

Abstract
De novo point mutations and chromosomal structural aberrations (CSA) detected in offspring of unaffected parents show a preferential paternal origin with higher risk for older fathers. Studies in rodents suggest that heritable mutations transmitted from the father can arise from either paternal or maternal misrepair of damaged paternal DNA, and that the entire spermatogenic cycle can be at risk after mutagenic exposure. Understanding the susceptibility and mechanisms of transmission of paternal mutations is important in family planning after chemotherapy and donor selection for assisted reproduction. We report that treatment of male mice with melphalan (MLP), a bifunctional alkylating agent widely used in chemotherapy, induces DNA lesions during male mouse meiosis that persist unrepaired as germ cells progress through DNA repair-competent phases of spermatogenic development. After fertilization, unrepaired sperm DNA lesions are mis-repaired into CSA by the egg's DNA repair machinery producing chromosomally abnormal offspring. These findings highlight the importance of both pre- and post-fertilization DNA repair in assuring the genomic integrity of the conceptus.
Advances in cancer therapies have dramatically improved the survival rates of afflicted children and young adults1. Successful cures into the reproductive years increase the opportunity for transmitting chemotherapy-induced germ-cell mutations resulting in pregnancy abnormalities and inherited diseases in the offspring of cancer survivors2. High fidelity DNA repair in germ cells is important for maintaining genetic integrity and for limiting the transmission of de novo mutations and chromosomal structural aberrations (CSA) to the offspring3,4. Animal studies have shown that DNA damage transmitted via sperm leads to a broad spectrum of abnormal reproductive outcomes including pregnancy loss, birth defects, and offspring with chromosomal abnormalities, neurological diseases, cancer and other genetic diseases5,6,7. Understanding the mechanisms of induction of DNA lesions in germ cells by chemotherapeutic agents and the role of DNA repair during gametogenesis and after fertilization is particularly important for the male germ line since de novo mutations and CSA show a preferential paternal origin and increase with the age of the father8,9,10.
DNA repair during spermatogenesis and maternal DNA repair in the zygote have distinctly different roles in how sperm DNA damage is repaired or misrepaired into de novo mutations and CSA in the zygote6,11. DNA repair is highly effective during male germ-cell mitosis and meiosis12, but the ability to repair DNA lesions declines dramatically towards the latter part of spermatogenesis4,12,13 as haploid spermatids undergo major nuclear chromatin remodelling into highly compacted sperm nuclei14,15. The last few weeks of sperm development prior to fertilization (~3 weeks in humans and ~2 weeks in mice) are highly susceptible to the accumulation of sperm DNA damage6,16 and damaged DNA is transmitted unrepaired into the egg where it is subject to maternal DNA repair machinery. Thus, errors in maternal DNA repair of sperm lesions result in zygotes with CSA16.
Mammalian oogenesis does not have a repair deficient window like spermatogenesis, and female germ cells are capable of repairing DNA damage throughout oogenesis. The egg genome codes for maternal DNA repair machinery to detect and initiate the repair of DNA damage of the fertilizing sperm within minutes after the sperm nucleus undergoes transformation into the male pronucleus17,18. Studies in mice have shown that the efficiency of maternal DNA repair of sperm lesions varies over 10 fold across females from different inbred stains19. Furthermore, genetic disruption of maternal DNA double strand break repair pathways in the zygote altered the repair of postmeiotic sperm DNA lesions, resulting in significant increases in both chromosome- and chromatid-type aberrations in the paternal genome at first cleavage metaphase20,21. These findings provided compelling evidence that CSA in paternal chromosomes are formed after fertilization rather than before and that both pre- and post-S phase repair mechanisms are operating in the zygote. Prior studies from our laboratory also suggest that zygotes with unbalanced CSA result in dead implants, and zygotes with balanced CSA produce offspring with reciprocal translocations22. These studies suggest that the risk of paternal transmission of heritable chromosomal damage depends on (a) susceptibility of the spermatogenic cell types at the time of paternal exposure, (b) the efficacy of paternal DNA repair, and (c) the efficacy of maternal DNA repair of remaining sperm lesions in the zygote.
Melphalan (MLP) is a nitrogen mustard that is commonly used, alone or in combination with other chemotherapeutic drugs, in the treatment of breast and ovarian cancers and multiple myelomas23. MLP is a direct-acting bifunctional alkylating agent that is carcinogenic in both humans and rodents23 and forms a variety of DNA adducts including interstrand crosslinks (ICLs) and several mono-adducts at guanine and adenine24,25. Unlike other nitrogen mustards, MLP induces very few intrastrand crosslinks26. MLP is an established male germ cell mutagen and an efficient inducer of mutations transmitted after exposure of male meiotic cells27,28,29. We used a mouse model to investigate the induction and transmission of CSA after paternal exposure to MLP during meiotic and post-meiotic windows of sperm development. We hypothesized that MLP lesions induced during paternal meiosis would be converted to CSA by meiotic repair, while MLP lesions induced during the repair-deficient window of spermatogenesis would be converted to CSA after fertilization by maternal DNA repair in the zygote. However, our findings point to the alternative hypothesis that exposure to MLP during male meiosis induced DNA lesions that persist in an unrepaired state as exposed germ cells progress through the normal DNA-repair-competent windows of meiosis, meiotic chromosome segregation and post-meiosis. After fertilization these unrepaired lesions in sperm DNA are mis-repaired into CSA by the female DNA repair machinery in the zygote.
Results
We used three mouse cytogenetic assays (spermatocyte metaphase cytogenetics, sperm CT8 FISH, and zygote cytogenetics) to characterize the differential sensitivity of premeiotic, meiotic and postmeiotic male germ cells to paternal MLP exposures. Our study design enabled us to determine whether MLP-induced DNA damage was converted into CSA before the end of meiosis, during postmeiosis, or after fertilization.
MLP induces paternal chromosomal damage at first-cleavage zygote metaphase after both meiotic and post-meiotic exposures
Male mice were treated with MLP and mated with untreated female at different times after exposure to investigate the effects of MLP on progressively less mature male germ cells. The percentages of successful matings were significantly (P < 0.001) reduced during the first week after exposure (Table 1). However, neither the percent of fertilized eggs nor the percent of fertilized eggs that reached the zygotic metaphase stage were significantly affected by MLP exposure at any of the time points investigated (data not shown). Despite the lack of effects on fertilization, paternal exposure to MLP significantly increased the frequencies of zygotes with CSA of paternal origin in matings that sampled meiotic and post-meiotic exposed germ cells, although the magnitude of the effect varied across time points. Specifically, MLP treatment of diplotene spermatocytes (day 23) yielded the highest frequency of zygotes with CSA (64.2%, 58-fold over sham controls, P < 0.0001, Table 1, Figure 1F–H) and the highest amount of chromosomal damage per zygote (1.7 CSA per metaphase, Table 1). Elevated frequencies of zygotes with CSA with respect to controls were also observed after MLP treatment of epididymal sperm (day 3, 10-fold, P < 0.001), testicular sperm (day 7, 16-fold, P < 0.001) and dividing spermatogonia (day 37, 5-fold, P < 0.01, Chi-Square). However, CSA in zygotes were not increased (P = 0.9) after treatment of spermatogonial stem cells (day 49).
Table 1. Chromosome structural aberrations (CSA) in first-cleavage (1-Cl) zygotes after exposure of male mice to 7.5 mg/kg MLP.
| DAPI Analysisc | PAINT Analysisc | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Treatment daya | No of. miceb | Stage at treatment | No. of zygotes analyzed | No. of zygotes with CSA | % ± S.E. | Chromosome-type | Chromatid-type | Cell equivalents | Total |
| Controls | 45 (91) | 282 | 3 | 1.1 ± 0.6 | 0.014 ± 0.008 | 0 | 166 | 0.018 ± 0.016 | |
| 3 | 30 (60*) | Epidydimal sperm | 139 | 15 | 10.8 ± 0.6* | 0.237 ± 0.153 | 0.007 ± 0.008 | 82 | 0.219 ± 0.096 |
| 7 | 53 (57*) | Testicular sperm | 230 | 41 | 17.8 ± 2.0# | 0.426 ± 0.124 | 0.004 ± 0.005 | 135 | 0.488 ± 0.179 |
| 23 | 52 (81) | Diplotene spermatocytes | 293 | 188 | 64.2 ± 3.8# | 1.683 ± 0.158 | 0.007 ± 0.007 | 170 | 1.929 ± 0.207 |
| 37 | 27 (89) | Dividing spermatogonia | 216 | 12 | 5.6 ± 1.1§ | 0.093 ± 0.008 | 0 | 127 | 0.064 ± 0.013 |
| 49 | 37 (92) | Stem cells | 167 | 2 | 1.2 ± 0.6 | 0.006 ± 0.007 | 0.006 ± 0.005 | 93 | 0 |
aPrior to mating.
bPercent of matings in parenthesis.
cNumber of CSA per metaphase (mean ± Standard Error).
*p < 0.001 versus controls (Chi-square).
#p < 0.0001 versus controls (Chi-square).
§p < 0.05 versus controls (Chi-square).
Figure 1. Photomicrographs of mouse spermatocytes, sperm and zygotes.
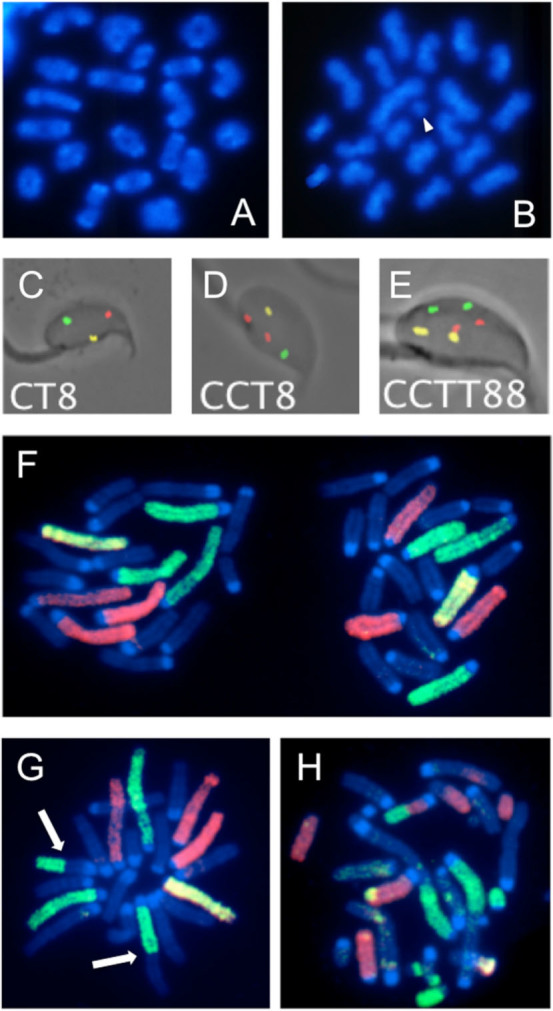
(A–C) Mouse MI and MII spermatocytes after DAPI staining. (A) Normal MI spermatocyte. (B) MII spermatocyte with on chromatid fragment (arrowhead). (C–E). Mouse sperm after hybridization with probe specific for the centromeric region of chromosome 2 labeled with FITC, the telomeric region of chromosome 2 labeled with rhodamine, and the subcentromeric region of chromosome 8 labeled with rhodamine and FITC. (C) Normal mouse sperm showing three spots each of a different color. (D). Sperm with the duplication of the centromeric region of chromosome 2 (two red spots). (E) Diploid sperm (two spots for each of the three probes). (F–h) Mouse zygotes after hybridization with chromosome-specific painting probes for chromosomes 1, 3, 5, X and Y labeled with FITC and chromosomes 2, 4, 6, X and Y labeled with biotin and signaled with Texas red. (F) Normal 1-Cl zygote metaphase with X-bearing sperm-derived chromosomes on the left. (G) Paternal chromosomes from a 1-Cl zygote showing a reciprocal translocation (arrows) involving one chromosome painted in green and one unpainted chromosome. (H) Paternal chromosome from a 1-Cl zygote with extensive chromosomal damage involving painted and unpainted chromosomes.
Chromosome-type aberrations represented most of the aberrations detected at zygotic metaphase (Table 1) regardless of the germ cell stage that was exposed to MLP, and chromatid-type aberrations were seldom observed. CSA were most frequent in the day 23 mating group (total number of CSA per zygotes was ~1.7–1.9 by DAPI and PAINT analyses). Acentric fragments were the most common CSA in all mating groups (by both staining methods), except at 23 days, when PAINT analysis showed that MLP exposure of diplotene spermatocytes resulted in higher rates of reciprocal translocations (supplemental Table 1).
Chromosomal aberrations in zygotes predict dominant lethality and heritable translocations
We previously reported a significant association between the frequencies of zygotes with unstable and stable aberrations and the frequencies of dead implants (dominant lethality, DL) and of live offspring with reciprocal translocations (heritable translocations, HT), respectively6,22. We now report that this prediction is also confirmed for zygotes with stable and unstable CSA after paternal exposure to MLP. We found a significant association (r = 0.977; P < 0.008) between the frequencies of zygotes with unstable aberrations and DL and that MLP induced DL throughout spermatogenesis with a peak induction in round spermatids and diplotene spermatocytes (Figure 2).
Figure 2. Comparison of the proportions of zygotes with unstable chromosomal aberrations (gray columns) vs proportions of dead implants (white columns).
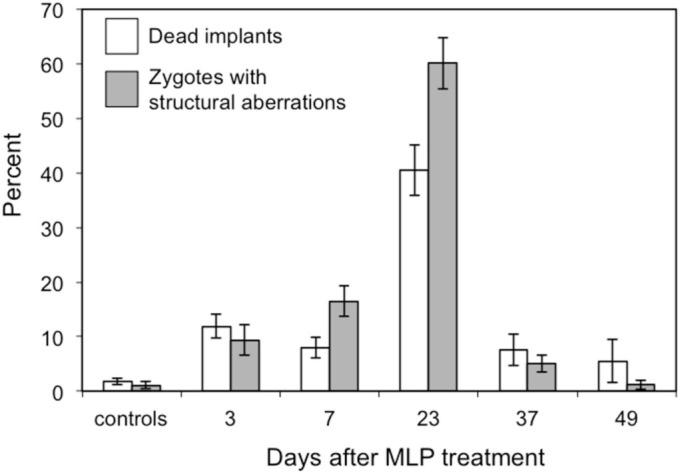
Bars represent the standard error.
Prior HT data showed that the frequencies of offspring with reciprocal translocations were 21.2% and 19.8% in matings occurring at 15–18 and 21–24 days, respectively29. At 23 days (diplotene exposure), we analyzed 170 zygotes by PAINT (Table 1), which corresponds to analyzing the complete genomes of 50 offspring when adjusted for the percentage of the genome painted by our probe combination and the number of zygotes with unstable aberrations that are expected to die in utero. Among these 50 genomes, we found 15 with reciprocal translocations for a HT frequency of 30.0 ± 6.5 (% ± S.E). Thus, our predicted HT frequency is not different (P = 0.15) from that observed in the standard HT test. Similar analyses conducted at the other mating times investigated in this study predict that MLP does not induce HT in the other germ cell types (data not shown).
Chromosomal aberrations are not observed in meiotic metaphases after exposure of diplotene spermatocytes
Next, we tested the hypothesis that lesions induced during meiosis would result in CSA at meiotic metaphase. MI and MII spermatocytes were analyzed 48 hr after exposure of male mice to MLP. This time point corresponds to the analysis of germ cells exposed during the diplotene phase of meiosis and allows a direct comparison with the analysis of zygotes at day 23 as both assays measure effects in cells that were at the same stage of spermatogenesis at the time of exposure. At variance with the high frequency of zygotes with CSA after exposure of diplotene spermatocytes (Table 1), the analysis of MI and MII spermatocytes failed to detect an increase in the frequencies of CSA in spermatocytes. Analysis of 227 MI spermatocytes identified only 6 metaphases (2.6 ± 1.5% versus 1.5 ± 0.7% in controls, P = 0.49) with CSA versus the expected 146 based on the frequency of zygotes with CSA at day 23 (64.2%). All aberrations at MI consisted of chromatid breaks. However, chromatid exchanges, the type of aberration expected to result in reciprocal translocations, were not observed. Similarly, the analysis of 139 MII metaphases showed only 3 (2.2 ± 1.1% versus 1.3 ± 0.7%, P = 0.75) metaphases with CSA, while 69 were predicted based on the results in zygotes. All CSA detected in MII spermatocytes consisted of chromatid breaks. Although we cannot exclude that some heavily damaged spermatocytes may be eliminated by apoptosis before reaching the metaphase stage, our findings suggest that exposure of male mice to MLP induced DNA lesions in diplotene cells that were not detected as CSA at the subsequent MI and MII metaphases, but were detected as CSA at the first zygotic metaphase after fertilization.
Chromosomal duplications and deletions are not observed in mature sperm
To confirm that reciprocal translocations were not induced during meiosis, we used the mouse sperm CT8 assay30 to investigate the frequencies of mature sperm with duplications and deletions, which are predicted to increase when meiotic reciprocal translocations are present in spermatocytes30. As shown in Table 2, no significant differences were found in the frequencies of sperm with structural or numerical abnormalities between MLP exposed mice and concurrent sham controls. Extrapolation of the sperm CT8 data for duplications and deletions across the entire genome, assuming that chromosome 2 represents ~6.4% of the male genome31, generated an overall frequencies of sperm with chromosomal structural aberrations of 0.7%. This frequency is clearly lower that the 64% of zygotes with CSA after mating of males 23 days after MLP exposure. Taken together with the results of meiotic cytogenetics, the CT8 assay findings show that MLP exposure during meiosis induced DNA lesions that are not converted into CSA in the time window between diplotene and fertilization.
Table 2. Frequencies of sperm with structural and numerical abnormalities in B6C3F1 mice after melphalan treatment of meiotic cellsa.
| Genotype | Fluorescent pattern | Controlsb | Melphalanb | P value |
|---|---|---|---|---|
| No. Mice | 3 | 4 | ||
| No. cells scored | 30,288 | 40,208 | ||
| Sperm with structural aberrations | ||||
| Centromeric duplication | CCT8 | 0.0 | 1.2 ± 0.5 | 0.05 |
| Centromeric deletion | OT8c | 0.0 | 1.0 ± 1.0 | 0.08 |
| Telomeric duplication | CTT8 | 2.6 ± 0.7 | 1.5 ± 1.2 | 0.28 |
| Telomeric deletion | CO8 | 3.0 ± 1.0 | 1.0 ± 0.7 | 0.06 |
| Total Structural | 5.6 ± 1.5 | 4.7 ± 1.5 | 0.61 | |
| Sperm with numerical abnormalities | ||||
| Disomy 2 | CCTT8 | 1.0 ± 0.0 | 0.7 ± 0.5 | 0.73 |
| Nullisomy 2 | OO8 | 1.0 ± 0.0 | 3.0 ± 0.8 | 0.07 |
| Disomy 8 | CT88 | 1.7 ± 0.7 | 3.7 ± 0.5 | 0.11 |
| Nullisomy 8 | CTO | 2.0 ± 1.0 | 2.5 ± 1.7 | 0.66 |
| Total | 5.6 ± 1.5 | 9.9 ± 1.5 | 0.04 | |
| Diploid sperm | CCTT88 | 6.6 ± 4.2 | 10.7 ± 2.2 | 0.07 |
| Sperm with complex abnormalities | 1.0 ± 0.6 | 0.7 ± 0.5 | 0.73 |
a23 days after treatment with melphalan.
bFrequencies per 10,000 sperm ± Standard Error.
c“O” indicates the absence of an expected signal.
Discussion
We report the unexpected finding that paternal exposure to MLP induced DNA damage in male meiotic cells that persisted unrepaired for three weeks as these cells transitioned through windows of spermatogenic development that are known to be DNA repair competent. These persisting lesions in fertilizing sperm are converted into CSA in paternal chromosomes by the maternal DNA repair machinery in the zygote. Furthermore, we report that: (i) paternal exposure to MLP produced zygotes carrying CSA with a peak of sensitivity after exposures to diplotene spermatocytes, with significant elevation after exposures to dividing spermatogonia and sperm, but not stem-cell spermatogonia; (ii) CSA were not detected in meiotic metaphases or mature sperm after exposure of diplotene spermatocytes; and (iii) reciprocal translocations in zygotes were induced only after exposure of meiotic cells. The significant increase in CSA after exposure of dividing spermatogonia suggest that chemotherapies containing MLP may put individuals at a higher risk for abnormal reproductive outcomes for over 3 months after the end of therapy.
Our findings highlight the exquisite sensitivity of meiotic germ cells to MLP and confirm the chromosomal origin of the embryonic lethality observed in DL studies (supplementary Figure S1). MLP is one of the few chemicals that affect male meiotic cells6,32. The present time course study of MLP-induced effects in male germ cells showed a peak of sensitivity in diplotene spermatocytes when over 60% of analyzed zygotes presented with paternally-transmitted CSA. These results are in good agreement with the known sensitivity of the various germ cell types to MLP and strengthen the correlation between the presence of CSA in zygotes and embryonic fate20,22. As shown in Figure 2, the frequencies of predicted embryonic lethality based on the frequencies of zygotes with unstable aberrations showed a good correlation, in both magnitude and time course, with the findings of the standard DL study29. Also, the analysis of reciprocal translocations in zygotes confirmed the findings of the HT test that MLP induced high frequencies of reciprocal translocations after exposure of diplotene spermatocyte29. Our results further predict that reciprocal translocations would not be induced in the other spermatogenic cell types tested.
Stem cell exposure did not significantly increase the frequencies of zygotes with CSA. This finding may seem at variance with the reported paternal age effect for the induction of de novo mutations9. It is possible that the paternal age effect is limited to point mutations rather than gross chromosomal abnormalities, which may affect survival of affected stem cells more than point mutations. More importantly, the sample size analyzed in our study is too small to observe a significant effect after stem cell exposure, especially for detecting reciprocal translocations, which would not be expected to affect stem cell survival.
Despite the extensive chromosomal damage detected in zygotes originated from treated diplotene spermatocytes, direct analysis of CSA in MI and MII spermatocytes failed to detect the presence of chromosomal damage in these cells. Similarly, despite the fact that PAINT analysis showed the presence of reciprocal translocations in 30% of zygotes, neither the cytogenetic analysis of spermatocyte metaphases nor sperm FISH detected reciprocal translocations after diplotene exposure. These would have manifested as chains in spermatocyte metaphases and as an increase in sperm with duplications and deficiencies in the CT8 assay30. These observations strongly suggest that the chromosomal damage observed in zygotes fertilized by sperm exposed to MLP as diplotene spermatocytes was created after fertilization. The surprising conclusion is that MLP-induced DNA damage in diplotene spermatocytes goes unrepaired during the two meiotic divisions and the three weeks of postmeiotic development that elapsed between exposure and fertilization. Only after fertilization, as the sperm undergoes remodeling into the male pronucleus, is the damage detected by the egg's repair machinery and its misrepair results in its fixation into CSA during the first round of DNA replication. Thus, both meiotic and postmeiotic MLP-induced DNA damage is transmitted as such by the sperm and is converted into CSA by maternal misrepair after fertilization.
The high incidence of reciprocal translocations in zygotes after diplotene exposure to MLP suggests that ICLs may represent the unrepaired DNA lesions that are transmitted by the sperm. Similarly to other cross-linking agents, ICLs compose only a small portion of the adducts formed by MLP33. However, they are considered the major determinants of the cytotoxicity of these anticancer drugs. ICLs interfere with DNA replication and transcription by preventing DNA strand separation33,34 and their repair require the coordinated interplay of multiple DNA repair pathways35. ICL misrepair results in high frequencies of chromosomal exchanges as demonstrated by in vitro and in vivo studies with systems that are deficient in the repair of this type of damage, such as the Fanconi Anemia pathway34,36. In mammalian cells, ICL repair occurs mostly during DNA replication and transcription at stalled replication forks33,34,35,37,38 and removal of MLP-induced ICLs appears to follow slow kinetics of repair in vivo39. Therefore, the repair of MLP-induced ICLs during spermatogenesis is expected to require DNA replication in order to occur. Consistent with this requirement, DNA repair proteins that are involved in ICLs tend to be highly expressed in spermatogonia and early spermatocytes40. Conversely, DNA repair mechanisms for MLP monoadducts, such as the nucleotide excision repair, are not dependent on DNA replication and operate throughout the cell cycle. In addition, these repair mechanisms are active during early postmeiosis4.
Figure 3 shows our working model for the induction of MLP lesions during spermatogenesis and whether these lesions are fixed into CSA before or after fertilization. The model takes into account that: (i) the last round of DNA synthesis occurs in B spermatogonia41; (ii) DNA repair declines in elongating spermatids4; and, (iii) histones are replaced by protamines in condensed spermatids14,15. The model further considers that the ability of MLP to induce ICLs is progressively reduced as protamines replace histones and compact the DNA. We propose that MLP-induced ICLs escape detection in diplotene spermatocytes and throughout postmeiois because the lack of DNA replication in these germ cell types does not provide an opportunity for the male germ cell repair machinery to detect and repair them. ICLs are then transmitted as such to the zygote and the egg's attempt to repair them results in the formation of CSA following fertilization, including the high frequencies of reciprocal translocations that are the hallmark of ICL misrepair36. We further propose that a similar mechanism can be invoked for the high sensitivity of early spermatids to the induction of reciprocal translocations after MLP exposure29. As postmeiotic germ cells progress through spermiogenesis, they replace histones with protamines and compact the DNA into the highly condensed state that is typical of maturing sperm14,15. This morphological change in the nature of the sperm chromatin limits the formation of ICLs after exposure to MLP in these more developmentally advanced postmeiotic germ cells and reduces the chance of forming reciprocal translocations following fertilization. Thus, the sensitivity of diplotene spermatocytes and early spermatids to the induction of reciprocal translocation29 is due to common cellular characteristics that include chromatin conformation and lack of DNA replication.
Figure 3. Working model for the induction of MLP lesions during spermatogenesis.

Schematic of mammalian spermatogenesis (on the left) with the mating times (days) used in this study and the first cell cycle in the zygote (on the right). The approximate periods of DNA replication, DNA repair efficiency, protamine deposition, MLP-induced lesions, and maternal DNA repair are shown. See text for further explanation. As: A single; Ap: A pair; Aal: A aligned; In: Intermediate; Pl: preleptotene; L: leptotene; Z: zygotene; P: pachytene; D: Diplotene; MI: metaphase I; MII: metaphase II; E: elongating; Cg: condensing; Cd: condensed.
Additional support for our hypothesis that meiotic ICLs represent the unrepaired sperm DNA damage responsible for the formation of reciprocal translocations in zygotes comes from studies in oogenesis, which demonstrate that DNA double strand breaks, but not ICLs, prevent progression through meiosis of fully-grown mouse oocytes42. It is of relevance to the proposed mechanism for the induction of CSA following exposure to MLP that, similarly to diplotene spermatocytes and spermatids, fully-grown oocytes are characterized by a lack of DNA synthesis. Together, these studies show that ICL damage sustained during gametogenesis is a type of DNA damage whose repair requires DNA replication. Therefore, ICL damage induced during the post DNA replication phases of spermatogenesis (which includes meiosis and post-meiosis) can persist for several weeks and has a high chance of being transmitted to the zygote where it manifests itself as CSA.
The results of this study are dramatically different from those observed with etoposide43, another chemotherapeutic agent that targets meiotic germ cells (supplementary Figure S2). Exposure of male meiotic germ cells with etoposide induces CSA in zygotes43. However, at variance with the present study, CSA and duplications and deficiencies were observed at high frequencies in both MI/MII spermatocytes43 and sperm44, respectively. Unlike MLP, etoposide induces mostly double strand breaks through interference with the ligation function of topoisomerase II45. As topoisomerase II activity is critical for proper condensation of chromosomes during meiosis46,47, etoposide exposure of diplotene spermatocytes results in extensive DNA breakage and chromosome fragmentation in meiotic metaphase43. However, etoposide is an ineffective inducer of reciprocal translocations because the creation of DNA breaks during chromosome condensation limits the opportunity for chromosomal exchanges. The differing results with etoposide and MLP highlight the complexity of germ cell mutagenesis and demonstrate that both the chemical's mechanism of action and the biological characteristics of the germ cell type exposed contribute to determining the nature of the damage that is transmitted by the sperm to the egg.
Our findings have notable implications for individuals who undergo chemotherapy with MLP and for assisted reproductive technology. First, the ability of MLP to induce heritable CSA in dividing spermatogonia suggests that individuals who undergo chemotherapy with this drug may be at increased risk for abnormal reproductive outcomes up to three months after the end of therapy, when the products of treated dividing spermatogonia appear in the ejaculate. Secondly, selection of sperm samples for in vitro fertilization using assays that assess sperm genetic integrity may greatly underestimate the amount of DNA damage that is actually present in the sperm and that can be transmitted to the egg.
In summary, we have identified a new class of meiotic DNA damage that can be transmitted unrepaired by the sperm and that requires maternal DNA repair to be converted into CSA in the zygote. Our findings highlight the importance of both pre- and post-fertilization DNA repair in assuring the genomic integrity of the conceptus after paternal exposure to chemical mutagens.
Methods
Animals and chemical treatment of males
B6C3F1 males (Harlan-Sprague-Dawley, Indianapolis, IN, USA) were maintained under a 12-h light and 12 hr dark photoperiod at room temperature of 21–23°C and relative humidity of 50 ± 5%. Animals were ~8 weeks of age at the beginning of the experiments and were provided with sterilized tap water and pelleted food at libitum. The Institutional Animal Care and Use Committees at Lawrence Livermore National Laboratory and Health Canada, the locations where animal work took place, approved the use of vertebrate animals in these experiments. All animal procedures were performed in accordance with US and Canadian guidelines and regulations.
Male mice received 7.5 mg/kg MLP (CAS 148-82-3; 4-[bis(2-chloroethyl)amino]-L-phenylalanine, MW 305.23; Sigma Aldrich) dissolved in 60% methanol or the diluent only for controls. This dose was selected to allow comparison with existing dominant lethality (DL) and heritable translocation (HT) data29. MLP and diluent were administered i.p. at the final volume of 0.1 ml/30 g of body weight. MLP-treated and control males were mated with untreated females 3, 7, 23, 37, and 49 days after exposure and zygotes collected from mated females were used to investigate the transmission of MLP-induced CSA at zygotic metaphase. These time points correspond to fertilization with sperm that were epididymal sperm, testicular sperm, diplotene spermatocytes, dividing spermatogonia or stem cells at the time of treatment6, respectively. Other groups of males were euthanized by CO2 inhalation 2 or 23 days after treatment for investigating MLP effects on diplotene spermatocytes through the analysis of CSA in metaphase I (MI) and metaphase II (MII) spermatocytes43 and through the analysis of chromosomal structural and numerical abnormalities in sperm using the sperm CT8 assay30, respectively.
Analysis of first-cleavage zygote metaphases
At various times after treatment (see above), groups of 12 male mice were mated with superovulated B6C3F1 females. For each time point, at least three matings were conducted, each using a different set of males. Female mice were superovulated by an i.p. injection of 7.5 and 5.0 units of pregnant's mare serum (PMS, Sigma) and human chorionic gonadotrophin (HCG) 44–50 hr apart, respectively. They were caged with males, one to one, 6 hr after HCG injection and left overnight. The following morning, females were checked for the presence of vaginal plugs, and mated females received an i.p. injection of 0.08 mg colchicine (CAS no. 64-86-8) in 0.2 ml of distilled water to prevent the union of the parental pronuclei and arrest development at the metaphase stage of the first cell cycle. Females were euthanized 6 hr later by CO2 inhalation and zygotes collected from the oviducts, pooled and processed using the mass harvest procedure48. Zygotic metaphases were hybridized with a probe combination that contained FITC-painting probes for chromosomes 1, 3, 5, X and Y, and biotin-painting probes for chromosomes 2, 4, 6, X and Y (CAMBIO, Cambridge, UK) and scored for the presence of CSA as previously described22.
Analysis of MI/MII spermatocytes
Testes were isolated 48 hrs after treatment from 4 males per dose and processed according to the standard method for making cytogenetic preparations of metaphase spermatocytes49. To increase the number of spermatocytes at the metaphase stage, males received an i.p. injection of colchicine (same dosage as for females) 2 hrs before euthanasia. Prepared slides were coded and stained with 4,6-diamidino-2-phenylindole (DAPI) at 0.25 μg/ml in Vectashield mounting medium (Vector Laboratories). A metaphase finder (Metafer 4.0, Metasystems GmbH, Altlussheim, Germany) installed on Zeiss Axio Imager 1.1 fluorescence microscope was used to localize MI and MII spermatocytes. Each metaphase was then visually analyzed for the presence of chromosomal abnormalities at ×100 magnification. For each timepoint, the data from 4 animals were combined and the mean plus the standard error of the mean was calculated.
Sperm CT8 assay
Epididymal sperm were isolated from 4 treated and 3 concurrent control animals 23 days after treatment as previously described30. Briefly, both epididymides were surgically removed, placed in 300 μl of 2.2% sodium citrate and partially incised with iris scissors. After 5 min at 32°C to allow sperm to swim out into the solution, both epididymides were removed from the sperm suspension. Seven μl of sperm suspension from each mouse were pipetted onto clean dry glass slides and let air dry overnight. The smears were then used for hybridization or stored at −20°C in nitrogen gas.
Semen smears were hybridized with a buffer that contained mouse Cot-1 DNA, herring sperm and probes for chromosome 2 centromere [C] and telomere [T] and chromosome 8 [8] (Figure 1C). Probe composition, probe labeling, sperm pretreatment and hybridization conditions were as previously described30,44. A single scorer analyzed coded slides using a Zeiss Axioplan epifluorescence microscope equipped with single, dual and triple bandpass filters for rhodamine, fluorescein and DAPI. On each slide, ~5,000 sperm were scored. Slides were recoded and a second set of 5,000 sperm was scored on a different area of the slides. For each mouse, the two sets of 5000 sperm were compared and if no difference was found, the data were combined. Only hook-shaped nuclei were scored. Strict scoring criteria30 were used to identify sperm with duplications and deletions of chromosome 2, or numerical abnormalities of chromosome 2 and/or 8.
Statistical analyses
A Chi-square test with adjustment for overdispersion50 was used for the analysis of the results in spermatocytes and zygotes. Linear regression analysis was performed to test the association between the frequencies of zygotes with unstable chromosomal aberrations and dominant lethality. For the sperm CT8 assay, a Cochran's test for equal proportion51 was used to compare the results of the first and second set of 5,000 sperm scored for each mouse. No significant differences were found between the two sets of data for all mice used in this study. Therefore, the two sets of data for each mouse were combined and the frequencies per 10,000 of each class of sperm abnormality identified by the CT8 assay were calculated. Comparisons between treated and control groups were made using the Mann-Whitney U-test.
Author Contributions
F.M., J.B. and A.J.W. designed the research. F.M. and J.G. performed the experiments. F.M., J.B. and A.J.W. analyzed the data. F.M. wrote the initial draft of the paper and all authors commented on the paper.
Supplementary Material
Marchetti_Suppl Materials.pdf
Acknowledgments
This work was funded by the National Institute of Environmental Health Sciences through a National Institute of Environmental Health Sciences/Department of Energy Interagency Agreement (Y01-ES-102-00) and the Canadian Regulatory System for Biotechnology. Work conducted in part under the auspices of the US Department of Energy by the Lawrence Livermore National Laboratory through contract W-7405-END-48 and by the Lawrence Berkeley National Laboratory under contract DE-AC02-05CH11231. We thank Debbie Cabreros for help with the CT8 assay.
References
- Jemal A. et al. Annual Report to the Nation on the Status of Cancer, 1975–2009, featuring the burden and trends in human papillomavirus (HPV)-associated cancers and HPV vaccination coverage levels. J. Natl. Cancer Inst. 105, 175–201 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson A. B. et al. Semen quality and spermatozoal DNA integrity in survivors of childhood cancer: a case-control study. Lancet 360, 361–367 (2002). [DOI] [PubMed] [Google Scholar]
- Ashwood M. J. & Edwards R. DNA repair by oocytes. Mol. Hum. Reprod. 2, 46–51 (1996). [DOI] [PubMed] [Google Scholar]
- Olsen A. K., Lindeman B., Wiger R., Duale N. & Brunborg G. How do male germ cells handle DNA damage? Toxicol. Appl. Pharmacol. 207, 521–531 (2005). [DOI] [PubMed] [Google Scholar]
- McFadden D. E. & Friedman J. M. Chromosome abnormalities in human beings. Mutat. Res. 396, 129–140 (1997). [DOI] [PubMed] [Google Scholar]
- Marchetti F. & Wyrobek A. J. Mechanisms and consequences of paternally-transmitted chromosomal abnormalities. Birth Defects Res. C Embryo Today 75, 112–129 (2005). [DOI] [PubMed] [Google Scholar]
- Hassold T. & Hunt P. To err (meiotically) is human: the genesis of human aneuploidy. Nat. Rev. Genet. 2, 280–291 (2001). [DOI] [PubMed] [Google Scholar]
- Crow J. F. The origins, patterns and implications of human spontaneous mutation. Nat. Rev. Genet. 1, 40–47 (2000). [DOI] [PubMed] [Google Scholar]
- Kong A. et al. Rate of de novo mutations and the importance of father's age to disease risk. Nature 488, 471–475 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J. X. et al. A direct characterization of human mutation based on microsatellites. Nat. Genet. 44, 1161–1165 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell L. B. Significance of the perigametic interval as a major source of spontaneous mutations that result in mosaics. Environ. Mol. Mutagen. 34, 16–23 (1999). [DOI] [PubMed] [Google Scholar]
- Baarends W. M., van der Laan R. & Grootegoed J. A. DNA repair mechanisms and gametogenesis. Reproduction 121, 31–39 (2001). [DOI] [PubMed] [Google Scholar]
- Sotomayor R. E. & Sega G. A. Unscheduled DNA synthesis assay in mammalian spermatogenic cells: an update. Environ. Mol. Mutagen. 36, 255–265 (2000). [PubMed] [Google Scholar]
- Meistrich M. L. [Histone and basic nuclear protein transitions in mammalian spermatogenesis]. Histones and other basic nuclear proteins, [Hnilica, L. S., Stein, G. S. & Stein, J. L. (eds.)][165–182] (CRC Press, Boca Raton, 1989). [Google Scholar]
- Wouters-Tyrou D., Martinage A., Chevaillier P. & Sautiere P. Nuclear basic proteins in spermiogenesis. Biochimie 80, 117–128 (1998). [DOI] [PubMed] [Google Scholar]
- Marchetti F. & Wyrobek A. J. DNA repair decline during mouse spermiogenesis results in the accumulation of heritable DNA damage. DNA Repair (Amst) 7, 572–581 (2008). [DOI] [PubMed] [Google Scholar]
- Derijck A. H. A. et al. gammaH2AX signalling during sperm chromatin remodelling in the mouse zygote. DNA Repair (Amst) 5, 959–971 (2006). [DOI] [PubMed] [Google Scholar]
- Barton T. S., Robaire B. & Hales B. F. DNA damage recognition in the rat zygote following chronic paternal cyclophosphamide exposure. Toxicol. Sci. 100, 495–503 (2006). [DOI] [PubMed] [Google Scholar]
- Generoso W. M., Cain K. T., Krishna M. & Huff S. W. Genetic lesions induced by chemicals in spermatozoa and spermatids of mice are repaired in the egg. Proc. Natl. Acad. Sci. U S A 76, 435–437 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti F., Essers J., Kanaar R. & Wyrobek A. J. Disruption of maternal DNA repair increases sperm-derived chromosomal aberrations. Proc. Natl. Acad. Sci. U S A 104, 17725–17729 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derijck A. H. A., van der Heijden G., Giele M., Philippens M. & de Boer P. DNA double-strand break repair in parental chromatin of mouse zygotes, the first cell cycle as an origin of de novo mutation. Hum. Mol. Genet. 17, 1922–1937 (2008). [DOI] [PubMed] [Google Scholar]
- Marchetti F., Bishop J. B., Cosentino L., Moore D. II & Wyrobek A. J. Paternally transmitted chromosomal aberrations in mouse zygotes determine their embryonic fate. Biol. Reprod. 70, 616–624 (2004). [DOI] [PubMed] [Google Scholar]
- IARC. Melphalan. IARC Monogr. Eval. Carcinog. Risk Chem. Man 100A, 107–117 (2012). [Google Scholar]
- Povirk L. F. & Shuker D. E. DNA damage and mutagenesis induced by nitrogen mustards. Mutat. Res. 318, 205–226 (1994). [DOI] [PubMed] [Google Scholar]
- Lawley P. D. & Phillips D. H. DNA adducts from chemotherapeutic agents. Mutat. Res. 355, 13–40 (1996). [DOI] [PubMed] [Google Scholar]
- Bauer G. B. & Povirk L. F. Specificity and kinetics of interstrand and intrastrand bifunctional alkylation by nitrogen mustards at a G-G-C sequence. Nucleic Acids Res. 25, 1211–1218 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell L. B., Hunsicker P. R., Cacheiro N. L. & Rinchik E. M. Genetic, cytogenetic, and molecular analyses of mutations induced by melphalan demonstrate high frequencies of heritable deletions and other rearrangements from exposure of postspermatogonial stages of the mouse. Proc. Natl. Acad. Sci. U S A 89, 6182–6186 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell L. B., Hunsicker P. R. & Shelby M. D. Melphalan, a second chemical for which specific-locus mutation induction in the mouse is maximum in early spermatids. Mutat. Res. 282, 151–158 (1992). [DOI] [PubMed] [Google Scholar]
- Generoso W. M. et al. Dominant lethal and heritable translocation tests with chlorambucil and melphalan in male mice. Mutat. Res. 345, 167–180 (1995). [DOI] [PubMed] [Google Scholar]
- Hill F. S. et al. A new FISH assay to simultaneously detect structural and numerical chromosomal abnormalities in mouse sperm. Mol. Reprod. Dev. 66, 172–180 (2003). [DOI] [PubMed] [Google Scholar]
- Disteche C. M., Carrano A. V., Ashworth L. K., Burkhart-Schultz K. & Latt S. A. Flow sorting of the mouse Cattanach X chromosome, T(X;7)1 Ct in an active or inactive state. Cytogenet. Cell Genet. 29, 189–197 (1981). [DOI] [PubMed] [Google Scholar]
- Russell L. B. Effects of male germ-cell stage on the frequency, nature, and spectrum of induced specific-locus mutations in the mouse. Genetica 122, 25–36 (2004). [DOI] [PubMed] [Google Scholar]
- Dronkert M. L. & Kanaar R. Repair of DNA interstrand cross-links. Mutat. Res. 486, 217–247 (2001). [DOI] [PubMed] [Google Scholar]
- Deans A. J. & West S. C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 11, 467–480 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S. & Canman C. E. REV1 and DNA polymerase zeta in DNA interstrand crosslink repair. Environ. Mol. Mutagen. 53, 725–740 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee Y. & D'Andrea A. D. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 24, 1680–1694 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Räschle M. et al. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell 134, 969–980 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legerski R. J. Repair of DNA interstrand cross-links during S phase of the mammalian cell cycle. Environ. Mol. Mutagen. 51, 540–551 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross W. E., Ewig R. A. & Kohn K. W. Differences between melphalan and nitrogen mustard in the formation and removal of DNA cross-links. Cancer Res. 38, 1502–1506 (1978). [PubMed] [Google Scholar]
- Holloway J. K. et al. Mammalian BTBD12(SLX4) protects against genomics instability during mammalian spermatogenesis. PLoS Genet 7, e1002094 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips B. T., Gassei K. & Orwig K. E. Spermatogonial stem cell regulation and spermatogenesis. Philos. Trans. R Soc. Lond. B. Biol. Sci. 365, 1663–1678 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen W. S., Merriman J. A., O'Bryan M. K. & Jones K. T. DNA double strand breaks but not interstrand crosslinks prevent progress through meiosis in fully grown mouse oocytes. PLoS One 7, e43875 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti F. et al. Etoposide induces heritable chromosomal aberrations and aneuploidy during male meiosis in the mouse. Proc. Natl. Acad. Sci. USA 98, 3952–3957 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti F., Pearson F. S., Bishop J. B. & Wyrobek A. J. Etoposide induces chromosomal abnormalities in mouse spermatocytes and stem cell spermatogonia. Hum. Reprod. 21, 888–895 (2006). [DOI] [PubMed] [Google Scholar]
- Wang J. C., Caron P. R. & Kim R. A. The role of DNA topoisomerases in recombination and genome stability: a double-edged sword? Cell 62, 403–406 (1990). [DOI] [PubMed] [Google Scholar]
- Poljak L. & Kas E. Resolving the role of topoisomerase II in chromatin tructure and function. Trends Cell Biol. 5, 348–354 (1995). [DOI] [PubMed] [Google Scholar]
- Moens P. B. Unravelling meiotic chromosomes: topoisomerase II and other proteins. J. Cell Sci. 97, 1–3 (1990) [DOI] [PubMed] [Google Scholar]
- Mailhes J. B. & Yuan Z. P. Cytogenetic technique for mouse metaphase II oocytes. Gamete Res 18, 77–83 (1987). [DOI] [PubMed] [Google Scholar]
- Evans E. P., Breckon G. & Ford C. E. An Air-Drying Method for Meiotic Preparations from Mammalian Testes. Cytogenetics 3, 289–294 (1964). [DOI] [PubMed] [Google Scholar]
- Collett D. Modeling binary data (Chapman and Hall, London, 1991). [Google Scholar]
- Snedecor G. W. & Cochran W. G. Statistical methods (Iowa State University Press, Ames, IA, 6th Ed, 1967). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Marchetti_Suppl Materials.pdf