

Abstract
The detection and inactivation of pathogenic strains of bacteria continues to be an important therapeutic goal. Hence, there is a need for materials that can bind selectively to specific microorganisms, for diagnostic or anti-infective applications, but which can be formed from simple and inexpensive building blocks. Here, we exploit bacterial redox systems to induce a copper-mediated radical polymerisation of synthetic monomers at cell surfaces, generating polymers in situ that bind strongly to the microorganisms which produced them. This ‘bacteria-instructed synthesis’ can be carried out with a variety of microbial strains, and we show that the polymers produced are self-selective binding agents for the ‘instructing’ cell types. We further expand on the bacterial redox chemistries to ‘click’ fluorescent reporters onto polymers directly at the surfaces of a range of clinical isolate strains, allowing rapid, facile and simultaneous binding and visualisation of pathogens.
The recognition and inactivation of pathogenic microorganisms remains a scientific challenge and a practical problem of enormous significance.1 Conventional antibiotics have been extremely successful in combating microbial infections, but the emergence of resistant strains of many pathogens is an increasing concern. New approaches to prevent bacterial infections are required that do not invoke the selection of resistant populations.2 Non-lethal means for targeting bacteria include inactivating their invasive pathways, for example by disrupting cell-cell signalling mechanisms known as Quorum Sensing within microbial populations,3-5 or, more simply, by sequestering bacteria away from an infective site.6 The latter route is attractive also from a diagnostic perspective,7 as the binding of a specific organism may facilitate detection of pathogens7,8 and also aid in choice of therapeutic. However, the selective binding of specific bacterial species and/or strains is difficult and in current practice requires expensive ‘cold-chain’ reagents such as antibodies and aptamers which precludes their use in non-hospital environments or in developing nations. Accordingly, we have been interested in developing a route to cell-binding agents that does not require delicate and expensive biological affinity agents, and which can be tailored to produce sequestrants for a wide range of biological targets with minimal changes in methodology.
Approaches to cell-binding agents have included soft-lithography, molecular imprinting, and multivalent carbohydrate-receptor mediated cell capture.9-15 While each technique has advantages and a key recent paper has shown the application of toxin-binding polymers in vivo16 nevertheless, there is no current platform concept which can be used to generate materials which might be adapted for different targets as desired. Of particular utility would be enhanced methods for generating polymeric agents which are hydrophilic, soluble and derived from accessible precursors, as such materials are already widely used in diagnostic assays. Hydrophilic polymers are of note too since nearly all bacteria produce complex macromolecules in the form of an Extra-Cellular Matrix (ECM). The ECM helps to support cell communities and to tailor niche environments to suit the bacterial population as a whole. It would be particularly advantageous for targeting bacteria if synthetic mimics of these ECM materials could be produced, ideally by a process which exploits natural metabolic processes. For example, bacteria adapt to their surroundings with a variety of redox enzyme cascades and metal-binding/efflux systems. Consideration of copper-homeostasis mechanisms in Escherichia coli (E. coli) and other bacteria17-19 suggested to us that a wholly synthetic ECM production process, i.e. free radical polymerisation catalysed by copper species, could be induced from cell populations by hijacking the copper-binding and redox pathways with synthetic monomers rather than biological precursors. In such a way, bacteria might be directed into producing a synthetic ECM rather than a natural one, and at the same time entrap themselves in an environment rather different to that intended in their normal surroundings,
We report here the first example wherein bacterial metabolic processes have been co-opted for the synthesis of acrylic polymers by copper catalysed ATRP, and in such a way that the polymers inherently grow from monomers bound at the bacterial cell surface. In this manner, the bacteria select their own binding agents, and grow different polymers at their surfaces compared to polymers formed in solution, or in the absence of bacteria. We show further that monomer composition for the resultant materials is affected by the templating process and leads to polymers that can be specific binding agents for the bacteria which templated them – a process we denote as “bacteria-instructed synthesis” (Figure 1) as in effect the bacteria direct the formation of polymers from their surfaces. The same methodology can be applied to similarly catalysed processes such as azide-alkyne cycloadditions, a process that provides a simple in-situ labelling protocol (Figure 1). In addition, we have shown that these protocols have application across a range of bacteria, including clinically relevant pathogenic strains.
Figure 1. Schematic of the ‘bacteria-instructed synthesis’ process.
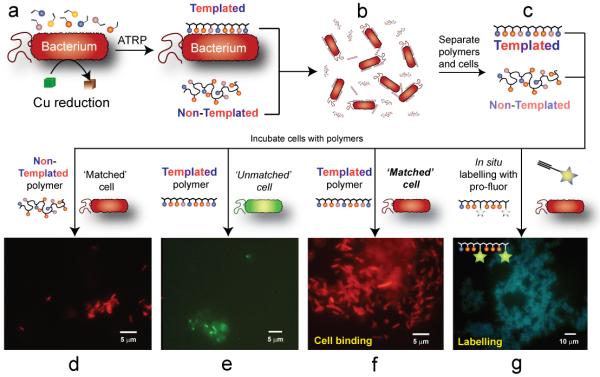
In (a) bacteria induce polymerisation in monomer / catalyst suspensions to generate a synthetic extra-cellular matrix of polymers (b). Recovery of polymers from the suspensions leads to two fractions (c), with polymer obtained from the aqueous phase suspension around the bacteria denoted as ‘non-templated’ and a second fraction obtained from a wash of the cell surfaces denoted as ‘templated’. Incubation of polymers with bacteria results in low binding of cells for which the polymer is non-templated (d), or where a polymer templated with one cell type (shown in orange) is incubated with a cell (shown in green) of another type (e). Addition of a polymer, templated by one cell type, with its own ‘matched’ cell population results in the formation of large polymer – cell clusters (g), as the templated polymers sequester the bacteria which ‘instructed’ their formation with high affinity. The same reducing environment at bacterial surfaces which aids the polymer synthesis can also be used to label the cells in situ (g) via pro-fluorescent markers, which react with cell-surface bound polymers containing ‘clickable’ residues.
The first part of our strategy involved development of a novel bacteria-mediated Atom Transfer Radical Polymerisation (b-ATRP) process. Key papers describing the mechanisms of ATRP and SET-LRP have shown that reduction of copper (II) species is critical in controlling the radical generation and regeneration processes that lead to pseudo-living polymerisations.20-25 It has also been established that certain bacterial strains ensure safe copper handling under varying environmental conditions by binding and redox cascades. For example, the Cu(I)-translocating P-type ATPase CopA, the central component in copper homeostasis, is responsible for removing excess Cu(I) from the cytoplasm.26 The multi-copper oxidase CueO and the multi-component copper transport system CusCFBA act to safeguard the periplasmic space from copper-induced toxicity. We reasoned therefore that the reducing activity of certain bacteria via respiratory chain components, for example in E. coli by quinones and, to a lesser degree, NADH dehydrogenase might be sufficient to generate the Cu(I) catalytic species required to initiate an ATRP-type polymerization.26,27
We chose two specific monomers for bacterial templating, a permanent cation, ([2-(methacryloyloxy)-N,N,N-trimethylethanaminium chloride-(TMAEMA), which we expected would bind strongly to negatively charged cell surfaces, and a zwitterionic sulfobetaine, [2-(N-3-Sulfopropyl-N,N-dimethyl ammonium)ethyl methacrylate, (MEDSA) which was intended to aid polymer solubility as well as act as a ‘spacer’ between binding cationic sections. Long-chain quaternised amines are toxic to bacteria, but polymers prepared from the shorter chain TMAEMA as well as the sulfobetaine derivative were found not to cause significant cell death during preliminary experiments (data not shown). Poly(2-TMAEMA) was found to be a potent sequestrant for a range of bacteria, while poly(MEDSA) showed a much lower ability to cluster the same cell types (ESI). These experiments thus defined the intended boundary conditions of polymer space, i.e. a highly potent binder for all bacteria (pTMAEMA) at one extreme, and a low-binding affinity, highly cell compatible polymer (pMEDSA) at the other.
Investigations into the redox potential of bacterial growth suspensions confirmed the presence of a highly reductive environment suitable for activating ATRP-type reactions across a range of cell types, including model strains of the clinically important species E. coli and P. aeruginosa (MG1655 and PAO1, respectively). In typical experiments with E. coli strains, the redox potential in suspensions after washing and concentrating the cells was Eh = −244 mV. We therefore considered that adaptations of the Activators Generated by Electron Transfer (AGET) ATRP and Single Electron Transfer methodologies, in which catalytically active Cu(I) catalytic species are continually regenerated under reducing conditions, should enable polymers to be prepared with very low amounts of added copper. Polymerisations were carried out with the TMAEMA and MEDSA monomers both in the absence and in the presence of bacteria, Cu(II)Br2, ATRP ligands and an ATRP-initiator and the resultant polymers recovered by successive wash and filter steps. As apparent from Figure 2, good control over polymerisation kinetics could be achieved through tailoring ratios of ATRP initiator: copper (II): ligand. The total Cu content in these reactions was 4.42 ng/mL, well below concentrations causing cytotoxic effects.
Figure 2. Generation of a reductive environment during bacteria-instructed synthesis and evaluation of the cell-binding properties of the resultant polymers.

Polymers grown in the presence of bacteria (‘templated’) exhibit different properties compared to those grown in the same conditions but without the cells (‘control’). In (A) are shown (i) the changes in redox potential of suspensions (red line) as E. coli cells proliferate and enter stationary phase (optical density at 600 nm, green line), and in (ii) the kinetics of polymer growth in E coli suspensions at different ratios of polymerisation initiator: copper (II): ligand. In (B) the templating process is shown schematically, while in (C), fluorescence microscopy shows clusters of mCherry-labelled bacteria in presence of templated polymer, indicating a high affinity of the polymer for the cell type by which it was templated. This is in contrast to the isolated cells observed after incubation with control polymer after 30 min (insets show phase contrast images from the selected sections to show depth of clustering). In (D) bacterial aggregation is quantified via Coulter counter analysis of polymer-bacteria clusters: in D(i) ~ 50 μm clusters are apparent within 15 min for the templated species in contrast to the lack of cluster formation (D(ii) for the control polymers. In (E) optical density measurements confirm rapid binding and sequestration of E. coli by polymers grown in the presence of E. coli and recovered from the cells by a high salt wash step (‘strongly-templated’ polymers).
Having established that the bacteria could indeed promote polymerisation, polymers were isolated and subsequently analysed in terms of monomer composition and their ability to cluster bacteria. Our working hypothesis was that monomers in closest proximity to the cell surface, i.e. those bound or closest to the bacterial outer membranes, should polymerise first, while those less strongly associated could form polymers also when they diffused into the ‘reductive zone’ nearest the bacteria, where a favourable ratio between Cu(I)/Cu(II) activating/deactivating catalytic species can be achieved. We thus expected that two predominant populations of polymers would be produced: the first population being polymers grown at and templated by the bacterial surface, and a second more random-sequenced polymer formed in the solution away from the cell surface. In turn, we expected the strongly-templated polymers (denoted hereafter as STPs) to have a sequence of monomers encoded in their structure that mirrored components of the bacterial surface, and hence a higher propensity to bind the cells that templated them, compared to those of the polymers formed more extensively in solution (denoted as Weakly-Templated Polymers, or WTPs). Furthermore, the polymers grown in the presence of bacteria were expected to differ from control polymers (CPs) grown under the same conditions using ascorbic acid as the reducing agent but in the absence of cells.
Initial experiments indicated that, as hypothesised, two populations of polymers could be recovered from polymerisations carried out in the presence of bacteria. Weakly bound WTPs, dissolved in the supernatant, were isolated by removal of the bacterial clusters by centrifugation. A second fraction of polymers (STP) was recovered by resuspension of the centrifuged bacteria in high ionic strength buffer NaCl (0.15M, aq) followed by a second centrifugation step. The polymers recovered from the higher ionic strength salt solution, i.e. the strongly-templated fraction showed much enhanced cell binding (as denoted by cluster formation, Figure 2D) than fractions of polymers obtained from bacterial culture supernatants after centrifugation (the WTP fraction). Not only were cell clusters formed to a greater extent with templated polymers, but also aggregation and sequestration of cells took place more quickly than with WTPs or CPs in the same concentrations and under the same experimental conditions (Fig 2D and E).
The ability of templated polymers to exhibit specificity for the cell surfaces on which they were grown immediately suggested that polymers templated by different bacteria might show different selectivities for the different cell types, even if the same monomers were utilised in the b-ATRP synthesis. Accordingly, polymers were grown in the presence of both E. coli MG1655 (mCherry) and P. aeruginosa PAO1 (pyocyanin-negative mutant ΔphzAG1 ΔphzAG2 of the Nottingham PAO1 strain (PAKR76)). Following the synthesis steps, bacteria were washed sequentially with water and NaCl (0.15M, aq) to yield distinct STP and WTP polymer populations which varied in their composition (Table 1). Marked differences were observed in co-monomer ratios in the templated polymer fractions and those in the control polymers, and these differences in composition were outside the range based on reactivity ratios of the monomers (Table 1 and Table S02, ESI).
Table 1.
Monomer feed composition, conversion and final composition for control and templated polymers
| Polymer | Feed compositiona | Conversion | Composition (NMR)a |
|---|---|---|---|
| CP | 1:1 | 35% | 1:0.82 ± 0.04 |
| E. coli MG1655 WTP | 1:1 | 32% | 1:0.27 ± 0.16 |
| E. coli MG1655 STP | 1:1 | 32% | 1:1.06 ± 0.18 |
| P. aeruginosa PAO1 WTP | 1:1 | 45% | 1:0.35 ± 0.18 |
| P. aeruginosa PAO1 STP | 1:1 | 45% | 1:1.05 ± 0.60 |
Compositions expressed as MEDSA:TMAEMA molar ratios.
The interaction between polymers and the bacteria was probed using microscopy, light scattering measurements of cell-polymer suspensions and turbidimetry assays. The ability of polymers to bind to particular cell types was extrapolated from their ability to form bacterial aggregates. The link between binding affinity and aggregate formation is not always quantitative as it includes a kinetic component which can vary with cell concentration. However, for the constant experimental conditions in our assays, aggregate quantitation via light scattering provided a rapid and convenient readout most closely analogous to the intended diagnostic microbiology setting. The aggregate size distribution and microscopy assays which illustrate the binding /cell clustering process are shown in Figure 3.
Figure 3. Demonstration of self-selective microbial binding by ‘bacteria-instructed polymers’.

In (A) the experimental design for the cell-selective polymer binding assays is shown. Bacteria (E. coli, depicted in orange, or P. aeruginosa, shown in green) were used to template bacterial-mediated synthesis to generate two polymer fractions, shown schematically below the cells. ‘Weakly-Templated Polymers’ (WTP) were obtained from the supernatant following cell centrifugation while the more tightly-bound ‘Strongly-Templated Polymers’ (STP), were recovered from the bacteria by a salt solution wash. In (B) are shown fluorescence micrographs of experiments where polymers templated with E coli MG1655 (expressing mCherry, orange-red) and P aeruginosa PAO1 (expressing GFP, green) were incubated with their ‘matched’ bacteria (i,e. fresh suspension of the cell strains used to template synthesis) and with ‘mismatched’ bacteria (i.e. P. aeruginosa suspensions added to polymers grown from E. coli and vice versa). Micrographs labelled ‘Control’ refer to incubations of cells with polymers grown in the absence of cells. The ‘mix and match’ combinations of bacteria, the polymers templated from them and the non-templated polymers are shown by connecting red arrows for E coli cells and green arrows for P. aeruginosa (images A to B). Cell aggregation by polymers, as shown in (B), is quantified in (C), with each graph in (C i-iv) relating to microscopy images of the ‘matched’ and ‘mis-matched’ combinations of bacteria and polymers shown in (B) above. The distributions of E. coli aggregates (C i. ‘matched’) are shifted to higher sizes with E. coli-templated polymers compared to P. aeruginosa clusters with the same polymer (C ii, ‘mis-matched’), while (C iv, ‘matched’) indicates P. aeruginosa-templated polymers induce greatest cluster sizes with P. aeruginosa compared to E. coli (C iii, ‘mis-matched’), confirming the enhanced binding of ‘bacteria-instructed polymers’.
As apparent from Figure 3, the E. coli-templated polymers rapidly generated larger aggregates with their ‘matched’ bacteria i.e. E coli than with the ‘mis-matched’ cells P. aeruginosa. In addition, the P. aeruginosa-templated polymers formed larger P. aeruginosa clusters than E. coli clusters i.e. the “matched” template (STP) polymer pairs in each case (Figure 3C(i) and (iv)). Significantly, the bacterial templating effect was confirmed through quantification of the NMR integrals, which indicated there were marked differences in overall monomer composition across the sets of polymers, with increased incorporation of the quaternary ammonium TMAEMA monomer in the STPs compared to the WTP and control polymers (Table 1). Intriguingly, although the proportion of the cationic TMAEMA units was higher overall in the P. aeruginosa-templated polymers than in the E. coli-templated polymers, the polymers formed in the presence of E. coli were better overall at cluster formation for both bacteria than those synthesised in the presence of P. aeruginosa (ESI).
This suggested that the cell-binding effects of templated polymers were due to subtle variations in monomer sequence and spacing across polymer chains grown from different cell surfaces compared to those formed extending further into solution. Indeed, while we concentrated our cell binding studies on zwitterionic and quaternary ammonium-functional monomers, this difference in final monomer composition through carrying out a polymerisation in the presence of bacteria was also observed for polymers incorporating non-charged diol-functionalised monomers (glycerol methacrylate) as well as the zwitterionic components (Table S01, ESI). The diol-functionalised polymers also displayed a templating effect, with enhanced binding of bacteria by polymers grown in the presence of cells compared to polymers with the same overall monomer composition but grown in the absence of bacteria (Figure S22, ESI). In turn, this implied that some bacterial surface properties such as charge and receptor spacing were encoded into the monomer sequences in the templated polymer structures, although sensitivity of detection in NMR of block sequences did not allow unambiguous confirmation of sequence effects. In gram-negative bacteria the outer membrane is composed of tightly packed lipopolysaccharides (LPS),28 which, for E. coli, occupy ~75% of the surface.29,30 The polysaccharides are directed outwards, extending up to 10 nm from the surface, while the presence of phosphate groups and anionic carbohydrates gives the bacterial membranes an overall negative charge. The density of these charges tends to be significantly higher than in mammalian cells, a feature that has been exploited in selective antimicrobial polymers.31 For the degrees of polymerisation for both control and templated polymers calculated from NMR, and the corresponding predicted chain lengths of the polymers (5-15 nm, dependent on local charge content near the bacterial surface, Table S03, ESI), it is quite plausible that charged monomers in the templated polymers spanned complementary functional groups at the bacterial surfaces. We also found that while the strongly-templated (STP) fraction represented no more than 22 % of the total yield of polymer in a ‘b-ATRP’ synthesis, the WTP fraction was nevertheless different in co-monomer ratio compared to polymers grown in the absence of bacteria (CPs) for all the monomer combinations tried. This indicated that b-ATRP was the dominant polymerisation process in these experiments, and that the templating process generated an inherently different polymer population than in the cell-free controls. Estimated molar masses from NMR, based on overall monomer conversion indicated small differences in calculated chain lengths for WTP and STP fractions (Table S03, ESI), and GPC indicated essentially similar Mw/Mn ratios. Further experiments showed that the change in monomer composition observed in templated co-polymers compared to control polymers was not due to selective adsorption of sub-compositions from within the total fraction of polymers grown in solution. Incubation of control polymers with E coli or P. aeruginosa and subsequent water and salt washes yielded identical monomer compositions within the adsorbed fractions to those of the parent control polymer, and no co-monomer compositions matching those of the templated polymers were observed.
Having demonstrated that two different species of bacteria could template the synthesis of polymeric sequestrants, we sought to expand the b-ATRP and ‘bacteria-instructed synthesis’ approach to more challenging and clinically important outcomes. Specifically, we aimed to template polymer growth from wild-type bacteria or clinical isolates, which exhibit rather different and variable cell surface components compared to laboratory strains.32 Preparation of polymers by this method might be an important platform method for generating cell binding or detecting agents as bacterial surface structures and capsules vary widely across clinical isolates, and sequestrants optimised to bind a lab strain might be ineffective against a newly emerging pathogenic strain.
A uropathogenic bacterium, the clinical isolate E. coli 539 (GFP-labelled), was selected and evaluated for b-ATRP polymer synthesis. The same set of template monomers was used as before, and polymers were formed rapidly in the presence of E coli 536 GFP. As apparent from Figure 4, polymers grown in the presence of the clinical strain were successful sequestrants of E coli 536 GFP, despite the presence of capsule surface components on this bacterium that were not present in the lab strains originally used to select monomers for cell binding.32,33 Thus even for the unoptimised monomer set for this cell type, the b-ATRP approach generated polymers able to bind strongly their templating bacteria.
Figure 4. Synthesis in presence of pathogen analogue bacterial strains and in situ labelling of clinical isolates.

Polymers were grown, as shown schematically in (A i), in suspensions of uropathogenic E. coli 536 GFP and the resultant polymer fractions were separated with salt washes as before to recover templated polymer (STP). Micrographs (A) of subsequent binding experiments with E. coli 536 GFP shows pronounced clustering of cells in fluorescence mode (A ii), with size and scale of aggregates apparent in the magnified phase contrast image (A iii). In (B) is shown schematically in situ labelling of cells via bacterial cell-instructed (“b-click”) chemistry. Incubation of non-fluorescent E.coli MG1655 with 3-azido-7-hydroxycoumarin (1) maintains dispersed suspension of cells (C i), while addition of cationic polymer (P1) results in cell clustering (C ii) Reduction of copper at the cell surface leads to no change in cell cluster state, but marked fluorescence in microscopy image (C iii). Efficiency of “b-click” shown in expanded merged phase contrast and fluorescence images (D i) and in the wells of assay plates (D ii). Image capture on a mobile phone camera (D iii) indicates ability to detect pathogenic bacteria including Escherichia coli, Clostridium difficile, Yersinia pseudotuberculosis, Helicobacter pylori and Campylobacter jejuni even in un-optimised conditions. White circles have been added to the image to aid identification of wells, control wells containing bacteria but no labelling components are shown above the wells containing in situ-labelled bacteria.
The culminating experiments sought to demonstrate not only a cell capture application for the b-ATRP but also a diagnostic component. The widely-utilised copper-catalysed Huisgen cycloaddition,34 an important member of the ‘click chemistry’ family of chemical transformations, was an obvious choice for further exploiting the bacterial redox cascades. We therefore prepared a pro-fluorescent marker compound, with a terminal azide, and a bacterial-binding cationic polymer with pendant acetylenic groups. Incubation of a non-labelled non-fluorescent E. coli MG1655 strain with mixtures of individual polymer, azide or copper(II) species yielded no fluorescence. By contrast, within 5 minutes of administration of E. coli suspensions to a mixture of the cell-binding acetylenic polymer, a highly fluorescent suspension resulted (Figure 4). These experiments also showed aggregation of the bacteria into large fluorescent clusters, indicating an experimentally simple dual sequestration and in-situ (‘b-click’) labelling of bacteria in a single step. For practical applications, the ability to label a range of cell types in a non cold-chain low-tech environment is advantageous, thus we used the same ‘b-click’ approach to label bacterial cultures directly in the plates. The strains used ranged across known pathogens such as Clostridium difficile, Yersinia pseudotuberculosis, Helicobacter pylori and Campylobacter jejuni, and fluorescence was apparent immediately on addition of labelling reagent. We deliberately did not optimise conditions for these tests to establish the feasibility of a simple diagnostic test, but noted that varying intensities of fluorescent output were obtained across the range of bacteria, suggesting that different polymers and in situ labelling species could be adopted to fine tune this assay for cell selectivity. Finally, we used a simple mobile phone camera to record the fluorescence output directly from the 96-well plates (Figure 4 C iii) in ambient conditions thus demonstrating the flexibility of the ‘b-click’ labelling methodology.
Taken together, the experiments show that bacterial metabolic processes are versatile enough not only to grow synthetic polymers by a controlled radical mechanism, but also to label polymer side-chains in situ via azide-alkyne chemistry. The analogies between the b-ATRP process and ‘click’ chemistries were readily demonstrated by the ‘b-click’ reactions of the pro-fluorescent markers at the surfaces of a range of cells. The implications of this work are therefore apparent in a detection setting wherein selective binding of one cell type be a specific polymer could be further amplified by a labelling reaction which can only occur at the surface of the desired bacterium to be detected. It is important to note that the cell-mediated chemistries employ readily-available materials which do not require cold-chain storage, and thus are adaptable for less-specialised laboratory settings. We also used a fractional sub-set of the many possible monomers for the b-ATRP reactions, suggesting that further refinement of binding specificity should be possible through judicious choice and variation of polymer components. We therefore believe this method could, if developed further, define a new platform of materials that can be adapted for a range of cell-specific diagnostic and therapeutic applications as desired.
Methods
Materials
Monomers were prepared according to literature procedures as detailed in Supporting Information. All other chemicals were purchased from Sigma-Aldrich® or Acros® and used without further purification. All solvents were HPLC grade, purchased from Sigma-Aldrich® or Fisher Scientific®, and used without further purification.
Escherichia coli MG1655 was obtained from stocks held within Nottingham University. Fluorescent E. coli MG1655 was generated using a plasmid obtained from the Tsien laboratories35 and the plasmid inserted using electroporation, before selecting the mutants which had taken up the plasmid using 100 μg/mL ampicillin LB plates and media. Fluorescence was increased by growing in the presence of 0.2% arabinose. The GFP-labelled strain used for fluorescent microscopy and rebinding studies was a P. aeruginosa PAO1 (Nottingham) wild type containing the pmE6032 GFP plasmid. The P. aeruginosa strain used for b-ATRP and click chemistry was a pyocyanin-negative mutant (ΔphzAG1 ΔphzAG2) of the Nottingham PAO1 strain (PAKR76).
Microbial templated polymers
p(TMAEMA-co-MEDSA) (P1)
Control polymer by conventional AGET ATRP
To a reaction flask, 144 mg (0.695 mmol) of TMAEMA, 194 mg (0.695 mmol) of MEDSA, 1.554 mg (5.6 μmol) of In1, 200 μL of a 0.069 M aqueous solution with CuBr2 and TPA, and 50 μL of DMSO were added. This mixture was deoxygenated with gentle nitrogen bubbling for 30 minutes over ice after which 270 μL of a degassed 1 mg/mL solution of ascorbic acid were added to begin the polymerisation. The polymerisation was monitored by 1H-NMR spectroscopy over time and when the desired conversion was reached (Table S01) the polymerisation was terminated by exposing to air. The polymers were obtained by dialysis against water for 3 days followed by freeze-drying to yield a white amorphous solid (CP). 1H-NMR (D2O, 400 MHz) δ (ppm): 1.0-2.0 (m, 6H, CH3), 2.28 (m, 2H, CH2SO3), 2.98 (m, 2H, CH2CH2SO3), 3.60 (m, 2H, CH2CH2CH2SO3), 3.2 (m, 15H, N(CH3)), 3.78 (m, 4H, NCH2), 4.49 (m, 4H, COCH2).
Microbial directed polymer synthesis by b-ATRP
To a reaction flask, 144 mg (0.695 mmol) of TMAEMA, 194 mg (0.695 mmol) of MEDSA, 1.55 mg (5.60 μmol) of the morpholine initiator In1 and 50 μL of DMSO were added. This mixture was mixed with bacteria as a 7 mL suspension with an optical density at 600 nm of 93.6 and deoxygenated for 30 minutes over ice after which 200 μL of a degassed 0.69 mM aqueous solution with CuBr2 and TPA were added to begin the polymerisation. The reaction was monitored by 1H-NMR spectroscopy when the desired conversion was reached (Table S01) the polymerisation was terminated by exposing to air. Polymers were obtained from the reaction by first washing the cells with deionised water (WTPs) (3 × 5 mL) followed by washing with a saturated solution of sodium chloride (0.15M aq) (STPs) (3 × 5 mL). These two separated solutions were then dialysed against water for 3 days followed by freeze drying to obtain the polymers as a white amorphous solid.
Details of fluorescent monomer and polymer syntheses for bacterial labelling experiments are given in the Electronic Supporting Information (ESI).
Aggregation Experiments
Bacterial aggregation by turbidimetry
The ability of the polymers to aggregate bacteria was initially evaluated by turbidimetry experiments. Briefly, polymer solutions were prepared at a concentration of 1 mg/mL in sterile deionised water. Bacteria were grown to an optical density at 600 nm (OD600) such that they were still in the exponential phase of their growth curve (OD600 around 0.4), at which point, they were washed once with PBS and twice with sterile deionised water. The cells were finally resuspended to a cell density such that when they were mixed with the polymer solutions the OD600 of the suspension was ≈ 1.9. The polymer solution (0.5 mL) was added to a UV cuvette followed by 1 mL of the bacteria suspension. The OD600 was quickly recorded (t0) and the change in OD600 was monitored with time.
Polymer-bacteria cluster measurements
Size distributions of bacterial clusters were determined under moderate stirring (default speed 5 setting) to the required concentration as indicated by the in-built display software. Particle size ranges were defined using PSS-Duke standards (Polymer Standard Service, Kromatek Ltd, Dunmow, UK). Particle size distribution was then determined as a function of the particle diffraction using the Coulter software (version 2.11a) and plotted as a function of the percentage of distribution volume.
In a typical experiment, 200 μL of a bacterial suspension with an OD600 of 1.9 were added to the flow cell (~ 14 mL) to obtain an obscuration of 8-12%. At this point the t0 population distribution was recorded with constant mixing. Then 100 μL of a 1 mg/mL polymer solution were added, the mixture was allowed to equilibrate and the population distribution was recorded after 15 and 30 minutes.
In order to determine the relative populations of individual bacteria, dimers and clusters, particle size distributions were deconvoluted using the peakfit.m command (http://terpconnect.umd.edu/~toh/spectrum/InteractivePeakFitter.htm#command) in MATLAB® R2012a package. The size of the clusters was then normalized to a single bacteria size (~ 1.5 μm), so that the relative population of unimers (~ 1.5 μm), dimers (~ 3 μm) and clusters (≥ 4.5 μm) could be plotted as a function of time.
Optical Microscopy
Aliquots (10μL) of the samples used to measure average cluster size were collected after 60 min, mounted on a glass slide with a cover slip on top and examined with an optical microscope.
Supplementary Material
Acknowledgements
We thank GlaxoSmithKline, the Biotechnology and Biological Sciences Research Council (BBSRC) and the Engineering and Physical Sciences Research Council (EPSRC) for funding (Grants BB/H53052X/1, EP/H005625/1, EP/G042462/1), Professor Miguel Camara, Drs Stephan Heeb and Karima Righetti for providing the pyocyanin-negative PAO1 strain and Chien-Yi Chang for the E. coli 536 GFP strain. We also thank Dr JP Magnusson for many helpful discussions.
Footnotes
Additional information
Supplementary information is available in the online version of the paper.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Bush K, et al. Tackling antibiotic resistance. Nature Rev. Microbiol. 2011;9:894–896. doi: 10.1038/nrmicro2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Little TJ, Allen JE, Babayan SA, Matthews KR, Colegrave N. Harnessing evolutionary biology to combat infectious disease. Nature Med. 2012;18:217–220. doi: 10.1038/nm.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Camilli A, Bassler BL. Bacterial small-molecule signaling pathways. Science. 2006;311:1113–1116. doi: 10.1126/science.1121357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Atkinson S, Williams P. Quorum sensing and social networking in the microbial world. J.Roy. Soc. Interface. 2009;6:959–978. doi: 10.1098/rsif.2009.0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lui LT, et al. Bacteria clustering by polymers induces the expression of quorum sense controlled phenotypes. Nature Chem. 2013;5:1058–1065. doi: 10.1038/nchem.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haldar J, An DQ, de Cienfuegos LA, Chen JZ, Klibanov AM. Polymeric coatings that inactivate both influenza virus and pathogenic bacteria. Proc. Natl Acad. Sci. USA. 2006;103:17667–17671. doi: 10.1073/pnas.0608803103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu T-Y, et al. Functionalized arrays of Raman-enhancing nanoparticles for capture and culture-free analysis of bacteria in human blood. Nature Commun. 2011;2 doi: 10.1038/ncomms1546. [DOI] [PubMed] [Google Scholar]
- 8.Smith EJ, et al. Lab-in-a-tube: ultracompact components for on-chip capture and detection of individual micro-/nanoorganisms. Lab Chip. 2012;12:1917–1931. doi: 10.1039/c2lc21175k. [DOI] [PubMed] [Google Scholar]
- 9.Qian XP, et al. Arrays of self-assembled monolayers for studying inhibition of bacterial adhesion. Anal. Chem. 2002;74:1805–1810. doi: 10.1021/ac011042o. [DOI] [PubMed] [Google Scholar]
- 10.Aherne A, Alexander C, Payne MJ, Perez N, Vulfson EN. Bacteria-Mediated Lithography of Polymer Surfaces. J. Am. Chem Soc. 1996;118:8771–8772. [Google Scholar]
- 11.Shepherd J, et al. Hyperbranched poly(NIPAM) polymers modified with antibiotics for the reduction of bacterial burden in infected human tissue engineered skin. Biomaterials. 2011;32:258–267. doi: 10.1016/j.biomaterials.2010.08.084. [DOI] [PubMed] [Google Scholar]
- 12.Gestwicki JE, Kiessling LL. Inter-Receptor Communication Through Arrays of Bacterial Chemoreceptors. Nature. 2002;415:81–84. doi: 10.1038/415081a. [DOI] [PubMed] [Google Scholar]
- 13.Krishnamurthy VM, et al. Promotion of opsonization by antibodies and phagocytosis of Gram-positive bacteria by a bifunctional polyacrylamide. Biomaterials. 2006;27:3663–3674. doi: 10.1016/j.biomaterials.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 14.Schillinger E, Moeder M, Olsson GD, Nicholls IA, Sellergren B. An Artificial Estrogen Receptor through Combinatorial Imprinting. Chem. Eur. J. 2012;18:14773–14783. doi: 10.1002/chem.201201428. [DOI] [PubMed] [Google Scholar]
- 15.Sellergren B. Molecularly Imprinted Polymers Shaping enzyme inhibitors. Nat. Chem. 2010;2:7–8. doi: 10.1038/nchem.496. [DOI] [PubMed] [Google Scholar]
- 16.Hoshino Y, et al. The rational design of a synthetic polymer nanoparticle that neutralizes a toxic peptide in vivo. Proc. Natl Acad. Sci. USA. 2012;109:33–38. doi: 10.1073/pnas.1112828109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gudipaty SA, Larsen AS, Rensing C, McEvoy MM. Regulation of Cu(I)/Ag(I) efflux genes in Escherichia coli by the sensor kinase CusS. FEMS Microbiol. Lett. 2012;330:30–37. doi: 10.1111/j.1574-6968.2012.02529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pontel LB, Soncini FC. Alternative periplasmic copper-resistance mechanisms in Gram negative bacteria. Mol. Microbiol. 2009;73:212–225. doi: 10.1111/j.1365-2958.2009.06763.x. [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto K, Ishihama A. Transcriptional response of Escherichia coli to external copper. Mol. Microbiol. 2005;56:215–227. doi: 10.1111/j.1365-2958.2005.04532.x. [DOI] [PubMed] [Google Scholar]
- 20.Ouchi M, Badi N, Lutz J-F, Sawamoto M. Single-chain technology using discrete synthetic macromolecules. Nat Chem. 2011;3:917–924. doi: 10.1038/nchem.1175. [DOI] [PubMed] [Google Scholar]
- 21.Kamigaito M, Ando T, Sawamoto M. Metal-Catalyzed Living Radical Polymerization. Chem. Rev. 2001;101:3689–3746. doi: 10.1021/cr9901182. [DOI] [PubMed] [Google Scholar]
- 22.McEwan KA, Haddleton DM. Combining catalytic chain transfer polymerisation (CCTP) and thio-Michael addition: enabling the synthesis of peripherally functionalised branched polymers. Polym. Chem. 2011;2:1992–1999. [Google Scholar]
- 23.Levere ME, et al. Assessment of SET-LRP in DMSO using online monitoring and Rapid GPC. Polym. Chem. 2010;1:1086–1094. [Google Scholar]
- 24.Matyjaszewski K, Tsarevsky NV. Nanostructured functional materials prepared by atom transfer radical polymerization. Nat. Chem. 2009;1:276–288. doi: 10.1038/nchem.257. [DOI] [PubMed] [Google Scholar]
- 25.Oh JK, Matyjaszewski K. Synthesis of poly(2-hydroxyethyl methacrylate) in protic media through atom transfer radical polymerization using activators generated by electron transfer. J.Polym. Sci. A-Polym. Chem. 2006;44:3787–3796. [Google Scholar]
- 26.Volentini SI, Farias RN, Rodriguez-Montelongo L, Rapisarda VA. Cu(II)-reduction by Escherichia coli cells is dependent on respiratory chain components. Biometals. 2011;24:827–835. doi: 10.1007/s10534-011-9436-3. [DOI] [PubMed] [Google Scholar]
- 27.Rensing C, Grass G. Escherichia coli mechanisms of copper homeostasis in a changing environment. FEMS Microbiol. Rev. 2003;27:197–213. doi: 10.1016/S0168-6445(03)00049-4. [DOI] [PubMed] [Google Scholar]
- 28.Silhavy TJ, Kahne D, Walker S. The Bacterial Cell Envelope. Cold Spring Harbor Perspectives in Biology. 2010;2 doi: 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caroff M, Karibian D. Structure of bacterial lipopolysaccharides. Carbohydrate Res. 2003;338:2431–2447. doi: 10.1016/j.carres.2003.07.010. [DOI] [PubMed] [Google Scholar]
- 30.Raetz CRH, Whitfield C. Lipopolysaccharide Endotoxins. Ann. Rev. Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lienkamp K, Madkour AE, Kumar K-N, Nuesslein K, Tew GN. Antimicrobial Polymers Prepared by Ring-Opening Metathesis Polymerization: Manipulating Antimicrobial Properties by Organic Counterion and Charge Density Variation. Chem. Eur. J. 2009;15:11715–11722. doi: 10.1002/chem.200900606. [DOI] [PubMed] [Google Scholar]
- 32.Liu D, Reeves PR. Escherichia-Coli K12 regains Its O-Antigen. Microbiol.-UK. 1994;140:49–57. doi: 10.1099/13500872-140-1-49. [DOI] [PubMed] [Google Scholar]
- 33.Schneider G, et al. The pathogenicity island-associated K15 capsule determinant exhibits a novel genetic structure and correlates with virulence in uropathogenic Escherichia coli strain 536. Infect. Immun. 2004;72:5993–6001. doi: 10.1128/IAI.72.10.5993-6001.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geng J, Lindqvist J, Mantovani G, Haddleton DM. Simultaneous copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC) and living radical polymerization. Angew. Chem.-Int. Ed. 2008;47:4180–4183. doi: 10.1002/anie.200800179. [DOI] [PubMed] [Google Scholar]
- 35.Shaner NC, et al. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp red fluorescent protein. Nature Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.