phys. Chem. B. 1997;101:7821–7826. [Google Scholar]
- 39.Yoshida N., Imai T., Hirata F. Molecular recognition in biomolecules studied by statistical-mechanical integral-equation theory of liquids. J. Phys. Chem. B. 2009;113:873–886. doi: 10.1021/jp807068k. [DOI] [PubMed] [Google Scholar]
- 40.Case, D. A., T. A. Darden, …, P. A. Kollman. 2012. AMBER 12. University of California, San Francisco.http://ambermd.org. Accessed August 2012.
- 41.Dolinsky T.J., Czodrowski P., Baker N.A. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007;35:W522–W525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dolinsky T.J., Nielsen J.E., Baker N.A. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H., Robertson A.D., Jensen J.H. Very fast empirical prediction and rationalization of protein pKa values. Proteins. 2005;61:704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 44.Olsson M.H.M., Sondergaard C.R., Jensen J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pK(a) Predictions. J. Chem. Theory Comput. 2011;7:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- 45.Bas D.C., Rogers D.M., Jensen J.H. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins. 2008;73:765–783. doi: 10.1002/prot.22102. [DOI] [PubMed] [Google Scholar]
- 46.Lazaridis T., Karplus M. Orientational correlations and entropy in liquid water. J. Chem. Phys. 1996;105:4294–4316. [Google Scholar]
- 47.Akasaka K., Li H., Woodward C.K. Pressure response of protein backbone structure. Pressure-induced amide 15N chemical shifts in BPTI. Protein Sci. 1999;8:1946–1953. doi: 10.1110/ps.8.10.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cooper A. Thermodynamic fluctuations in protein molecules. Proc. Natl. Acad. Sci. USA. 1976;73:2740–2741. doi: 10.1073/pnas.73.8.2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H., Yamada H., Akasaka K. Effect of pressure on the tertiary structure and dynamics of folded basic pancreatic trypsin inhibitor. Biophys. J. 1999;77:2801–2812. doi: 10.1016/S0006-3495(99)77112-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kamatari Y.O., Yamada H., Smith L.J. Response of native and denatured hen lysozyme to high pressure studied by (15)N/(1)H NMR spectroscopy. Eur. J. Biochem. 2001;268:1782–1793. [PubMed] [Google Scholar]
- 51.Kitahara R., Yokoyama S., Akasaka K. NMR snapshots of a fluctuating protein structure: ubiquitin at 30 bar-3 kbar. J. Mol. Biol. 2005;347:277–285. doi: 10.1016/j.jmb.2005.01.052. [DOI] [PubMed] [Google Scholar]
- 52.Wagner G. Activation volumes for the rotational motion of interior aromatic rings in globular proteins determined by high resolution 1H NMR at variable pressure. FEBS Lett. 1980;112:280–284. doi: 10.1016/0014-5793(80)80198-0. [DOI] [PubMed] [Google Scholar]
- 53.Silva J.L., Foguel D., Royer C.A. Pressure provides new insights into protein folding, dynamics and structure. Trends Biochem. Sci. 2001;26:612–618. doi: 10.1016/s0968-0004(01)01949-1. [DOI] [PubMed] [Google Scholar]
- 54.Koradi R., Billeter M., Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. 29–32. [DOI] [PubMed] [Google Scholar]
- 55.Ando N., Barstow B., Gruner S.M. Structural and thermodynamic characterization of T4 lysozyme mutants and the contribution of internal cavities to pressure denaturation. Biochemistry. 2008;47:11097–11109. doi: 10.1021/bi801287m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Collins M.D., Hummer G., Gruner S.M. Cooperative water filling of a nonpolar protein cavity observed by high-pressure crystallography and simulation. Proc. Natl. Acad. Sci. USA. 2005;102:16668–16671. doi: 10.1073/pnas.0508224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu L., Quillin M.L., Matthews B.W. Use of experimental crystallographic phases to examine the hydration of polar and nonpolar cavities in T4 lysozyme. Proc. Natl. Acad. Sci. USA. 2008;105:14406–14411. doi: 10.1073/pnas.0806307105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Imai T., Hiraoka R., Hirata F. Locating missing water molecules in protein cavities by the three-dimensional reference interaction site model theory of molecular solvation. Proteins. 2007;66:804–813. doi: 10.1002/prot.21311. [DOI] [PubMed] [Google Scholar]
- 59.Yokogawa D., Sato H., Sakaki S. The position of water molecules in Bacteriorhodopsin: a three-dimensional distribution function study. J. Mol. Liq. 2009;147:112–116. [Google Scholar]
- 60.Sindhikara D.J., Yoshida N., Hirata F. Placevent: an algorithm for prediction of explicit solvent atom distribution-application to HIV-1 protease and F-ATP synthase. J. Comput. Chem. 2012;33:1536–1543. doi: 10.1002/jcc.22984. [DOI] [PubMed] [Google Scholar]
- 61.Gekko K., Hasegawa Y. Compressibility-structure relationship of globular proteins. Biochemistry. 1986;25:6563–6571. doi: 10.1021/bi00369a034. [DOI] [PubMed] [Google Scholar]
- 62.Kuwata K., Li H., Akasaka K. High pressure NMR reveals a variety of fluctuating conformers in beta-lactoglobulin. J. Mol. Biol. 2001;305:1073–1083. doi: 10.1006/jmbi.2000.4350. [DOI] [PubMed] [Google Scholar]
- 63.Bridgman P.W. Water, in the liquid and five solid forms, under pressure. Proc. Am. Acad. Arts Sci. 1912;47:441–558. [Google Scholar]
- 64.Kovalenko A., Hirata F. Three-dimensional density profiles of water in contact with a solute of arbitrary shape: A RISM approach. Chem. Phys. Lett. 1998;290:237–244. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.