

Abstract
Alemtuzumab is a humanized monoclonal antibody therapy that has recently been approved in over 30 countries for patients with active relapsing-remitting multiple sclerosis. It acts by targeting CD52, an antigen primarily expressed on T and B lymphocytes, resulting in their depletion and subsequent repopulation. The alemtuzumab clinical development program used an active comparator, subcutaneous interferon beta-1a, to show that alemtuzumab is a highly efficacious disease-modifying therapy, with benefits on relapses, disability outcomes, and freedom from clinical disease and magnetic resonance imaging activity. The safety profile was consistent across studies and no new safety signals have emerged during follow-up in the extension study. Infusion-associated reactions are common with alemtuzumab, but rarely serious. Infection incidence was elevated with alemtuzumab in clinical studies; most infections were mild or moderate in severity. Autoimmune adverse events occurred in approximately a third of patients, manifesting mainly as thyroid disorders, and less frequently as immune thrombocytopenia or nephropathy. A comprehensive monitoring program lasting at least 4 years after the last alemtuzumab dose allows early detection and effective management of autoimmune adverse events. Further experience with alemtuzumab in the clinic will provide needed long-term data.
Keywords: alemtuzumab, disease-modifying therapy, efficacy, mechanism of action, multiple sclerosis, safety
Introduction
Alemtuzumab is a humanized monoclonal antibody therapy for relapsing-remitting multiple sclerosis (RRMS). It was granted licensing approval by the European Medicines Agency (EMA) in September 2013. This was followed soon afterwards by approval from regulatory authorities in several other countries. The indication varies across jurisdictions, being approved for the treatment of active RRMS defined by clinical or imaging features, for the treatment of active RRMS with inadequate response to interferon beta (IFNB) or other disease-modifying therapies (DMTs), or the treatment of relapsing forms of multiple sclerosis (MS). Approval by the US Food and Drug Administration (FDA) was initially denied owing to concerns about the design of the pivotal studies. Because patients were not blinded to treatment assignment, the FDA determined that the data were insufficient to demonstrate that the benefits of the treatment outweighed the risks [Coles and Compston, 2014]. Effective patient blinding was not possible due to the high incidence of infusion-associated reactions (IARs) associated with the drug, but all efficacy assessments were performed by blinded neurologists. The application was resubmitted for consideration with additional data analyses. In November 2014, alemtuzumab was approved by the FDA for relapsing forms of MS, generally reserved for patients who have had an inadequate response to 2 or more drugs indicated for the treatment of MS [Genzyme Corporation, 2014]. It will be available through a restricted distribution program.
Multiple sclerosis
The pathogenesis of MS is not fully understood, but is associated with activation of autoreactive lymphocytes, which infiltrate the central nervous system (CNS) and mediate demyelination. Demyelination leaves axons susceptible to injury from the inflammatory environment [Compston and Coles, 2008; Keough and Yong, 2013]. Ultimately, axonal transection or neural death results in irreversible functional deficits.
Various lymphocyte populations have been implicated in demyelination. Interleukin (IL)-17–producing T cells have been observed in active MS lesions in the CNS. Under experimental conditions, they have been associated with breaking down the blood–brain barrier, killing neurons, interfering with neural stem cell proliferation and enhancing oligodendrocyte apoptosis [Kebir et al. 2007; Paintlia et al. 2011; Yamout et al. 2013]. Regulatory T cells function to suppress autoreactive T-cell proliferation in healthy individuals through cytokine production and contact with effector T cells or antigen-presenting cells [Zozulya and Wiendl, 2008]. In patients with MS, this suppressive function is impaired [Viglietta et al. 2004; Fletcher et al. 2009]. B lymphocytes also play a role in MS pathology. Clonally expanded B lymphocytes have been observed in MS lesions and normal-appearing white matter [Baranzini et al. 1999]. The precise function of B cells in MS pathogenesis is unknown but likely involves antigen presentation, cytokine production and/or immunoglobulin synthesis [Krumbholz et al. 2012].
Alemtuzumab pharmacodynamics and mechanism of action
Alemtuzumab targets CD52, an antigen of unknown function that is expressed on lymphocytes, monocytes, some dendritic cell populations and, to a lesser degree, on natural killer (NK) cells and other leukocytes (Figure 1) [Rao et al. 2012]. Alemtuzumab primarily depletes circulating T and B lymphocytes via antibody-dependent cytolysis and complement-dependent cytolysis. Antibody-dependent cytolysis predominates in the mouse model and is mediated by neutrophils and NK cells [Hu et al. 2009]. Human lymphocytes are also susceptible to complement-dependent cytolysis after alemtuzumab exposure, at least in vitro [Rao et al. 2012].
Figure 1.

Alemtuzumab proposed mechanism of action.
NK, natural killer.
Depletion is followed by lymphocyte repopulation, which begins within weeks. B-lymphocyte counts typically return to baseline by 6 months post-treatment, whereas in clinical trials, mean T-cell counts approached normal (but not baseline) levels by 12 months post-treatment [Kovarova et al. 2012; Kasper et al. 2013]. CD4+ T-cell repopulation is particularly delayed. In a long-term follow-up of 37 patients who had received alemtuzumab treatment in the 1990s for MS, median recovery time to normal levels was 8.4 months for B cells, 20 months for CD8+ T cells and 12 years for CD4+ T cells [Hill-Cawthorne et al. 2012]. It should be noted that many of these patients received a single treatment course of 100 mg over 5 infusion days, which is higher than the approved dose (60 mg over 5 days for the initial course, and 36 mg over 3 days for subsequent courses). T-lymphocyte repopulation is accomplished through proliferation of mature lymphocytes that escaped depletion (i.e. ‘homeostatic’ proliferation) as well as new production from precursors in the thymus [Cox et al. 2005; Jones et al. 2013].
Despite profound depletion of circulating lymphocytes, animal studies have shown that lymphocyte numbers in primary and secondary lymphoid organs are maintained [Hu et al. 2009]. Other aspects of the immune system are also unaffected by alemtuzumab, including innate immune cells, some T-cell subsets (tissue-resident effector memory T cells), plasma cells and serum immunoglobulin levels [Coles et al. 1999b; Clark et al. 2012; Turner et al. 2013].
The therapeutic effect of alemtuzumab is likely not solely a consequence of lymphocyte depletion, but also of repopulation features. Patients with and without breakthrough disease activity after alemtuzumab treatment did not differ in the kinetics of lymphocyte repopulation, suggesting that the nature of the repopulating lymphocytes is as important as lymphocyte numbers [Kousin-Ezewu et al. 2014]. During repopulation, the relative proportions of regulatory T cells and memory-phenotype T cells are increased, and the proportion of naive T cells is decreased [Cox et al. 2005; Zhang et al. 2013]. These effects are most marked at month 1 and the cells generally return to baseline proportions by month 12. Similarly, the relative proportions of B-cell subsets are also shifted after alemtuzumab treatment [Hartung et al. 2012; Kasper et al. 2013]. The proportion of B cells with a mature naive phenotype was reduced after treatment, whereas the immature cell fraction increased. By month 6, these proportions approached their baseline levels. The serum cytokine profile is also altered for at least 6 months post-treatment, with marked decreases in IL-17 and cytokines that promote IL-17 production, including IL-21 and IL-23 [Zhang et al. 2013]. There are also decreases in the proinflammatory cytokines IFN-γ, IL-12 and IL-27.
Development of alemtuzumab for MS
Alemtuzumab was initially developed as a treatment for B-cell chronic lymphocytic leukemia (B-CLL), and was approved for that use by the FDA and EMA in 2001. The dose used in the setting of B-CLL (30 mg/day 3 times weekly for 12 weeks) is considerably higher than that used for MS [Genzyme Corporation, 2007; Genzyme Europe, 2007].
The initial trial of alemtuzumab in MS focused on patients (n = 28) with secondary progressive disease [Coles et al. 1999b]. Despite effective suppression of inflammation, more than half the patients had a sustained increase in disability measured by the Expanded Disability Status Scale (EDSS) and/or had further brain volume loss during the 18-month follow-up period. These observations led to the hypothesis that, although axonal degeneration in patients with secondary progression occurs largely in the absence of inflammation, it is conditioned by the amount of prior inflammation-driven disease activity. The focus therefore shifted to treating patients earlier in their disease course. Patients with relapsing-remitting disease were targeted in the phase II and III studies.
Efficacy
Phase II
CAMMS223 [ClinicalTrials.gov identifier: NCT00050778] was a randomized, rater-blinded, active-controlled, head-to-head trial of alemtuzumab versus subcutaneous (SC) IFNB-1a [CAMMS Trial Investigators et al. 2008]. Patients had early, active MS, defined as fulfilling the 2001 McDonald criteria [McDonald et al. 2001], ⩾2 relapses in the prior 2 years and ⩾1 gadolinium (Gd)-enhancing lesion, baseline EDSS score ⩽3.0, MS symptom onset within 3 years, and no prior immunotherapy for MS other than steroids. Alemtuzumab was administered by intravenous infusion on 5 consecutive days at baseline and on 3 consecutive days 12 months later (Figure 2). A third course at month 24 was available at the treating physician’s discretion if the CD4+ T-cell count was ⩾100 × 106 cells/l. All patients received prophylaxis for IARs consisting of methylprednisolone 1 g/day on the first 3 days of infusion of each course; antihistamines and antipyretics were also permitted. Almost 3 years after the study start, alemtuzumab dosing was suspended after three reports of immune thrombocytopenia (ITP), including the fatal index case, but safety and efficacy assessments continued. Coprimary efficacy outcomes were the time to sustained accumulation of disability (SAD) (⩾1.5-point increase in EDSS score in patients with baseline score of 0 and ⩾1-point increase for patients with baseline score of ⩾1.0) confirmed over 6 months, and relapse rate.
Figure 2.
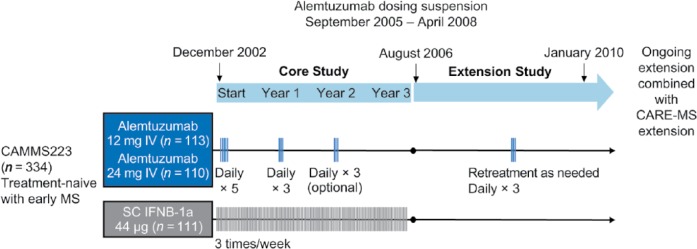
CAMMS223 phase II study design. Alemtuzumab was infused intravenously (IV) on 5 consecutive days at baseline and on 3 consecutive days at year 1. The third treatment course at year 2 was given at the discretion of the investigator if CD4+ T-cell counts were ⩾100 × 106 cells/l. In the extension study, patients originally randomized to SC IFNB-1a were not eligible for alemtuzumab treatment, but could take other disease-modifying therapies. Patients originally randomized to alemtuzumab could receive alemtuzumab retreatment at any point during the extension after the dosing suspension was lifted and ⩾12 months after the previous treatment course. Only the 12-mg dose was used for retreatment.
CARE-MS, Comparison of Alemtuzumab and Rebif® Efficacy in Multiple Sclerosis; IFNB, interferon beta; MS, multiple sclerosis; SC, subcutaneous.
Primary efficacy endpoints were met and have been reviewed elsewhere [Menge et al. 2014]. Post hoc analyses showed that at year 3, 73% of patients treated with alemtuzumab 12 mg were free of clinical disease activity, defined as an absence of 6-month SAD and relapse, compared with 43% in the SC IFNB-1a group (HR, 0.33; p < 0.0001) [Coles et al. 2011]. More alemtuzumab-treated patients also experienced a sustained reduction in disability, defined as a ⩾1-point decrease in EDSS score sustained over a 6-month period in patients with baseline EDSS score ⩾2.0 (45% versus 27% at year 3; p = 0.01) [Coles et al. 2011]. Median brain volume change from baseline to year 3 was −1.8% with SC IFNB-1a versus −0.9% with alemtuzumab 12 mg (p = 0.16).
Phase III
The Comparison of Alemtuzumab and Rebif® Efficacy in Multiple Sclerosis (CARE-MS) studies [ClinicalTrials.gov identifier: NCT00530348, NCT00548405] were 2-year, phase III, randomized, active-controlled, head-to-head trials (Figure 3) [Cohen et al. 2012; Coles et al. 2012b]. Although both CARE-MS studies enrolled patients with RRMS fulfilling the 2005 McDonald criteria [Polman et al. 2005] and active disease (defined as ⩾2 relapses in the prior 2 years and ⩾1 relapse in the prior year), their main point of differentiation was the treatment history of the target populations. In CARE-MS I, eligible patients had never received DMT, had a baseline EDSS score ⩽3.0 and MS symptom onset within 5 years. In contrast, CARE-MS II patients were required to have relapsed on prior IFNB or glatiramer acetate treatment after receiving that therapy for ⩾6 months (prior treatment with other therapies, including natalizumab, was also permitted). Additionally, they had to have baseline EDSS score ⩽5.0 and MS symptom onset within 10 years. In both studies, patients were randomized 2:1 to two annual treatment courses of alemtuzumab 12 mg/day or SC IFNB-1a 44 µg three times weekly, with corticosteroid premedication on the first 3 days of each treatment course for IAR prophylaxis. In CARE-MS II, there was an additional alemtuzumab 24-mg treatment arm; however, randomization into this arm was discontinued early to increase enrollment in the 12-mg arm and it was deemed exploratory for statistical purposes.
Figure 3.

CARE-MS phase III study design. CARE-MS studies were 2-year, phase III, randomized, active-controlled, head-to-head trials. Both studies enrolled patients with active relapsing-remitting multiple sclerosis (defined as ⩾2 relapses in the prior 2 years and ⩾1 relapse in the prior year). In the extension study, patients originally randomized to subcutaneous interferon beta-1a (SC IFNB-1a) received two annual courses of alemtuzumab 12 mg. Patients originally randomized to alemtuzumab could receive alemtuzumab retreatment if they fulfilled magnetic resonance imaging (MRI) or relapse criteria (⩾1 protocol-defined relapse in the previous year or ⩾2 unique MRI lesions on brain or spinal cord). Retreatment could occur at any point during the extension ⩾12 months after the previous treatment course. Only the 12-mg dose was used for retreatment.
CARE-MS, Comparison of Alemtuzumab and Rebif® Efficacy in Multiple Sclerosis; IV, intravenous.
The coprimary efficacy outcomes were relapse rate and time to 6-month SAD. Patient characteristics and primary efficacy results have been reviewed elsewhere [Menge et al. 2014]. Although the SAD endpoint was met in CARE-MS II, a statistically significant difference between alemtuzumab and SC IFNB-1a was not detectable in CARE-MS I. The inability to detect a treatment difference stemmed from the unexpectedly low rate of SAD in the SC IFNB-1a group. Power calculations for the study were based on CAMMS223 data; 20% of patients were expected to attain 6-month SAD with SC IFNB-1a rather than the observed 11%. In both studies, alemtuzumab 12 mg was superior to SC IFNB-1a in reducing relapses and increasing the proportion of patients who were free of clinical disease (absence of relapses and SAD), and the proportion free of magnetic resonance imaging (MRI) activity (Gd-enhancing and new/enlarging T2 lesions) and clinical disease (Figure 4).
Figure 4.

Efficacy of alemtuzumab in core phase III clinical trials compared with patients who received subcutaneous interferon beta-1a (SC IFNB-1a).
ARR, annualized relapse rate; CARE-MS, Comparison of Alemtuzumab and Rebif® Efficacy in Multiple Sclerosis; MRI, magnetic resonance imaging.
Long-term efficacy
All patients completing the phase II and III trials were eligible to continue in an ongoing extension study [ClinicalTrials.gov identifier: NCT00930553] in which they could receive as-needed alemtuzumab retreatment. Retreatment rates in the phase III program were 26−31% over 4 years of follow-up [Coles et al. 2014; Hartung et al. 2014]. In several long-term investigator-led studies (‘the Cambridge cohort’; n = 87), this figure rose to 48% over up to 12 years of follow-up [Tuohy et al. 2014].
In a 5-year follow-up of the CAMMS223 extension, the risk of SAD from baseline to year 5 was reduced by 69% (p = 0.0005) in the alemtuzumab 12-mg group relative to the SC IFNB-1a group [Coles et al. 2012a]. The EDSS score improved or remained stable in 74% of alemtuzumab 12-mg patients from baseline to year 5 compared with 54% of SC IFNB-1a patients (p = 0.014), and relapses were reduced by 66% (p < 0.0001). From year 3 to year 5, there was a 56% relative reduction in relapse rate, but this failed to reach significance (p = 0.09).
The phase III extension study currently has preliminary data up to 4 years after alemtuzumab initiation [Coles et al. 2014; Hartung et al. 2014]. In this study, 349 patients enrolled from the CARE-MS I alemtuzumab group and 393 patients enrolled from the CARE-MS II alemtuzumab 12-mg group. The annualized relapse rate (ARR) over 4 years was 0.16 in treatment-naive patients and 0.24 in patients who had relapsed on prior therapy; ARRs in years 3 and 4 were similar to that of the core studies. The extension study is ongoing and planned to continue for at least 5 years of total follow-up.
Safety
Long-term safety data for alemtuzumab are not yet available. However, safety data up to 4 years (CARE-MS extension), 7 years (CAMMS223 extension) or up to 12 years (Cambridge cohort) have thus far been consistent with what was observed in the core clinical trials. Whether any new safety signals emerge beyond this point remains to be seen.
In the core phase II and III studies, adverse events (AEs) were reported in 96−100% of patients treated with alemtuzumab 12 mg [CAMMS Trial Investigators et al. 2008; Cohen et al. 2012; Coles et al. 2012b]. The rate of AEs (number per patient per year) ranged from 7.2 to 8.7 across studies. With the 12-mg dose, serious AEs were reported in 18–22% of patients across studies; the rate was 0.1–0.16 events per patient per year. During the core studies, two deaths occurred in CAMMS223 (cardiovascular disease and ITP), one in CARE-MS I (automobile accident) and two in CARE-MS II (automobile accident and aspiration pneumonia following brainstem relapse).
Infusion-associated reactions
IARs were the most common AEs in all three studies and those associated with alemtuzumab are thought to be mainly attributable to cytokine-release syndrome [Genzyme Therapeutics, 2013]. Cytokine release occurs as a result of target cell lysis and recruitment of inflammatory cells [Breslin, 2007; Maggi et al. 2011]. In the alemtuzumab studies, IARs were defined as any AE that occurred during the infusion or within 24 hours after infusion, regardless of causality. The incidence of IARs was ⩾90% across studies [CAMMS Trial Investigators et al. 2008; Cohen et al. 2012; Coles et al. 2012b]. Serious IARs were reported in up to 1−3% of patients in each alemtuzumab treatment group. Most common IARs and their incidence with alemtuzumab 12 mg were as follows: headache (43−56%); rash (39−89%); pyrexia (16−36%); nausea (14−20%); urticaria (11−26%); pruritus (10−28%); flushing (8−11%); insomnia (10−19%); fatigue (7−24%); chills (7−18%); chest discomfort (6−14%); and dyspnea (6−13%). There were no cases of anaphylaxis and no IARs resulted in death in the core studies. The IARs were most frequent during the first treatment course (85%), and decreased during course 2 (69%) and course 3 (63%) [Mayer et al. 2014]. Few patients (2−6.6% per infusion day) required infusion interruption or infusion rate adjustment.
Infections
As would be expected with any lymphocyte-depleting agent, infections were more common with alemtuzumab 12 mg than with SC IFNB-1a in clinical trials (CAMMS223: 66% versus 47%; CARE-MS I: 67% versus 45%; CARE-MS II: 77% versus 66%). However, most were mild or moderate in severity [CAMMS Trial Investigators et al. 2008; Cohen et al. 2012; Coles et al. 2012b]. Serious infections were rare, but slightly elevated with alemtuzumab versus SC IFNB-1a (CAMMS223: 2.8% versus 1.9%; CARE-MS I: 2% versus 1%; CARE-MS II: 4% versus 1%). The most common infections were those of the respiratory tract and urinary tract. Herpetic infections, including mucocutaneous herpes simplex and herpes zoster, were increased with alemtuzumab in the CARE-MS studies, but declined after the introduction of acyclovir prophylaxis as a study protocol amendment [Cohen et al. 2012; Coles et al. 2012b; Wray et al. 2013a, 2013b].
Follow-up for up to 7 years in CAMMS223 showed that infection incidence did not increase with each course of alemtuzumab [Coles et al. 2012a]. For all alemtuzumab-treated patients, infections peaked in year 1, at 47%, and declined thereafter. By year 7, infection incidence was 10% among patients in the 12-mg arm. Most infections (96%) were mild or moderate over the follow-up period and upper respiratory tract infections were most common.
Alemtuzumab has not been associated with any cases of progressive multifocal leukoencephalopathy in studies of patients with MS. There have been several case reports of progressive multifocal leukoencephalopathy in patients treated with alemtuzumab for transplant rejection or for CLL [Waggoner et al. 2009; Isidoro et al. 2014]. In these cases, however, patients had been heavily treated with immunosuppressive therapies before receiving alemtuzumab.
Malignancy
Of 1486 alemtuzumab-treated patients in the clinical development program, 29 have been diagnosed with a malignancy, six of which were thyroid carcinomas [Miller et al. 2014]. The apparently high rate of thyroid cancer is related to increased surveillance of thyroid function; at least four cases were detected as incidental findings during treatment for Graves’ disease. A case report of thyroid carcinoma after alemtuzumab in a patient with otherwise normal thyroid function has also been published [Ibitoye and Wilkins, 2014]. Other malignancies occurring in >1 alemtuzumab-treated patient included basal cell carcinoma (n = 6), breast (n = 5) and malignant melanoma (n = 4).
Autoimmune AEs
Autoimmune AEs represent the most important risk associated with alemtuzumab treatment. Autoimmune sequelae are thought to arise from the way in which lymphocyte repopulation proceeds [Jones et al. 2013]. T lymphocytes repopulate via two mechanisms: the ‘homeostatic’ proliferation of mature lymphocytes that escaped depletion by alemtuzumab; and generation of new T cells in the thymus. Homeostatic proliferation predominates in patients who go on to develop autoimmune AEs; in contrast, patients without autoimmune AEs tend to generate relatively more new T lymphocytes in the thymus. The relative contribution of these repopulation mechanisms has implications for the clonal diversity of the resulting T-cell pool and autoimmunity is associated with reduced lymphocyte diversity.
With alemtuzumab, autoimmune AEs most commonly affect the thyroid, but rare cases of ITP and antiglomerular basement membrane disease have also been described; these events spurred the need for active monitoring after alemtuzumab treatment. A case of type 1 diabetes after alemtuzumab treatment has recently been described in the literature [Malmeström et al. 2014], but it is unknown whether this is an isolated case or represents another form of autoimmune AE that should be monitored. Because autoimmune AEs may develop years after the last dose of alemtuzumab, the potential for as-yet unrecognized AEs remains.
Thyroid
In the 5-year follow-up of CAMMS223, thyroid autoimmune AEs occurred in 39% of patients treated with alemtuzumab 12 mg and 29% of those who received the 24-mg dose [Daniels et al. 2014]. In total, 102 episodes were reported in 73 alemtuzumab patients. Graves’ hyperthyroidism (65.8%), hypothyroidism (20.5%) and subacute thyroiditis (12.3%) were most common. Onset ranged from 6 to 61 months after the first treatment course [Coles et al. 2012a]. Incidence peaked at year 3 and declined in subsequent years [Daniels et al. 2014]. Thyroid disorders responded to conventional therapy with antithyroid drugs, radioactive iodine, thyroid hormone or surgery. Most patients (85%) who developed thyroid disorders were negative for thyroid peroxidase antibodies at baseline. At the time of thyroid dysfunction, antithyroid-stimulating hormone receptor antibodies were present in 70% of episodes.
The CARE-MS studies have shown similar thyroid outcomes as observed in the phase II study. In pooled data from 4-year follow-up, the incidence of thyroid AEs was highest in year 3 (20.9%) and declined in year 4 (12.4%) [Twyman et al. 2014], consistent with the 5-year CAMMS223 results [Coles et al. 2012a] and 12-year results from the Cambridge cohort [Tuohy et al. 2014]. Overall, thyroid AEs were reported in 36% of patients and serious thyroid AEs in 3.8% of patients over 4 years of follow-up in the CARE-MS studies [Twyman et al. 2014]. Hyperthyroidism, hypothyroidism, goiter and thyroiditis were most common.
ITP
Alemtuzumab is associated with a unique form of ITP characterized by delayed onset, responsiveness to conventional ITP therapies, and prolonged remission [Cuker et al. 2011]. The index case of ITP occurred during CAMMS223, when a patient presented with intracranial hemorrhage and died. This prompted the implementation of a risk-management plan that comprised patient and physician education, monthly complete blood count (CBC) and monthly symptom surveys offset from the CBC by 2 weeks [Cuker et al. 2014]. During clinical trials, ITP was defined in the study protocols as normal hemoglobin, white blood cell count, and peripheral smear except for a decrease in platelets without clumping; absence of splenomegaly; and either a confirmed platelet count between 50 and 100 × 109/l on ⩾2 consecutive occasions over a period of at least 1 month, or a confirmed platelet count <50 × 109/l without clumping documented on two or more consecutive occasions over any period of time [Cuker et al. 2014]. The incidence of protocol-defined alemtuzumab-induced ITP was 2.0% (n = 30/1486) across the clinical development program, including the ongoing extension study [Cuker et al. 2014]. A further 21 patients did not meet the protocol definition but had platelet counts below normal limits. ITP onset occurred a mean of 16 months (range: 1−34) after the last dose of alemtuzumab. Most patients responded to first-line therapy with corticosteroids, intravenous immunoglobulin or platelet transfusion. Other therapies were rituximab (n = 4) and splenectomy (n = 1), and several events (n = 2) resolved spontaneously.
Nephropathy
Nephropathy has been described in clinical studies of alemtuzumab in RRMS. In pilot studies, two patients developed antiglomerular basement membrane (anti-GBM) disease that ultimately required renal transplant [Meyer et al. 2012]. In phase II and III trials, four cases of glomerulonephritis occurred among 1486 patients treated with alemtuzumab (0.3%), including 1 case of anti-GBM disease, 1 case of glomerulonephritis with positive anti-GBM antibody, and 2 cases of membranous glomerulonephritis [Cohen et al. 2012; Coles et al. 2012b; Wynn et al. 2013]. Onset ranged from 4 to 39 months after the last dose of alemtuzumab. Each case responded to medical treatment.
Pregnancy and fertility
Alemtuzumab is in the pregnancy category C, with no controlled studies on its effect on pregnant women or the developing fetus [Genzyme Corporation, 2007]. Immunoglobulin G molecules can cross the placental barrier and potentially affect the fetus. Women of childbearing potential are advised to use contraception during a course of alemtuzumab and for 4 months afterwards. Although contraception was required during clinical studies, 139 pregnancies in 104 patients occurred after exposure to alemtuzumab as of October 2013 [McCombe et al. 2014]. These pregnancies resulted in 67 live births, 14 elective abortions, 24 spontaneous abortions, one stillbirth, and 33 ongoing or with unknown outcome. Serious AEs occurred in 11 fetuses/infants, including two fetuses with abnormal development (cystic hygroma and hypoplastic heart; anembryonic gestation) and one intrauterine death due to nuchal cord.
CD52 is expressed in the male reproductive system, including the epididymis and seminal vesicle, sperm and seminal fluid, posing a theoretical risk of alemtuzumab for male fertility [Hale et al. 1993]. However, developing sperm are negative for CD52 expression, and although mature sperm express CD52, an abundance of CD52 antigen in seminal plasma effectively competes with sperm for alemtuzumab binding, making an adverse impact on male fertility unlikely. A limited data set (n = 13) showed that at baseline, and 1, 3 and 6 months post alemtuzumab treatment, there was no evidence of defects in sperm motility or morphology, and no patient had a consistently depressed sperm count [Margolin et al. 2013].
Managing safety risks associated with alemtuzumab
Every MS drug has a unique side effect profile that requires active management. Although alemtuzumab treatment is associated with safety risks, those risks are manageable in most patients. To enable early detection and treatment of autoimmune AEs, patients undergo a program of monthly testing for 4 years after the last alemtuzumab dose (Table 1). Patients must be made aware of this commitment before embarking on treatment and understand that monitoring is necessary even if no alemtuzumab retreatment occurs during this time. Unwillingness to comply with the risk-management program disqualifies a patient from receiving alemtuzumab treatment, as it presents health risks for the patient and a risk of legal liability for the physician. Serum creatinine, CBC and urinalysis are recommended at monthly intervals and thyroid function tests are recommended every 3 months [Genzyme Therapeutics, 2013]. The clinical trial experience has demonstrated the effectiveness of this monitoring program in detecting autoimmune AEs to allow for their successful treatment.
Table 1.
Risk management plan for alemtuzumab-treated patients.
| Potential AE | Laboratory assessment | Timing |
|---|---|---|
| Thyroid disorders | Thyroid function tests such as TSH level | Prior to alemtuzumab initiation and then every 3 months |
| ITP | CBC with differential | Prior to alemtuzumab initiation and then monthly |
| Nephropathies | Serum creatinine and urinalysis with microscopy | Prior to alemtuzumab initiation and then monthly |
AE, adverse event; CBC, complete blood count; ITP, immune thrombocytopenia; TSH, thyroid-stimulating hormone.
The ability to predict those patients who will develop autoimmune AEs would inform physician and patient decision-making when choosing a DMT. Preliminary work has suggested that serum levels of the cytokine IL-21 may be able to predict the development of autoimmune AEs [Jones et al. 2009]. IL-21 is a cytokine that promotes T-cell proliferation and apoptosis, and is thought to be a key driver of homeostatic proliferation [King et al. 2004]. Patients with autoimmune AEs had a two-fold increase in pretreatment serum IL-21 levels compared with those without autoimmune AEs [Jones et al. 2009]. Although this may hold promise, currently no commercially available reagents exist that have predictive utility for autoimmune AEs post-alemtuzumab treatment [Azzopardi et al. 2014]. More work is needed in determining the utility of IL-21 as a biomarker for predicting autoimmune sequelae to alemtuzumab treatment, and in identifying further biomarkers for this purpose.
Choosing the right patient for alemtuzumab
Alemtuzumab is not for every patient with MS and the decision to use it should be carefully considered. Study data suggest that alemtuzumab may be ineffective in patients with secondary progression [Coles et al. 1999a]. Although the safety profile is generally manageable for the right patient, risks probably outweigh benefits in patients with mild disease or clinically isolated syndrome, and there are no available data on these populations. The EMA label indicates use in patients with active RRMS defined by clinical or imaging features; this indication stresses that alemtuzumab should be reserved for patients with active inflammatory disease and aligns with the immunomodulatory mechanism of action. In the CARE-MS studies, active disease was defined as ⩾1 relapse in the past year and ⩾2 relapses in the past 2 years, plus the presence of MRI lesions; these criteria may help guide clinicians in practice. Patients with active disease despite treatment with other therapies demonstrated benefits with alemtuzumab in the CARE-MS II study, and are clearly candidates for more powerful therapy. For patients who meet criteria for active disease but are early in their disease course and treatment-naive, CARE-MS I and CAMMS223 have shown clinical and MRI benefits with alemtuzumab. Although some clinicians may be hesitant to use alemtuzumab as first-line therapy, the current lack of established neuroprotective or neuroregenerative therapies leaves only the option of preserving existing neural tissue before extensive pathology occurs. In patients with early MS who are clearly on a path of rapidly advancing disease, high-efficacy anti-inflammatory therapy must be considered.
Choosing an appropriate DMT for each patient must take into account the risks and relative benefits of each available therapy (Table 2). The alemtuzumab clinical studies were unique in that they used high-dose SC IFNB-1a as the comparator, whereas most other therapies were approved based on placebo-controlled studies. Fingolimod was tested in a head-to-head trial with lower-dose IFNB-1a [Cohen et al. 2010]. Although head-to-head data represent valuable information in comparing newer drugs with a platform therapy, the lack of placebo-controlled studies for alemtuzumab further adds to the pitfalls of comparing therapies across studies, which also vary in the endpoints used and patient populations studied. Head-to-head studies comparing alemtuzumab efficacy with other highly effective therapies are currently lacking, so choosing between these must rely on other factors including the patient’s medical history (e.g. John Cunningham virus antibody status, cardiac history), mode and frequency of administration, and willingness to comply with safety monitoring associated with each drug.
Table 2.
Efficacy results summary of disease-modifying therapies.
| Drug | Alemtuzumab 12 mg |
Natalizumab 300 mg |
Fingolimod 0.5 mg |
Dimethyl fumarate 240 mg (twice daily) |
Teriflunomide 14 mg |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Study | CARE-MS I [Cohen et al. 2012] n = 581 | CARE-MS II [Coles et al. 2012b] n = 667 | AFFIRM [Polman et al. 2006] n = 942 | FREEDOMS [Kappos et al. 2010] n = 1272 | TRANSFORMS [Cohen et al. 2010] n = 1292 | CONFIRM [Fox et al. 2012] n = 1430 | DEFINE [Gold et al. 2012] n = 1237 | TEMSO [O’Connor et al. 2011] n = 1088 | TOWER [Confavreux et al. 2014] n = 1169 | TENERE [Vermersch et al. 2013] n = 324 |
| Comparator | SC IFNB-1a | SC IFNB-1a | Placebo | Placebo | IFNB-1a IM | Placebo | Placebo | Placebo | Placebo | SC IFNB-1a |
| ARR, relative reduction versus comparator | 55% p < 0.0001 | 49% p < 0.0001 | 68% p < 0.001 | 54% p < 0.001 | 38% p < 0.001 | 44% p < 0.001 | 53% p < 0.001 | 32% p < 0.001 | 36% p = 0.0001 | None |
| HR of 3-month confirmed disability versus comparator (95% CI) | NA | 0.72 p = 0.08 | 0.58 (0.43−0.77) p < 0.001 | 0.70 (0.52−0.96) p = 0.02 | No significant difference in time to disability progression or proportion of patients with confirmed progression | 0.79 (0.52−1.19) p = 0.25 | 0.62 (0.44−0.87) p = 0.005 | 0.70 (0.51−0.97) | 0.68 (0.4−1.00) p = 0.0442 | NA |
| HR of 6-month confirmed disability versus comparator (95% CI) | 0.70 (0.40−1.23) p = 0.22 | 0.58 (0.38−0.87) p = 0.0084 | 0.46 (0.33−0.64) p < 0.001 | 0.63 (0.44−0.90) p = 0.01 | NA | NA | NA | NA | NA | |
ARR, annualized relapse rate; CI, confidence interval; HR, hazard ratio; IFNB-1a, interferon beta-1a; IM, intramuscular; NA, not available; SC, subcutaneous.
The decision to re-treat with alemtuzumab
The issue of retreatment with alemtuzumab beyond the initial two courses of therapy will become important as the second anniversary of its approval in Europe approaches. Labeling guidelines do not currently address whether or how retreatment with alemtuzumab should be undertaken. In phase III studies, alemtuzumab treatment beyond the initial two courses was offered if a relapse occurred or if a patient accumulated ⩾2 unique MRI lesions (either Gd-enhancing lesions and/or new or enlarging T2 lesions). Lymphocyte reconstitution rate does not appear to predict breakthrough disease activity, and use of lymphocyte counts as a biomarker to indicate retreatment has been discouraged [Kousin-Ezewu et al. 2014]. In clinical practice, it may be appropriate to monitor alemtuzumab efficacy by MRI. MRI scans were performed annually in clinical trials and helped to guide the decision to re-treat during the extension study. The maximal number of alemtuzumab courses that may be safely given is unknown. Some have suggested the use of a platform therapy after alemtuzumab [Menge et al. 2014], but there are currently no data addressing this approach.
Summary
Alemtuzumab is a highly effective therapy for patients with active RRMS who are either treatment-naive or who have had breakthrough disease on DMT. The available safety data from the phase II and III clinical studies span about 5 years and are consistent with the 12-year data from the Cambridge cohort. The safety profile is manageable with continued monitoring. Further experience with alemtuzumab in the clinic will provide the needed long-term data.
Footnotes
Funding: E.H., D.H. and I.K. have been supported by the Czech Ministry of Education (PRVOUK-P26/LF1/4). Editorial support for this manuscript was provided by Valerie P. Zediak, PhD, Evidence Scientific Solutions, and funded by Genzyme, a Sanofi company. The authors were responsible for all content and editorial decisions, and received no honoraria related to the preparation of this article.
Conflict of interest statement: E.H. has received honoraria and consulting fees from Bayer, GlaxoSmithKline, Genzyme, Biogen Idec, Sanofi, Merck Serono, Roche, Teva and Novartis for consulting services, speaking and serving on scientific advisory boards. D.H. has received speaker honoraria and consultant fees from Biogen Idec, Novartis, Merck Serono, Teva and Bayer Schering, and financial support for research activities from Biogen Idec. I.K. has received speaker honoraria and consultant fees from Biogen Idec, Novartis, Merck Serono, Teva and Bayer Schering, and financial support for research activities from Biogen Idec.
Contributor Information
Eva Havrdova, Department of Neurology and Center for Clinical Neuroscience, First Medical Faculty, Charles University in Prague, Katerinska 30, Prague, 120 00, Czech Republic.
Dana Horakova, Department of Neurology and Center for Clinical Neuroscience, First Medical Faculty, Charles University in Prague, Prague, Czech Republic.
Ivana Kovarova, Department of Neurology and Center for Clinical Neuroscience, First Medical Faculty, Charles University in Prague, Prague, Czech Republic.
References
- Azzopardi L., Thompson S., Harding K., Cossburn M., Robertson N., Compston A., et al. (2014) Predicting autoimmunity after alemtuzumab treatment of multiple sclerosis. J Neurol Neurosurg Psychiatry 85: 795–798. [DOI] [PubMed] [Google Scholar]
- Baranzini S., Jeong M., Butunoi C., Murray R., Bernard C., Oksenberg J. (1999) B cell repertoire diversity and clonal expansion in multiple sclerosis brain lesions. J Immunol 163: 5133–5144. [PubMed] [Google Scholar]
- Breslin S. (2007) Cytokine-release syndrome: overview and nursing implications. Clin J Oncol Nurs 11: 37–42. [DOI] [PubMed] [Google Scholar]
- CAMMS Trial Investigators, Coles A., Compston D., Selmaj K., Lake S., Moran S., et al. (2008) Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N Engl J Med 359: 1786–1801. [DOI] [PubMed] [Google Scholar]
- Clark R., Watanabe R., Teague J., Schlapbach C., Tawa M., Adams N., et al. (2012) Skin effector memory T cells do not recirculate and provide immune protection in alemtuzumab-treated CTCL patients. Sci Transl Med 4: 117ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J., Barkhof F., Comi G., Hartung H., Khatri B., Montalban X., et al. (2010) Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med 362: 402–415. [DOI] [PubMed] [Google Scholar]
- Cohen J., Coles A., Arnold D., Confavreux C., Fox E., Hartung H., et al. (2012) Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet 380: 1819–1828. [DOI] [PubMed] [Google Scholar]
- Coles A., Arnold D., Cohen J., Fox E., Giovannoni G., Hartung H., et al. (2014) Efficacy and safety of alemtuzumab in treatment-naive patients with relapsing-remitting MS: four-year follow-up of the CARE-MS I study. Presented at 2014 Joint Meeting of Americas Committee for Treatment and Research in Multiple Sclerosis and European Committee for Treatment and Research in Multiple Sclerosis, September, Boston, MA. [Google Scholar]
- Coles A., Compston A. (2014) Product licences for alemtuzumab and multiple sclerosis. Lancet 383: 867–868. [DOI] [PubMed] [Google Scholar]
- Coles A., Fox E., Vladic A., Gazda S., Brinar V., Selmaj K., et al. (2011) Alemtuzumab versus interferon beta-1a in early relapsing-remitting multiple sclerosis: post-hoc and subset analyses of clinical efficacy outcomes. Lancet Neurol 10: 338–348. [DOI] [PubMed] [Google Scholar]
- Coles A., Fox E., Vladic A., Gazda S., Brinar V., Selmaj K., et al. (2012a) Alemtuzumab more effective than interferon beta-1a at 5-year follow-up of CAMMS223 clinical trial. Neurology 78: 1069–1078. [DOI] [PubMed] [Google Scholar]
- Coles A., Twyman C., Arnold D., Cohen J., Confavreux C., Fox E., et al. (2012b) Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet 380: 1829–1839. [DOI] [PubMed] [Google Scholar]
- Coles A., Wing M., Molyneux P., Paolillo A., Davie C., Hale G., et al. (1999a) Monoclonal antibody treatment exposes three mechanisms underlying the clinical course of multiple sclerosis. Ann Neurol 46: 296–304. [DOI] [PubMed] [Google Scholar]
- Coles A., Wing M., Smith S., Coraddu F., Greer S., Taylor C., et al. (1999b) Pulsed monoclonal antibody treatment and autoimmune thyroid disease in multiple sclerosis. Lancet 354: 1691–1695. [DOI] [PubMed] [Google Scholar]
- Compston A., Coles A. (2008) Multiple sclerosis. Lancet 372: 1502–1517. [DOI] [PubMed] [Google Scholar]
- Confavreux C., O’Connor P., Comi G., Freedman M., Miller A., Olsson T., et al. (2014) Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol 13: 247–256. [DOI] [PubMed] [Google Scholar]
- Cox A., Thompson S., Jones J., Robertson V., Hale G., Waldmann H., et al. (2005) Lymphocyte homeostasis following therapeutic lymphocyte depletion in multiple sclerosis. Eur J Immunol 35: 3332–3342. [DOI] [PubMed] [Google Scholar]
- Cuker A., Coles A., Sullivan H., Fox E., Goldberg M., Oyuela P., et al. (2011) A distinctive form of immune thrombocytopenia in a phase 2 study of alemtuzumab for the treatment of relapsing-remitting multiple sclerosis. Blood 118: 6299–6305. [DOI] [PubMed] [Google Scholar]
- Cuker A., Palmer J., Oyuela P., Margolin D., Bass A. (2014) Successful detection and management of immune thrombocytopenia in alemtuzumab-treated patients with active relapsing-remitting multiple sclerosis Neurology 82: P2.198. [Google Scholar]
- Daniels G., Vladic A., Brinar V., Zavalishin I., Valente W., Oyuela P., et al. (2014) Alemtuzumab-related thyroid dysfunction in a phase 2 trial of patients with relapsing-remitting multiple sclerosis. J Clin Endocrinol Metab 99: 80–89. [DOI] [PubMed] [Google Scholar]
- Fletcher J., Lonergan R., Costelloe L., Kinsella K., Moran B., O’Farrelly C., et al. (2009) CD39+Foxp3+ regulatory T cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J Immunol 183: 7602–7610. [DOI] [PubMed] [Google Scholar]
- Fox R., Miller D., Phillips J., Hutchinson M., Havrdova E., Kita M., et al. (2012) Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med 367: 1087–1097. [DOI] [PubMed] [Google Scholar]
- Genzyme Corporation (2007) Campath (alemtuzumab). Full prescribing information. Cambridge, MA: Available at http://www.campath.com/pdfs/Full_Prescribing_Information_Brochure.pdf [accessed 15 April 2014]. [Google Scholar]
- Genzyme Europe (2007) MabCampath. Summary of product characteristics. Naarden, The Netherlands: Genzyme Europe BV. [Google Scholar]
- Genzyme Therapeutics (2013) LEMTRADA summary of product characteristics. Oxford, UK: Genzyme Therapeutics Ltd. [Google Scholar]
- Genzyme Corporation (2014) LEMTRADA (alemtuzumab). Full prescribing information. Cambridge, MA: Genzyme Corporation; Available at http://products.sanofi.us/lemtrada/lemtrada.pdf [accessed 2 December 2014]. [Google Scholar]
- Gold R., Kappos L., Arnold D., Bar-Or A., Giovannoni G., Selmaj K., et al. (2012) Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med 367: 1098–1107. [DOI] [PubMed] [Google Scholar]
- Hale G., Rye P., Warford A., Lauder I., Brito-Babapulle A. (1993) The glycosylphosphatidylinositol-anchored lymphocyte antigen CDw52 is associated with the epididymal maturation of human spermatozoa. J Reprod Immunol 23: 189–205. [DOI] [PubMed] [Google Scholar]
- Hartung H., Arnold D., Cohen J., Coles A., Confavreux C., Fox E., et al. (2012) Lymphocyte subset dynamics following alemtuzumab treatment in the CARE-MS I study. Presented at 28th Congress of the European Committee for Research and Treatment in Multiple Sclerosis, October, Lyon, France. [Google Scholar]
- Hartung H., Arnold D., Cohen J., Coles A., Fox E., Giovannoni G., et al. (2014) Efficacy and safety of alemtuzumab in patients with relapsing-remitting MS who relapsed on prior therapy: four-year follow-up of the CARE-MS II study. Presented at 2014 Joint Meeting of Americas Committee for Treatment and Research in Multiple Sclerosis and European Committee for Treatment and Research in Multiple Sclerosis, September, Boston, MA. [Google Scholar]
- Hill-Cawthorne G., Button T., Tuohy O., Jones J., May K., Somerfield J., et al. (2012) Long term lymphocyte reconstitution after alemtuzumab treatment of multiple sclerosis. J Neurol Neurosurg Psychiatry 83: 298–304. [DOI] [PubMed] [Google Scholar]
- Hu Y., Turner M., Shields J., Gale M., Hutto E., Roberts B., et al. (2009) Investigation of the mechanism of action of alemtuzumab in a human CD52 transgenic mouse model. Immunology 128: 260–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibitoye R., Wilkins A. (2014) Thyroid papillary carcinoma after alemtuzumab therapy for MS. J Neurol 261: 1828–1829. [DOI] [PubMed] [Google Scholar]
- Isidoro L., Pires P., Rito L., Cordeiro G. (2014) Progressive multifocal leukoencephalopathy in a patient with chronic lymphocytic leukaemia treated with alemtuzumab. BMJ Case Rep. 8 January 2014. [Epub ahead of print]. DOI: 10.1136/bcr-2013-201781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones J., Phuah C., Cox A., Thompson S., Ban M., Shawcross J., et al. (2009) IL-21 drives secondary autoimmunity in patients with multiple sclerosis, following therapeutic lymphocyte depletion with alemtuzumab (Campath-1H). J Clin Invest 119: 2052–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones J., Thompson S., Loh P., Davies J., Tuohy O., Curry A., et al. (2013) Human autoimmunity after lymphocyte depletion is caused by homeostatic T-cell proliferation. Proc Natl Acad Sci U S A 110: 20200–20205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappos L., Radue E., O’Connor P., Polman C., Hohlfeld R., Calabresi P., et al. (2010) A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med 362: 387–401. [DOI] [PubMed] [Google Scholar]
- Kasper L., Arnold D., Coles A., Hartung H., Havrdova E., Selmaj K., et al. (2013) Lymphocyte subset dynamics following alemtuzumab treatment in the CARE-MS II study. P531. Presented at 29th Congress of the European Committee for Research and Treatment in Multiple Sclerosis, October, Copenhagen, Denmark. [Google Scholar]
- Kebir H., Kreymborg K., Ifergan I., Dodelet-Devillers A., Cayrol R., Bernard M., et al. (2007) Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med 13: 1173–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keough M., Yong V. (2013) Remyelination therapy for multiple sclerosis. Neurotherapeutics 10: 44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King C., Ilic A., Koelsch K., Sarvetnick N. (2004) Homeostatic expansion of T cells during immune insufficiency generates autoimmunity. Cell 117: 265–277. [DOI] [PubMed] [Google Scholar]
- Kousin-Ezewu O., Azzopardi L., Parker R., Tuohy O., Compston A., Coles A., et al. (2014) Accelerated lymphocyte recovery after alemtuzumab does not predict multiple sclerosis activity. Neurology 82: 2158–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovarova I., Arnold D., Cohen J., Coles A., Confavreux C., Fox E., et al. (2012) Alemtuzumab pharmacokinetics and pharmacodynamics in Comparison of Alemtuzumab and Rebif® Efficacy in Multiple Sclerosis I. Presented at 23rd Meeting of the European Neurological Society, June, Prague, Czech Republic. [Google Scholar]
- Krumbholz M., Derfuss T., Hohlfeld R., Meinl E. (2012) B cells and antibodies in multiple sclerosis pathogenesis and therapy. Nat Rev Neurol 8: 613–623. [DOI] [PubMed] [Google Scholar]
- Maggi E., Vultaggio A, Matucci A. (2011) Acute infusion reactions induced by monoclonal antibody therapy. Expert Rev Clin Immunol 7: 55–63. [DOI] [PubMed] [Google Scholar]
- Malmeström C., Andersson B., Lycke J. (2014) First reported case of diabetes mellitus type 1 as a possible secondary autoimmune disease following alemtuzumab treatment in MS. J Neurol 261: 2016–2018. [DOI] [PubMed] [Google Scholar]
- Margolin D., Rizzo M., Smith G., Arnold D., Coles A., Hartung H., et al. (2013) Alemtuzumab treatment has no adverse impact on sperm quality, quantity, or motility: A CARE-MS substudy. Presented at 21st World Congress of Neurology, September, Vienna, Austria. [Google Scholar]
- Mayer L., Casady L., Clayton G., Oyuela P., Margolin D. (2014) Alemtuzumab infusion-associated reactions and management in multiple sclerosis. J Infus Nurs 37: 250–258. [Google Scholar]
- McCombe P., Achiron A., Giovannoni G., Brinar V., Margolin D., Palmer J., et al. (2014) Pregnancy outcomes in the alemtuzumab multiple sclerosis clinical development program. Presented at 2014 Joint Meeting of Americas Committee for Treatment and Research in Multiple Sclerosis and European Committee for Treatment and Research in Multiple Sclerosis, September, Boston, MA. [Google Scholar]
- McDonald W., Compston A., Edan G., Goodkin D., Hartung H., Lublin F., et al. (2001) Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the Diagnosis of Multiple Sclerosis. Ann Neurol 50: 121–127. [DOI] [PubMed] [Google Scholar]
- Menge T., Stüve O., Kieseier B., Hartung H. (2014) Alemtuzumab: the advantages and challenges of a novel therapy in MS. Neurology 83: 87–97. [DOI] [PubMed] [Google Scholar]
- Meyer D., Coles A., Oyuela P., Purvis A., Margolin D. (2012) Case report of anti-glomerular basement membrane disease following alemtuzumab treatment of relapsing-remitting multiple sclerosis. Mult Scler Rel Disord 2: 60–63. [DOI] [PubMed] [Google Scholar]
- Miller T., Habek M., Coles A., Selmaj K., Margolin D., Palmer J., et al. (2014) Analysis of data from RRMS alemtuzumab-treated patients in the clinical program to evaluate incidence rates of malignancy. Presented at 2014 Joint Meeting of Americas Committee for Treatment and Research in Multiple Sclerosis and European Committee for Treatment and Research in Multiple Sclerosis, September, Boston, MA. [Google Scholar]
- O’Connor P., Wolinsky J., Confavreux C., Comi G., Kappos L., Olsson T., et al. (2011) Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med 365: 1293–1303. [DOI] [PubMed] [Google Scholar]
- Paintlia M., Paintlia A., Singh A., Singh I. (2011) Synergistic activity of interleukin-17 and tumor necrosis factor-alpha enhances oxidative stress-mediated oligodendrocyte apoptosis. J Neurochem 116: 508–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polman C., O’Connor P., Havrdova E., Hutchinson M., Kappos L., Miller D., et al. (2006) A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 354: 899–910. [DOI] [PubMed] [Google Scholar]
- Polman C., Reingold S., Edan G., Filippi M., Hartung H., Kappos L., et al. (2005) Diagnostic criteria for multiple sclerosis: 2005 revisions to the ‘McDonald Criteria’. Ann Neurol 58: 840–846. [DOI] [PubMed] [Google Scholar]
- Rao S., Sancho J., Campos-Rivera J., Boutin P., Severy P., Weeden T., et al. (2012) Human peripheral blood mononuclear cells exhibit heterogeneous CD52 expression levels and show differential sensitivity to alemtuzumab mediated cytolysis. PLoS One 7: e39416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuohy O., Costelloe L., Hill-Cawthorne G., Bjornson I., Harding K., Robertson N., et al. (2014) Alemtuzumab treatment of multiple sclerosis: long-term safety and efficacy. J Neurol Neurosurg Psychiatry. 21 May 2014. [Epub ahead of print]. DOI: 10.1136/jnnp-2014-307721. [DOI] [PubMed] [Google Scholar]
- Turner M., Lamorte M., Chretien N., Havari E., Roberts B., Kaplan J., et al. (2013) Immune status following alemtuzumab treatment in human CD52 transgenic mice. J Neuroimmunol 261: 29–36. [DOI] [PubMed] [Google Scholar]
- Twyman C., Oyuela P., Palmer J., Margolin D., Dayan C. (2014) Thyroid autoimmune adverse events in patients treated with alemtuzumab for relapsing-remitting multiple sclerosis: four-year follow-up of the CARE-MS studies Neurology 82: P2.199. [Google Scholar]
- Vermersch P., Czlonkowska A., Grimaldi L., Confavreux C., Comi G., Kappos L., et al. (2013) Teriflunomide versus subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis: a randomised, controlled phase 3 trial. Mult Scler 20: 705–716. [DOI] [PubMed] [Google Scholar]
- Viglietta V., Baecher-Allan C., Weiner H., Hafler D. (2004) Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med 199: 971–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waggoner J., Martinu T., Palmer S. (2009) Progressive multifocal leukoencephalopathy following heightened immunosuppression after lung transplant. J Heart Lung Transplant 28: 395–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray S., Arnold D., Cohen J., Coles A., Confavreux C., Fox E., et al. (2013a) Comparison of infection risk with alemtuzumab and SC IFNB-1a in patients with multiple sclerosis who experienced disease activity while on prior therapy (CARE-MS II). Neurology 80: P01.172. [Google Scholar]
- Wray S., Arnold D., Cohen J., Coles A., Fox E., Hartung H., et al. (2013b) Herpes infection risk reduced with acyclovir prophylaxis after alemtuzumab. Annual Meeting of the Consortium of Multiple Sclerosis Centers (CMSC), May–June, 2013, Orlando, FL, Poster DX60. [Google Scholar]
- Wynn D., Arnold D., Coles A., Hartung H., Havrdova E., Selmaj K., et al. (2013) Detection, incidence, and management of glomerulonephritis in the alemtuzumab clinical development program. Presented at 29th Congress of the European Committee for Research and Treatment in Multiple Sclerosis, October, Copenhagen, Denmark. [Google Scholar]
- Yamout B., Issa Z., Herlopian A., El Bejjani M., Khalifa A., Ghadieh A., et al. (2013) Predictors of quality of life among multiple sclerosis patients: a comprehensive analysis. Eur J Neurol 20: 756–764. [DOI] [PubMed] [Google Scholar]
- Zhang X., Tao Y., Chopra M., Ahn M., Marcus K., Choudhary N., et al. (2013) Differential reconstitution of T cell subsets following immunodepleting treatment with alemtuzumab (anti-CD52 monoclonal antibody) in patients with relapsing-remitting multiple sclerosis. J Immunol 191: 5867–5874. [DOI] [PubMed] [Google Scholar]
- Zozulya A., Wiendl H. (2008) The role of regulatory T cells in multiple sclerosis. Nat Clin Pract Neurol 4: 384–398. [DOI] [PubMed] [Google Scholar]