

Abstract
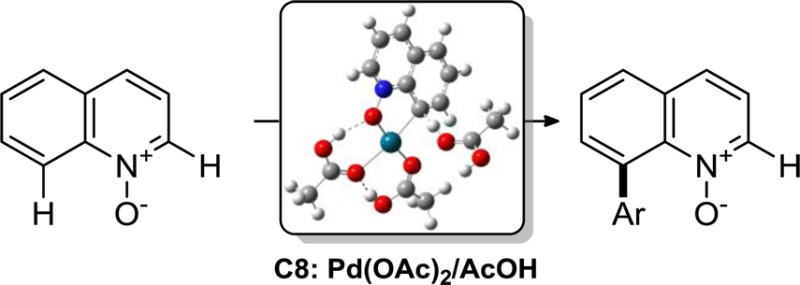
We report herein a palladium-catalyzed C–H arylation of quinoline N-oxides that proceeds with high selectivity in favor of the C8 isomer. This site selectivity is unusual for palladium, since all of the hitherto described methods of palladium-catalyzed C–H functionalization of quinoline N-oxides are highly C2 selective. The reaction exhibits a broad synthetic scope with respect to quinoline N-oxides and iodoarenes and can be significantly accelerated to subhour reaction times under microwave irradiation. The C8-arylation method can be carried out on a gram scale and has excellent functional group tolerance. Mechanistic and density functional theory (DFT) computational studies provide evidence for the cyclopalladation pathway and describe key parameters influencing the site selectivity.
Keywords: C−H activation, quinolines, C−C coupling, palladium, site selectivity, solvent effects
Introduction
Transition-metal-catalyzed C–H functionalization has emerged as powerful general strategy for the synthesis of complex synthetic targets.1 Due to the abundance of C–H bonds in organic molecules, one of the key challenges in this area of catalysis is to gain an understanding of the factors that govern the regioselectivity of competing C–H functionalization pathways.2 Two strategies have been used to achieve the desired control over regioselectivity. While the first strategy is based on the use of directing groups, the second strategy exploits the inherent steric and electronic biases imposed by substrate functional groups.3 Advantageously, directing groups can be tailored to guide the catalyst to specific C–H bonds within the substrate.4 However, they have to be installed and then removed from the product, thus potentially reducing their synthetic utility. This shortcoming can be attenuated if the directing groups are converted into other desired functional groups of the synthetic target after the C–H functionalization step.
8-Substituted quinolines are important structural motifs with applications in the areas of drug discovery,5 materials science,6 and catalysis.7 A significant number of methods exist for catalytic functionalization of unsubstituted quinolines in the C2 position with Pd,8,9 Cu,10 Ni,11 Rh,12 Ru,13 and Ag14 as catalysts.
In contrast, very few methods are known for functionalization of the C8 position in the quinoline (Figure 1). In a pioneering study, Chang developed a direct arylation of quinolines in the C8 position catalyzed by a dirhodium NHC complex.15 Shibata recently reported on the cationic Rh-catalyzed C8 alkenylation of quinoline N-oxides,16 while an Ir-catalyzed C8 borylation reaction was developed by Steel, Marder, and Sawamura.17 Recently, Chang utilized an Ir-based catalyst for C8 amidation with sulfonyl azides and a Rh-based catalyst for a C8-selective iodination of quinoline N-oxides.18 In addition, an example of Rh-catalyzed C–C/C–O coupling with alkynes in the C8 position of quinoline N-oxides was disclosed by Li,19 and Rh-catalyzed C8 alkynylation and alkylation with α-diazo esters were reported by Chang.20
Figure 1.
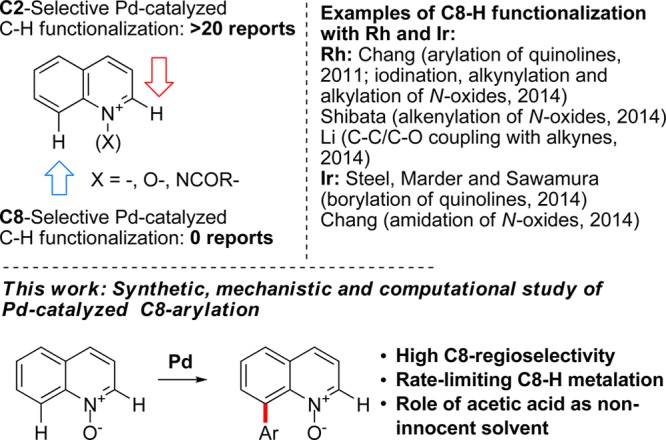
Current approaches to C2- and C8-selective C–H functionalization of quinolines.
Since the first report by Fagnou, azine N-oxides have been employed as substrates for a variety of highly C2 selective C–H functionalizations.9 The original method developed by Fagnou relied on the use of palladium complexes with sterically demanding phosphines (e.g., di-tert-butylphenylphosphine and tri-tert-butylphosphine) as catalysts. Initial mechanistic investigations supported the inner-sphere concerted metalation–deprotonation (CMD) pathway that did not involve precoordination of the N-oxide oxygen atom to palladium.9h On the other hand, a recent study by Hartwig provided strong evidence that the cyclometalated complex [Pd(OAc)(tBu2PCMe2CH2)] is initially formed, when tri-tert-butylphosphine is used as a ligand.21 This complex subsequently serves as a catalyst for the arylation of pyridine N-oxide. Intriguingly, in none of the examples of the Pd-catalyzed C–H functionalization of quinoline N-oxides was any C8-functionalization product reported, even as a minor isomer. This is surprising, considering the proximity and the parallel alignment of the C8–H bond and the N–O bond that can potentially guide palladium in the C8 position. Although the origins of the C2 selectivity for quinoline N-oxides have not been studied, in the case of pyridine N-oxides the high C2 preference of palladium was explained by the increased acidity22 and the increased strength23 of the C2–H bond effected by the adjacent electron-withdrawing N–O group. This report outlines the development of the palladium-catalyzed C8-selective arylation of quinoline N-oxides, as well as the mechanistic details pertaining to the divergent site selectivity of the palladium/quinoline N-oxide couple and the crucial roles of additives and solvents.
Results and Discussion
Initial Studies
Quinoline N-oxide was selected to study the site selectivity of Pd-catalyzed arylation under ligand-free conditions. Interestingly, the Pd-catalyzed C2–H arylation reactions of quinoline N-oxides described to date have typically been carried out either with phosphine-bound palladium catalysts or under phosphine-free conditions24 in neutral solvents (e.g., DMF and dioxane). Addition of substoichiometric amounts of pivalic or acetic acids was found to improve the yields but had no effect on the site selectivity. On the other hand, recent studies point toward an important role of polar acidic solvents in the acceleration of cyclopalladation processes.25 Thus, the influence of acetic acid as a solvent (Table 1) was investigated first. The reaction proceeds with a high C8 selectivity (12:1 C8/C2 ratio) and in ∼9% yield with 5 mol % of Pd(OAc)2. Addition of silver acetate improved the conversion without affecting the C8/C2 ratio (entry 2). The crucial role of acetic acid became evident when it was replaced with dimethylformamide and tert-butyl alcohol (entries 3 and 4): in both cases the C2-aryl isomer 3 was formed as a predominant product (1:7 and 1:6 C8/C2 ratios, respectively). Other palladium salts (entries 5 and 6) caused undesired side reactions, as can be seen from the lower recovery of unreacted substrate 1. A survey of silver salts showed that the carbonate is less effective than the acetate. On the other hand, phosphate dramatically improved the C8/C2 ratio and the conversion (entry 8). A reaction carried out with 0.5 equiv of silver phosphate proceeded with >30:1 C8/C2 ratio and a 73% conversion to 2. Further improvement was achieved using 30 equiv of acetic acid (entry 9). In this case the C8/C2 ratio was >30:1, and the yield improved to 78%. Water had a notable accelerating effect (entry 10), leading to a clean reaction and nearly complete conversion, while providing a high C8/C2 ratio of 23:1. The reaction also proved to give excellent results when carried out under microwave irradiation26 at 180 °C (entries 11 and 12). A comparable conversion and C8/C2 ratio were obtained within 45 min with 5.5 equiv of water, and 65% yield was obtained within 10 min with 40 equiv of water. Aryl iodides proved to be significantly more reactive than bromides, while no reaction was observed with chlorides.27 This pattern of reactivity indicates that bromo and chloro groups can be compatible with the conditions of the C8 arylation method.
Table 1. Reaction Development for the Palladium-Catalyzed C8 Arylation of Quinoline N-Oxidesa.
| yield
(%) |
|||||||
|---|---|---|---|---|---|---|---|
| entry | catalyst (5 mol %) | AgX (amt (equiv)) | additive (amt (equiv)) | 1 | 2 (C8) | 3 (C2) | C8/C2 (2/3) |
| 1 | Pd(OAc)2 | AcOH (10) | 91 | 8 | 0.6 | 12:1 | |
| 2 | Pd(OAc)2 | AgOAc (3) | AcOH (10) | 38 | 39 | 3 | 13:1 |
| 3 | Pd(OAc)2 | AgOAc (3) | DMF (10) | 29 | 5 | 36 | 1:7 |
| 4 | Pd(OAc)2 | AgOAc (3) | tBuOH (10) | 50 | 7 | 41 | 1:6 |
| 5 | PdCl2 | AgOAc (3) | AcOH (10) | 7 | 17 | 2 | 9:1 |
| 6 | Pd(TFA)2 | AgOAc (3) | AcOH (10) | 27 | 40 | 11 | 4:1 |
| 7 | Pd(OAc)2 | Ag2CO3 (0.5) | AcOH (10) | 29 | 23 | 3 | 8:1 |
| 8 | Pd(OAc)2 | Ag3PO4 (0.5) | AcOH (10) | <2 | 73 | <2 | >30:1 |
| 9 | Pd(OAc)2 | Ag3PO4 (0.5) | AcOH (30) | 8 | 78 | <2 | >30:1 |
| 10 | Pd(OAc)2 | Ag3PO4(0.5) | AcOH (30)/H2O (5.5) | 1 | 95 | 4 | 23:1 |
| 11b | Pd(OAc)2 | Ag3PO4(0.5) | AcOH (30)/H2O (5.5) | 6 | 90 | 3 | 30:1 |
| 12c | Pd(OAc)2 | Ag3PO4(0.5) | AcOH (30)/H2O (40) | 28 | 65 | <2 | >30:1 |
Yields were determined by 1H NMR analysis with 1,4-dimethoxybenzene as an internal standard added prior to workup. Reaction conditions: 1 (0.2 mmol), 4 (3 equiv), with the catalyst and the additives under Ar for 12 h at 120 °C.
Reaction was run under microwave irradiation at 180 °C for 45 min.
Reaction was run under microwave irradiation at 180 °C for 10 min.
Synthetic Scope
The reaction tolerates a number of functional groups in the quinoline system under the optimized conditions using conventional heat and microwave irradiation (Table 2). Methyl ester, methoxy, nitro, and bromo substituents have been found to be compatible with the C8-selective catalytic system. Similarly, iodoarenes bearing fluoro, bromo, chloro, trifluoromethyl, trifluoromethoxy, and methoxy groups afforded the corresponding 8-arylquinoline N-oxides in good to excellent yields. The C8-substitution pattern was confirmed by means of NMR spectroscopy and, in the case of product 2, by single-crystal X-ray crystallography. Thermal and microwave conditions were found to provide C8-arylated products in comparable yields with the advantage of significant reduction of reaction time in the latter case. The microwave-assisted reactions were generally complete within 1 h at 180 °C. The C8/C2 ratio was >20:1 in most of the cases, and simple purification by flash chromatography afforded pure C8 isomer. The reaction proceeded more slowly with N-oxides bearing electron-withdrawing groups in the C6 position (e.g., nitro and ester groups), resulting in somewhat lower yields of the corresponding products. 2-Substituted quinoline N-oxides reacted cleanly and afforded 8-arylated derivatives as the sole products. Gratifyingly, no α-arylation at the methyl group was observed in the case of 2-methylquinoline N-oxide. The reaction is amenable to gram-scale operations. For example, the synthesis of 8-phenylquinoline N-oxide (5) was successfully carried out with 2.1 g of 1 and afforded 2.6 g of 5 (82% yield).
Table 2. Scope of the C8–H Arylation of Quinoline N-Oxides with Iodoarenes under Thermal (T) and Microwave (MW) Conditionsa.
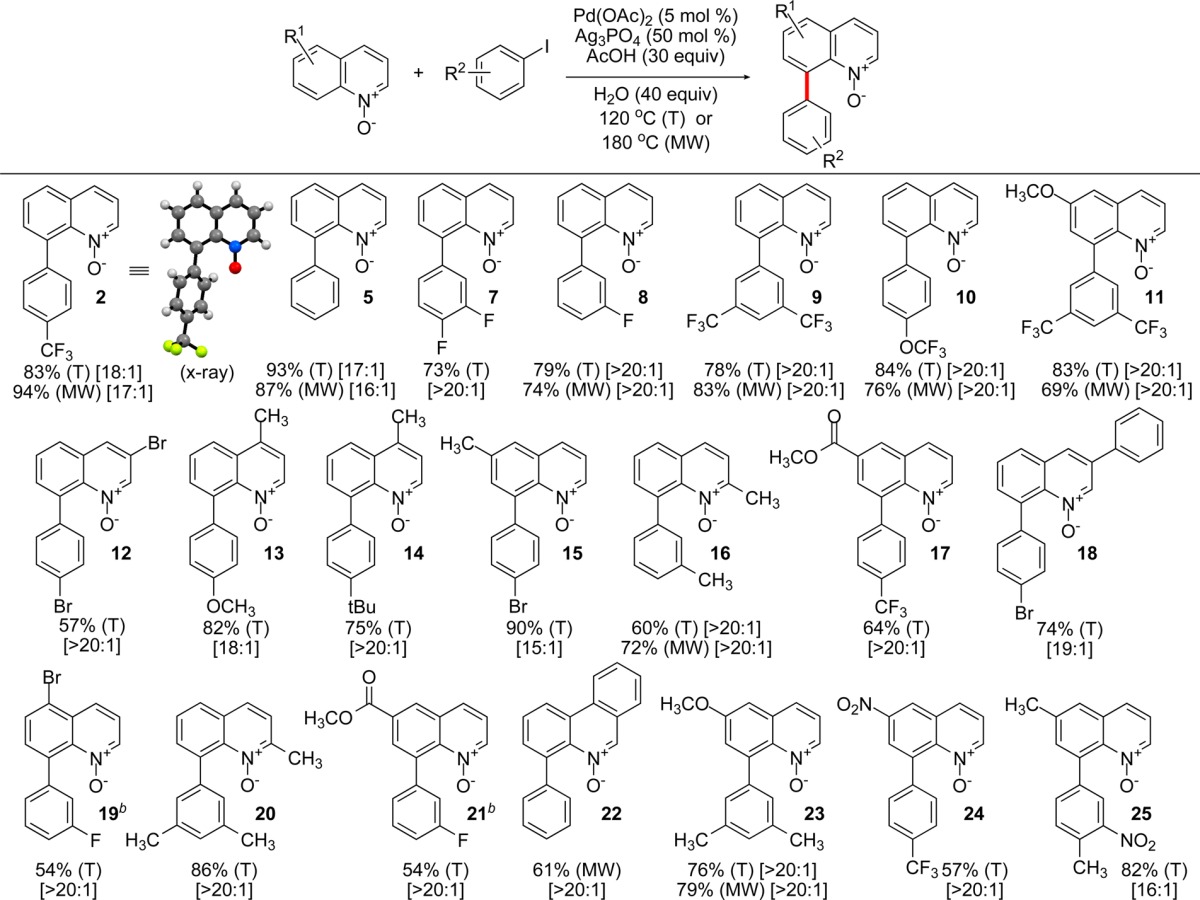
The reactions were carried out with 0.5–1 mmol of the substrate for 16 h (T) or 50 min (MW). The yields are reported for isolated 8-arylquinoline N-oxides. The C8/C2 ratios are reported in brackets for the crude product before chromatographic purification.
5.5 equiv H2O was used.
In order to bypass the isolation steps en route to 8-arylquinolines, a practical one-pot protocol for the synthesis of 8-arylquinolines from quinoline under microwave irradiation was developed (Figure 2). N-oxidation of quinoline with hydrogen peroxide in the presence of acetic acid for 40 min at 180 °C was followed by the Pd-catalyzed C8 arylation with 4-bromoiodobenzene.
Figure 2.
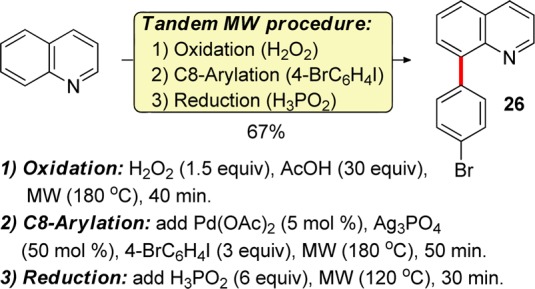
8-Arylquinoline synthesis by a tandem microwave-accelerated procedure.
Subsequent reduction of the N–O bond by hypophosphorous acid at 120 °C afforded 8-(4-bromophenyl)quinoline (26) in a 67% yield from quinoline. Due to significant acceleration of each of the three reactions under microwave irradiation, the one-pot procedure can be completed within 3 h. Heterocyclic N-oxides are synthetically versatile intermediates, due to their ease of preparation28 and the variety of methods that can be used to convert the N-oxide moiety into a number of other functional groups, in particular in the C2 position.29,30 For example, treatment of N-oxide 9 with tert-butylamine and p-toluenesulfonic anhydride31 gave rise to 2-N-tert-butylaminoquinoline (27) in 70% yield (Figure 3). Similarly, a reaction32 with p-toluenesulfonyl chloride and methanol afforded 2-methoxy-8-phenylquinoline (28) in 83% yield. 2-Quinolone 29 was readily prepared by a trifluoroacetic anhydride mediated rearrangement33 of 5 in 92% yield.
Figure 3.
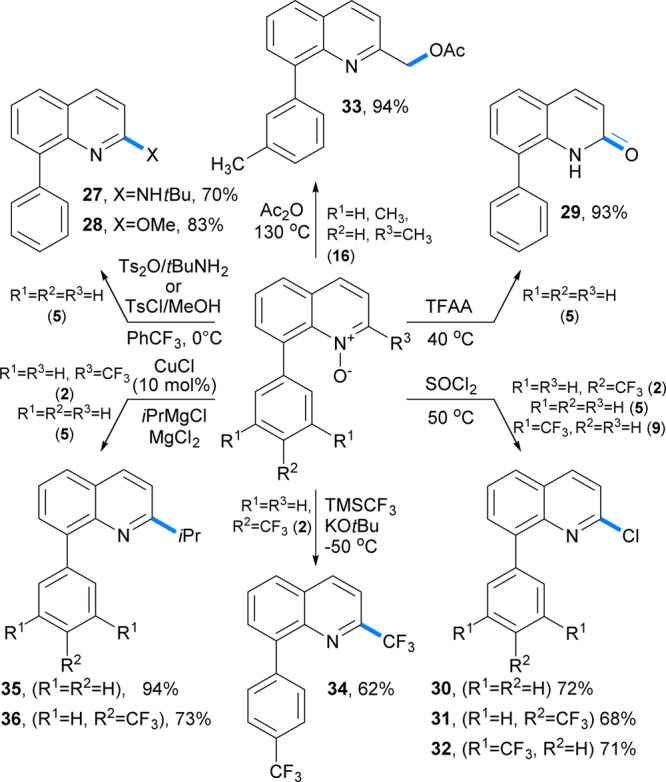
Derivatization of C8-arylation products (TFAA = trifluoroacetic anhydride).
Further, the N–O moiety was used to install a trifluoromethyl group in the C2 position by means of a base-mediated reaction34 with the Ruppert–Prakash reagent. A regioselective conversion of 8-aryl-substituted quinoline N-oxides to the corresponding 2-chloroquinolines 30–32 was accomplished by a reaction with thionyl chloride in 68–72% yields. Copper-catalyzed deoxygenative C2 arylation and alkylation10e of the N-oxide products was also effected by Grignard reagents in the presence of magnesium chloride. In addition, a reaction of the 8-arylquinaldine N-oxide 16 with acetic anhydride led to clean transposition of the oxy functionality to the α-carbon and afforded α-acetoxy-substituted quinaldine 33 in 94% yield.
Mechanistic Studies
The speciation of palladium(II) acetate in solution strongly depends on the solvent, temperature, and presence of water. Facile partial hydrolysis of the trimer in organic solvents in the presence of water was observed by Cotton and Murillo.35
Kinetic studies by Vargaftik36 and Hii37 showed that water promotes dissociation of the cyclotrimer, and linear trimeric and monomeric species were suggested as intermediates in aqueous acetic acid and tetrahydrofuran. While the cyclic trimeric structure Pd3(OAc)6 is largely retained in chloroform, benzene, methanol, and acetic acid at room temperature, linear trimers and monomeric species have also been detected in chloroform–acetic acid and in N-methyl-2-pyrrolidinone (NMP),38 respectively. We examined the influence of water on the C8 arylation under increasing dilution and constant-volume conditions. In the first case addition of water leads to acceleration of the reaction even as the concentration of the reagents decreases at high water loadings.39 A conversion of 74% is reached with 40 equiv of water but remains nearly constant as the amount of added water increases. Since the reaction rates can be influenced by changes in the concentrations due to dilution, a second set of experiments was carried out in the acetic acid–water system by varying the water content and keeping the reaction volume constant.40 The rate acceleration is observed in this case as well; however, a maximum is achieved at ∼40% water. The reaction rate then decreases as water becomes the major component of the mixture, presumably due to the decreasing solubility of the reactants in the predominantly aqueous solutions. The accelerating effect of water that is especially strong at lower water concentrations may be due to the faster dissociation of the oligomeric species of palladium acetate to the more reactive acyclic and monomeric forms. In addition, N-oxide complexes of palladium, although stable under anhydrous conditions, are labile in the presence of water;41 hence, water may facilitate the turnover of the palladium catalyst.
Several functional group guided Pd-catalyzed C–H arylation reactions have previously been shown to require acidic solvents (AcOH or TFA), and a Pd(II)/Pd(IV) catalytic cycle has been implicated.42 While the role of the acidic solvents has not been studied in detail, it is conceivable that the current system proceeds by a similar mechanistic pathway.
We further examined the behavior of the present catalytic system in deuterated solvents (Figure 4).
Figure 4.
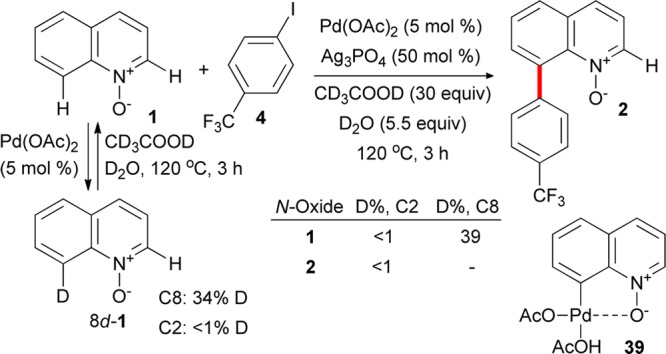
H/D exchange in C2 and C8 positions of recovered unreacted N-oxide 1 and product 2.
Preliminary experiments showed that no H/D exchange occurred in the absence of palladium acetate in CD3COOD/D2O at 120 °C in 3 h with and without silver phosphate present. Conversely, a reversible H/D exchange in the C8 position was observed in the presence of palladium acetate (5 mol %). No deuterium incorporation was detected in the C2 position of quinoline N-oxide. Next, the C8-arylation reaction was carried out in CD3COOD/D2O, and the extent of the H/D exchange was determined for the recovered unreacted quinoline N-oxide and the product 2. While no deuterium incorporation was observed at the C2 position of both the recovered substrate 1 and the arylation product 2, 39% deuteration was detected in the C8 position of the recovered N-oxide 1.
These results indicate that a palladacycle, e.g. as represented by structure 39 or a (hetero)dimeric analogue, can be formed reversibly under the reaction conditions by an N–O group guided activation of the C8–H bond. Attempts at observing palladacycle 39 (or any structural or (hetero)dimeric analogue) by means of NMR at temperatures below 100 °C or isolating it were not successful.43 These results suggest that the putative palladacycle is kinetically labile under the reaction conditions and that the protodepalladation is very fast. This behavior is in line with the aforementioned lability of palladium(II) complexes of N-oxides in the presence of water. The H/D exchange does not take place at temperatures below 100 °C.
The C2-H/D exchange is promoted by base under aqueous conditions.10c Although acetic acid may prevent the base-mediated C2 metalation by protonating the base, the C8 selectivity of C–H activation in acetic acid cannot be explained by this effect. Experiments with Pd salts show that the rate of C2–H/D exchange (10 mol % PdX2, 1 in AcOH at 120 °C, 3 h) increases with less basic anions (Pd(OAc)2 C8–D 39%, C2–D <1%; Pd(O2CCF3)2 C8–D 93%, C2–D 25%; Pd(CH3CN)4(OTf)2 C8–D 94%, C2–D 13%), pointing to the electrophilic character of the C–H activation. Similarly, the rate of C2–H/D exchange catalyzed by Ag salts (10 mol % of AgX, 1 in AcOH at 120 °C, 3 h), increases in the following order: OAc (26% C2–D) < OTf (30% C2–D) < SbF6 (59% C2–D), with no appreciable C8–H/D exchange. These results indicate that the C2– and C8–H activation processes in acetic acid are primarily electrophilic and the C8/C2 selectivity is determined by the nature of the metal and the solvent. The crucial role of acetic acid is further supported by the results of the H/D-exchange experiments in DMF (10 mol % of PdX2, DMF, 5.5 equiv of D2O, 120 °C, 3 h). In this case, a much slower and nonregioselective C2/C8–H activation was observed for Pd(OAc)2 (C8–D 8%, C2–D 17%) and Pd(O2CCF3)2 (C8–D 13%, C2–D 14%).
Kinetic isotope experiments have become a useful tool for mechanistic studies of C–H functionalization processes.44 Toward this end, the primary kinetic isotope effect for the C8 arylation of quinoline oxide was determined in an parallel competition experiment with 1 and 2,8-d2-1 in CH3CO2H/H2O and CD3CO2D/D2O, respectively, to exclude the influence of the H/D exchange (eq 1). The observed KIE (kH/kD = 2.0) indicated that C8–H bond cleavage may be involved in the turnover-determining step or precede it.
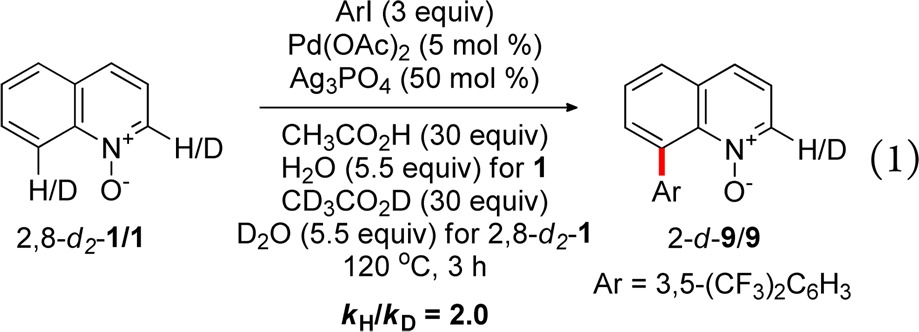 |
1 |
We next investigated the electronic effect of substituents in the 5- and 6-positions of quinoline N-oxide on the rate of the H/D exchange and the rate of the arylation (Figure 5). The Hammett plots indicate that both reactions are accelerated by electron-donating substituents in the 5- and 6-positions of the quinoline system. A large negative ρ value for the H/D exchange (ρ = −2.98) is indicative of considerable cationic character in the transition state of the C–H bond activation step and points to a significant electrophilic character of the reacting palladium catalyst.
Figure 5.
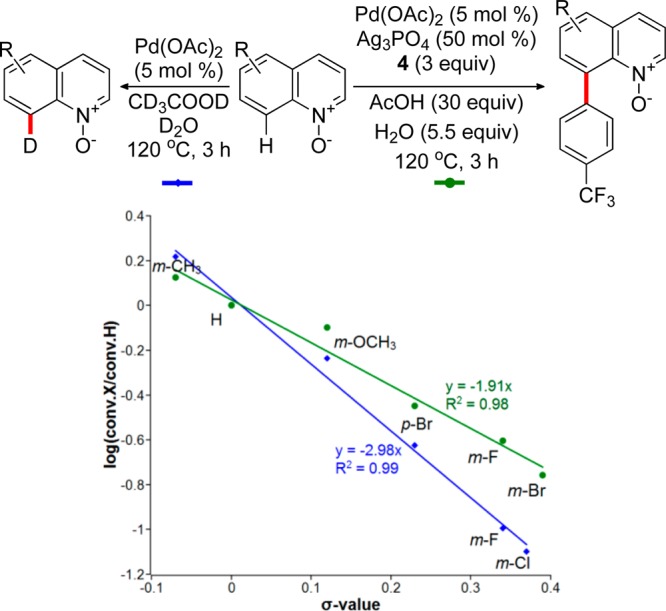
Hammett plots for the C8–H arylation of substituted quinoline N-oxides (green line) and for the H/D exchange (blue line).
The ρ value for the C–H arylation was also large and negative, suggesting that the palladacycle formation is an important contributor to the overall kinetics of the reaction, further supporting the results of the kinetic isotope effect experiments. For comparison, a positive ρ value (+1.53) was obtained for the Pd/tBu3P-catalyzed C2-arylation of pyridine N-oxides,9h which was interpreted as an indication of a buildup of the negative charge in the TS along the CMD pathway and is in line with the electron-rich nature of the phosphine-bound Pd catalyst.
The influence of substituents in the iodoarene on the rate of the C8–H arylation was also investigated (Figure 6). The Hammett analysis suggests that the reaction is accelerated by electron-withdrawing substituents in the iodoarene.
Figure 6.
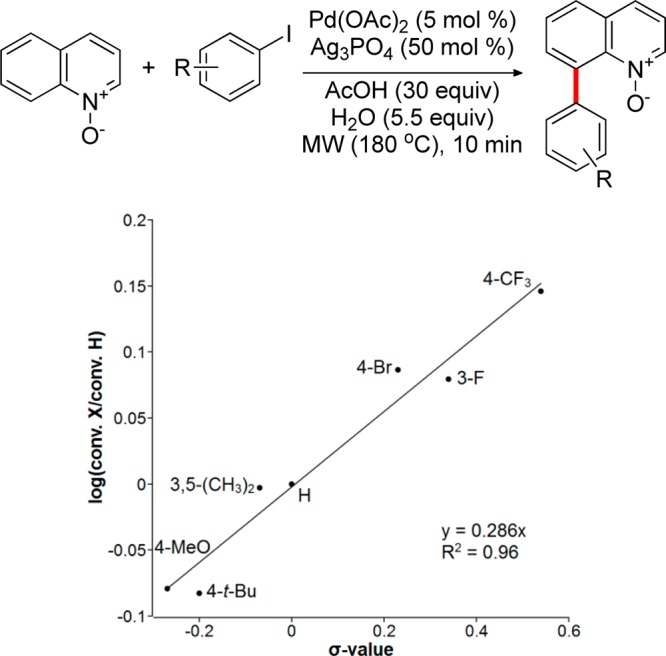
Hammett plot for the C8–H arylation with substituted iodoarenes.
The ρ value (+0.29) is substantially smaller than Hammett reaction constants observed for oxidative additions to Pd. For example, ρ values for rate-determining oxidative additions to Pd0 are typically within the range of +0.6 to +2.5 for iodoarenes,45 while Sanford reported ρ = +1.7 for the rate-limiting oxidative addition to PdII for the direct C–H arylation with diaryliodonium salts.46 Substantially smaller ρ values have been interpreted as an indication that the oxidative addition is not a turnover-limiting step.47 Further, it was found that the rate of conversion of 1 to 2 after 3 h at 120 °C is independent of the concentration of iodoarene 4 within the 0.5–2.5 equiv range of 4. Furthermore, no reaction was observed when Pd2(dba)3 and PhPd(tmeda)I were used as catalysts, indicating that cyclopalladation likely precedes oxidative addition. Together with the data of the Hammett study of electronic effects of substituents in the quinoline core and kinetic isotope effect data, these results point to C8 cyclopalladation as a turnover-limiting step in the catalytic cycle.
Computational Studies
In an effort to explain the C8 selectivity of the arylation reaction, we have examined several mechanistic scenarios for the palladium-catalyzed C–H cleavage by means of density functional theory (DFT) calculations and compared the C8- and C2-metalation pathways. In particular, we focused on (i) the role of acetic acid as a noninnocent solvent in the catalytic process and (ii) the disparity in the site selectivity of metalation between palladium acetate and the phosphine-bound Pd metal center. Previous experimental and computational studies31,48 have shown that the mechanisms involving Pd(OAc)2, Pd(OAc)(HOAc), and Pd(HOAc)3 as the active palladium species can be operative, when acetic acid is used as a solvent. We also studied the C–H activation of quinoline N-oxide by Pd(PMe3)Ph, as this species shows exclusive C–H activation at the C2 position. Hence, the following four mechanistic scenarios were investigated.
In mechanism 1a (Figure 7), cyclometalation is effected by a dicationic Pd metal center with three acetic acid ligands, whereas mechanisms 1b (Figure 8) and 1c (Figure 9) describe cyclopalladation by monocationic and neutral Pd metal centers, respectively. Mechanism 2 (Figure 10) describes metalation by the phosphine-bound Pd center in a fashion similar to what has been proposed for pyridine N-oxides.9h Acetic acid complexes of palladium similar to those examined in mechanisms 1a–c were implicated in the acid-assisted C–H bond activation by Pd in oxazolone ligands in neat acetic acid solutions.49 All of the C–H bond cleavage transition states connect the C–H bond cleavage products to starting structures, in which quinoline N-oxide coordinates to palladium through its oxygen atom.9d
Figure 7.
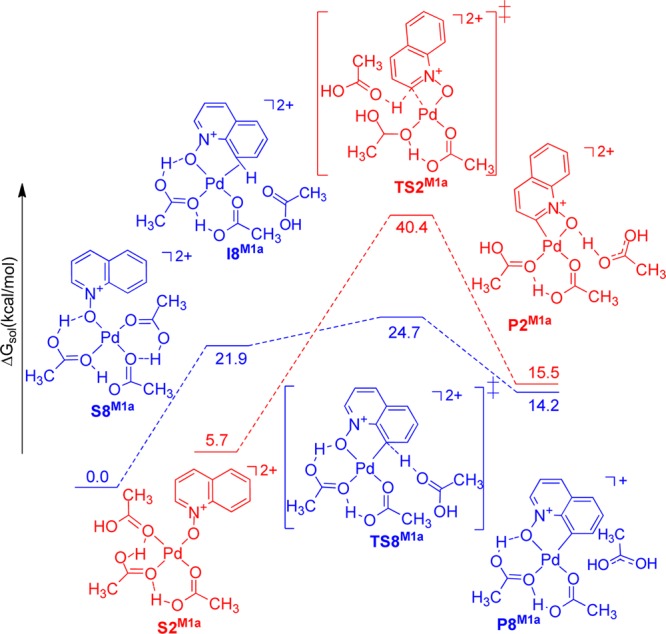
Solvation Gibbs free energy diagram (ΔG in kcal mol–1, at 298 K, in acetic acid) for the relevant intermediates, transition states, and products in mechanism 1a.
Figure 8.
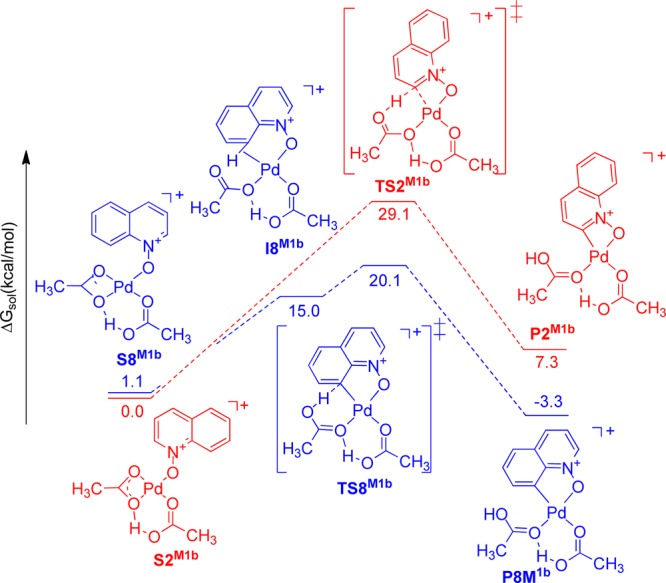
Solvation Gibbs free energy diagram (ΔG in kcal mol–1, at 298 K, in acetic acid) for the relevant intermediates, transition states, and products in mechanism 1b.
Figure 9.
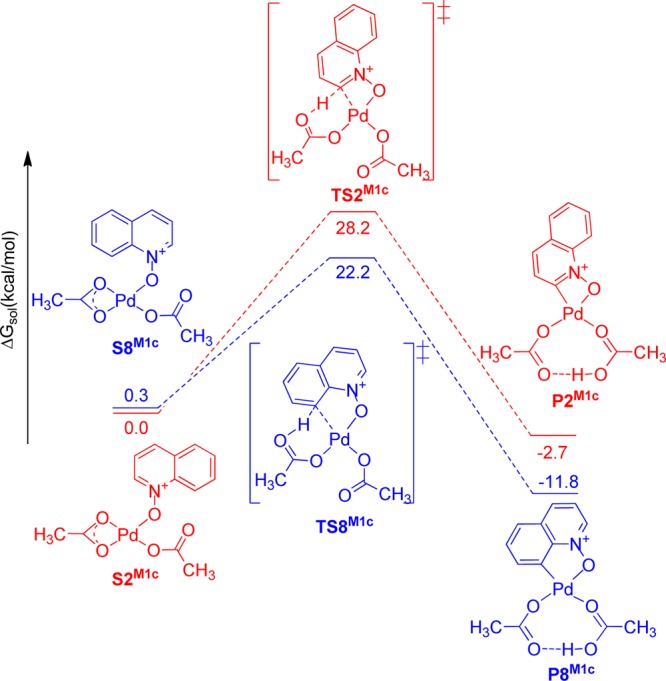
Solvation Gibbs free energy diagram (ΔG in kcal mol–1, at 298 K, in acetic acid) for the relevant intermediates, transition states, and products in mechanism 1c.
Figure 10.
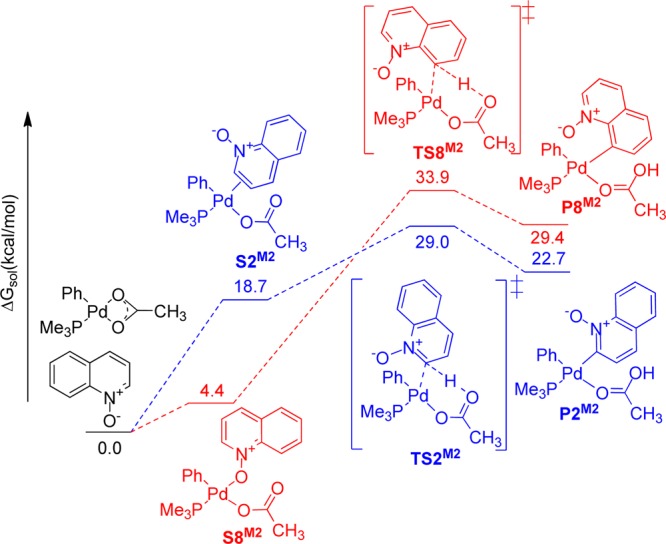
Solvation Gibbs free energy diagram (ΔG in kcal mol–1, at 298 K, in DMF) for the relevant intermediates, transition states, and products in mechanism 2.
In mechanism 1a, the initial formation of the dicationic [Pd(HOAc)3(κ1-Oquinoline N-oxide)]2+ complex containing three ligated acetic acid molecules was considered due to the high acetic acid concentration.25,50 In mechanisms 1b and 1c, the initial formation of monocationic [Pd(κ2-OAc)(HOAc)(κ1-Oquinoline N-oxide)]+ complex, with only one of the acetate ligands being protonated, and the neutral [Pd(κ2-OAc)(HOAc)(κ1-Oquinoline N-oxide)] complex was considered, respectively. In mechanism 2, the reaction of quinoline N-oxide with (Ph)(PMe3)Pd(κ2-OAc) starting complex was examined for the purpose of comparing the regioselectivity of C–H activation of quinoline N-oxide with those of other N-heteroarene substrates widely studied experimentally and computationally.9h,23,51
Solvation Gibbs free energies were calculated for the relevant intermediates, transition states, and products involved in mechanisms 1a–c relative to each of the most stable Pd(κ1-Oquinoline N-oxide) starting complexes. In mechanism 1a (Figure 7), the transition state structure for the C–H bond activation of the quinoline N-oxide at the C8 position, TS8M1a, connects the C–H bond activation product, P8M1a, to a shallow intermediate, I8M1a, having an agostic C–H interaction with the palladium center. This intermediate is only 2.8 kcal/mol lower in energy than TS8M1a and 21.9 kcal/mol higher in energy than the [Pd(HOAc)3(κ1-Oquinoline N-oxide)]2+ starting complex, S8M1a. Formation of an agostic C–H intermediate followed by proton abstraction by an acetic acid molecule is consistent with the reported mechanism for the C–H activation reactions of oxazolone.48 In contrast, the C2–H activation pathway does not involve such an intermediate.
The transition state, TS2M1a, connects the [Pd(HOAc)3(κ1-Oquinoline N-oxide)]2+ starting complex, S2M1a, directly to the C–H bond activation product, P2M1a, via a CMD pathway, in which the Pd–C bond formation occurs concurrently with the cleavage of the C–H bond. The 15.7 kcal/mol energy difference between the transition states TS2M1a and TS8M1a results from the formation of a four-membered palladacycle in the case of C2–H activation versus a more stable five-membered palladacycle in the C8–H activation.52
Both C2–H and C8–H activation pathways proceed through the dissociation of one of the three acetic acid molecules coordinated to the palladium center in S2M1a and S8M1a followed by an outer-sphere metalation and deprotonation by the external acetic acid molecule.
The free energy diagram for mechanism 1b (Figure 8) is similar to that of mechanism 1a. The C2–H activation pathway proceeds through the six-membered inner-sphere CMD transition state structure TS2M1b, where the acetate ligand accepts the hydrogen with simultaneous formation of a Pd–C bond to generate the palladium aryl species P2M1b.
The C8–H activation pathway proceeds through the formation of the agostic C–H intermediate I8M1b followed by proton abstraction by the acetate ligand to give P8M1b. The transition state TS8M1b is 9.0 kcal/mol lower in energy than TS2M1b.
In mechanism 1c (Figure 9), both C2–H and C8–H activation reactions proceed through an inner-sphere CMD pathway. Both C–H activation products P2M1c and P8M1c are more stable than the starting palladium complexes S2M1c and S8M1c. C–H activation product P8M1c is 12.1 kcal/mol lower in energy than S8M1c.
The experimentally observed reversibility of the C–H activation step and the short lifetime of the C–H activation products indicate that the C–H activation products have higher energies in comparison to the starting structures leading to their formation. Among the three mechanistic scenarios investigated, mechanism 1a is the most plausible on the basis of the endothermicity of the C–H bond cleavage step. Natural bond order (NBO)53 atomic charge analysis was also conducted to determine the atomic charges on the Pd and carbon atoms involved in the C–H activation step in mechanisms 1a–c (see Table S7, Supporting Information). In all three mechanisms, the C8 atom was found to be more electron rich than the C2 atom (−0.443 vs −0.204 in mechanism 1a, −0.383 vs −0.12 in mechanism 1b, and −0.350 vs −0.084 in mechanism 1c), while the charge on Pd was around 0.6 in all cases. These results indicate that the preference of the electrophilic Pd2+ center for the C8–H bond can be ascribed to the greater nucleophilicity of C8 in comparison to that of C2. All three of the mechanisms discussed above are consistent with the experimentally observed regioselectivity of the C–H bond activation reactions of quinoline N-oxide at the C8 position.
Solvation Gibbs free energies of the relevant intermediates, transition states, and products involved in mechanism 2 relative to the total solvation Gibbs free energies of (Ph)(PMe3)Pd(κ2-OAc) and quinoline N-oxide are shown in Figure 10. The trans geometry between the phenyl group and the acetate ligand has been found to have a lower energy than the cis isomer.50 The previously reported optimized transition state structure for the C2–H bond activation of pyridine N-oxide was used as an initial estimate for both C2–H and C8–H bond cleavage transition states of quinoline N-oxide.
The transition state structure TS8M2 connects the (Ph)(PMe3)Pd(κ1-OAc)(κ1-Oquinoline N-oxide) starting complex, S8M2, directly to the C–H bond activation product, P8M2, via a CMD pathway. In the C2–H activation pathway, the transition state structure TS2M2 connects the C–H bond activation product, P2M2, to a Pd η2 π-complex intermediate, S2M2. With this structure as a starting point, a stepwise decrease of the interatomic distance between the quinoline N-oxide oxygen and palladium metal center resulted in the dissociation of quinoline N-oxide and formation of the palladium κ2-bound acetate intermediate, (PMe3)(Ph)Pd(κ-acetate), prior to the formation of the palladium κ1-bound quinoline N-oxide intermediate. Transition state TS2M2 is 14.9 kcal/mol lower in energy than transition state TS8M2. The C2–H bond activation product, P2M2, is 6.7 kcal/mol lower in energy than P8M2. These results are in agreement with the kinetic–thermodynamic connection: a thermodynamically more stable C–H bond activation product results from the lowest energy transition state.23 In summary, the detailed DFT calculations accurately predict the observed regioselectivity of the C–H bond activation reactions of quinoline N-oxide at the C8 position with palladium acetate in acetic acid solution. This can be explained on the basis of the relative stabilities of the five- and four-membered palladacycles formed from the C–H bond activation of quinoline N-oxide at the C8 and C2 positions, respectively, and by the higher nucleophilicity of the C8 position. The role of acetic acid solvent was evaluated by considering its coordination to the palladium metal center. All of the mechanistic pathways for the C–H bond cleavage step at the C2 position proceed through a CMD pathway. For the activation at the C8 position, mechanisms 1a and 1b proceed through an agostic C–H intermediate, while mechanisms 1c and 2 proceed through a CMD pathway. Computational studies on the complete catalytic cycle are currently underway and will be reported separately.
Conclusions
In summary, this paper describes synthetic, mechanistic, and computational studies of the palladium-catalyzed regioselective C8 arylation of quinoline N-oxides. The current method tolerates a number of functional groups in quinolines and iodoarenes and can be carried out under thermal or microwave conditions on a gram scale. Mechanistic studies indicate the key role of the C–H bond cleavage step. Computational studies by means of DFT point to the C8 cyclopalladation as the lower energy pathway under phosphine-free conditions with acetic acid as a noninnocent solvent/ligand and explain the reversal of site selectivity to C2 with phosphine-bound Pd.
Acknowledgments
Financial support by the Welch Foundation (AX-1788), the NIGMS (SC3GM105579), the Max and Minnie Tomerlin Voelcker Fund, and UTSA is gratefully acknowledged. J.L.-B. is supported by the IFARHU, National Secretariat for Science Technology and Innovation (SENACYT), and Ministry of Economic and Finance (DIPRENA-DPIP-10866-2013) of Panama. The authors acknowledge the Texas Advanced Computing Center at UT Austin for providing high-performance computing resources. Mass spectroscopic analysis was supported by a grant from the NIMHD (G12MD007591).
Supporting Information Available
The following file is available free of charge on the ACS Publications website at DOI: 10.1021/cs501813v.
Experimental, computational, and spectral details for all new compounds and all reactions reported (PDF)
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Shilov A. E.; Shul’pin G. B. Chem. Rev. 1997, 97, 2879–2932 10.1021/cr9411886. [DOI] [PubMed] [Google Scholar]; b Daugulis O.; Do H.-Q.; Shabashov D. Acc. Chem. Res. 2009, 42, 1074–1086 10.1021/ar9000058. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Engle K. M.; Mei T.-S.; Wasa M.; Yu J.-Q. Acc. Chem. Res. 2011, 45, 788–802 10.1021/ar200185g. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Hickman A. J.; Sanford M. S. Nature 2012, 484, 177–185 10.1038/nature11008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Vetter A. J.; Flaschenriem C.; Jones W. D. J. Am. Chem. Soc. 2005, 127, 12315–12322 10.1021/ja042152q. [DOI] [PubMed] [Google Scholar]; b Beck E. M.; Grimster N. P.; Hatley R.; Gaunt M. J. J. Am. Chem. Soc. 2006, 128, 2528–2529 10.1021/ja058141u. [DOI] [PubMed] [Google Scholar]; c Clot E.; Eisenstein O.; Jones W. D. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 6939–6944 10.1073/pnas.0609454104. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Neufeldt S. R.; Sanford M. S. Acc. Chem. Res. 2012, 45, 936–946 10.1021/ar300014f. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Wagner A. M.; Hickman A. J.; Sanford M. S. J. Am. Chem. Soc. 2013, 135, 15710–15713 10.1021/ja408112j. [DOI] [PubMed] [Google Scholar]; f Green A. G.; Liu P.; Merlic C. A.; Houk K. N. J. Am. Chem. Soc. 2014, 136, 4575–4583 10.1021/ja411699u. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Shi L.; Ding Y. J. Am. Chem. Soc. 2011, 133, 20218–20229 10.1021/ja205294y. [DOI] [PubMed] [Google Scholar]
- a Chen X.; Engle K. M.; Wang D.-H.; Yu J.-Q. Angew. Chem., Int. Ed. 2009, 48, 5094–5115 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lyons T. W.; Sanford M. S. Chem. Rev. 2010, 110, 1147–1169 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Baudoin O. Chem. Soc. Rev. 2011, 40, 4902–4911 10.1039/c1cs15058h. [DOI] [PubMed] [Google Scholar]; d Wencel-Delord J.; Dröge T.; Liu F.; Glorius F. Chem. Soc. Rev. 2011, 40, 4740–4761 10.1039/c1cs15083a. [DOI] [PubMed] [Google Scholar]
- a Burgin A. B.; Magnusson O. T.; Singh J.; Witte P.; Staker B. L.; Bjornsson J. M.; Thorsteinsdottir M.; Hrafnsdottir S.; Hagen T.; Kiselyov A. S.; Stewart L. J.; Gurney M. E. Nat. Biotechnol. 2010, 28, 63–70 10.1038/nbt.1598. [DOI] [PubMed] [Google Scholar]; b Denny W. A.; Cain B. F.; Atwell G. J.; Hansch C.; Panthananickal A.; Leo A. J. Med. Chem. 1982, 25, 276–315 10.1021/jm00345a015. [DOI] [PubMed] [Google Scholar]
- a Yang G.; Si Y.; Su Z. Org. Biomol. Chem. 2012, 10, 8418–8425 10.1039/c2ob26374b. [DOI] [PubMed] [Google Scholar]; b Hughes G.; Bryce M. R. J. Mater. Chem. 2005, 15, 94–107 10.1039/b413249c. [DOI] [Google Scholar]
- a Nakajima M.; Saito M.; Shiro M.; Hashimoto S.-i. J. Am. Chem. Soc. 1998, 120, 6419–6420 10.1021/ja981091r. [DOI] [Google Scholar]; b Denmark S. E.; Fan Y. J. Am. Chem. Soc. 2002, 124, 4233–4235 10.1021/ja025670e. [DOI] [PubMed] [Google Scholar]; c Saito M.; Nakajima M.; Hashimoto S. Chem. Commun. 2000, 19, 1851–1852 10.1039/b005994n. [DOI] [Google Scholar]
- a Wen P.; Li Y.; Zhou K.; Ma C.; Lan X.; Ma C.; Huang G. Adv. Synth. Catal. 2012, 354, 2135–2140 10.1002/adsc.201200195. [DOI] [Google Scholar]; b Liu B.; Huang Y.; Lan J.; Songa F.; You J. Chem. Sci. 2013, 4, 2163–2167 10.1039/c3sc50348h. [DOI] [Google Scholar]
- a Campeau L. C.; Rousseaux S.; Fagnou K. J. Am. Chem. Soc. 2005, 127, 18020–18021 10.1021/ja056800x. [DOI] [PubMed] [Google Scholar]; b Leclerc J.-P.; Fagnou K. Angew. Chem., Int. Ed. 2006, 45, 7781–7786 10.1002/anie.200602773. [DOI] [PubMed] [Google Scholar]; c Kanyiva K. S.; Nakao Y.; Hiyama T. Angew. Chem., Int. Ed. 2007, 46, 8872–8874 10.1002/anie.200703758. [DOI] [PubMed] [Google Scholar]; d Cho S. H.; Hwang S. J.; Chang S. J. Am. Chem. Soc. 2008, 130, 9254–9256 10.1021/ja8026295. [DOI] [PubMed] [Google Scholar]; e Larivee A.; Mousseau J. J.; Charette A. B. J. Am. Chem. Soc. 2008, 130, 52–54 10.1021/ja710073n. [DOI] [PubMed] [Google Scholar]; f Campeau L. C.; Stuart D. R.; Leclerc J.-P.; Bertrand-Laperle M.; Villemure E.; Sun H. Y.; Lasserre S.; Guimond N.; Lecavallier M.; Fagnou K. J. Am. Chem. Soc. 2009, 131, 3291–3306 10.1021/ja808332k. [DOI] [PubMed] [Google Scholar]; g Wu J.; Cui X.; Chen L.; Jiang G.; Wu Y. J. Am. Chem. Soc. 2009, 131, 13888–13889 10.1021/ja902762a. [DOI] [PubMed] [Google Scholar]; h Sun H.-Y.; Gorelsky S. I.; Stuart D. R.; Campeau L.-C.; Fagnou K. J. Org. Chem. 2010, 75, 8180–8189 10.1021/jo101821r. [DOI] [PubMed] [Google Scholar]; i Ackermann L.; Fenner S. Chem. Commun. 2011, 47, 430–432 10.1039/c0cc02360d. [DOI] [PubMed] [Google Scholar]; j Xiao B.; Liu Z.-J.; Liu L.; Fu Y. J. Am. Chem. Soc. 2013, 135, 616–619 10.1021/ja3113752. [DOI] [PubMed] [Google Scholar]; k Liu W.; Li Y.; Wang Y.; Kuang C. Org. Lett. 2013, 15, 4682. 10.1021/ol4019776. [DOI] [PubMed] [Google Scholar]; l Kaneko E.; Matsumoto Y.; Kamikawa K. Chem.—Eur. J. 2013, 19, 11837. 10.1002/chem.201302083. [DOI] [PubMed] [Google Scholar]; m Willis N. J.; Smith J. M. RSC Adv. 2014, 4, 11059. 10.1039/c3ra44411b. [DOI] [Google Scholar]; n Liu W.; Yu X.; Liab Y.; Kuang C. Chem. Commun. 2014, 50, 9291. 10.1039/C4CC04129A. [DOI] [PubMed] [Google Scholar]; o Gao Q.; Gu D.-W.; You S.-L. ACS Catal. 2014, 4, 2741. 10.1021/cs500813z. [DOI] [Google Scholar]; p Suresh R.; Muthusubramanian S.; Kumaran R. S.; Manickam G. Asian J. Org. Chem. 2014, 3, 604–611 10.1002/ajoc.201400013. [DOI] [Google Scholar]; q Kianmehr E.; Rezaeefard M.; Rezazadeh Khalkhali M.; Khan K. M. RSC Adv. 2014, 4, 13764–13767 10.1039/c4ra01034e. [DOI] [Google Scholar]
- a Do H.-Q.; Kashif Khan R. M.; Daugulis O. J. Am. Chem. Soc. 2008, 130, 15185–15192 10.1021/ja805688p. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Xiao Q.; Ling L.; Ye F.; Tan R.; Tian L.; Zhang Y.; Li Y.; Wang J. J. Org. Chem. 2013, 78, 3879–3885 10.1021/jo4002883. [DOI] [PubMed] [Google Scholar]; c Wu Z. Y.; Song H. Y.; Cui X. L.; Pi C.; Du W. W.; Wu Y. J. Org. Lett. 2013, 15, 1270–1273 10.1021/ol400178k. [DOI] [PubMed] [Google Scholar]; d Shen Y.; Chen J.; Liu M.; Ding J.; Gao W.; Huang X.; Wu H. Chem. Commun. 2014, 50, 4292–4295 10.1039/c3cc48767a. [DOI] [PubMed] [Google Scholar]; e Larionov O. V.; Stephens D.; Mfuh A.; Chavez G. Org. Lett. 2014, 16, 864–867 10.1021/ol403631k. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Shen Y.; Chen J.; Liu M.; Ding J.-c.; Gao W.; Huang X.; Wu H. Chem. Commun. 2014, 50, 4292–4295 10.1039/c3cc48767a. [DOI] [PubMed] [Google Scholar]
- Nakao Y.; Kanyiva K. S.; Hiyama T. J. Am. Chem. Soc. 2008, 130, 2448–2449 10.1021/ja710766j. [DOI] [PubMed] [Google Scholar]
- Lewis J. C.; Bergman R. G.; Ellman J. A. J. Am. Chem. Soc. 2007, 129, 5332–5333 10.1021/ja070388z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue D.; Jia Z.-H.; Zhao C.-J.; Zhang Y.-Y.; Wang C.; Xiao J. Chem. Eur. J. 2014, 20, 2960–2965 10.1002/chem.201304120. [DOI] [PubMed] [Google Scholar]
- Seiple I. B.; Su S.; Rodriguez R. A.; Gianatassio R.; Fujiwara Y.; Sobel A. L.; Baran P. S. J. Am. Chem. Soc. 2010, 132, 13194–13196 10.1021/ja1066459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak J.; Kim M.; Chang S. J. Am. Chem. Soc. 2011, 133, 3780–3783 10.1021/ja111670s. [DOI] [PubMed] [Google Scholar]
- Shibata T.; Matsuo Y. Adv. Synth. Catal. 2014, 7, 1516–1520 10.1002/adsc.201400223. [DOI] [Google Scholar]
- Konishi S.; Kawamorita S.; Iwai T.; Steel P. G.; Marder T. D.; Sawamura M. Chem. Asian J. 2014, 9, 434–438 10.1002/asia.201301423. [DOI] [PubMed] [Google Scholar]
- Hwang H.; Kim J.; Jeong J.; Chang S. J. Am. Chem. Soc. 2014, 136, 10770–10776 10.1021/ja5053768. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Qi Z.; Li X. Angew. Chem., Int. Ed. 2014, 53, 10794–10798 10.1002/anie.201406747. [DOI] [PubMed] [Google Scholar]
- Jeong J.; Patel P.; Hwang H.; Chang S. Org. Lett. 2014, 16, 4598–4601 10.1021/ol502173d. [DOI] [PubMed] [Google Scholar]
- Tan Y.; Barrios-Landeros F.; Hartwig J. F. J. Am. Chem. Soc. 2012, 134, 3683–3686 10.1021/ja2122156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelsky S. I. Coord. Chem. Rev. 2013, 257, 153–164 10.1016/j.ccr.2012.06.016. [DOI] [Google Scholar]
- Petit A.; Flygare J.; Miller A. T.; Winkel G.; Ess D. H. Org. Lett. 2012, 14, 3680–3683 10.1021/ol301521n. [DOI] [PubMed] [Google Scholar]
- Zhao D.; Wang W.; Lian S.; Yang F.; Lan J.; You J. Chem. Eur. J. 2009, 15, 1337–1340 10.1002/chem.200802001. [DOI] [PubMed] [Google Scholar]
- Granell J.; Martínez M. Dalton Trans. 2012, 41, 11243–11258 10.1039/c2dt30866e. [DOI] [PubMed] [Google Scholar]
- Sharma A.; Vacchani D.; Van der Eycken E. Chem. Eur. J. 2013, 19, 1158–1168 10.1002/chem.201201868. [DOI] [PubMed] [Google Scholar]
- See Table S1 in the Supporting Information.
- a Coperet C.; Adolfsson H.; Khuong T.-A. W.; Yudin A. K.; Sharpless K. B. J. Org. Chem. 1998, 63, 1740–1741 10.1021/jo9723467. [DOI] [Google Scholar]; b Larionov O. V.; Stephens D.; Mfuh A. M.; Arman H. D.; Naumova A. S.; Chavez G.; Skenderi B. Org. Biomol. Chem. 2014, 12, 3026–3036 10.1039/c4ob00115j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Paudler W. W.; Jovanovic M. V. J. Org. Chem. 1983, 48, 1064–1069 10.1021/jo00155a027. [DOI] [Google Scholar]; b Wengryniuk S. E.; Weickgenannt A.; Reiher C.; Strotman N. A.; Chen K.; Eastgate M. D.; Baran P. S. Org. Lett. 2013, 15, 792–795 10.1021/ol3034675. [DOI] [PubMed] [Google Scholar]
- a Keith J. M. J. Org. Chem. 2010, 75, 2722–2725 10.1021/jo1001017. [DOI] [PubMed] [Google Scholar]; b Londregan A. T.; Jennings S.; Wei L. Q. Org. Lett. 2010, 12, 5254–5257 10.1021/ol102301u. [DOI] [PubMed] [Google Scholar]; c Londregan A. T.; Jennings S.; Wei L. Org. Lett. 2011, 13, 1840–1843 10.1021/ol200352g. [DOI] [PubMed] [Google Scholar]
- Yin J.; Xiang B.; Huffman M. A.; Raab C. E.; Davies I. W. J. Org. Chem. 2007, 72, 4554–4557 10.1021/jo070189y. [DOI] [PubMed] [Google Scholar]
- Shichiri K.; Funakoshi K.; Saeki S.; Hamana M. Chem. Pharm. Bull. 1980, 28, 493–499 10.1248/cpb.28.493. [DOI] [Google Scholar]
- Konno K.; Hashimoto K.; Shirahama H.; Matsumoto T. Heterocycles 1986, 24, 2169–2172 10.3987/R-1986-08-2169. [DOI] [Google Scholar]
- Stephens D. E.; Chavez G.; Valdes M.; Dovalina M.; Arman H.; Larionov O. V. Org. Biomol. Chem. 2014, 12, 6190–6199 10.1039/C4OB01088D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhmutov V. I.; Berry J. F.; Cotton F. A.; Ibragimov S.; Murillo C. A. Dalton Trans. 2005, 1989–1992 10.1039/b502122g. [DOI] [PubMed] [Google Scholar]
- Akhmadullina N. S.; Cherkashina N. V.; Kozitsyna N. Y.; Stolarov I. P.; Perova E. V.; Gekhman A. E.; Nefedov S. E.; Vargaftik M. N.; Moiseev I. I. Inorg. Chim. Acta 2009, 362, 1943–1951 10.1016/j.ica.2008.09.042. [DOI] [Google Scholar]
- Adrio L. A.; Nguyen B. N.; Guilera G.; Livingston A. G.; Hii K. K. Catal. Sci. Technol. 2012, 2, 316–323 10.1039/c1cy00241d. [DOI] [Google Scholar]
- Evans J.; O’Neill L.; Kambhampati V. L.; Rayner G.; Turin S.; Genge A.; Dent A. J.; Neisius T. J. Chem. Soc., Dalton Trans. 2002, 2207–2212 10.1039/b200617k. [DOI] [Google Scholar]
- See Figure S1 in the Supporting Information.
- See Figure S2 in the Supporting Information.
- Karayannis N. M.; Pytlewski L. L.; Mikulski C. M. Coord. Chem. Rev. 1973, 11, 93–159 10.1016/S0010-8545(00)82007-X. [DOI] [Google Scholar]
- a Kalyani D.; Deprez N. R.; Desai L. V.; Sanford M. S. J. Am. Chem. Soc. 2005, 127, 7330–7331 10.1021/ja051402f. [DOI] [PubMed] [Google Scholar]; b Daugulis O.; Zaitsev V. G. Angew. Chem., Int. Ed. 2005, 44, 4046–4048 10.1002/anie.200500589. [DOI] [PubMed] [Google Scholar]; c Chiong H. A.; Pham O.-N.; Daugulis O. J. Am. Chem. Soc. 2007, 129, 9879–9884 10.1021/ja071845e. [DOI] [PubMed] [Google Scholar]
- Molecular ions that may correspond to 39 were detected by ESI-MS. Less than 2% C8–H/D exchange was detected below 100 °C, indicating that cyclopalladation at lower temperatures is slow.
- Simmons E. M.; Hartwig J. F. Angew. Chem., Int. Ed. 2012, 51, 3066–3072 10.1002/anie.201107334. [DOI] [PubMed] [Google Scholar]
- a Puri M.; Gatard S.; Smith D. A.; Ozerov O. V. Organometallics 2011, 30, 2472–2482 10.1021/om1008956. [DOI] [Google Scholar]; b Dong Z.-B.; Manolikakes G.; Shi L.; Knochel P.; Mayr H. Chem. Eur. J. 2010, 16, 248–253 10.1002/chem.200902132. [DOI] [PubMed] [Google Scholar]; c Consorti C. S.; Ebeling G.; Flores F. R.; Rominger F.; Dupont J. Adv. Synth. Catal. 2004, 346, 617–624 10.1002/adsc.200303228. [DOI] [Google Scholar]; d Stille J. K.; Lau K. S. Y. Acc. Chem. Res. 1977, 10, 434–442 10.1021/ar50120a002. [DOI] [Google Scholar]
- a Deprez N. R.; Sanford M. S. J. Am. Chem. Soc. 2009, 131, 11234–11241 10.1021/ja904116k. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Canty A. J.; Ariafard A.; Sanford M. S.; Yates B. F. Organometallics 2013, 32, 544–555 10.1021/om301013w. [DOI] [Google Scholar]
- Weissman H.; Milstein D. Chem. Commun. 1999, 1901–1904 10.1039/a906246g. [DOI] [Google Scholar]
- Roiban G. D.; Serrano E.; Soler T.; Aullón G.; Grosu I.; Cativiela C.; Martínez M.; Urriolabeitia E. P. Inorg. Chem. 2011, 50, 8132–8143 10.1021/ic200564d. [DOI] [PubMed] [Google Scholar]
- Roiban G. D.; Serrano E.; Soler T.; Aullón G.; Grosu I.; Cativiela C.; Martínez M.; Urriolabeitia E. P. Inorg. Chem. 2011, 50, 8132–8143 10.1021/ic200564d. [DOI] [PubMed] [Google Scholar]
- a Estevan F.; Lahuerta P.; Peris E.; Ubeda M. A.; Garcia-Granda S.; Gómez-Beltrán F.; Pérez-Carreño E.; González G.; Martínez M. Inorg. Chim. Acta 1994, 218, 189–193 10.1016/0020-1693(94)03799-X. [DOI] [Google Scholar]; b Garcia-Granda S.; Lahuerta P.; Latorre J.; Martínez M.; Peris E.; Sanau M.; Ubeda M. J. Chem. Soc., Dalton Trans. 1994, 539–544 10.1039/dt9940000539. [DOI] [Google Scholar]; c González G.; Lahuerta P.; Martínez M.; Peris E.; Sanau M. J. Chem. Soc., Dalton Trans. 1994, 545–550 10.1039/dt9940000545. [DOI] [Google Scholar]; d Estevan F.; González G.; Lahuerta P.; Martínez M.; Peris E.; van Eldik R. J. Chem. Soc., Dalton Trans. 1996, 1045–1050 10.1039/dt9960001045. [DOI] [Google Scholar]; e Gómez M.; Granell J.; Martínez M. J. Chem. Soc., Dalton Trans. 1998, 37–43 10.1039/a705435a. [DOI] [Google Scholar]; f Gómez M.; Granell J.; Martínez M. Eur. J. Inorg. Chem. 2000, 217–224. [DOI] [Google Scholar]; g Favier I.; Gómez M.; Granell J.; Martínez M.; Font-Bardía M.; Solans X. Dalton Trans. 2005, 123–132 10.1039/b415613g. [DOI] [PubMed] [Google Scholar]; h Aullón G.; Chat R.; Favier I.; Font-Bardía M.; Gómez M.; Granell J.; Martínez M.; Solans X. Dalton Trans. 2009, 8292–8300 10.1039/b905134a. [DOI] [PubMed] [Google Scholar]; i Engle K. M.; Wang D. H.; Yu J.-Q. J. Am. Chem. Soc. 2010, 132, 14137–14151 10.1021/ja105044s. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Ackermann L. Chem. Rev. 2011, 111, 1315–1345 10.1021/cr100412j. [DOI] [PubMed] [Google Scholar]; k Esteban J.; Martínez M. Dalton Trans. 2011, 40, 2638–2644 10.1039/c0dt01228a. [DOI] [PubMed] [Google Scholar]
- a Gorelsky S. I.; Laponite D.; Fagnou K. J. Org. Chem. 2012, 77, 658–668 10.1021/jo202342q. [DOI] [PubMed] [Google Scholar]; b Gorelsky S. I.; Laponite D.; Fagnou K. J. Am. Chem. Soc. 2008, 130, 10848–10849 10.1021/ja802533u. [DOI] [PubMed] [Google Scholar]; c Potavathri S.; Pereira K. C.; Gorelsky S. I.; Pike A.; LeBris A. P.; DeBoef B. J. Am. Chem. Soc. 2010, 132, 14676–14681 10.1021/ja107159b. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Gorelsky S. I. Organometallics 2012, 31, 794–797 10.1021/om2012612. [DOI] [Google Scholar]
- Dupont J.; Consorti C. S.; Spencer J. Chem. Rev. 2005, 105, 2527–2571 10.1021/cr030681r. [DOI] [PubMed] [Google Scholar]
- Glendening E. D.; Reed A. E.; Carpenter J. E.; Weinhold F.. NBO Version 3.1.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.