

Abstract
The availability of purified and active protein is the starting point for the majority of in vitro biomedical, biochemical, and drug discovery experiments. The use of polyhistidine affinity tags has resulted in great increases of the efficiency of the protein purification process, but can negatively affect structure and/or activity measurements. Similarly, buffer molecules may perturb the conformational stability of a protein or its activity. During the determination of the structure of a Gcn5-related N-acetyltransferase (GNAT) from Pseudomonas aeruginosa (PA4794), we found that both HEPES and the polyhistidine affinity tag bind (separately) in the substrate-binding site. In the case of HEPES, the molecule induces conformational changes in the active site, but does not significantly affect enzyme activity. In contrast, the uncleaved His-tag does not induce major conformational changes but acts as a weak competitive inhibitor of peptide substrate. In two other GNAT enzymes, we observed that the presence of the His-tag had a strong influence on the activity of these proteins. The influence of protein preparation on functional studies may affect the reproducibility of experiments in other laboratories, even when changes between protocols seem at first glance to be insignificant. Moreover, the results presented here show how critical it is to adjust the experimental conditions for each protein or family of proteins, and investigate the influence of these factors on protein activity and structure, as they may significantly alter the effectiveness of functional characterization and screening methods. Thus, we show that a polyhistidine tag and the buffer molecule HEPES bind in the substrate-binding site and influence the conformation of the active site and the activity of GNAT acetyltransferases. We believe that such discrepancies can influence the reproducibility of some experiments and therefore could have a significant “ripple effect” on subsequent studies.
Keywords: reproducibility of biomedical experiments, acetyltransferase, polyhistidine affinity tag, His-tag, HEPES, GNAT
Introduction
For nearly 3 decades, polypeptide fusion affinity tags have been used to easily and efficiently produce large quantities of highly purified recombinant proteins. Affinity tags are produced by altering the sequence of a gene to add specific polypeptide sequences at the N- and/or C-terminus of an expressed protein that provides a “handle” for a specific purification.1 Among the most widely used types of affinity tags is the polyhistidine tag (His-tag), which allows the expressed protein to be purified through binding to immobilized metal cations such as Ni2+ or Co2+.2,3
Typically, a His-tag contains six or more consecutive histidine residues at either the N- or C-terminus of the protein. The polyhistidine sequence of the tag may be removed via proteolytic cleavage4 if a specific endoprotease recognition sequence is also introduced between the tag and the main polypeptide sequence. This also permits additional “subtractive” purification by passing cleaved protein through a metal affinity column and collecting the flowthrough. However, tag removal is sometimes impossible or inefficient as cleavage of the tag requires additional purification steps, and thus the tag is sometimes left uncut (particularly in high-throughput protein purification). Additionally, leaving a His-tag is sometimes desirable because it improves protein solubility or alters crystal packing.
Since His-tags are significantly smaller than most other types of affinity tags, they are often thought to only minimally perturb protein structure and function. However, there is evidence that suggests this is not always true.5 For instance, the tag may interfere with the protein’s ability to bind ligands, cause the protein to aggregate, or change its solubility.4 In particular, purification of metalloproteins using polyhistidine is often problematic, as both the adsorption matrix and the affinity tag itself may potentially strip metal ions from the protein.1
Many examples of how a His-tag can affect protein structure, activity, or binding affinity have been described in the literature. For instance, the rat corticotropin-releasing factor receptor type 2a adopts two different disulfide bond patterns at the N-terminus when the His-tag is at either the N- or C-terminus. However, the tag did not significantly influence the binding affinities of the ligands used in the study.6 On the other hand, the His-tag placement on the tumor-associated single-chain Fv protein significantly affects its ability to bind to the epitope.7 Finally, the substrate specificity, kinetic constants, or protein dynamics may also be affected by His-tag placement in certain proteins. Some of these include the enantioselectivity of some lipases from Staphylococcus8 and the protein dynamics of myoglobin.9
These changes can be also observed in crystal structures. Over 16,000 protein structures determined by X-ray diffraction have a polyhistidine tag reported in their SEQRES records, but only a small fraction (over 1000, or about 6%) have all or part of the His-tag in the final refined model. In many cases, the His-tag is indeed too flexible to be modeled, but in some cases it is not modeled, even though clear electron density for it is present (see Results). Sometimes large conformation changes are observed at sites distant from the His-tag, as at the DNA-binding site of the gene regulatory protein AreA from Aspergillus nidulans.10 The His-tag can also mimic interactions between proteins and/or affect the oligomeric state. For example, the structure of the Archaeoglobus fulgidus AF0173 redox maturation protein shows that the His-tag mimics a portion of the twin-arginine motif that binds to this protein.11
In addition to polypeptide fusion affinity tags, another factor that may perturb protein stability is buffer composition. The choice of buffer is crucial, not only for hydrogen ion buffering capacity at the desired pH, but also because of other unwanted chemical effects.12 Buffers have been shown to interfere with proteins by reacting with protein substrates or acting as inhibitors of enzymatic reactions.13,14
Buffer molecules are also frequently observed in crystal structures. Ordered HEPES buffer molecules, for example, are found in hundreds of protein structures and have yielded a variety of information about substrate binding and protein conformational changes that occur during catalysis. For instance, HEPES bound in the structure of an endoglucanase mimics the substrate, which changes the conformation of the protein to the active state.15 Other structures that have HEPES bound in the substrate-binding site and mimics the native substrate include an l-serine dehydrogenase PA0743 from Pseudomonas aeruginosa16 and an aminoglycoside 6′-N-acetyltransferase from Salmonella typhimurium, where HEPES acts as a transition state analog of the reaction.17 While nonphysiological buffers are typically needed to maintain protein stability for in vitro studies, these artificial components can sometimes interact with the system being studied.
In this work, we describe the impact of both polyhistidine affinity tags and buffer molecules on GNAT activity and structure. The very large GNAT superfamily comprises proteins that acetylate a variety of small molecule substrates like glucosamine-6-phosphate and aminoglycoside antibiotics, as well as large substrates like proteins and histones.18,19 GNAT homologs are found in all kingdoms of life, and many organisms have multiple paralogs. Here we show and analyze the structures of the GNAT protein PA4794 from Pseudomonas aeruginosa20 in complex with either HEPES or the His-tag in the substrate-binding site. We discuss the influence of those factors on PA4794 conformation and activity, and the influence of His-tags on the activity of two other GNATs—spermidine/spermine acetyltransferase SpeG from Vibrio cholerae, and SACOL1063, an N-terminal protein acetyltransferase from Staphylococcus aureus.21
Results
His-tag and HEPES binding
Currently, there are several hundred three-dimensional structures of GNATs in the Protein Data Bank (PDB). Nearly one quarter of these were crystallized in the presence of a poly-histidine tag and many have ligands bound, including HEPES. To determine the influence of the His-tag or HEPES on the structure of a GNAT from Pseudomonas aeruginosa (PA4794) we produced and analyzed two crystal structures—one of His-tagged apo-protein and one of the protein:HEPES complex. We previously established that PA4794 has acetylation activity, produced complexes of the protein with products of the reaction (CoA and acetylated peptide), and identified three residues important for PA4794 activity: Tyr128, Cys117, and Cys29.20
The apo-PA4794 protein with and without the His-tag, as well as PA4794 complexed with HEPES, crystallized in the orthorhombic crystal system (space group P21212) with a monomer in the asymmetric unit. The PA4794:HEPES complex structure was the first structure obtained, which contained the HEPES buffer molecule bound in the substrate-binding site [Fig. 1(B)]. The complex was produced using protein purified and crystallized in HEPES buffer in the absence of substrates. The residues that interact with the HEPES molecule include the following: (1) the sulfonate group of HEPES is coordinated through several interactions, including the Nε and Nω atoms of Arg141, the Oω atom of Tyr68 through a water molecule, and several additional water molecules, (2) the oxygen of the hydroxyethyl group of HEPES interacts with the main chain oxygen of Tyr28, Met81, and (3) the oxygen of the hydroxyethyl group and the nitrogen atom of the piperazine ring of HEPES both interact with main chain oxygen atoms of Cys29 and Gly79 through the same water molecule.
Figure 1.
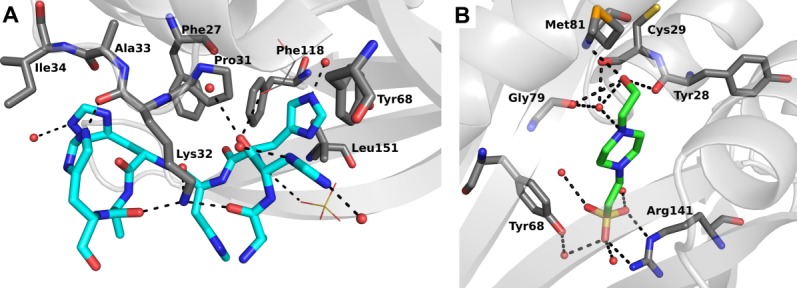
Structures of PA4794 with either His-tag (A) or HEPES (B) bound to the substrate-binding site.
The structure of PA4794 bound to HEPES also shows that HEPES binding induces a conformational change between the apo-form of PA4794 and the HEPES-bound form (Fig. 2). The differences are located in the active site area, involving both the main-chain and the side chains. Interestingly, the most distinct change is a flip of the main chain of Gly79 and Asn80. We have not observed this conformational change in any of the structures of PA4794 previously reported,20 and thus its biological relevance is unclear. On the opposite side of the active site there is a shift of main chain residues 28 and 29 and a conformational change of the Cys29 side chain. This shows that the buffer-bound structure cannot be treated as a truly apo-form due to its changed conformation, and it may also be of limited use for interpreting function.
Figure 2.
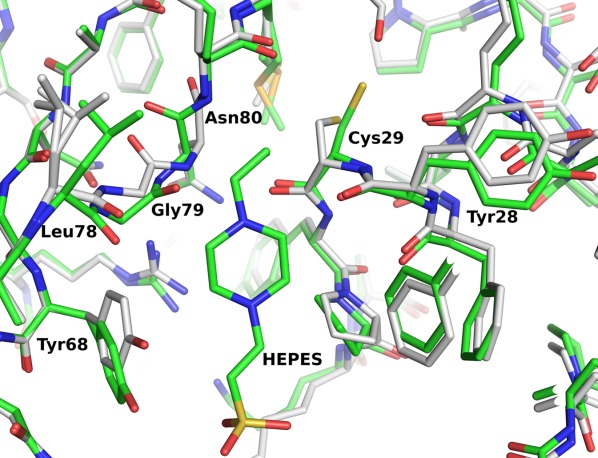
Conformational change of PA4794 upon binding HEPES. The apo structure is shown in gray, and the HEPES-bound structure is shown in green. Upon binding of HEPES we observe a flip of the main chain of Gly79 and Asn80, a shift in the main chain of residues 28 and 29, and a conformational change of the Cys29 side chain.
The protein that was used for the second structure was purified in Tris-HCl buffer instead of HEPES, and crystallization trials were set in conditions that did not contain HEPES. However, the second structure revealed additional electron density for the His-tag. The analysis of the symmetry-related molecules in the crystal shows that the tag of one molecule is bound in the substrate-binding cleft of the protein molecule in the adjacent asymmetric unit, presumably blocking substrate binding [Fig. 1(A)]. The binding of the tag is driven by several interactions: (1) polar interactions through Lys32, (2) hydrophobic interactions including Phe118, Leu151, Phe27, Pro31, Tyr68, and Ile34, and (3) numerous hydrogen-bonding interactions with the protein backbone as well as the side chains. These include interactions of the His-tag backbone and side chains with the main chain oxygen atoms of Pro31 and Ala33, and the main chain and side chain atoms of Lys32. The interaction with Lys32 seems to be most relevant in the context of peptide binding, as it includes hydrogen bonds with three main chain oxygen atoms of the His-tag. This would give strong, but sequence independent, interaction with the peptide, while other residues may be responsible for the substrate specificity.
To determine whether PA4794 structures in the presence of HEPES or the His-tag provide relevant information regarding substrate binding, we compared them with PA4794 structures containing either the acetylated N-phenylacetyl-Gly-Lys (NPAcGK) substrate or AcCoA20 (Fig. 3). The comparison of the HEPES structure with the acetylated NPAcGK structure shows that the position of both NPAcGK peptide and HEPES are very similar. When the structures of PA4794 that are in complex with either HEPES or AcCoA are superimposed, the acetyl group of AcCoA is very close to the hydroxyethyl group of HEPES. This confirms that HEPES is positioned similarly to the potential substrate, and thus may be mimicking true substrate binding [Fig. 3(B)]. A comparison of the His-tag and acetylated NPAcGK structures shows how the His-tag may compete with NPAcGK during catalysis by blocking the peptide-binding site if the tag is not removed from the protein before kinetic characterization. Although a portion of the His-tag mimics how a peptide may bind, it is not inserted as deeply into the active site as NPAcGK [Fig. 3(A)].
Figure 3.
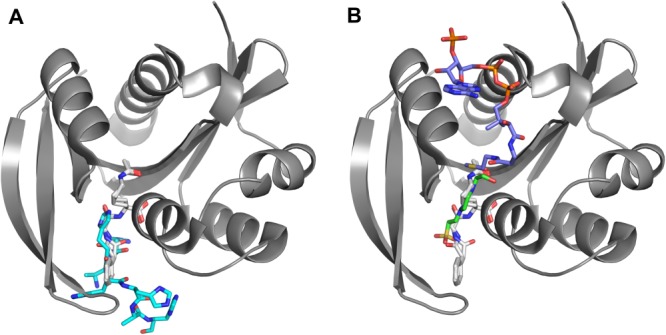
Superposition of PA4794 with different ligands bound. (A) product—NPAcGAcK (in gray) and His-tag (in cyan), (B) product—NPAcGAcK (in gray), AcCoA (in violet) and HEPES (in green). An interactive view is available in the electronic version of the article.
Influence of the polyhistidine tag on kinetic activity
We analyzed the kinetics of the activity of PA4794 in the presence of AcCoA and the NPAcGK peptide to determine if the presence of the His-tag affected the function of PA4794. As expected, the predominant effect of the His-tag, which binds in the acetyl group acceptor binding site, was to decrease the enzyme’s affinity for the acceptor—NPAcGK peptide (Fig. 4). Alternatively, the tag had relatively little effect on the enzyme’s maximal velocity or affinity for AcCoA (which binds in a different binding site that is inaccessible to the His-tag; Fig. 4). Contrary to the effects of the His-tag, HEPES did not significantly affect the activity of the enzyme even at very high millimolar (>200) concentrations.
Figure 4.
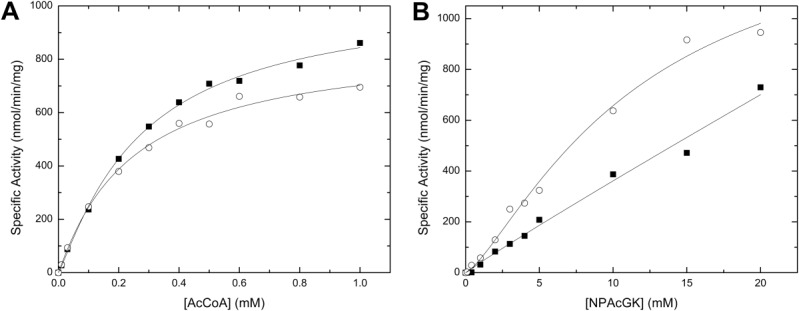
Effect of the His-tag on the kinetic activity of PA4794. A comparison of the kinetic curves for AcCoA (A) or NPAcGK (B) is shown for the enzyme with (filled squares) or without (open circles) a His-tag. Kinetic curves for AcCoA were performed using a set concentration of NPAcGK (10 mM) (A), and curves for NPAcGK were determined in the presence of 1 mM AcCoA (B). All kinetic assays were performed using 2.6 μM enzyme. The kinetic parameters for the PA4794 enzyme for AcCoA were the following: Vmax (1040 ± 52 (SD) (His-tagged) and 865 ± 67 (SD) (untagged) nmol/min/mg), S0.5 (272 ± 29 (SD) (His-tagged), and 244 ± 44 (SD) (untagged) μM). The kinetic parameters for the untagged PA4794 enzyme for NPAcGK were: Vmax (1460 ± 282 (SD) nmol/min/mg) and S0.5 (11.6 ± 3.8 (SD) mM). The NPAcGK curve did not reach saturation for the His-tagged enzyme, so we were unable to accurately determine its kinetic parameters.
To investigate the possible effect of the His-tag on other types of GNATs, we also analyzed the kinetic parameters of two other proteins: a spermidine/spermine acetyltransferase SpeG from Vibrio cholerae and an N-terminal protein acetyltransferase SACOL1063 from Staphylococcus aureus. Both tagged SpeG and SACOL1063 enzymes exhibit a decreased activity or affinity for AcCoA compared to the untagged enzyme (Figs. 5 and 6). For SpeG, the activity of the untagged enzyme was significantly higher (8-fold) than that of the His-tagged enzyme, and both Vmax and S0.5 were significantly affected (Fig. 5). On the other hand, when the substrate saturation curves for AcCoA were compared for both His-tagged and untagged SACOL1063 enzyme, we observed that the presence of the His-tag mainly decreased the enzyme’s affinity for substrate (Fig. 6). Compared to PA4794, where the His-tag primarily affected the enzyme’s affinity for the NPAcGK peptide, SpeG and SACOL1063 showed that the His-tag is also capable of affecting the enzymes’ affinities for AcCoA and/or their maximal velocities. We were not able to obtain crystals of SACOL1063 in the presence of the His-tag. For SpeG, the tag was not visible in the electron density, and although it has a strong influence on enzyme activity, it may still be relatively flexible in the crystal. Our results show that the kinetic parameters for members of the GNAT superfamily can be affected by the presence of the His-tag in a number of ways; therefore, the His-tag should be removed from all GNATs before functional studies.
Figure 5.
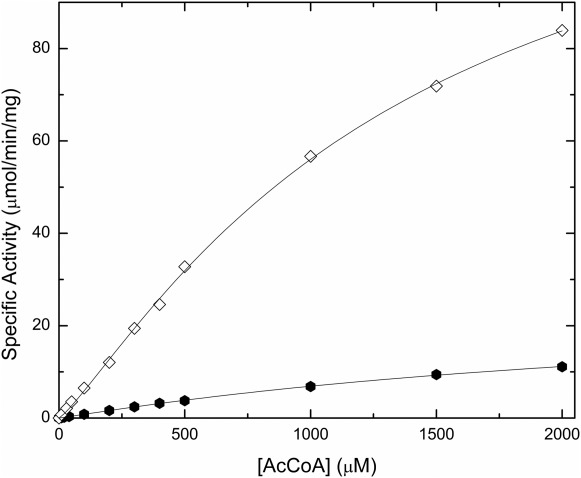
Polyhistidine tag effect on SpeG activity. The activity of the His-tagged (filled hexagons, 0.6 μM enzyme) and untagged (open diamonds, 0.044 μM enzyme) SpeG enzyme at 3 mM spermine and varying concentrations of AcCoA (0–2 mM) is shown. The kinetic parameters for the His-tagged enzyme were not able to be determined because the curve did not reach saturation. The Vmax and concentration of substrate at half the maximal velocity (S0.5) for the untagged enzyme were 140 ± 9 (SD) μmol/min/mg and 1370 ± 150 (SD) μM, respectively.
Figure 6.
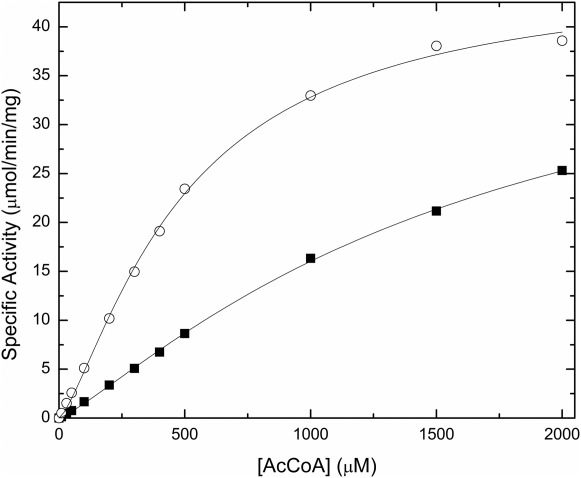
Polyhistidine tag effect on SACOL1063 activity. The activity of the His-tagged (filled squares, 0.32 μM enzyme) and untagged (open circles, 0.12 μM enzyme) SACOL1063 enzyme at 20 mM l-threonine and varying concentrations of AcCoA (0–2 mM). The Vmax for the His-tagged and untagged enzyme is 45.4 ± 2.5 (SD) and 45.4 ±1.4 (SD) μmol/min/mg, respectively. The concentration of substrate at half the maximal velocity (S0.5) for the His-tagged and untagged enzyme is 1660 ± 152 (SD) and 492 ± 31 (SD) μM, respectively.
Effects of buffers and His-tags are underestimated
We found that in more than 18% of structures containing HEPES, the buffer molecule is bound in close proximity to the active site of proteins, but often is not mentioned in accompanying publications. Similarly, many structures of proteins with His-tags have density for the affinity tag in the proximity of active sites, but are either not discussed or are omitted altogether from the model, regardless of crystallographic evidence of its presence. For example, in structure 2QHA, despite the fact that there was clear density for the His-tag in the active site, this motif was not only not modeled, but in the accompanying publication it was also claimed that the motif was not visible in the density map.22 With manual building of the six histidine residues (Fig. 7) and a few cycles of refinement with Refmac, we were able to decrease the R/Rfree of this structure from 16.0%/19.0% to 12.2%/16.4%, respectively.
Figure 7.
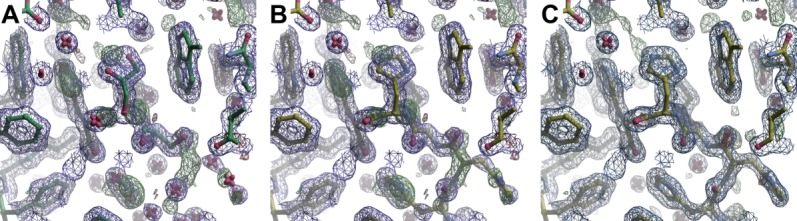
Polyhistidine tag modeled in the structure of 2QHA. (A) 2QHA as originally modeled with glycerol and waters, (B) with histidines modeled into the original electron density, and (C) after refinement with the modeled histidines.
Since many proteins bind the tag with relatively high affinity in their active sites or between oligomers, difficulties may arise once tag cleavage is attempted and may lead to further unexpected behaviors during experiments. For example, a putative redox-enzyme maturation protein (REMP) from Archaeoglobus fulgidus (AF0173) crystal structure shows that the tag allows the protein to form dimers. In doing so, it causes the TEV protease recognition site to become inaccessible and therefore unable to be cleaved proteolytically.11 The presence and position of the tag has also been shown to affect protein stability and can change the substrate specificity of enzymes. For example, the stability of the Msed_1072 carboxylesterase from Metallosphaera sedula varies depending upon tag placement and presence or absence of detergents or organic solvents.23 Additionally, the Escherichia coli thioesterase I enzyme substrate specificity shifted its preference towards more hydrophilic substrates when a C-terminal His-tag was added.24 At times, the presence of a His-tag may be deleterious to protein activity, as was shown for Rhodobacter sphaeroides YedY reductase.25 Many buffers used in biological research have a strong influence in thermodynamics studies due to ionization reactions.26
Discussion
The results demonstrated that both the polyhistidine affinity tag and the buffer HEPES bind to the active site of PA4794, and the binding of HEPES induced structural changes not observed in the structure of apo-protein. However, the His-tag also inhibited the kinetic activity of the enzyme. It is reasonable to conclude from the structural and kinetic data that the His-tag binds in the substrate site and acts as a weak competitive inhibitor for the peptide. Competitive inhibition of AcCoA by the His-tag was also observed for two other GNAT enzymes, SpeG and SACOL1063, indicating that the effects of the His-tag may be widespread for members of the GNAT superfamily. Most certainly, this will also be true for many other classes of proteins, especially enzymes. Moreover, PA4794 is a very promiscuous enzyme. It is becoming clear that the number of promiscuous enzymes is higher than previously acknowledged,27 and it is tempting to speculate that the capability of promiscuous enzymes to interact with a range of molecules might make these enzymes more prone to unwanted tag or buffer influence.
Often the presence of an affinity tag can actually aid in protein crystallization, especially when the tag is well ordered and interacts with the protein (for example, see structures with PDB codes 1ZKP, 1ZSW, and 2AUA). The presence of the tag has been reported to either improve or deteriorate the resolution of protein structures. The effect might depend on the localization of the tag and the way it interacts with the protein. For example, binding of the tag in the ligand binding site might stabilize the whole protein, while flexible tag binding to the surface of the protein in disparate ways might increase the overall flexibility and thus have a deteriorating effect on the quality and resolution of the crystals and thereby resolution. Because affinity tags are commonplace for protein production, their potential effects on the structure and function of protein are sometimes overlooked. In the case of PA4794, the His-tag does not influence crystallization of the protein, but it does bind to the substrate-binding site of the protein, with an affinity sufficient to act as a weak competitive inhibitor of substrate. Thus, the presence of a His-tag may influence both the activity and binding affinity for ligands because it blocks the substrate binding site in the crystal and competes for the substrate-binding site in solution.
The presence of nonphysiological small molecules in buffers used in purification and crystallization screening methods can yield important clues as to the types of ligands (e.g., substrates, inhibitors, cofactors, etc.) that may bind to the protein, and analysis of residues involved in these interactions can help with understanding protein function. The crystallization screens often contain molecules that represent a larger class of compounds, but might not be the strongest binder in a particular class (for example, the cefmetazole included in the crystallization ligand screen was the first cephalosporin discovered to bind to PA4794, but further experiments revealed that other cephalosporins bind and inhibit more strongly20). Since many of these molecules may compete with binding of biologically relevant ligands, unambiguous identification of physiologically relevant ligands may not be obvious. The conformational changes in protein structure may also give clues for determining the catalytic or regulatory mechanisms of a given protein. However, these components can also interfere with physiological mechanisms. Clearly it is critical to select buffer conditions optimized for a particular protein to ensure and verify that the nonphysiological components of the buffer do not influence the results of biochemical studies in artificial ways.
While the potential effects of both affinity tags and sulfonyl buffers, especially HEPES, on biological activity have been known and observed for many years, these effects are rarely discussed in the published literature. The conditions of protein expression and purification can play a significant role in the outcome of protein functional and structure-function studies. In particular, if these nonphysiological compounds are observed in structures that are used for in silico screening, this can negatively affect in silico binding and screening analyses by occluding binding sites and/or inducing artificial conformational changes in the proteins. This may cause a “ripple effect” as subsequent studies are based on questionable data. To properly interpret results of structural and functional studies, it is essential to pay attention to these types of details. Our results have shown the importance of taking these factors into account for members of the GNAT superfamily, but they may also lend a cautionary word of advice for interpreting results obtained for other proteins in different families. In our opinion, recognition, identification, and clarification of artifacts due to buffers or affinity tags should be described and evaluated in order to truly understand a protein’s function and accurately characterize the enzyme of interest.
Materials and Methods
Cloning, expression, and purification
The PA4794 gene was cloned into the p11 pET expression vector (Structural Genomics Consortium), and the SpeG and SACOL1063 genes were cloned into the pMCSG7 expression vector (Midwest Center for Structural Genomics).28 The proteins that are produced using these vectors contain an N-terminal poly-6-histidine affinity tag followed by a spacer and a tobacco etch virus (TEV) protease cleavage site. The sequence Gly-His (in the case of the p11 pET vector) or Ser-Asn-Ala (in the case of the pMCSG7 vector) remains on the N-terminus of each protein after tag cleavage with recombinant TEV protease. The PA4794 protein used for the 4M3S structure was overexpressed and purified as described.29 This protein was stored in a buffer containing 500 mM NaCl and 10 mM HEPES at pH 7.5. The PA4794 fusion protein used for the 3KKW structure and the SpeG and SACOL1063 fusion proteins used for the kinetic assays were overexpressed and purified as described.20,21 Both His-tagged and cleaved proteins were purified and stored in a buffer containing 500 mM NaCl and 10 mM Tris-HCl at pH 7.5. The PA4794 proteins were concentrated to 9 (PDB ID: 3KKW) and 10 (PDB ID: 4M3S) mg/mL using an Ultracel-10K concentrator (Millipore) and used for crystallization trials.
Enzyme kinetic assays
Assays were performed using previously described procedures.20,21 To determine the effect of the polyhistidine tag on enzyme activity, we measured the kinetic parameters of the enzymes toward different substrates in a 50 μL reaction volume. Substrate saturation curves were produced by holding one substrate at a constant concentration while the other one was varied.30 The kinetic parameters for PA4794 were determined for AcCoA (0–1 mM) at 10 mM NPAcGK or for NPAcGK (0–20 mM) at 1 mM AcCoA in Bicine buffer pH 9.0 with 2.6 μM enzyme (His-tagged or untagged). Kinetic parameters for AcCoA (0–2 mM) for SpeG and SACOL1063 were determined by holding the concentration of spermine at 3 mM (SpeG) or the concentration of l-threonine at 20 mM (SACOL1063). SpeG was assayed in Bicine buffer pH 9.0 and SACOL1063 was assayed in Tris-HCl buffer pH 8.0. The activity of SpeG was determined using 0.6 μM His-tagged enzyme and 0.044 μM untagged enzyme, whereas the activity of SACOL1063 was determined using 0.32 and 0.12 μM His-tagged and untagged enzyme, respectively. All measurements were performed in duplicate and the data shown is the average of the two individual trials. One unit of activity is defined as the amount of enzyme that produces 1 μmol of CoA per min under the described reaction conditions. All kinetic curves were fitted using a modified Hill equation in the program Origin v.8.1 as described previously.20
Crystallization
Tracking and analysis of the crystallization experiments were performed with the Xtaldb system.31 The crystals for 4M3S were grown using vapor diffusion and sitting drop setup. The crystallization drops were composed of a 1:1 µL mixture of the protein solution to precipitant solution (100 mM HEPES-Na pH 7.5, 2% PEG400, 2M (NH4)2SO4). The crystals for 3KKW were grown using a vapor diffusion and hanging drop setup. The crystallization drops were composed of a 1:1 µL mixture of the protein solution to precipitant solution (2M (NH4)2SO4, 100 mM Bis-Tris pH 6.5). Crystals were grown overnight at 16°C. Prior to data collection, the crystal was transferred to a solution containing a 2:1 mixture of well solution and ethylene glycol and was immediately cryo-cooled in liquid nitrogen.
Data collection, structure determination and refinement
Data collection for PA4794 was performed at the 19-BM beamline of the Structural Biology Center32 at the Advanced Photon Source (APS, Argonne National Laboratory). Data were collected at 100 K and processed with HKL-2000.33 Structure determination for 4M3S was performed using single-wavelength anomalous diffraction and HKL-3000,34 which is integrated with SHELXC/D/E,35 MLPHARE,36 DM,37 ARP/wARP,38 CCP4,39 and SOLVE and RESOLVE.40 That structure was then used as a model to solve the structure of PA4794 with the His-tag by molecular replacement (MR), using HKL-300034 coupled with MOLREP.41 Refinement was performed using HKL-3000 coupled with REFMAC5,42 COOT,43,44 and selected programs from the CCP4 package.39 The B-factors were refined in each case using translation/libration/screw (TLS) groups assigned using the TLSMD server.45 Validation of the structures was performed using MOLPROBITY46 and ADIT.47 The coordinates, structure factors, and intensities were deposited in the PDB (PDB IDs: 4M3S, 3KKW). Statistics describing crystallographic data collection and refinement are summarized in Table 1.
Table 1.
Crystallographic Data Collection and Refinement Statistics
| Complex | His-tag | HEPES |
|---|---|---|
| PDB code | 3KKW | 4M3S |
| Data collection | ||
| Space group | P21212 | P21212 |
| Unit cell | ||
| a, b, c (Å) | 57.53, 76.14, 39.45 | 56.95, 75.51, 39.48 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Resolution (Å)a | 1.41 (1.41–1.43) | 1.30 (1.30–1.32) |
| No. reflections | 32398 | 42543 |
| Rmerge | 0.047 (0.391) | 0.103 (0.779) |
| I/σ(I)a | 47.2 (4.2) | 37.2 (2.1) |
| Completeness (%)a | 99.9 (100.0) | 99.9 (100.0) |
| Redundancy | 6.5 (5.7) | 8.8 (8.1) |
| Refinement | ||
| Rwork (%)/Rfree (%)b | 15.7/17.9 | 13.5/16.9 |
| No. atoms | ||
| Protein | 1299 | 1245 |
| Ligand/Ion | 78 | 29 |
| Water | 210 | 318 |
| B factors | ||
| Wilson B factor | 14.6 | 12.9 |
| Protein | 21 | 15 |
| Ligand/Ion | 29 | 19 |
| Water | 32 | 32 |
| Structure quality | ||
| R.m.s. deviations | ||
| bond length (Å) | 0.02 | 0.02 |
| bond angles (°) | 1.8 | 1.7 |
| Ramachandran Statistics | ||
| Favored (%) | 99.38 | 98.73 |
| Allowed (%) | 0.62 | 1.27 |
Highest resolution shell values are shown in parentheses.
About 5% of reflections were randomly assigned to the Rfree set.
Acknowledgments
We would like to thank Matthew D. Zimmerman, Alex Wlodawer, Thomas Terwilliger, and Zbigniew Dauter for numerous discussion and suggestions.
References
- Terpe K. Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol. 2003;60:523–533. doi: 10.1007/s00253-002-1158-6. [DOI] [PubMed] [Google Scholar]
- Porath J, Carlsson J, Olsson I, Belfrage G. Metal chelate affinity chromatography, a new approach to protein fractionation. Nature. 1975;258:598–599. doi: 10.1038/258598a0. [DOI] [PubMed] [Google Scholar]
- Hochuli E, Dobeli H, Schacher A. New metal chelate adsorbent selective for proteins and peptides containing neighbouring histidine residues. J Chromatogr. 1987;411:177–184. doi: 10.1016/s0021-9673(00)93969-4. [DOI] [PubMed] [Google Scholar]
- Kapust RB, Waugh DS. Controlled intracellular processing of fusion proteins by TEV protease. Protein Expr Purif. 2000;19:312–318. doi: 10.1006/prep.2000.1251. [DOI] [PubMed] [Google Scholar]
- Carson M, Johnson DH, McDonald H, Brouillette C, Delucas LJ. His-tag impact on structure. Acta Cryst D. 2007;63:295–301. doi: 10.1107/S0907444906052024. [DOI] [PubMed] [Google Scholar]
- Klose J, Wendt N, Kubald S, Krause E, Fechner K, Beyermann M, Bienert M, Rudolph R, Rothemund S. Hexa-histidin tag position influences disulfide structure but not binding behavior of in vitro folded N-terminal domain of rat corticotropin-releasing factor receptor type 2a. Protein Sci. 2004;13:2470–2475. doi: 10.1110/ps.04835904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel A, Colcher D, Koo JS, Booth BJ, Pavlinkova G, Batra SK. Relative position of the hexahistidine tag effects binding properties of a tumor-associated single-chain Fv construct. Biochim Biophys Acta. 2000;1523:13–20. doi: 10.1016/s0304-4165(00)00086-6. [DOI] [PubMed] [Google Scholar]
- Sayari A, Mosbah H, Verger R, Gargouri Y. The N-terminal His-tag affects the enantioselectivity of staphylococcal lipases: a monolayer study. J Colloid Interface Sci. 2007;313:261–267. doi: 10.1016/j.jcis.2007.04.053. [DOI] [PubMed] [Google Scholar]
- Thielges MC, Chung JK, Axup JY, Fayer MD. Influence of histidine tag attachment on picosecond protein dynamics. Biochemistry. 2011;50:5799–5805. doi: 10.1021/bi2003923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chant A, Kraemer-Pecore CM, Watkin R, Kneale GG. Attachment of a histidine tag to the minimal zinc finger protein of the Aspergillus nidulans gene regulatory protein AreA causes a conformational change at the DNA-binding site. Protein Expr Purif. 2005;39:152–159. doi: 10.1016/j.pep.2004.10.017. [DOI] [PubMed] [Google Scholar]
- Kirillova O, Chruszcz M, Shumilin IA, Skarina T, Gorodichtchenskaia E, Cymborowski M, Savchenko A, Edwards A, Minor W. An extremely SAD case: structure of a putative redox-enzyme maturation protein from Archaeoglobus fulgidus at 3.4 A resolution. Acta Cryst D. 2007;63:348–354. doi: 10.1107/S0907444906055065. [DOI] [PubMed] [Google Scholar]
- Good NE, Winget GD, Winter W, Connolly TN, Izawa S, Singh RM. Hydrogen ion buffers for biological research. Biochemistry. 1966;5:467–477. doi: 10.1021/bi00866a011. [DOI] [PubMed] [Google Scholar]
- Rej R, Vanderlinde RE. Effects of buffers on aspartate aminotransferase activity and association of the enzyme with pyridoxal phosphate. Clin Chem. 1975;21:1585–1591. [PubMed] [Google Scholar]
- Desmarais WT, Bienvenue DL, Bzymek KP, Holz RC, Petsko GA, Ringe D. The 1.20 A resolution crystal structure of the aminopeptidase from Aeromonas proteolytica complexed with tris: a tale of buffer inhibition. Structure. 2002;10:1063–1072. doi: 10.1016/s0969-2126(02)00810-9. [DOI] [PubMed] [Google Scholar]
- Crennell SJ, Hreggvidsson GO, Nordberg Karlsson E. The structure of Rhodothermus marinus Cel12A, a highly thermostable family 12 endoglucanase, at 1.8 A resolution. J Mol Biol. 2002;320:883–897. doi: 10.1016/s0022-2836(02)00446-1. [DOI] [PubMed] [Google Scholar]
- Tchigvintsev A, Singer A, Brown G, Flick R, Evdokimova E, Tan K, Gonzalez CF, Savchenko A, Yakunin AF. Biochemical and structural studies of uncharacterized protein PA0743 from Pseudomonas aeruginosa revealed NAD+-dependent l-serine dehydrogenase. J Biol Chem. 2012;287:1874–1883. doi: 10.1074/jbc.M111.294561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice F, Broutin I, Podglajen I, Benas P, Collatz E, Dardel F. Enzyme structural plasticity and the emergence of broad-spectrum antibiotic resistance. EMBO Rep. 2008;9:344–349. doi: 10.1038/embor.2008.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyda F, Klein DC, Hickman AB. GCN5-related N-acetyltransferases: a structural overview. Annu Rev Biophys Biomol Struct. 2000;29:81–103. doi: 10.1146/annurev.biophys.29.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetting MW, SdC LP, Yu M, Hegde SS, Magnet S, Roderick SL, Blanchard JS. Structure and functions of the GNAT superfamily of acetyltransferases. Arch Biochem Biophys. 2005;433:212–226. doi: 10.1016/j.abb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Majorek KA, Kuhn ML, Chruszcz M, Anderson WF, Minor W. Structural, functional and inhibition studies of a GNAT superfamily protein PA4794: a new C-terminal lysine protein acetyltransferase from Pseudomonas aeruginosa. J Biol Chem. 2013;288:30223–30235. doi: 10.1074/jbc.M113.501353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn ML, Majorek KA, Minor W, Anderson WF. Broad-substrate screen as a tool to identify substrates for bacterial Gcn5-related N-acetyltransferases with unknown substrate specificity. Protein Sci. 2013;22:222–230. doi: 10.1002/pro.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan XX, An XM, Gui LL, Liang DC. From structure to function: insights into the catalytic substrate specificity and thermostability displayed by Bacillus subtilis mannanase BCman. J Mol Biol. 2008;379:535–544. doi: 10.1016/j.jmb.2008.03.068. [DOI] [PubMed] [Google Scholar]
- Killens-Cade R, Turner R, MacInnes C, Grunden A. Characterization of a thermostable, recombinant carboxylesterase from the hyperthermophilic Archaeon metallosphaera sedula DSM5348. Advan Enzyme Res. 2014;2:1–13. [Google Scholar]
- Lee YL, Su MS, Huang TH, Shaw JF. C-terminal His-tagging results in substrate specificity changes of the thioesterase I from Escherichia coli. J Am Oil Chem Soc. 1999;76:1113–1118. [Google Scholar]
- Sabaty M, Grosse S, Adryanczyk G, Boiry S, Biaso F, Arnoux P, Pignol D. Detrimental effect of the 6 His C-terminal tag on YedY enzymatic activity and influence of the TAT signal sequence on YedY synthesis. BMC Biochem. 2013;14:28. doi: 10.1186/1471-2091-14-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg RN, Kishore N, Lennen RM. Thermodynamic quantities for the ionization reactions of buffers. J Phys Chem Ref Data. 2002;31:231–370. [Google Scholar]
- Khersonsky O, Tawfik DS. Enzyme promiscuity: a mechanistic and evolutionary perspective. Annu Rev Biochem. 2010;79:471–505. doi: 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr Purif. 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- Zhang RG, Skarina T, Katz JE, Beasley S, Khachatryan A, Vyas S, Arrowsmith CH, Clarke S, Edwards A, Joachimiak A, Savchenko A. Structure of Thermotoga maritima stationary phase survival protein SurE: a novel acid phosphatase. Structure. 2001;9:1095–1106. doi: 10.1016/s0969-2126(01)00675-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland RA. Enzymes: a practical introduction to structure, mechanism, and data analysis. New York: Wiley; 2000. [Google Scholar]
- Zimmerman MD, Chruszcz M, Koclega KD, Otwinowski Z, Minor W. The Xtaldb system for project salvaging in high-throughput crystallization. Acta Cryst A. 2005;61:c178–c179. [Google Scholar]
- Rosenbaum G, Alkire RW, Evans G, Rotella FJ, Lazarski K, Zhang RG, Ginell SL, Duke N, Naday I, Lazarz J, Molitsky MJ, Keefe L, Gonczy J, Rock L, Sanishvili R, Walsh MA, Westbrook E, Joachimiak A. The Structural Biology Center 19ID undulator beamline: facility specifications and protein crystallographic results. J Synchrotron Radiat. 2006;13:30–45. doi: 10.1107/S0909049505036721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Methods in enzymology: macromolecular crystallography, part A. New York: Academic Press; 1997. Processing of X-ray diffraction data collected in oscillation mode; pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Cryst D. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- Sheldrick GM. A short history of SHELX. Acta Cryst A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. Warrington, UK: 1991. Isomorphous replacement and anomalous scattering. Daresbury Laboratory. [Google Scholar]
- Cowtan KD, Main P. Improvement of macromolecular electron-density maps by the simultaneous application of real and reciprocal space constraints. Acta Cryst D. 1993;49:148–157. doi: 10.1107/S0907444992007698. [DOI] [PubMed] [Google Scholar]
- Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- Collaborative Computional Project N. The CCP4 suite: programs for protein crystallography. Acta Cryst D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Terwilliger T. SOLVE and RESOLVE: automated structure solution, density modification and model building. J Synchrotron Radiat. 2004;11:49–52. doi: 10.1107/s0909049503023938. [DOI] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
- Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Cryst D. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Cryst D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter J, Merritt EA. TLSMD web server for the generation of multi-group TLS models. J Appl Cryst. 2006;39:109–111. [Google Scholar]
- Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, III, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Guranovic V, Dutta S, Feng Z, Berman HM, Westbrook JD. Automated and accurate deposition of structures solved by X-ray diffraction to the Protein Data Bank. Acta Cryst D. 2004;60:1833–1839. doi: 10.1107/S0907444904019419. [DOI] [PubMed] [Google Scholar]