

Summary
The ruminant developmental transition from late foetus to lamb is associated with marked changes in skeletal muscle structure and function that reflect programming for new physiological demands following birth. To determine whether epigenetic changes are involved in this transition, we investigated the genomic architecture of the chromatin modification, histone 3 lysine 27 trimethylation (H3K27me3), which typically regulates early life developmental processes; however, its role in later life processes is unclear. Chromatin immunoprecipitation coupled with next‐generation sequencing was used to map H3K27me3 nucleosomes in ovine longissimus lumborum skeletal muscle at 100 days of gestation and 12 weeks post‐partum. In both states, H3K27me3 modification was associated with genes, transcription start sites and CpG islands and with transcriptional silencing. The H3K27me3 peaks consisted of two major categories, promoter specific and regional, with the latter the dominant feature. Genes encoding homeobox transcription factors regulating early life development and genes involved in neural functions, particularly gated ion channels, were strongly modified by H3K27me3. Gene promoters differentially modified by H3K27me3 in the foetus and lamb were enriched for gated ion channels, which may reflect changes in neuromuscular function. However, most modified genes showed no changes, indicating that H3K27me3 does not have a large role in late muscle maturation. Notably, promyogenic transcription factors were strongly modified with H3K27me3 but showed no differences between the late gestation foetus and lamb, likely reflecting their lack of involvement in the myofibre fusion process occurring in this transition. H3K27me3 is a major architectural feature of the epigenetic landscape of ruminant skeletal muscle, and it comments on gene transcription and gene function in the context of late skeletal muscle development.
Keywords: ChIP‐Seq, chromatin, epigenetics, muscle
Introduction
Development is underpinned by hierarchical programmes of gene expression changes orchestrated by epigenetic modifications to the genome (Ringrose & Paro 2007; Schuettengruber et al. 2007). These programmes are conserved in evolutionary lineages but also are exquisitely tuned to define the uniqueness of each species (Feinberg 2010; Feinberg & Irizarry 2010). The molecular bases of the epigenetic events underpinning development have only recently begun to be elucidated. Polycomb‐group (PcG) and trithorax‐group proteins and their associated combinatorial histone modifications, particularly PcG2‐mediated histone 3 lysine 27 trimethylation (H3K27me3), are core components of this epigenetic machinery (Sparmann & van Lohuizen 2006; Ringrose & Paro 2007; Schuettengruber et al. 2007). However, little is known about the roles of these chromatin modifications in later life transitions, particularly at the major physiological transition between foetal and post‐natal animal life.
Ruminants are born in an advanced developmental state compared with other mammals, as they can stand and run within hours of birth. Their muscles must accommodate the new physiological demands of locomotion and support against gravity after birth, which is associated with major changes in myofibre oxidative status, size (hypertrophy) and gene expression (Byrne et al. 2010). Mammalian skeletal muscle undergoes three stages of development. In the sheep foetus, primary muscle fibres first appear at day 32 of gestation (birth is at 147 days) and secondary muscle fibres use the primary fibres as scaffolds for their deposition starting around day 38. Tertiary fibres first develop on the secondary fibres by gestation days 62–76, and these contribute to the substantial muscle mass present in large mammals, as there is no net increase in the number of muscle fibres after birth in these animals, unlike in small mammals (Stickland 1978; Bonnet et al. 2010).
The PcG2 complex catalyses the methylation of histone 3 lysine 27 at specific sites in the genome, thereby hierarchically repressing developmentally important genes (Boyer et al. 2006). The genomic landscape of this histone modification markedly changes during embryonic development at the juncture of stem cell commitment and differentiation; however, its role in later life developmental events is unclear (Bernstein et al. 2006; Pauler et al. 2009; Zhou et al. 2011a). We hypothesised that changes in H3K27me3 may underlie the programmed physiological adaptations occurring in ovine skeletal muscle during the transition between late foetus and lamb states. To investigate this epigenetic modification, we employed chromatin immunoprecipitation followed by next‐generation sequencing to provide detailed maps of H3K27me3 nucleosomes across the genome.
Materials and methods
Details for all methods are listed in Appendix S1, and only brief descriptions are given below.
Biological samples
Foetal (100 days of gestation) and lamb (12 weeks of age) longissimus lumborum (LL) skeletal muscle samples were obtained from a flock of Dorset/Suffolk/Rambouillet cross‐bred sheep raised at Utah State University (Vuocolo et al. 2007; Byrne et al. 2010). Animals were reared and euthanised in accordance with the animal ethics guidelines of Utah State University (Logan, Utah, USA). The same animals were used for all experiments.
Chromatin immunoprecipitation followed by sequencing
Enrichment of H3K27me3 at genomic loci was measured using chromatin immunoprecipitation followed by next‐generation sequencing (ChIP‐Seq) for the foetal (n = 3) and lamb (n = 3) LL samples (Wagschal et al. 2007). Input nucleosomal DNA from the three animals in each group was used as a control.
Mapping of sequence reads to the bovine genome and peak detection
Sequence reads were mapped to the Btau_4.0 bovine genome, as an annotated high‐quality ovine assembly was unavailable (Bovine Genome Sequencing and Analysis Consortium et al. 2009; Tellam et al. 2009). Uniquely mapped reads were used to identify genomic regions of H3K27me3 nucleosome enrichment for each sample relative to the corresponding input control (CisGenome (Ji et al. 2008)). The reads were extended to 147 bp, that is, the length of the input nucleosomal DNA. False discovery rate (FDR) estimates were based on the level of enrichment compared to the input nucleosome control using a conditional binomial model (FDR ≤ 0.1; Ji et al. 2008). To examine the H3K27me3 architecture across the genome within each developmental state, the unique reads from the replicates within each group were pooled and compared to their age‐matched input control for peak detection using cisgenome. The X chromosome was excluded from analysis. These data were used to identify correlations between H3K27me3 peaks and transcriptional start sites (TSS), CpG islands (CGIs) and genes (CDS) as well as to define genes with promoters modified by H3K27me3.
Nucleotide composition analysis for nucleosomal DNA
The G+C content and nucleotide profile of the 147‐bp H3K27me3 nucleosomes located in peaks by cisgenome, as well as the input nucleosomal DNA, were determined for each biological state. Similar analysis was performed using one million 147‐bp sequences randomly extracted from the repeat masked bovine genome sequence. Differences in G+C contents were identified using the Mann–Whitney U‐test.
Visualisation of ChIP‐Seq peaks using Hilbert space filling curves
H3K27me3 peaks identified for each developmental state were visualised on each autosome using a Hilbert space filling curve, which is continuous fractal space filling curve maintaining locality information and adapted for the representation of genomic data (Anders 2009).
Association of sequence reads within H3K27me3 peaks with genome architecture
Sequence reads within H3K27me3 peaks in both developmental states were examined for association with TSS (10 891 RefFlat genes), the first exon of each gene (29 027 RefSeq genes) and 37 595 CGIs. For each gene set, the distance from the gene feature start site to each H3K27me3 peak was calculated and the number of sequence reads within 100‐bp windows derived to a maximum distance of 10 kb in either direction determined. The H3K27me3 peaks overlapping gene promoters were identified using a 2.5‐kb window (2 kb upstream and 500 bp downstream of the first nucleotide of each RefSeq gene).
Association of H3K27me3 in RefSeq gene promoters with gene expression
Bovine Affymetrix microarrays were used for gene expression measurement (Fleming‐Waddell et al. 2007; Vuocolo et al. 2007; Byrne et al. 2010). H3K27me3 peaks were detected in 4070 and 3384 RefSeq gene promoters for the foetal and lamb samples respectively. Of the genes containing H3K27me3 promoter peaks, gene expression signals were correspondingly measured for 1953 and 1466 Affymetrix probe sets, corresponding to 1430 and 1107 annotated and expressed genes respectively. Gene expression was partitioned into four categories: no, low (log2‐scaled averages <4), medium (log2‐scaled averages ≥4 and <8) and high expression (log2‐scaled averages ≥8). A Behrens–Fischer test was used to measure the difference between the level of H3K27me3 promoter reads in each gene expression category compared with the ‘no’ expression category. For the scatter plots, a minimum log2‐scaled average of 4 for gene expression was used.
Differentially modified gene promoters between the foetus and lamb
The number of reads in the promoters of RefSeq genes for each of the biological replicates (n = 3/group) was determined. After normalisation for library size, the read count for each gene was corrected for the input control. The data were then subjected to quantile normalisation to allow for the identification of significant differences in H3K27me3 promoter reads between the two developmental states using the statistical tools in the CLCBIO package (CLCBIO). A FDR‐corrected P‐value ≤ 0.1 was considered significant.
Gene function enrichment analyses
To identify functional themes associated with gene lists, the Database for Annotation, Visualization and Integrated Discovery (DAVID) was used (Huang da et al. 2009).
Results
H3K27me3 genome profiles during late development of ovine skeletal muscle
ChIP‐Seq, using a specific antibody to H3K27me3, was performed on ovine LL skeletal muscle (n = 3/gp) sampled at 100 days of gestation (foetus) and 12 weeks of age (lamb) (Bock et al. 2010; Egelhofer et al. 2011). Input nucleosomal DNA, prepared by micrococcal nuclease (MNase) digestion of isolated chromatin, was characterised by a ~150‐bp band representing MNase‐protected DNA (147 bp) wrapped around the histone octamer (Fig. S1) (Luger et al. 1997). H3K27me3 nucleosomes were isolated by immunoabsorption of the digested chromatin and the associated DNA sequenced. Input nucleosomal DNA was used as the control. A total of 95.8 million 65‐bp reads (6.2 Gb) were generated and mapped to the non‐repeat‐masked bovine genome sequence (Btau_4.0) (Bovine Genome Sequencing and Analysis Consortium et al. 2009). Uniquely mapped reads were then extended to 147 bp. The ChIP‐Seq processing statistics are summarised in Table S1. Genomic regions enriched with H3K27me3 nucleosomes relative to the input control for each sample were identified by using cisgenome (Ji et al. 2008).
G+C content of H3K27me3 nucleosomal DNA
The mean G+C content of the lamb H3K27me3 nucleosomal DNA (57.7 ± 9.0%, SD = 1) was greater than the genome‐wide figure (41.7%) (Bovine Genome Sequencing and Analysis Consortium et al. 2009) and different from the mean value of 40.3 ± 9.1% for one million randomly selected 147‐bp sequences (P < 0.0001). The higher G+C content is typical for nucleosomal DNA (Hughes & Rando 2009). The H3K27me3 nucleosomal DNA G+C content was not different from the input nucleosomal DNA (54.3 ± 7.1%; P > 0.05). The corresponding foetal data gave essentially the same results. There were no differences between the foetal and lamb H3K27me3 nucleosomal DNA G+C content or the foetal and lamb input nucleosomal DNA G+C content (both P > 0.05).
Distinct categories of H3K27me3 nucleosomes
Representative genomic tracks highlighting H3K27me3 enrichment for each sample that exemplify two major categories of gene‐associated H3K27me3 enrichment, that is, the focussed promoter peak and broad regional modification, are shown in Fig. 1. The former reflects enrichment confined to the promoter (defined as −2 kb to +0.5 kb from the TSS or from the first base of exon 1 depending on gene data set). This was typified by RASGRF1 (Ras protein‐specific guanine nucleotide‐releasing factor 1). Regional enrichment was exemplified by the HOXD (homeobox D cluster) locus, where the modification extended over a broad region of ~ 60 kb encompassing multiple HOXD genes and their intergenic regions. The regional enrichment did not include all HOXD genes. Similar regional H3K27me3 landscapes were also apparent at the HOXA, HOXB and HOXC loci (Fig. S2). The enrichment of H3K27me3 across broad segments of the HOX gene loci is a consistent finding for all mammalian species and most tissues (Bernstein et al. 2007; Rinn et al. 2007; Wang et al. 2009). Moreover, regional enrichment also is the dominant H3K27me3 characteristic in other mammalian genomes (Pauler et al. 2009). The HOX loci also highlighted the concordance between the foetal and lamb samples, indicating that there were few large changes at these loci during late skeletal muscle development. Also shown in Fig. S2 is a representation of the H3K27me3 read counts in the promoters of HOX genes for each of the four HOX gene loci, using pooled data for lamb [panel (e)]. At all four loci, there were strong trends towards greater modification of HOX genes located at the 5′ end of each of these loci. Thus, regional H3K27me3 nucleosomes marked large territories of the four HOX loci and showed distinctive patterns collinear with chromosomal gene order. Very similar profiles were obtained using the pooled foetal data (result not shown) and human skeletal muscle (Zhou et al. 2011b).
Figure 1.
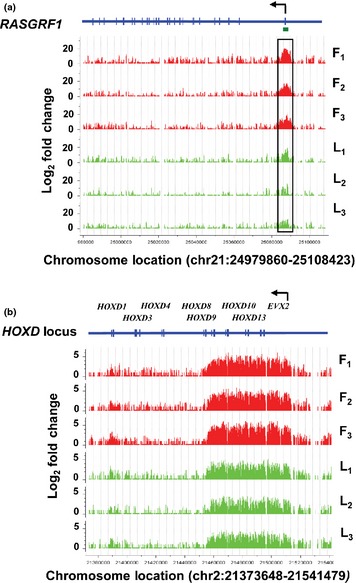
Representative H3K27me3 nucleosome profiles. Examples of gene promoter‐specific and regional H3K27me3 nucleosome profiles identified using cisgenome (Ji et al. 2008). The H3K27me3 landscape at two loci in the bovine genome (Btau_4.0) for each of the three foetal and three lamb skeletal muscle samples is shown: (a) RASGRF1 (BTA21:24,979,860‐25,108,423), (b) HOXD locus (BTA2:21,373,648‐21,541,479). The top track in each panel shows RefSeq genes with vertical marks denoting exons. Arrows denote the direction of transcription. Green bar, CpG island; black box, promoter‐specific enrichment of H3K27me3 nucleosomes. Data are displayed for individual animals (foetal, F1–F3, red; lamb, L1–L3, green). The ordinate shows the normalised log2 fold change enrichment for the H3K27me3 nucleosomes compared with the age‐matched input nucleosomal DNA.
There was near chromosome‐wide H3K27me3 modification on the X chromosome of females (Fig. 2). The lamb samples were all male, whereas the foetal samples were derived from one male and two females. Direct comparison of female with male samples, regardless of developmental stage, revealed means of 370 883 and 40 471 uniquely mapped H3K27me3 reads respectively on the X chromosome. This near 10‐fold difference (P < 0.0001) contrasts with the autosomes, which showed no gender‐specific chromosomal differences of total mapped reads (P > 0.05). H3K27me3 is strongly associated with X chromosome inactivation in female placental mammals (King et al. 2008; Escamilla‐Del‐Arenal et al. 2011). This dosage compensation mechanism largely silences one of the two female X chromosomes, ensuring equivalence of transcriptional activity with the single X chromosome in males. The X chromosome data also provide an internal positive control demonstrating the specificity of the H3K27me3 ChIP‐Seq analyses.
Figure 2.
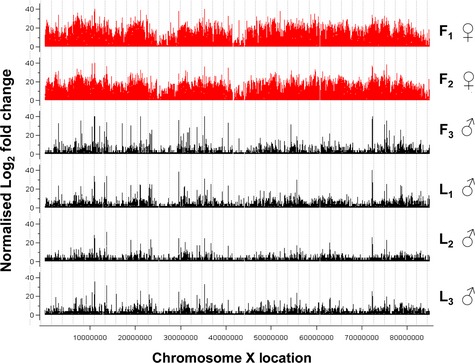
Distribution of H3K27me3 nucleosomal peaks on the bovine X chromosome. The H3K27me3‐enriched landscape across the bovine X chromosome for the foetal (F1–F3, red) and lamb (L1–L3, green) samples was determined using cisgenome. The ordinate shows the normalised log2 fold change enrichment for the H3K27me3 nucleosome data compared to the age‐matched input control. The gender of each animal also is shown. The window size for visualisation of H3K27me3 peaks across the X chromosome was 500 bp.
Two analysis strategies were subsequently undertaken and, in each case, only autosomes were considered. The first analysis examined the relationship between H3K27me3 modification and major architectural features of the genome, whereas the second identified gene promoters that were differentially modified in the foetal‐to‐lamb transition (n = 3/gp). To increase the power of the first within‐genome analysis, the primary data sets for the biological replicates within each developmental state were combined and reanalysed using cisgenome to identify H3K27me3 peak enrichments across the genome relative to the corresponding input control.
Visualisation of H3K27me3 nucleosomes
Maps of the H3K27me3 modification for foetal and lamb muscle samples were generated for each autosome using Hilbert curves, which provide enhanced 2‐D visual presentation of enrichment regions (Fig. S3) (Anders 2009). A Hilbert curve is a continuous fractal space filling curve formed by folding a line, representing each autosome, whilst maintaining locality relationships. In particular, these curves highlight regional effects within a chromosome. Hilbert curves for the distribution of H3K27me3 on bovine chromosome 4 for the foetal and lamb samples are shown in Fig. 3. In this example, significant regional enrichments of H3K27me3 were associated with three homeobox‐encoding gene loci, that is, (i) DLX5 (distal‐less homeobox 5) and DLX6 (distal‐less homeobox 6); (ii) EN2 (engrailed homeobox 2); and (iii) HOXA9 (homeobox A9) and HOXA10 (homeobox A10), as well as the SP8 (SP8 transcription factor) locus. Overall, there were striking similarities between the foetal and lamb profiles for all autosomes. The genomic coordinates of all peaks are provided in Table S2 and S3 respectively.
Figure 3.
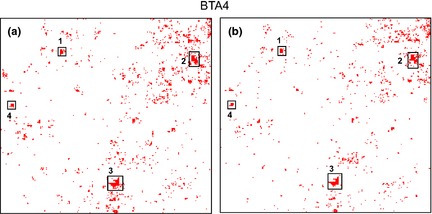
Visualisation of H3K27me3 enrichment on bovine chromosome 4 using Hilbert curves. Hilbert curves were used to visualise H3K27me3 peaks along BTA 4 for each of the combined foetal and lamb samples. This fractal method folded the chromosome and its associated H3K27me3 features into two‐dimensional space whilst maintaining locality relationships. (a) Foetal, (b) lamb. Examples of regional enrichments are indicated by black boxes for three homeobox encoding gene loci: (1) the DLX loci [DLX5 (distal‐less homeobox 5), DLX6 (distal‐less homeobox 6)]; (2) EN2 (engrailed homeobox 2); (3) the HOXA loci (HOXA9, HOXA10) as well as (4) SP8 (SP8 transcription factor). All autosomal Hilbert curves for foetus and lamb are shown in Fig. S3, and genome coordinates for H3K27me3 peaks are reported in Table S2 and S3 respectively.
Association of H3K27me3 peaks with genome architecture
The sequence reads in H3K27me3 peaks for each biological state were correlated with architectural features in the genome including genes (CDS; beginning of exon 1 of RefFlat genes), TSS (first nucleotide of RefSeq genes) and CGIs in both developmental states (Fig. 4). The half‐peak widths for all gene features were ~2 kb, but in each case there were substantial trailing edges extending beyond 10 kb of the feature reflecting contributions from regional modifications. For each feature, at the resolution of the analysis, the peaks were approximately symmetrical and broadly centred on the feature. Thus, there were strong associations of H3K27me3 nucleosomes with gene start features. H3K27me3 enrichment was identified in 1944 (17.8%) and 1577 (14.5%) RefFlat gene promoters for the foetal and lamb states respectively. Promoters were defined as a 2.5‐kb region consisting of 2 kb upstream and 500 bp downstream of the TSS (Mikkelsen et al. 2007). For the more comprehensive though less well‐annotated RefSeq genes, H3K27me3 enrichment was identified in 2654 (9.1%) and 3319 (11.24%) autosomal RefFlat gene promoters respectively. H3K27me3 enrichment was identified for 14 942 (39.7%) and 13 380 (35.6%) bovine CGI for the foetal and lamb samples respectively. There were no differences between the foetus and lamb in the distributions of H3K27me3 modifications associated with these genomic features.
Figure 4.
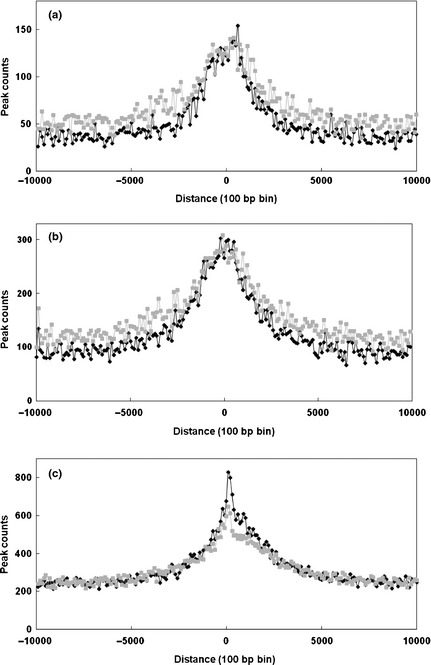
Association of H3K27me3 with genome features. The distribution of read counts in H3K27me3 peaks in each of the combined foetal and lamb samples was measured relative to: (a) transcriptional start sites (TSS) defined by 10 891 bovine full‐length cDNA sequences (RefFlat), (b) the first nucleotide of 29 027 annotated bovine genes (RefSeq) and (c) 35,595 bovine CpG islands (CGIs). For each gene set, the distance from the gene start coordinate to each H3K27me3 peak was calculated. The number of sequence reads in 100‐bp windows of H3K27me3 peaks was derived to 10 kb in either direction from the feature. For CGIs, the boundary coordinates were used. If a peak overlapped within the CGI coordinates, the distance was collapsed to zero; otherwise, the minimum distance to the CGI was plotted using a 100‐bp window. Grey, foetal samples; black, lamb.
H3K27me3 nucleosomes correlate with repression of gene expression
The relationship between the number of sequence reads in H3K27me3 nucleosome peaks within autosomal gene promoters for 1430 and 1107 RefSeq genes and their corresponding gene expression levels (Byrne et al. 2010) for the foetal and lamb samples respectively are shown in Fig. 5. Gene expression was measured by microarray analysis and then divided into four categories: no expression, low expression (log2‐scaled averages <4) and medium expression (≥4 and <8) and high expression (≥8). For the foetal samples, there was a decreased level of H3K27me3 modification in the promoters of the high and medium gene expression categories compared with non‐expressed genes (P = 8.2E‐15, P = 2.5E‐3 respectively; Behrens–Fischer test). The low expression category showed no difference (P < 0.05; B‐F test) from the ‘no’ expression category. These results are consistent with previous studies (Boyer et al. 2006). Very similar results were obtained for the lamb data. There were no differences in mean H3K27me3 promoter read count between the lamb and foetal samples for any of the corresponding gene expression categories (P > 0.05; B‐F test). Scatter plots for these data, which highlight a group of intensively modified genes with low levels of gene expression and the low level of H3K27me3 modification associated with highly expressed genes, also are shown in Fig. 5.
Figure 5.
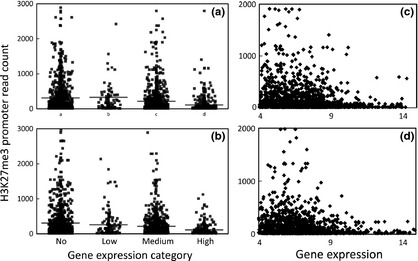
H3K27me3 nucleosomes correlate with repression of gene expression. Plots show the relationship between four gene expression categories and H3K27me3 sequence reads in the promoters of genes for the foetal (a) and lamb (b) samples. The levels of H3K27me3 in the promoters of bovine RefSeq genes were measured in enriched peaks after normalisation for library size and subtraction of the normalised input control. A promoter was defined as a window 2 kb 5′ plus 500 bp 3′ to the beginning of each gene. Gene expression was measured in the same samples by microarray analysis (Byrne et al. 2010). Expression categories consisted of: no, low [log2 <4 (scaled average)], medium (log2 >4 and <8) and high expression (log2 ≥8). Corresponding scatter plots are shown in Fig. 5(c) and (d). A minimum log2 expression value of 4 was used.
Functional enrichment analysis of genes with promoters modified by H3K27me3
Autosomal RefSeq genes with promoters enriched for H3K27me3 were examined for functional enrichment using DAVID (Huang da et al. 2009). Functional annotation clustering was initially performed as it provided a high‐level perspective of multiple enriched gene function terms. The analyses identified 58 and 60 clusters [enrichment score (ES) ≥1.3 and ≥5 genes/term] for the foetal and lamb modified genes respectively (Table S4). As many strongly enriched clusters were functionally related, the analysis was summarised into related functional themes (Table 1). The foetal and lamb analyses showed strikingly common themes often composed of near‐identical clusters with similar scores, for example homeobox genes (HOX genes), which are key transcription factors involved in body patterning and early developmental processes, were the highest scoring clusters for both the foetal and lamb samples (ES = 20.9 and 18.3 respectively), whereas aspects of gated ion channel function and structure were common next highest ranking clusters (ES = 15.3 and 15.3 respectively). There were strong enrichments for functional themes broadly described as development, transcriptional regulation and neural structure and function. The first two themes are strongly linked through homeobox encoding gene functions. Likewise, the enrichment for the theme gated ion channel, ion transport and G‐coupled receptors has strong overlaps with neural structure and function. Three foetal and four lamb clusters were weakly enriched for aspects of muscle development. Collectively, these data demonstrate: (i) striking similarities in the H3K27me3 gene promoter modifications for both the foetus and lamb, (ii) strong enrichments for a number of functional themes indicating high gene function specificity for H3K27me3, (iii) strong representation of themes related to development/HOX genes and gated ion channels/neural function and (iv) weak H3K27me3 enrichment for genes involved in muscle development including transforming growth factor β structure and function (ES = 4.4 and 3.4 respectively) (Table S4). The latter pathway regulates myogenesis and muscle hypertrophy (Kollias & McDermott 2008). Inspection of the data revealed that the promyogenic transcription factors MYOD, MYF5 and PAX3 were all strongly modified in both the foetal and lamb samples. Independent analysis using the PFAM protein structure database in DAVID identified three dominant protein structures common to both developmental states, that is, homeobox domains (typified by: PF00046: homeobox, Padj = 1.59E‐19 and 2.1E‐16 respectively), gated ion channels (typified by: PS00520, Padj = 5.4E‐4 and 4.2E‐4 respectively) and TGF‐beta (PF00019, Padj = 0.001 and 0.01 respectively).
Table 1. DAVIDa functional annotation clusters arranged into related functional themes for foetal and lamb RefSeq genes with promoters enriched for H3K27me3.
| Functional themeb | Foetal functional annotation cluster number (enrichment score)c | Lamb functional annotation cluster number (enrichment score) |
|---|---|---|
| Homeobox genes | 1 (20.9) | 1 (18.3) |
| Development | 1 (20.9), 12 (4.9), 16 (4.6), 21 (3.9), 22 (3.9), 35 (2.4), 38 (2.3), 41 (2.0), 44 (1.9), 52 (1.6) | 10 (5.5), 15 (5.0), 16 (4.8), 18 (3.9), 20 (3.8), 28 (2.8), 28 (2.8), 50 (1.6), 52 (1.6), 56 (1.5), 59 91.4) |
| Transcriptional regulation | 1 (20.9), 3 (11.4), 14 (4.8), 34 (2.4), 46 (1.9) | 1 (18.3), 3 (11.6), 21 (3.8), 32 (2.4) |
| Gated ion channel, ion transport, G‐protein‐coupled receptors | 2 (15.3), 4 (10.9), 5 (8.2), 6 (7.5), 7 (7.4), 8 (6.9), 10 (5.6), 11 (5.2), 15 (4.7), 17 (4.5), 25 (3.1), 33 (2.4), 39 (2.2), 43 (2.0), 47 (1.8), 48 (1.7), 51 (1.7), 58 (1.3) | 2 (15.3), 4 (10.4), 5 (8.1), 6 (7.5), 8 (6.6), 11 (5.3), 12 (5.2), 13 (5.0), 27 (2.9), 30 (2.7), 34 (2.4), 35 (2.3), 38 (2.3), 40 (2.1), 43 (1.9), 45 (1.7), 47 (1.7), 51 (1.6) |
| Neural structure and function | 11 (5.2), 12 (4.9), 21 (3.9), 22 (3.9), 40 (2.2), 43 (2.0) | 8 (6.6), 16 (4.8), 20 (3.8), 36 (2.3), 40 (2.1), 50 (1.6), 51 (1.6), 52 (1.6) |
| Muscle development | 18 (4.4), 26 (2.9), 38 (2.3) | 10 (5.5), 24 (3.4), 34 (2.4), 39 (2.1), 56 (1.5) |
| Calcium signalling | 8 (6.9), 25 (3.1), 33 (2.4), 47 (1.8) | 25 (3.2), 37 (2.3), 45 (1.7) |
DAVID functional annotation clustering (Huang da et al. 2009).
Functional themes were defined by manual identification of related functional annotation clusters.
Cluster number (enrichment score). Enrichment scores (ES) ≥ 1.3 and gene number/term ≥ 5 were considered significant. ES is defined as – log10 (mean P‐value for cluster terms). Some themes contain the same cluster number. Cluster numbers for foetus and lamb are unrelated. Full details are available in Table S4.
Enriched KEGG pathways largely reiterated the molecular functions described above and the strong similarities between the developmental states, that is, all 16 enriched pathways for foetus were present in the 19 pathways enriched for lamb. The two highest ranking pathways for the foetus and lamb were both neuroactive ligand–receptor interaction (bta04080; Padj = 3.4E‐23 and 9.4E‐23 respectively) and calcium signalling pathway (bta04020, Padj = 1.0E‐11 and 3.9E‐10 respectively). Other common pathways included four that commented on hypertrophic muscle development, albeit mainly in a cardiac context, as well as a number of signalling pathways known to be involved in regulating muscle development [e.g. TGF‐beta (lamb), Wnt (foetus and lamb) and Hedgehog (foetus and lamb)] (Kollias & McDermott 2008; Otto et al. 2008; Straface et al. 2009).
Stratification of H3K27me3 gene promoter density identifies different enriched gene functions
Autosomal RefSeq genes containing H3K27me3 peaks within their promoters were ranked on read count within their promoters for each developmental state. Histograms of log10 H3K27me3 read count normalised to the top ranked gene (nominated value = 100) as a function of rank are shown in Fig. 6. Both the foetal and lamb plots showed two distinct components to these histograms. The first corresponded with the approximate top ranked 300–500 genes, which are clearly distinguished by substantially greater modification. These data signify stratification of H3K27me3 promoter density. The top ranked 500 genes (1–500) and the next top ranked 500 genes (501–1000) were independently assessed for enriched Gene Ontology (GO) molecular functions using DAVID (Fig. 6). The top ranked 1–500 genes for both the foetal and lamb samples were exclusively characterised by four strongly enriched GO molecular function terms, all of which related to transcription factor binding; underlying this is the enrichment of homeobox encoding genes. Thus, the most intense H3K27me3 promoter modification was highly selective for this class of genes. In contrast, the top ranked 501–1000 genes showed exclusion of the transcription factor binding theme but enrichment for functions relating to gated ion channels for both biological states. Functional annotation cluster analysis and Pfam analyses revealed similar information (results not shown). Thus, the density of H3K27me3 modification in the promoters of genes was strongly stratified, and it discriminated between two specific classes of gene functions.
Figure 6.
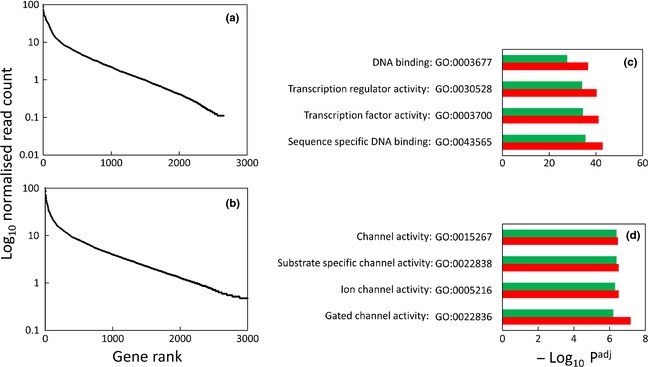
Stratification of H3K27me3 density in gene promoters. Autosomal RefSeq genes containing H3K27me3 peaks within their promoters were ranked according to the level of H3K27me3 reads normalised (×100) to the highest ranked gene. (a) and (b) show histograms of the distribution of normalised H3K27me3 promoter read count as a function of gene rank for the foetal and lamb samples respectively. Enriched gene ontology molecular function terms identified using DAVID for the top ranked 1–500 genes (c) and top ranked 501–1000 genes (d) were plotted against –log10 Padj (Benjamini–Hochberg‐corrected). Red (foetal); green (lamb).
Identification of gene promoters with differences in H3K27me3 density in foetal and lamb skeletal muscle
Changes in H3K27me3 RefSeq gene promoter density between the foetal and lamb samples (n = 3/gp; Padj < 0.1) were identified using clc bio (CLC BIO) (Table S5). Only 86 gene promoters were differentially modified of the total of 4467 unique gene promoters modified in either foetus or lamb. DAVID functional annotation clustering revealed weak enrichment for three interrelated functional themes, plasma membrane, cell signalling and synapse, which were largely reiterated in independent analyses of the GO cellular compartment and biological process categories (Tables 2 and 3 and S5). There was also weak enrichment for the regulation of transcription revealed by GO biological process analysis. A number of the differentially modified genes contributing to the enriched terms plasma membrane, cell signalling and synapse may reflect aspects of restructuring and innervation of neuromuscular junctions. CACNB4 encodes the beta 4 subunit of a voltage‐dependent calcium channel (2.04‐fold more modification in lamb), which is present in myotubes, possibly at the neuromuscular junction (Pagani et al. 2004; Subramanyam et al. 2009). Similarly, SCN5 (also known as VPS45) encodes a voltage‐gated sodium channel subunit expressed in myotubes (1.47‐fold more modification in lamb); SCN5 gene expression was changed by muscle fibre denervation or limb immobilisation (Martinez‐Marmol et al. 2007; Morel et al. 2010; Rannou et al. 2011). MC4R encodes the melanocortin 4 receptor (a G‐protein‐coupled receptor) (1.8‐fold less modification in lamb), which has been linked with obesity in several human studies but also is crucial for motor and sensory neuron regeneration after injury (Tanabe et al. 2007; Xi et al. 2012). GABRA5 encodes a ligand‐gated chloride channel [gamma‐aminobutyric acid (GABA) A receptor alpha 5] (10.55‐fold less modification in lamb). Specific ligands binding to GABRA5 mediate muscle relaxation (Milic et al. 2012). Some of the identified transcriptional regulators have roles in myofibre type specification (PRDM1) and fatty acid and glucose metabolism (PRDM1, MLXIPL, LIN28A) (Wang et al. 2011; Herman et al. 2012; Takeshita et al. 2012; Shinoda et al. 2013). The promyogenic transcription factors MYOD, MYF5, and PAX3 were not differentially modified. One possible exception was MYOD (P = 0.015; not significant after multiple testing correction, Padj = 0.32).
Table 2. DAVIDa functional annotation clusters for genes with promoters differentially modified by H3K27me3.
| Category | Term | Enrichment scoreb |
|---|---|---|
| Cluster 1 | ||
| SP_PIR_KEYWORDS | GO:0005886~plasma membrane | 3.1 |
| GOTERM_CC_FAT | Cell membrane | |
| SP_PIR_KEYWORDS | Membrane | |
| Cluster 2 | ||
| GOTERM_BP_FAT | GO:0003001~ signal involved in cell–cell signalling | 1.4 |
| GOTERM_BP_FAT | Signalling | |
| GOTERM_BP_FAT | GO:0046903~secretion | |
| GOTERM_BP_FAT | GO:0007267~cell–cell signalling | |
| Cluster 3 | ||
| GOTERM_CC_FAT | GO:0030054~cell junction | 1.1 |
| SP_PIR_KEYWORDS | Synapse | |
| GOTERM_CC_FAT | GO:0045202~synapse | |
| SP_PIR_KEYWORDS | Cell junction | |
DAVID functional annotation clustering (Huang da et al. 2009).
Enrichment scores ≥1.0 were considered significant. Full details are available in Table S5.
Table 3. Gene Ontology term enrichments for genes with differential H3K27me3 promoter modification between the foetal and lamb statesa.
| Term | P‐value |
|---|---|
| GO biological process | |
| GO:0003001~generation of a signal involved in cell–cell signalling | 0.01 |
| GO:0007610~behaviour | 0.03 |
| GO:0007264~small GTPase‐mediated signal transduction | 0.03 |
| GO:0045449~regulation of transcription | 0.03 |
| GO:0007242~intracellular signalling cascade | 0.04 |
| GO:0007517~muscle organ development | 0.04 |
| GO:0007214~gamma‐aminobutyric acid signalling pathway | 0.04 |
| GO cellular component | |
| GO:0005886~plasma membrane | 5.30E‐05 |
| GO:0044459~plasma membrane part | 0.01 |
| GO:0019898~extrinsic to membrane | 0.03 |
DAVID analysis. Full details are available in Table S5. There were no significant terms for GO molecular function.
Discussion
H3K27me3 is a repressive chromatin mark associated with a distinct group of approximately 4500 genes in foetal and lamb skeletal muscle. There are three types of modification, that is, (i) broad regional modification encompassing gene body and often multiple genes, (ii) promoter‐specific peaks and (iii) X chromosome inactivation in females. Stratification of H3K27me3 promoter density, reflecting differences between the first and second type of modification, strongly identified two gene function groups. The most intensively modified autosomal genes were nearly exclusively enriched for homeobox encoding transcription factors or pioneering transcriptional regulators important in early life development. This group likely represents genes that were transcriptionally active during very early development, but in the foetal and lamb states these genes were silenced by H3K27me3. The modification of these genes may present a barrier to their reactivation in skeletal muscle, thereby preventing differentiation into non‐mesodermal lineages. The second group is enriched for gated ion channels and G‐protein‐coupled receptors, often with neural functions. H3K27me3 modification in this group probably indicates the silencing of genes in skeletal muscle that are transcriptionally active in other tissues, particularly neural tissues. Notably, there are also examples of individual highly modified genes that were strongly transcribed and vice versa, suggesting that H3K27me3 is likely necessary but not sufficient for transcriptional repression (He et al. 2012).
Although stem cells and satellite cells (quiescent mononucleated myogenic cells) will be present in the samples, it is unlikely that they have major influence on the H3K27me3 signal due to their small numbers, and moreover, very similar H3K27me3 results were also obtained using adipose tissue and white blood cells (results not shown). One pattern of chromatin modification not investigated was related to ‘poised’ or bivalent chromatin typically found in embryonic stem cells, characterised by simultaneous modification with the repressive H3K27me3 mark and the activating H3K3me3 mark (Bernstein et al. 2006). Genes of this type are silent but marked for subsequent activation during cell commitment and differentiation. The skeletal muscle samples investigated in the current study used highly differentiated foetal and lamb muscle cells, and hence, it is unlikely that poised modification is a dominant feature of the H3K27me3 results for these late developmental stages.
During the muscle transition between late foetal gestation and lamb at 4 months of age, there are considerable gene expression and physiological changes (Byrne et al. 2010). In contrast, most H3K27me3 modifications, particularly the regional modification type, showed little change, indicating that H3K27me3 does not have a pivotal role in the late development of skeletal muscle. Hence, H3K27me3 is likely to be primarily identifying genes that were active in very early embryonic and early foetal development but are no longer required in the late gestational foetus or lamb or, alternatively, tissue‐specific genes silenced in skeletal muscle. For example, the promyogenic factors are functional in early embryogenesis, when there is considerable myogenesis, but not in the differentiated muscle cells present in the late gestation foetus and lamb muscles. It is likely therefore that these promyogenic transcription factors are silenced by H3K27me3 in both the foetus and lamb and are not involved in the myofibre fusion process occurring in this latter transition. The relatively small number of genes showing changed H3K27me3 promoter modification during the late foetal‐to‐lamb transition was enriched for gated ion channels, some of which have been implicated in skeletal muscle function. One possibility is that there is changing usage of the encoded proteins in developing neuromuscular junctions, which undergo structural and innervation changes in response to movement during this developmental transition. Another possibility is that these proteins may be involved in the process of myofibrillar hypertrophy and cell fusion accompanying the transition.
Conclusions
H3K27me3 is a repressive chromatin mark with striking associations with chromosome architecture and gene function. Stratification of H3K27me3 density in gene promoters distinguishes between different gene functions. H3K27me3 probably marks the silencing of genes that were active in very early development or the silencing of genes active in other tissues, particularly neural tissues. During the transition from late foetus to lamb, there are marked changes in the structure and function of skeletal muscle but few changes in H3K27me3 gene promoter modification. The identified changes suggest the involvement of genes contributing to neuromuscular junctions and aspects of muscle hypertrophy in response to locomotion in the lamb.
Supplementary Material
Figure S1. Mononucleosomal DNA obtained from lamb longissimus lumborum muscle.
Figure S2. Regional H3K27me3 enrichment at four HOX gene loci.
Figure S3. Hilbert curve visualisation of chromosome specific H3K27me3 profiles for the fetal and lamb samples.
Table S1. Processing statistics for each sequenced library.
Table S2. Foetal H3K27me3 peaks.
Table S3. Lamb H3K27me3 peaks.
Table S4. DAVID functional analysis of RefSeq genes with promoters enriched with H3K27me3.
Table S5. Gene promoters with differential H3K27me3 read counts in the fetal and lamb developmental states.
Appendix S1. Supplemental materials and methods.
Acknowledgements
We are grateful to Tracy Hadfield and Dave Forrester (Utah State University) for sample collection, Weiwei Deng (CSIRO) for advice on ChIP‐Seq methods, Wes Barris and Vicki Whan (CSIRO) for advice regarding bioinformatics and the statistical package r. The research was supported by CSIRO Transformational Biology.
References
- Bernstein B.E., Mikkelsen T.S., Xie X.H.et al (2006) A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–26. [DOI] [PubMed] [Google Scholar]
- Bernstein B.E., Meissner A. & Lander E.S. (2007) The mammalian epigenome. Cell 128, 669–81. [DOI] [PubMed] [Google Scholar]
- Bock I., Dhayalan A., Kudithipudi S., Brandt O., Rathert P. & Jeltsch A. (2010) Detailed specificity analysis of antibodies binding to modified histone tails with peptide arrays. Epigenetics, 6, 256–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet M., Cassar‐Malek I., Chilliard Y. & Picard B. (2010) Ontogenesis of muscle and adipose tissues and their interactions in ruminants and other species. Animal 4, 1093–109. [DOI] [PubMed] [Google Scholar]
- Bovine Genome Sequencing and Analysis Consortium , Elsik C.G. & Tellam R.L. (2009) The genome sequence of taurine cattle: a window to ruminant biology and evolution, Science, 324, 522–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer L.A., Plath K., Zeitlinger J.et al (2006) Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441, 349–53. [DOI] [PubMed] [Google Scholar]
- Byrne K., Vuocolo T., Gondro C., White J.D., Cockett N.E., Hadfield T., Bidwell C.A., Waddell J.N. & Tellam R.L. (2010) A gene network switch enhances the oxidative capacity of ovine skeletal muscle during late fetal development. BMC Genomics 11, 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- clc bio . CLC Genomics Workbench 6. URL http://www.clcbio.com.
- Egelhofer T.A., Minoda A., Klugman S.et al (2011) An assessment of histone‐modification antibody quality. Nature Structural & Molecular Biology 18, 91–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escamilla‐Del‐Arenal M., da Rocha S.T. & Heard E. (2011) Evolutionary diversity and developmental regulation of X‐chromosome inactivation. Human Genetics 130, 307–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg A.P. (2010) Epigenomics reveals a functional genome anatomy and a new approach to common disease. Nature Biotechnology 28, 1049–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg A.P. & Irizarry R.A. (2010) Evolution in health and medicine Sackler colloquium: stochastic epigenetic variation as a driving force of development, evolutionary adaptation, and disease. Proceedings of the National Academy of Sciences of the United States of America 107(Suppl 1), 1757–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming‐Waddell J.N., Wilson L.M., Olbricht G.R., Vuocolo T., Byrne K., Craig B.A., Tellam R.L., Cockett N.E. & Bidwell C.A. (2007) Analysis of gene expression during the onset of muscle hypertrophy in callipyge lambs. Animal Genetics 38, 28–36. [DOI] [PubMed] [Google Scholar]
- He Y., Yu Y., Zhang Y., Song J., Mitra A., Zhang Y., Wang Y., Sun D. & Zhang S. (2012) Genome‐wide bovine H3K27me3 modifications and the regulatory effects on genes expressions in peripheral blood lymphocytes. PLoS ONE 7, e39094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman M.A., Peroni O.D., Villoria J., Schon M.R., Abumrad N.A., Bluher M., Klein S. & Kahn B.B. (2012) A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 484, 333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W., Sherman B.T. & Lempicki R.A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Hughes A. & Rando O.J. (2009) Chromatin ‘programming’ by sequence ‐ is there more to the nucleosome code than %GC? Journal of Biology 8, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H., Jiang H., Ma W., Johnson D.S., Myers R.M. & Wong W.H. (2008) An integrated software system for analyzing ChIP‐chip and ChIP‐seq data. Nature Biotechnology 26, 1293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King W.A., Coppola G., Pinton A., Joudrey E.M. & Basrur P.K. (2008) Spatial distribution of histone isoforms on the bovine active and inactive x chromosomes. Sexual Development 2, 12–23. [DOI] [PubMed] [Google Scholar]
- Kollias H.D. & McDermott J.C. (2008) Transforming growth factor‐beta and myostatin signaling in skeletal muscle. Journal of Applied Physiology 104, 579–87. [DOI] [PubMed] [Google Scholar]
- Luger K., Mader A.W., Richmond R.K., Sargent D.F. & Richmond T.J. (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–60. [DOI] [PubMed] [Google Scholar]
- Martinez‐Marmol R., David M., Sanches R.et al (2007) Voltage‐dependent Na+ channel phenotype changes in myoblasts. Consequences for cardiac repair. Cardiovascular Research 76, 430–41. [DOI] [PubMed] [Google Scholar]
- Mikkelsen T.S., Ku M., Jaffe D.B.et al (2007) Genome‐wide maps of chromatin state in pluripotent and lineage‐committed cells. Nature 448, 553–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milic M., Divljakovic J., Rallapalli S., van Linn M.L., Timic T., Cook J.M. & Savic M.M. (2012) The role of alpha1 and alpha5 subunit‐containing GABAA receptors in motor impairment induced by benzodiazepines in rats. Behavioural Pharmacology 23, 191–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel J., Rannou F., Talarmin H., Giroux‐Metges M.A., Pennec J.P., Dorange G. & Gueret G. (2010) Sodium channel Na(V)1.5 expression is enhanced in cultured adult rat skeletal muscle fibers. Journal of Membrane Biology 235, 109–19. [DOI] [PubMed] [Google Scholar]
- Otto A., Schmidt C., Luke G., Allen S., Valasek P., Muntoni F., Lawrence‐Watt D. & Patel K. (2008) Canonical Wnt signalling induces satellite‐cell proliferation during adult skeletal muscle regeneration. Journal of Cell Science 121, 2939–50. [DOI] [PubMed] [Google Scholar]
- Pagani R., Song M., McEnery M., Qin N., Tsien R.W., Toro L., Stefani E. & Uchitel O.D. (2004) Differential expression of alpha 1 and beta subunits of voltage dependent Ca2 + channel at the neuromuscular junction of normal and P/Q Ca2 + channel knockout mouse. Neuroscience 123, 75–85. [DOI] [PubMed] [Google Scholar]
- Pauler F.M., Sloane M.A., Huang R., Regha K., Koerner M.V., Tamir I., Sommer A., Aszodi A., Jenuwein T. & Barlow D.P. (2009) H3K27me3 forms BLOCs over silent genes and intergenic regions and specifies a histone banding pattern on a mouse autosomal chromosome. Genome Research 19, 221–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rannou F., Pennec J.P., Morel J., Gueret G., Leschiera R., Droguet M., Gioux M. & Giroux‐Metges M.A. (2011) Na v1.4 and Na v1.5 are modulated differently during muscle immobilization and contractile phenotype conversion. Journal of Applied Physiology 111, 495–507. [DOI] [PubMed] [Google Scholar]
- Ringrose L. & Paro R. (2007) Polycomb/Trithorax response elements and epigenetic memory of cell identity. Development 134, 223–32. [DOI] [PubMed] [Google Scholar]
- Rinn J.L., Kertesz M., Wang J.K.et al (2007) Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129, 1311–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettengruber B., Chourrout D., Vervoort M., Leblanc B. & Cavalli G. (2007) Genome regulation by polycomb and trithorax proteins. Cell 128, 735–45. [DOI] [PubMed] [Google Scholar]
- Shinoda G., Shyh‐Chang N., de Soysa T.Y.et al (2013) Fetal deficiency of Lin28 Programs life‐long aberrations in growth and glucose metabolism. Stem Cells 31, 1563–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparmann A. & van Lohuizen M. (2006) Polycomb silencers control cell fate, development and cancer. Nature Reviews Cancer 6, 846–56. [DOI] [PubMed] [Google Scholar]
- Stickland N.C. (1978) A quantitative study of muscle development in the bovine foetus (Bos indicus). Anatomia Histologia and Embryologia 7, 193–205. [DOI] [PubMed] [Google Scholar]
- Straface G., Aprahamian T., Flex A.et al (2009) Sonic hedgehog regulates angiogenesis and myogenesis during post‐natal skeletal muscle regeneration. Journal of Cellular and Molecular Medicine 13, 2424–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanyam P., Obermair G.J., Baumgartner S., Gebhart M., Striessnig J., Kaufmann W.A., Geley S. & Flucher B.E. (2009) Activity and calcium regulate nuclear targeting of the calcium channel beta4b subunit in nerve and muscle cells. Channels (Austin, TX) 3, 343–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita S., Suzuki T., Kitayama S., Moritani M., Inoue H. & Itakura M. (2012) Bhlhe40, a potential diabetic modifier gene on Dbm1 locus, negatively controls myocyte fatty acid oxidation. Genes & Genetic Systems 87, 253–64. [DOI] [PubMed] [Google Scholar]
- Tanabe K., Gamo K., Aoki S., Wada K. & Kiyama H. (2007) Melanocortin receptor 4 is induced in nerve‐injured motor and sensory neurons of mouse. Journal of Neurochemistry 101, 1145–52. [DOI] [PubMed] [Google Scholar]
- Tellam R.L., Lemay D.G., Van Tassell C.P., Lewin H.A., Worley K.C. & Elsik C.G. (2009) Unlocking the bovine genome. BMC Genomics 10, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuocolo T., Byrne K., White J., McWilliam S., Reverter A., Cockett N.E. & Tellam R.L. (2007) Identification of a gene network contributing to hypertrophy in callipyge skeletal muscle. Physiological Genomics 28, 253–72. [DOI] [PubMed] [Google Scholar]
- Wagschal A., Delaval K., Pannetier M., Arnaud P. & Feil R. (2007) Chromatin immunoprecipitation (ChIP) on unfixed chromatin from cells and tissues to analyze histone modifications. CSH Protocols 2007, pdb.prot4767. [DOI] [PubMed] [Google Scholar]
- Wang K.C., Helms J.A. & Chang H.Y. (2009) Regeneration, repair and remembering identity: the three Rs of Hox gene expression. Trends in Cell Biology 19, 268–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Ono Y., Tan S.C., Chai R.J., Parkin C. & Ingham P.W. (2011) Prdm1a and miR‐499 act sequentially to restrict Sox6 activity to the fast‐twitch muscle lineage in the zebrafish embryo. Development 138, 4399–404. [DOI] [PubMed] [Google Scholar]
- Xi B., Chandak G.R., Shen Y., Wang Q. & Zhou D. (2012) Association between common polymorphism near the MC4R gene and obesity risk: a systematic review and meta‐analysis. PLoS ONE 7, e45731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou V.W., Goren A. & Bernstein B.E. (2011a) Charting histone modifications and the functional organization of mammalian genomes. Nature Reviews Genetics 12, 7–18. [DOI] [PubMed] [Google Scholar]
- Zhou X., Maricque B., Xie M.et al (2011b) The human epigenome browser at Washington University. Nature Methods 8, 989–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Mononucleosomal DNA obtained from lamb longissimus lumborum muscle.
Figure S2. Regional H3K27me3 enrichment at four HOX gene loci.
Figure S3. Hilbert curve visualisation of chromosome specific H3K27me3 profiles for the fetal and lamb samples.
Table S1. Processing statistics for each sequenced library.
Table S2. Foetal H3K27me3 peaks.
Table S3. Lamb H3K27me3 peaks.
Table S4. DAVID functional analysis of RefSeq genes with promoters enriched with H3K27me3.
Table S5. Gene promoters with differential H3K27me3 read counts in the fetal and lamb developmental states.
Appendix S1. Supplemental materials and methods.