

Abstract
The majority of melanomas demonstrate constitutive activation of the RAS-RAF-MEK-MAPK pathway. AZD6244 is a selective MEK1/2 inhibitor which markedly reduces tumor P-MAPK levels, but it produced few clinical responses in melanoma patients. An improved understanding of the determinants of resistance to AZD6244 may lead to improved patient selection and effective combinatorial approaches. The effects of AZD6244 on cell growth and survival were tested in a total of 14 Braf-mutant and 3 wild-type human cutaneous melanoma cell lines. Quantitative assessment of phospho-protein levels in the Braf-mutant cell lines by reverse phase protein array (RPPA) analysis showed no significant association between P-MEK or P-MAPK levels and AZD6244 sensitivity, but activation-specific markers in the PI3K-AKT pathway correlated with resistance. We also identified resistant cell lines without basal activation of the PI3K-AKT pathway. RPPA characterization of the time-dependent changes in signaling pathways revealed that AZD6244 produced durable and potent inhibition of P-MAPK in sensitive and resistant Braf-mutant cell lines, but several resistant lines demonstrated AZD6244-induced activation of AKT. In contrast, sensitive cell lines demonstrated AZD6244 treatment-induced upregulation of PTEN protein and mRNA expression. Inhibition of AKT, TORC1/2, or IGF1R blocked AZD6244-induced activation of AKT and resulted in synergistic cell killing with AZD6244. These findings identify basal and treatment-induced regulation of the PI3K-AKT pathway as a critical regulator of AZD6244 sensitivity in Braf-mutant cutaneous melanoma cells, the novel regulation of PTEN expression by AZD6244 in sensitive cells, and suggest new combinatorial approaches for patients.
Keywords: AZD6244, AZD8055, MEK, BRAF, MAPK, AKT, PTEN, melanoma
Introduction
Melanoma is the most aggressive form of skin cancer. Patients with metastatic disease have a median survival of less than one year, and outcomes are not improved with chemotherapy or immunotherapy (1). A review of the SEER database from 1950 to 2000 showed that the incidence and mortality from melanoma are increasing at a greater rate than for any other cancer. Thus, there is a tremendous need for more effective therapeutic approaches for this disease. The majority of melanomas harbor an activating mutation in the RAS-RAF-MAPK signaling pathway. Braf mutations are detected in approximately 50% of melanomas (2). Over 90% of these mutations affect the V600 residue, and they result in constitutive activation of the BRAF protein (3). In addition, approximately 20% of melanomas have an activating Nras mutation (2). Mutations in Nras also activate the RAS-RAF-MAPK pathway (4). As Braf and Nras mutations are mutually exclusive (5, 6), approximately 70% of melanomas harbor a mutation in the RAS-RAF-MAPK pathway. Many melanomas without identifiable mutations in the pathway also demonstrate constitutive MAPK activation (7). Thus, activation of the RAS-RAF-MAPK pathway is likely a critical event in melanoma, and it may be an important therapeutic target.
AZD6244 (ARRY-142996, Selumetinib) is a potent, highly selective, uncompetitive inhibitor of MEK1/2 (8). AZD6244 has an in vitro IC50 of 14 nM against purified MEK, whereas minimal inhibition was seen in more than 40 kinases at 10 μM. AZD6244 inhibited ERK phosphorylation (P-ERK) at ∼40 nM in cells growing in vitro, and in xenografts growing in mice. Initial testing demonstrated that many different cancer types, including melanoma, colon, breast, and pancreatic cancer with Braf or Nras mutations were sensitive to the inhibitory effects of AZD6244, although the degree of growth suppression varied even among cell lines with those mutations (9). Phase I clinical testing in patients demonstrated that AZD6244 was well-tolerated (10). Paired biopsies from 20 patients with assessable tissue demonstrated a mean reduction in tumor P-ERK of 79% with AZD6244 treatment. A subsequent randomized Phase II trial in metastatic melanoma compared AZD6244 to temozolomide, a standard cytotoxic agent used in melanoma (11). Even among the patients with Braf mutations (n=67), which in vitro generally correlated with sensitivity to AZD6244, surprisingly there was only a 12% clinical response rate, which was not superior to temozolomide.
The clinical experience with AZD6244 demonstrates that doses can be achieved in patients that significantly inhibit the RAS-RAF-MAPK pathway. There is also evidence that clinical responses can be achieved in some patients. However, the rate of clinical response, particularly among Braf-mutant patients, is lower than predicted by preclinical testing, and less than that reported with the BRAF inhibitor PLX4032/RG7204. (12). The future clinical use of AZD6244 would be enhanced by an improved understanding of the factors that predict and regulate sensitivity to the drug. Previous testing of other MEK inhibitors in melanoma cell lines demonstrated that P-ERK levels, both at baseline and with treatment, did not correlate with the degree of cell growth inhibition that was achieved (13). Studies are currently ongoing to further investigate a possible threshold level of in vivo pathway inhibition that correlates with clinical responses to RAS-RAF-MAPK pathway inhibitors, particularly PLX4032. However, studies with other targeted therapies have demonstrated that inhibition of the intended target often results in unexpected activation of other pathways through feedback regulation of signaling networks (14, 15). Understanding these changes can identify combinatorial approaches that overcome these effects and improve efficacy. We have developed reverse phase protein arrays (RPPA) to perform efficient and quantitative assessment of signaling pathways in cancer (16). We have used RPPA to perform integrated analysis of kinase signaling pathways with clinical and molecular features in a number of cancers, including melanoma (17-19). We have also used RPPA to identify time- and dose-dependent changes in kinase signaling networks in cancer cells in response to growth factors and targeted therapies (20- 21).
To improve our understanding of the regulators of treatment response in melanoma, we have analyzed a panel of Braf-mutant human cutaneous melanoma cell lines for their sensitivity to growth and survival inhibition by AZD6244. We compared these effects to the baseline activation status of signaling pathways in the cells, and to AZD6244 treatment-induced changes in signaling networks. These studies have identified the PI3K-AKT pathway as a critical regulator of AZD6244's efficacy in Braf-mutant melanomas, including in cells without baseline activation of the pathway. We have also identified and validated combinatorial approaches that increase the cytocidal effects of AZD6244 in Braf-mutant melanoma cell lines, which suggest strategies for testing in patients.
Materials and Methods
Cell lines and inhibitors
All human melanoma cell lines were authenticated by STR fingerprinting, as previously described (17). Mutation status of Braf, Nras, c-Kit, PIK3CA, and Akt1/2/3 was determined by mass-spectroscopy based genotyping, and has been reported previously (17, 22, 23). Cells were maintained in RPMI media in 5% fetal bovine serum (FBS) (Gemini Bioproducts, Sacramento, CA) at 37°C supplemented with 5% CO2. AZD6244 and AZD8055 were obtained under a material transfer agreement with Astra Zeneca (Alderly Park, UK). Rapamycin was from Cell Signaling Technology (Danvers, MA). Recombinant IGF-1 was from Invitrogen (Camarillo, CA).
Cell viability assay
Cells were seeded in 96 well plates overnight and treated with increasing concentrations of drugs or DMSO (vehicle). The DMSO concentrations were maintained at 0.02% in all wells. After 48 h incubation, cell viability was determined using Cell Titer Blue Cell Viability Assay (Promega, Madison, WI). Data was analyzed and graphed using MS-Excel.
Cell cycle analysis
Cells seeded in 6 well plates were treated with inhibitors and/or siRNA as described. After the indicated treatment periods, cells were trypsinized, fixed in 70 % ethanol, and stained with propidium iodide [BD Biosciences, Franklin Lakes, NJ]. Cell cycle analysis was performed using a BCI XL4 flow cytometer (Beckman Coulter Inc., Miami, FL), the data was analyzed using MultiCycle AV software (Phoenix Flow Systems, San Diego, CA), and graphs were generated using MS-Excel.
Apoptosis Assay
Cells were plated in 96 well plates overnight and treatments were performed as described. Apoptosis was determined by the cytoplasmic histone-associated DNA fragment method using the Cell Death Detection ELISA Plus Kit (Roche Applied Science, Indianapolis, IN).
Reverse phase protein array and Western blotting analysis
Cells were seeded in 100 mm tissue culture plates overnight, treated with AZD6244 or vehicle (DMSO) in triplicate, and harvested at the indicated time points. Protein lysate preparation for RPPA analysis has been described previously (16-20). RPPA analysis was performed by the University of Texas M. D. Anderson Functional Proteomics Core Facility. Samples were analyzed for the expression of 94 protein markers using RPPA-validated antibodies (Table S1). Results were normalized as previously described (17), and were reported as log2 values. After correcting for sample loading differences, for each cell line and timepoint the protein expression levels were normalized against those of DMSO-treated cells. Clustering analysis was performed using Cluster 2.1 and was visualized using Treeview software1. Western blotting analysis was performed using lysates prepared using the same methods, using standard western blotting procedures (16). Antibodies used for western blotting are also listed in Table S1. Those not listed in the table are GAPDH (Ambion, Austin, TX), FOXO3A, P-FOXO3A(S318), IGF1R, P-IGF1R (Y1131) and P-IGF1R (Y1135) (Cell Signaling Technology, Danvers, MA).
siRNA transfections
Cells seeded in 6-well plates were transfected with 20 nM of siRNA, using Xtremegene (Roche Applied Science), or Dharmafect 1 (Dharmacon, Lafayette, CO) transfection reagents. Inhibitors were added after 48 h, and cells were harvested after 24h or 48 h for western blotting and cell cycle analysis respectively. The siRNAs used were siControl RISC-free, siPTEN, siAKT (mix of AKT1, AKT2 and AKT3 siRNAs), siIGF1R, (Dharmacon), and siIGF1 (Ambion, Austin, TX).
RNA analysis
Cells seeded in 10 cm plates were treated with AZD6244 or vehicle (DMSO). After 24 h incubation, RNA was isolated using the miRNeasy kit (Qiagen, Valencia, CA). RNA was quantified using Nanodrop. Reverse transcription and PCR amplification were performed using the Taqman One-Step RT-PCR assay reagent using PTEN or GAPDH primer-probe mixes from Applied Biosystems Inc. (Branchburg, NJ). Fold changes in the treated samples compared to controls were determined using the formula– fold change = efficiencyδCt, where δCt is the threshold cycle difference between control and treated samples normalized against GAPDH expression.
Measurement of secreted IGF-1
Cells (105) were seeded in 6 well plates overnight, replenished with 0.5 ml fresh media and treated with AZD6244 for 24h. siRNA treated cells were replenished with 0.5 ml of fresh media 48h after transfection and then were treated with AZD6244 for 24 h. Secreted IGF-1 in the media was quantified using the Quantikine human IGF-1 Immunoassay kit (R&D Systems, Minneapolis, MN).
Results
Comparison of baseline signaling pathways to growth inhibitory effects of AZD6244
We initially determined the relative sensitivity of 11 Braf-mutant cutaneous melanoma cell lines to growth inhibition by AZD6244 (Figure 1A). This panel included 4 cell lines (WM2664, WM1799, UCSD-354L, WM1552) with concurrent PTEN loss, and one cell line with concurrent Akt3 E17K mutation (WM46). We also tested 2 cell lines that were wild-type for Braf and Nras (Mewo, HS294T). Overall, the cell lines organized in 3 groups, which we termed Group 1 (high sensitivity), Group 2 (intermediate sensitivity), and Group 3 (low sensitivity). Of the wild-type cell lines, the HS294T were not sensitive to AZD6244 (Group 3), but the Mewo were (Group 1). To further investigate the observed heterogeneity in sensitivity to AZD6244, cell cycle analysis was performed on cell lines from each group. The Group 1 cell lines WM35 (Figure 1B), A375, and UACC257 (Figure S1) demonstrated a G1 cell-cycle arrest with 40 nM AZD6244, and a marked sub-G1 accumulation with higher doses (i.e. >60% sub-G1 with 360 nM). AZD6244 treatment of the Group 2 cell lines SKMEL5 (Figure 1C), MEL624, UCSD354L, and WM1799 (Figure S1) induced G1-cell cycle arrest, but no significant cell death (sub-G1) was observed up to 3 μM. The highly resistant Group 3 cells Hs294T (Figure 1D) did not undergo significant G1 or sub-G1 accumulation up to 1 μM AZD6244 (Figure 1D). Representative cell cycle profile histograms of AZD6244 treated cells belonging to each group are shown in Figure S2. A cytoplasmic histone-DNA-fragment-based apoptosis assay confirmed that the Group 1 cells underwent apoptosis, while the Group 2 and Group 3 cells did not (Figure S3). Time-course cell cycle analysis of the sensitive cell lines found that cell death was observed after 48 hours with AZD6244 treatment, whereas resistant cells showed no significant cell death for at least 72 hours (Figure S4). Drug washout experiments with the WM35 and A375 demonstrated that 24-48 hours drug exposure was required to induce cell death (data not shown).
Figure 1. Growth and survival inhibition by AZD6244.


(A) Growth inhibition in human melanoma cell lines treated with AZD6244 for 48 h. X-axis, concentration of AZD6244 (nM); Y-axis, % growth inhibition. Groups of cells with high (“1”), moderate (“2”), and low (“3”) sensitivity are indicated. Cells with data points connected by dotted lines are wild-type for Braf, all other cell lines have Braf mutations. Data points are the average of 3 replicates; error bars, standard deviation. (B) Cell cycle analysis of WM35 cells (Group 1) after 72 h treatment AZD6244. X-axis, concentration of AZD6244 (nM); Y-axis, % of cell population. The Sub-G1 (dead cells), G1, S, and G2/M phases of the cell cycle are indicated by black, dark grey, light grey and white bars respectively. Each bar is the average of two replicates; error bars, standard deviation. (C) Cell cycle analysis of SKMEL5 cells (Group 2). (D) Cell cycle analysis of HS294T cells (Group 3).
To determine if activation of specific protein signaling networks correlated with resistance to AZD6244 in the Braf-mutant cell lines, we compared the IC50 data (Table S2) for the cell lines to the expression of 99 protein features determined by RPPA (17, 24). Consistent with previous experiments with other MEK inhibitors (13), we did not observe a significant correlation between the basal levels of P-P44/42 MAPK (p = 0.22) or P-MEK1/2 (p =0.88) and the degree of growth inhibition. Among all of the proteins assessed, P-AKT_Thr308 (Pearson r =0.65, p = 0.02) showed the highest correlation with resistance to AZD6244. P-AKT_Ser473 had a similar correlation (r = 0.53, p = 0.08), as did the AKT substrates P-TSC2 (r =0.50) and P-GSK3α/β (r = 0.42), supporting that basal activation of the PI3K-AKT pathway predicts resistance to AZD6244 in Braf-mutant melanoma cells (Table 1). The Braf-mutant cell lines with low sensitivity to AZD6244 (Group 2) included the two cell lines with the highest levels of P-AKT in the panel (WM1799, UCSD-354L). However, the Group 2 cell lines SKMEL5 and MEL624 demonstrated similar resistance to AZD6244 with normal PTEN expression and low P-AKT. Additional experiments were performed to determine the mechanism of resistance in these cell lines.
Table 1.
The ten proteins with the highest correlation with resistance (increased IC50) to AZD6244 in Braf-mutant human melanoma cell lines. Protein expression levels were determined for the cell lines growing under normal tissue culture conditions by RPPA. r2, Pearson correlation; p determined by the t-statistic.
| Protein | Protein Expression vs IC50 (r2) | p value |
|---|---|---|
| P-AKT_Thr308 | 0.65 | 0.02 |
| C-RAF | 0.55 | 0.06 |
| IGF1R beta | 0.55 | 0.07 |
| P-AKT_Ser473 | 0.53 | 0.08 |
| eIF4E | 0.51 | 0.09 |
| P-TSC2_Thr1462 | 0.50 | 0.10 |
| P27 | 0.43 | 0.16 |
| P-GSK3α/β_Ser21/9 | 0.42 | 0.18 |
| Cyclin E | 0.41 | 0.19 |
| AMPK | 0.40 | 0.20 |
Treatment-induced changes in signaling pathways by AZD6244
To identify early and late signaling events that correlate with cell death induction by AZD6244, we performed RPPA analysis on A375 and WM35 cells treated with 360 nM AZD6244 for 0.5, 3, 6, 12, 24, and 48 h. Identical experiments were performed with the SKMEL5 and MEL624 cells, which undergo G1 phase growth arrest only in response to AZD6244, and the highly resistant HS294T, which do not die or arrest. Triplicate protein lysates were analyzed, and protein expression levels for each cell line were normalized to DMSO-treated controls.
We initially analyzed the effects of AZD6244 on the expression of P-P44/42 MAPK. We observed a similar degree (≥90%) and duration (≥48 hours) of inhibition of P44/42 MAPK phosphorylation in the sensitive and resistant Braf-mutant cell lines (Figure S5). The HS294T cells had much lower baseline levels of P-MAPK, but this activity was inhibited by treatment with AZD6244, suggesting that their resistance was not due to lack of intracellular drug accumulation (Figure S5E-F). In order to analyze the role of other pathways, we performed hierarchical clustering analysis (by time) of the RPPA data for each of the cell lines (Figure 2). Comparison to the completely resistant HS294T cells identified a group of proteins that were similarly decreased in the other 4 cell lines, including P-MAPK, P-YB1, Cyclin D1, and P-S6. The HS294T cells demonstrated a marked increase in P-MEK levels upon treatment with AZD6244, which was unique compared to the other cell lines.
Figure 2. Time-dependent effects of AZD6244 on protein expression and activation.

RPPA analysis of WM35, A375, MEL624, and SKMEL5 Braf-mutant melanoma cells, and wild-type HS294T cells. Lysates were harvested after treatment with 360 nM AZD6244 at the time points indicated on the right side. Cell lines are organized by their sensitivity (Groups 1, 2 and 3) indicated at the left side of the heatmap. Proteins analyzed are indicated at the top of the heatmap. Results for each cell line were normalized against vehicle-treated controls. Red box, group of proteins showing similar inhibition pattern in Group1/2 versus Group 3; Orange box, proteins with treatment-induced increase in Group 1 (cytocidal effect) versus Group 2 (cytostatic effect); Blue box, proteins with treatment induced increase in Group 2 versus Group 1.
Patterns of protein expression in the Group 1 versus the Group 2 cells were compared to identify signaling events that might determine cell death induction versus cytostasis with AZD6244. The MEL624 and SKMEL5 cells, which undergo G1-arrest but not cell death with AZD6244, showed a time-dependent increase in the expression of P-AKT_Ser473 and P-AKT_Thr308 that was not observed in the WM35 or A375 cells (Figure 2). In contrast, the WM35 and A375 cells showed upregulation of PTEN protein expression, as well as Cleaved Caspase 7 (apoptosis marker), and p27. Western blotting confirmed the time-dependent increase in P-AKT in the moderately resistant SKMEL5 and, MEL624 (Figure 3A). In contrast, the highly sensitive WM35, A375 (Figure 3A), and UACC257 (Figure S6) showed no induction of P-AKT. The WM35 showed a marked increase in PTEN levels, which was also detected, but was less dramatic, in the A375 and UACC257. To determine if these effects were specific to these cells, western blotting was performed on additional cells recently identified to be resistant to AZD6244. (25). The Braf/Nras-wild type D24, which were similar to Group 2 cells in their relative sensitivity to growth inhibition and G1 cell cycle arrest (Figure S7), demonstrated increased P-AKT with AZD6244 treatment (Figure S6). The Braf-mutant PTEN-expressing D29, which by growth inhibition and cell cycle were similar to the HS294T (Figure S7), did not demonstrate increased P-AKT, but did have increased P-MEK (Figure S8). The Braf-mutant PTEN-expressing MM595 and A2 also were similar to the HS294T in their growth resistance (Figure S7), but they did not demonstrate an increase in P-AKT or P-MEK (Figure S8).
Figure 3. PTEN expression and function in AZD6244-mediated cell death.
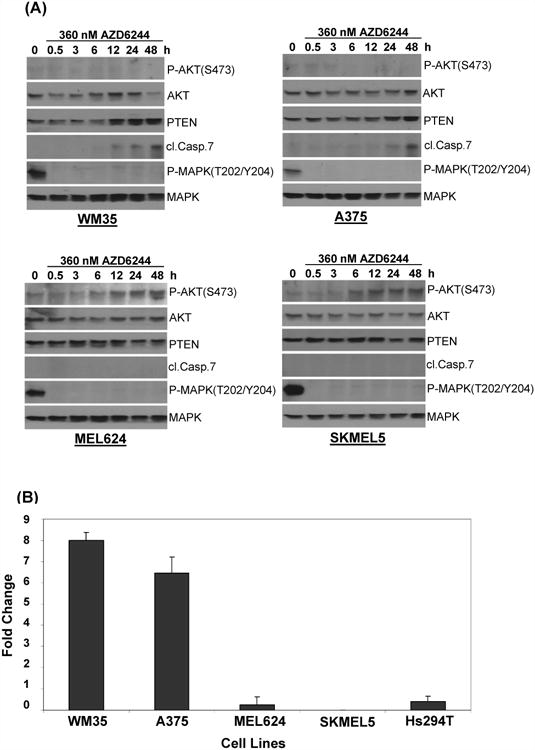
(A) Western blotting analysis of Group1 (sensitive) WM35 and A375 (top) and Group 2 (resistant) MEL624 and SKMEL5 (bottom) following treatment with AZD6244. Time point is indicated above the P-AKT results (hours). (B) PTEN mRNA expression following AZD6244 treatment. Fold change versus baseline in PTEN mRNA as measured by qRT-PCR after 24 h treatment with 360 nM of AZD6244. X-axis, cell line; Y-axis, relative fold-change in PTEN transcripts. Bars, average of 3 samples; error bars, standard deviation. (C) Western blot of WM35 cells showing effects of RISC-free siRNA and PTEN siRNA on P-AKT, P-GSK3, p27 and Cleaved Caspase 7. Cells were transfected with siRNAs for 48 h, followed by 24 h treatment with 360 nM AZD6244 or vehicle. (D) Effect of PTEN knockdown on AZD6244-induced cell death. Cells were transfected with RISC-free or PTEN siRNA. After 48 h, cells were treated with AZD6244 360 nM or vehicle for 48 h, and cell cycle analysis was performed. (E) WM35 cells were treated and incubated as above in 96 well plates and subjected to cytoplasmic histone DNA fragment-analysis based apoptosis assay. The absorbance values were plotted as shown. Bars, average of three replicates; error bars, standard deviation.
We further investigated the observed increase in PTEN protein expression in the sensitive cell lines. RT-PCR analysis showed a >6 fold increased in PTEN transcript levels in the WM35 and A375 following AZD6244 treatment, whereas no significant change was observed in the SKMEL5, MEL624, or HS294T (Figure 3B). To test the functional significance of PTEN induction, WM35 cells were transfected with siRNA against PTEN. Knockdown of PTEN increased the basal levels of P-AKT and reduced the AZD6244-induced cleavage of caspase 7 (Figure 3C). PTEN knockdown also reduced AZD6244-induced cell killing as measured by both FACS analysis (Figure 3D) and apoptosis assay (Figure 3E).
Inhibition of the PI3K/AKT pathway sensitizes melanoma cells to AZD6244
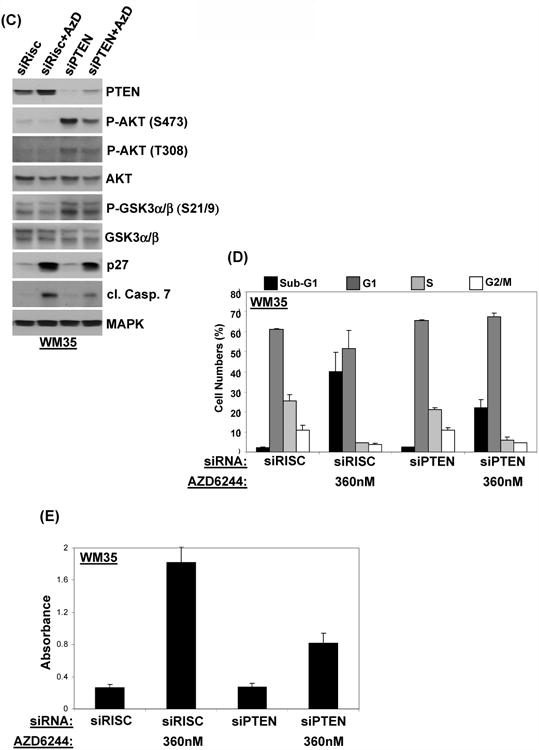
To test if activation of AKT mediated resistance to AZD6244-induced cell death, SKMEL5 cells were transfected with siRNA against AKT. Greater than 90% knockdown of AKT was achieved; decreased expression of P-GSK3α/β and P-FOXO3a confirmed functional inhibition of AKT activity (Figure 4A). Compared to transfection with control siRNA, knockdown of AKT resulted in a slight increase in cell death (12.5% versus 3.3%), as assessed by FACS analysis (Figure 4B). Knockdown of AKT markedly sensitized the SKMEL5 to AZD6244-induced cell death, with 40% sub-G1 accumulation as compared to 5% with AZD6244 alone (Figure 4B), which was confirmed as apoptosis (Figure 4C).
Figure 4. Effects of AKT signaling on sensitivity to AZD6244.
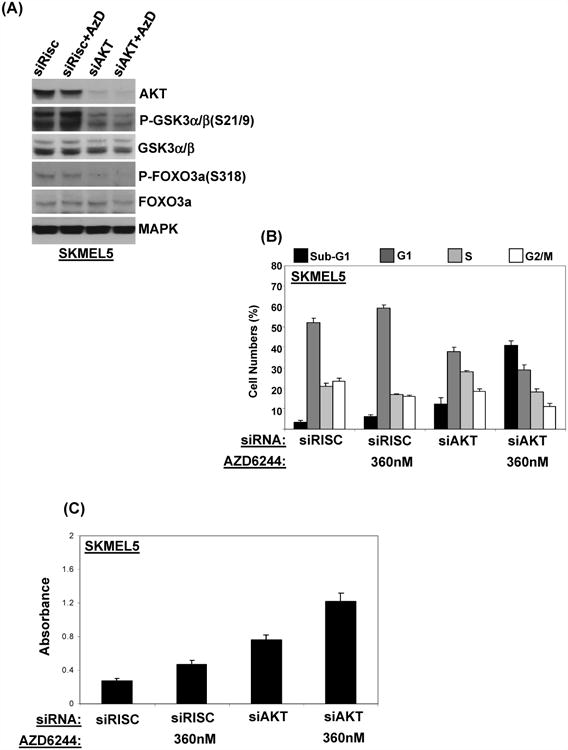
(A) Western blotting of SKMEL5 cells transfected with RISC-free control siRNA (30nM) or a mixture of siAKT1, siAKT2 and siAKT3 siRNAs (10 nM each) for 48h, followed by 24h treatment with 360nM of AZD6244 or vehicle. (B) FACS analysis of SKMEL5 cells transfected with indicated siRNAs, followed by AZD6244 or vehicle for an additional 48 h. (C) Apoptosis assay results for the same treatments. Bars, average of triplicates; errors bars, standard deviations.
Previous studies in human breast cancer cell lines indicated that activation of AKT following MEK inhibition may be mediated through the epidermal growth factor receptor (14). However, like most melanomas, both the SKMEL5 and MEL624 do not express detectable EGFR protein by RPPA (data not shown). The insulin-like growth factor 1 (IGF-1) and its receptor (IGF1R) have been shown to play an important role in promoting melanoma growth through activation of the PI3K-AKT pathway (26- 28). AZD6244-resistant cells showed high correlation (Pearson r = 0.55, p = 0.07) with basal levels of IGF1R protein in the RPPA analysis (Table 1) and western blotting (Figure S9). The levels of IGF1R protein were the highest in the group 2 cells MEL624 and SKMEL5, when compared to the other melanoma cell lines (Figure S9). Knockdown of IGF1R expression by siRNA abrogated AZD6244-induced P-AKT expression in the SKMEL5 cells (Figure 5A). IGF1R inhibition alone resulted in minimal cell death, but increased cell death in combination with AZD6244 treatment (Figure 5B, C). AZD6244 treatment also induced an increase of IGF-1 growth factor secretion into the medium by the moderately resistant cells, and knockdown of IGF-1 by siRNA blocked AZD6244 treatment-induced increase in P-AKT (Figure S10). Conversely, treatment of the sensitive WM35 cells with recombinant IGF-1 induced AKT activation (Figure S11A) and reduced AZD6244-induced apoptosis, but it did not alter the induction of a G1 phase arrest (Figure 5 and S11B). This suggests that IGF1R signaling is a critical determinant of AZD6244-induced cell death, but not the cytostatic effects of the drug. Of note, we did not detect changes in the total IGF1R levels or phosphorylation of IGF1R at residues Y1131 and Y1135 following AZD6244 treatment (data not shown), but this does not preclude the possibility of differential effects on other phosphorylation sites or other regulators of this pathway.
Figure 5. Effects of IGF1R signaling on AZD6244-induced activation of AKT.
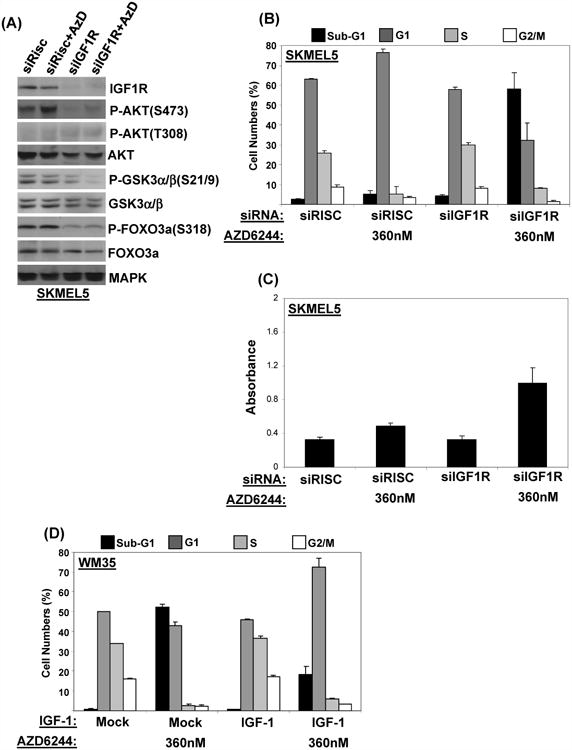
(A) Western blotting of SKMEL5 cells transfected with RISC-free or IGF1R siRNA. After 48 hours cells were treated with AZD6244 360 nM or vehicle, then were harvested after 24 hours. (B) FACS analysis and (C) apoptosis induction of SKMEL5 cells transfected as in A. Cells were harvested for analysis after 48 hours drug exposure. (D) Effect of IGF-1 on survival of AZD6244-treated WM35 cells. WM35 were treated with vehicle or AZD6244 360 nM, in the absence or presence of 100ng/ml IGF-1. Cells were harvested after 48 h, and FACS analysis was performed. Bars, average of triplicates; errors bars, standard deviations.
AZD8055 is a novel TOR kinase inhibitor which inhibits the activity of both the TORC1 and TORC2 complexes and therefore inhibits AKT activation (29). Treatment of the SKMEL5 cells with AZD8055 inhibited the AZD6244-induced increase in P-AKT (S473), while P-AKT (T308) was mostly unchanged (Figure 6A). Treatment with rapamycin, which inhibits the TORC1 but not the TORC2 complex, resulted in increased P-AKT at both residues. FACS cell cycle analysis demonstrated that treatment of the SKMEL5 cells with AZD8055 alone did not result in significant cell death, but it did when combined with AZD6244 (Figure 6B). Rapamycin did not induce cell death, either as a single agent or when combined with AZD6244. Similar results were observed with the apoptosis assay (Figure S12-A). Treatment of MEL624 cells with AZD8055 also inhibited P-AKT (S473) expression, while the levels of P-AKT (T308) were slightly increased (Figure 6C). FACS analysis (Figure 6D) and apoptosis assay (Figure S12-B) both demonstrated cell death induction following combined treatment with AZD6244 and AZD8055.
Figure 6. Signaling and cell cycle effects of AZD8055 treatment with AZD6244.
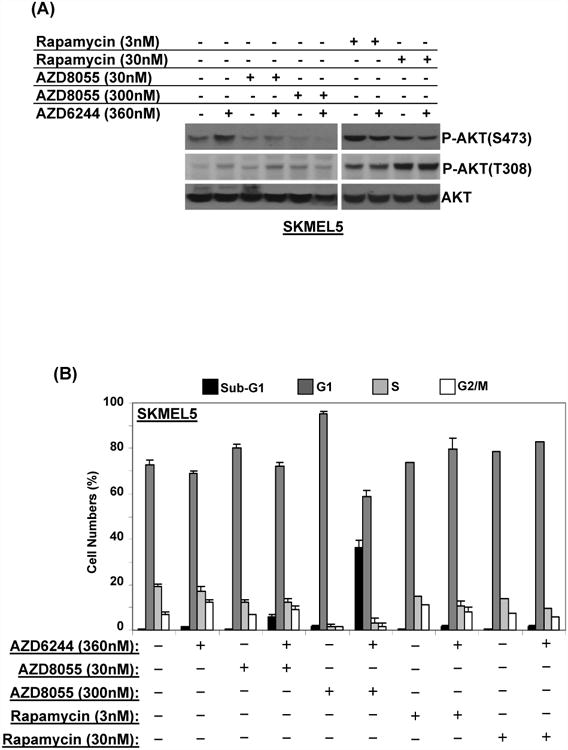
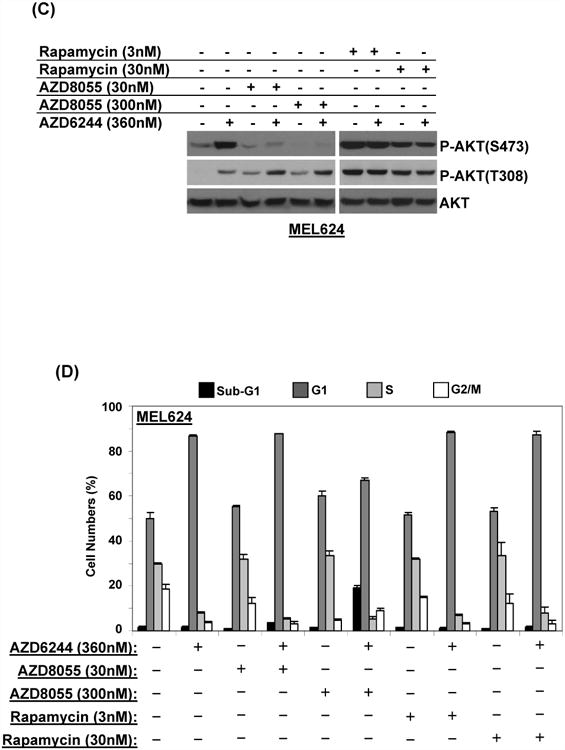
(A) Western blotting analysis of SKMEL5 cells treated with the indicated inhibitors for 24 h. (B) FACS of SKMEL5 cells treated for 24 h with the indicated combinations. Bars and replicates as in Figure 1. Identical experiments were performed in the MEL624 cell line for the effects on signaling proteins (C) and cell death (D).
Discussion
The RAS-RAF-MAPK signaling pathway is an attractive target for therapy development for melanoma due to the high prevalence of activation of this pathway. Despite the demonstration of potent inhibition of this pathway, AZD6244 had low activity as a single agent in melanoma, even in patients with Braf mutations. As Braf mutations are the most common activating mutation in cutaneous melanomas, and as most evidence supports that they primarily act through the activation of MEK-MAPK signaling, we have performed a proteomic analysis to explore the role of kinase signaling pathways in resistance to AZD6244.
Initial testing of the effects of AZD6244 on growth and survival segregated the Braf-mutant melanoma cell lines into two groups: Group 1 demonstrated marked growth inhibition and significant cell death induction with nanomolar concentrations of AZD6244, whereas Group 2 was less growth inhibited and exhibited G1-cell cycle arrest but no significant cell death. We also identified cells (HS294T) that were not growth inhibited significantly by AZD6244 up to micromolar doses, which was therefore used as a model for effects of AZD6244 in the absence of any growth inhibitory effects. Previously, we have performed RPPA analysis of melanoma cell lines for a variety of proteins in signaling pathways (17). Analysis of basal protein expression levels identified weak correlation between levels of P-MEK or P-44/42 MAPK and AZD6244 IC50 values, similar to previous experiments in melanoma cell lines with the MEK inhibitor U0126 (13) In contrast, a number of activation-specific markers of the PI3K-AKT pathway correlated with resistance to AZD6244. While this study is the first to report a significant correlation of increased PI3K-AKT activity with resistance to MEK inhibitors in melanoma, these results mirror the results recently reported in a study of AZD6244 in lung cancer cell lines (30). We also observed a positive correlation for CRAF expression and AZD6244 IC50 values, although it did not quite reach statistical significance (Table 1). Increased CRAF expression has also been associated with resistance to BRAF inhibitors (31)
It is not surprising that Braf-mutant melanoma cells with concurrent activation of the PI3K-AKT pathway have increased resistance to the growth inhibitory effects of MEK inhibition. While the clinical trials with AZD6244 have not reported an analysis of the activation status of the PI3K-AKT pathway in treated patients, we and others have demonstrated that genetic mutations and constitutive activation of the PI3K-AKT pathway are present in only a subset of Braf-mutant melanomas (17, 23, 32-35). Thus, basal-activation of the PI3K-AKT pathway is likely not sufficient to fully explain or predict resistance to AZD6244 in melanoma. We noted that while the resistant Group 2 included two cell lines with PTEN loss and very high levels of P-AKT, it also included 2 cell lines (SKMEL5, MEL624) with normal PTEN and low basal P-AKT. We therefore performed further testing to determine possible mechanisms of resistance in these cell lines by comparing the effects of AZD6244 treatment on their signaling pathways to effects in sensitive cell lines. While all 4 of these Braf-mutant cell lines demonstrated similar degree and duration of MAPK inhibition and several other proteins, the resistant cell lines increased their P-AKT levels following exposure to AZD6244, which was not observed in the sensitive cell lines. The functional significance of AKT activation is supported by the fact that inhibition of AKT activity, either by AKT knockdown or concurrent treatment with the TORC1/2 inhibitor AZD8055, resulted in synergistic cell killing in the resistant cell lines. These findings give support for the upcoming clinical trial testing the effects of AZD6244 and MK22062, a small molecule inhibitor of AKT, as well as further exploration of the effects of AZD6244 and AZD8055. We also tested signaling effects in additional cell lines recently characterized for resistance to AZD6244 by another laboratory (25). We found that several of these cells lines exhibited similar effects to those observed in our panel. However, the differential results in the MM595 and A2 cells support the likely existence of additional resistance mechanisms.
Previous studies in breast cancer cell lines demonstrated that MEK inhibition resulted in cross-activation of the EGFR tyrosine growth factor receptor (36). As this receptor is not frequently expressed in melanoma, nor in the cell lines used in this experiment, we hypothesized that other growth factor receptors could mediate this effect. We tested the role of the IGF-1 pathway, as it previously has been implicated in AKT activation in melanoma (26-28). Inhibition of IGF1R did not kill SKMEL5 cells, but it blocked the induction of P-AKT by AZD6244 and induced cell death in combination with AZD6244. AZD6244 treatment induced a slight increase of IGF-1 secretion by the cells, and knockdown of IGF-1 also blocked P-AKT induction by AZD6244. Supporting a specific role for the pathway in cell survival, recombinant IGF-1 treatment blocked AZD6244-induced cell death, but not growth arrest, in the sensitive WM35. These results support the need for further testing the effects of combining AZD6244 with inhibition of IGF1R-mediated signaling. Further work is also merited to better understand the mechanism of cross-talk to this pathway, to help identify and assess optimal combinatorial approaches. Additional testing will also be needed to determine if this combinatorial approach is most effective in melanomas with elevated IGF1R expression.
Our finding that MEK inhibitor treatment can induce the upregulation of PTEN transcript and protein is unprecedented. The mechanism(s) of the transcriptional regulation of PTEN remains poorly understood. While several miRNAs have been shown to regulate PTEN transcript stability (37-39), our profiling of eight miRNAs that recognize sites in the PTEN message (miR-10a, -26a, -29a, -30a-3p, -30e-3p, -183, -221, -519d) did not demonstrate changes or differential effects that would explain the observed differences in PTEN expression (data not shown). Understanding these effects may require more comprehensive miRNA profiling, or functional analysis of the PTEN promoter. Identification of molecular features that predict the upregulation of PTEN expression with MEK inhibition may indicate patients who would selectively benefit from treatment with these agents. Such markers would need to be validated in larger panels of cell lines and patients.
These studies have provided new insights into the regulation of the growth inhibitory effects of AZD6244 in melanoma. We specifically focused these studies on Braf-mutant melanoma cells as these mutations are predominant in the common cutaneous melanomas, and ongoing clinical trials with AZD6244 require Braf mutation for patient enrollment. However, there is a clear need to determine if similar effects and combinations are important in other melanoma genotypes, including Nras-mutants and melanomas in which activating kinase mutations are not detected. Further, while RPPA is a powerful technology that can be used to investigate both the expression and the activation states of proteins, we recognize that its scope is limited to investigations of targets for which validated antibodies are available. Whole-genomic approaches, such as mRNA or miRNA profiling, may suggest additional pathways that regulate sensitivity to AZD6244 in Braf-mutant melanomas. At the protein level, there remains a strong rationale to determine if the differential clinical outcomes seen with RAS-RAF-MAPK pathway inhibitors in vivo correlates with a threshold level of pathway inhibition. However, our functional testing of off-target pathways that are activated by, and correlate with resistance to, AZD6244 supports performing similar investigations with other agents. We believe this approach will identify combinatorial approaches with increased efficacy for this highly aggressive disease.
Supplementary Material
Acknowledgments
Supported in part by AstraZeneca. The University of Texas M. D. Anderson Functional Proteomics Core Facility is supported by an NCI Cancer Center Support Grant (CA-16672).
Footnotes
References
- 1.Tsao H, Atkins MB, Sober AJ. Management of cutaneous melanoma. N Engl J Med. 2004;351:998–1012. doi: 10.1056/NEJMra041245. [DOI] [PubMed] [Google Scholar]
- 2.Hocker T, Tsao H. Ultraviolet radiation and melanoma: a systematic review and analysis of reported sequence variants. Hum Mutat. 2007;28:578–88. doi: 10.1002/humu.20481. [DOI] [PubMed] [Google Scholar]
- 3.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 4.Chudnovsky Y, Adams AE, Robbins PB, Lin Q, Khavari PA. Use of human tissue to assess the oncogenic activity of melanoma-associated mutations. Nat Genet. 2005;37:745–9. doi: 10.1038/ng1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goel VK, Lazar AJ, Warneke CL, Redston MS, Haluska FG. Examination of mutations in BRAF, NRAS, and PTEN in primary cutaneous melanoma. J Invest Dermatol. 2006;126:154–60. doi: 10.1038/sj.jid.5700026. [DOI] [PubMed] [Google Scholar]
- 6.Tsao H, Goel V, Wu H, Yang G, Haluska FG. Genetic Interaction Between NRAS and BRAF Mutations and PTEN//MMAC1 Inactivation in Melanoma. J Investig Dermatol. 2004;122:337–41. doi: 10.1046/j.0022-202X.2004.22243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haass NK, Sproesser K, Nguyen TK, et al. The mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in melanoma cells and tumor regression when combined with docetaxel. Clin Cancer Res. 2008;14:230–9. doi: 10.1158/1078-0432.CCR-07-1440. [DOI] [PubMed] [Google Scholar]
- 8.Yeh TC, Marsh V, Bernat BA, et al. Biological Characterization of ARRY-142886 (AZD6244), a Potent, Highly Selective Mitogen-Activated Protein Kinase Kinase 1/2 Inhibitor. Clinical Cancer Research. 2007;13:1576–83. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- 9.Davies BR, Logie A, McKay JS, et al. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases:mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Molecular Cancer Therapeutics. 2007;6:2209–19. doi: 10.1158/1535-7163.MCT-07-0231. [DOI] [PubMed] [Google Scholar]
- 10.Adjei AA, Cohen RB, Franklin W, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26:2139–46. doi: 10.1200/JCO.2007.14.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dummer R, Robert C, Chapman P, et al. AZD6244 (ARRY-142886) vs temozolomide (TMZ) in patients with advanced melanoma: an open-label, randomized, multicenter, phase II study. J Clin Oncol. 2008;26:9033. [Google Scholar]
- 12.Flaherty K, Puzanov I, Sosman J, et al. Phase I study of PLX4032: Proof of concept of V600E BRAF mutation as a therapeutic target in human cancer. J Clin Oncol. 2009;27:15s. Suppl; abstr 9000. [Google Scholar]
- 13.Smalley KS, Contractor R, Haass NK, et al. Ki67 expression levels are a better marker of reduced melanoma growth following MEK inhibitor treatment than phospho-ERK levels. Br J Cancer. 2007;96:445–9. doi: 10.1038/sj.bjc.6603596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mirzoeva OK, Das D, Heiser LM, et al. Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition. Cancer Res. 2009;69:565–72. doi: 10.1158/0008-5472.CAN-08-3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–8. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tibes R, Qiu Y, Lu Y, et al. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther. 2006;5:2512–21. doi: 10.1158/1535-7163.MCT-06-0334. [DOI] [PubMed] [Google Scholar]
- 17.Davies MA, Stemke-Hale K, Lin E, et al. Integrated Molecular and Clinical Analysis of AKT Activation in Metastatic Melanoma. Clin Cancer Res. 2009;15:7538–46. doi: 10.1158/1078-0432.CCR-09-1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68:6084–91. doi: 10.1158/0008-5472.CAN-07-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vasudevan KM, Barbie DA, Davies MA, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009;16:21–32. doi: 10.1016/j.ccr.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amit I, Citri A, Shay T, et al. A module of negative feedback regulators defines growth factor signaling. Nat Genet. 2007;39:503–12. doi: 10.1038/ng1987. [DOI] [PubMed] [Google Scholar]
- 21.Hennessy BT, Lu Y, Poradosu E, et al. Pharmacodynamic Markers of Perifosine Efficacy. Clin Cancer Res. 2007;13:7421–31. doi: 10.1158/1078-0432.CCR-07-0760. [DOI] [PubMed] [Google Scholar]
- 22.Thomas RK, Baker AC, Debiasi RM, Winckler W, et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39:347–51. doi: 10.1038/ng1975. [DOI] [PubMed] [Google Scholar]
- 23.Davies MA, Stemke-Hale K, Tellez C, et al. A novel AKT3 mutation in melanoma tumours and cell lines. Br J Cancer. 2008;99:1265–8. doi: 10.1038/sj.bjc.6604637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park ES, Rabinovsky R, Carey M, et al. Integrative Analysis of Proteomic Signatures, Mutations, and Drug Responsiveness in the NCI 60 Cancer Cell Line Set. Mol Cancer Ther. 2010;9:257–67. doi: 10.1158/1535-7163.MCT-09-0743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dry JR, Payey S, Pratilas CA, et al. Transcriptional pathway signatures predict MEK addiction and response to selumetinib (AZD6244) Cancer Res. 2010;70:2264–73. doi: 10.1158/0008-5472.CAN-09-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanter-Lewensohn L, Dricu A, Girnita L, Wejde J, Larsson O. Expression of insulin-like growth factor-1 receptor (IGF-1R) and p27Kip1 in melanocytic tumors: a potential regulatory role of IGF-1 pathway in distribution of p27Kip1 between different cyclins. Growth Factors. 2000;17:193–202. doi: 10.3109/08977190009001068. [DOI] [PubMed] [Google Scholar]
- 27.Satyamoorthy K, Li G, Vaidya B, Patel D, Herlyn M. Insulin-like growth factor-1 induces survival and growth of biologically early melanoma cells through both the mitogen-activated protein kinase and beta-catenin pathways. Cancer Res. 2001;61:7318–24. [PubMed] [Google Scholar]
- 28.Hilmi C, Larribere L, Giuliano S, et al. IGF1 promotes resistance to apoptosis in melanoma cells through an increased expression of BCL2, BCL-X(L), and survivin. J Invest Dermatol. 2008 Jun;128:1499–505. doi: 10.1038/sj.jid.5701185. [DOI] [PubMed] [Google Scholar]
- 29.Chresta CM, Davies BR, Hickson I, et al. AZD8055 Is a Potent, Selective, and Orally Bioavailable ATP-Competitive Mammalian Target of Rapamycin Kinase Inhibitor with In vitro and In vivo Antitumor Activity. Cancer Res. 2009;70:288–298. doi: 10.1158/0008-5472.CAN-09-1751. [DOI] [PubMed] [Google Scholar]
- 30.Meng J, Peng H, Dai B, et al. High level of AKT activity is associated with resistance to MEK inhibitor AZD6244 (ARRY-142886) Cancer Biol Ther. 2009;8:2073–80. doi: 10.4161/cbt.8.21.9844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montagut C, Sharma SV, Shioda T, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68:4853–61. doi: 10.1158/0008-5472.CAN-07-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsao H, Zhang X, Benoit E, Haluska FG. Identification of PTEN/MMAC1 alterations in uncultured melanomas and melanoma cell lines. Oncogene. 1998;16:3397–402. doi: 10.1038/sj.onc.1201881. [DOI] [PubMed] [Google Scholar]
- 33.Zhou XP, Gimm O, Hampel H, Niemann T, Walker MJ, Eng C. Epigenetic PTEN silencing in malignant melanomas without PTEN mutation. Am J Pathol. 2000;157:1123–8. doi: 10.1016/S0002-9440(10)64627-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Curtin JA, Stark MS, Pinkel D, Hayward NK, Bastian BC. PI3-kinase subunits are infrequent somatic targets in melanoma. J Invest Dermatol. 2006;126:1660–3. doi: 10.1038/sj.jid.5700311. [DOI] [PubMed] [Google Scholar]
- 35.Omholt K, Krockel D, Ringborg U, Hansson J. Mutations of PIK3CA are rare in cutaneous melanoma. Melanoma Res. 2006;16:197–200. doi: 10.1097/01.cmr.0000200488.77970.e3. [DOI] [PubMed] [Google Scholar]
- 36.Yoon YK, Kim HP, Han SW, et al. Combination of EGFR and MEK1/2 inhibitor shows synergistic effects by suppressing EGFR/HER3-dependent AKT activation in human gastric cancer cells. Mol Cancer Ther. 2009;8:2526–36. doi: 10.1158/1535-7163.MCT-09-0300. [DOI] [PubMed] [Google Scholar]
- 37.Kato M, Putta S, Wang M, et al. TGF-beta activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat Cell Biol. 2009;11:881–9. doi: 10.1038/ncb1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huse JT, Brennan C, Hambardzumyan D, et al. The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev. 2009;23:1327–37. doi: 10.1101/gad.1777409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poliseno L, Salmena L, Riccardi L, et al. Identification of the miR-106b∼25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Sci Signal. 2010;3:ra29. doi: 10.1126/scisignal.2000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.