

Abstract
PI3Kα remains an attractive target for the development of anticancer targeted therapy. A number of p110α crystal structures in complex with the nSH2-iSH2 fragment of p85 regulatory subunit have been reported, including a few small molecule co-crystal structures, but the utilization of this crystal form is limited by low diffraction resolution and a crystal packing artifact that partially blocks the ATP binding site. Taking advantage of recent data on the functional characterization of the lipid binding properties of p110α, we designed a set of novel constructs allowing production of isolated stable p110α subunit missing the Adapter Binding Domain and lacking or featuring a modified C-terminal lipid binding motif. While this protein is not catalytically competent to phosphorylate its substrate PIP2, it retains ligand binding properties as indicated by direct binding studies with a pan-PI3Kα inhibitor. Additionally, we determined apo and PF-04691502 bound crystal structures of the p110α (105-1048) subunit at 2.65 and 2.85 Å, respectively. Comparison of isolated p110α(105-1048) with the p110α/p85 complex reveals a high degree of structural similarity, which validates suitability of this catalytically inactive p110α for iterative SBDD. Importantly, this crystal form of p110α readily accommodates the binding of noncovalent inhibitor by means of a fully accessible ATP site. The strategy presented here can be also applied to structural studies of other members of PI3KIA family.
Keywords: crystal structure, lipid kinase activity, ATPase activity, surface plasmon resonance, isothermal titration calorimetry
Introduction
PI3Kα is the best characterized Class I PI3K isoform comprising a catalytic p110α subunit and p85α regulatory subunit. The p110α catalytic subunit, similar to the other members of Class I PI3K, consists of an N-terminal adaptor binding domain (ADB), a RAS-binding domain (RBD), a C2 domain (C2), a helical domain, and C-terminal bilobal kinase domain. The p85α regulatory subunit contains two Src homology (SH2) domains, nSH2 and cSH2, connected by a coiled–coiled domain iSH2, which binds the ABD of the p110α subunit. The nSH2-iSH2-cSH2 unit is preceded in p85α by Src homology 3 domain, a Bar cluster region homology domain (BH), and two proline-rich regions.
PI3Kα activation can be achieved via binding of the nSH2 regulatory subunit to phosphopeptides derived from the cytoplasmic region of receptor tyrosine kinases (RTKs) (for a review see Ref. 1). Following phosphopeptide binding, the regulatory nSH2 dissociates from the helical domain of the p110α subunit resulting in enzyme activation. Additionally, the nSH2 domain contacts with iSH2 residues 571–598, maintaining an inhibitory conformation of PI3Kα. Truncation of p85 at residue 572 abolishes inhibition of p110α, leading to a constitutively active PI3K. Furthermore, a minimal regulatory unit consisting of nSH2 and iSH2 (niSH2) was shown to sufficient to inhibit p110α subunit activity and was shown to be activated by phosphopeptides.2 Activated RAS proteins bind directly to an N-terminal RBD on p110α, acting synergistically with the input from tyrosine-phosphorylated proteins to optimally activate lipid kinase activity (for a review see Ref. 1). Following their activation by RTK and RAS proteins, PI3Kα phosphorylates phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-trisphosphate (PIP3), which recruits protein kinase B (AKT), and PDK1 to the plasma membrane. Following activation by PDK1 and mTORC2, AKT promotes growth, metabolism, and tumorogenesis via phosphorylation of many target proteins involved in those cellular functions.3
The role of PI3Kα in cancer was first highlighted by the identification of the PTEN tumor suppressor as a PIP3-phosphatase, explaining how frequent inactivation of PTEN in cancer leads to constitutive activation of the PI3K pathway.4 Cancer-specific mutations in the catalytic subunit of phosphatidylinositol 3-kinase (PI3K) p110α occur in diverse tumors in frequencies that can exceed 30% (for a review see Ref. 5). These mutations are found throughout the p110α subunit with the majority of them mapping to three hotspots. The E542K and E545K mutations are located in the helical domain while H1047R is in the C-terminal region of the p110α kinase domain. Mutations in the p85 subunit have also been identified, which reduce inhibitory contacts with p110α subunit.6 Most of these cancer-specific mutations induce a gain of function resulting in elevated lipid kinase activity and constitutive signaling through the kinases AKT and TOR (for review see Ref. 7). The high prevalence of PI3Kα mutations in various human tumors and recent progress in the development of personalized medicines in oncology makes PI3Kα an attractive target for drug discovery.
Recently, a number of crystal structures have became available for a minimal PI3Kα complex consisting of a full length p110α subunit (wild type PDB ID: 2RDO and H1047R mutation PDB ID: 3HIZ) and the niSH2 region of p85 subunit.8–10 The structures provide mechanistic understanding of critical interactions governing activation of p110α and how different classes of oncogenic mutants result in the activation of lipid kinase activity of the PI3Kα complex (for review see Ref. 11). The structural analysis also provided a foundation for studies of the dynamics of PI3Kα activation as well as understanding of the mechanism and determinants involved in membrane and lipid binding.10,12,13 In addition, there are three crystal structures of PI3Kα with small molecule inhibitors, two with covalent inhibitors Wortmannin (PDB ID:3HHM)8 and CNX-1351 (PDB ID: 3ZIM)14 and one with the noncovalent PIK-108 (PDB ID: 4A55)10 inhibitor identified by Shokat and colleagues.15 The latter study mentions that a number of more potent and specific PI3Kα inhibitors were tried in co-crystallization trials, but only PIK-108 complex with p110α/niSH2 produced crystals with reasonable diffraction resolution [10]. Our in-house experience also supports that only a few PI3Kα inhibitor chemotypes could be co-crystallized with the p110α/niSH2 complex (Ya-Li Deng, unpublished data). We hypothesized that occlusion of the ATP binding site of the p110α subunit by the RBD loop from an adjacent molecule in the asymmetric unit is the restraint on co-crystallization.9 Supporting this, a number of published PI3Kα inhibitor studies relied on PI3Kγ crystal structure lacking ABD as a surrogate to drive SAR. Unlike p110α, which is dependent on p85 for it stability,2 PI3Kγ belongs to the Class IB family and unlike like Class IA isoforms is stable without the p85 inhibitory subunit.
Based on the recent advances in understanding of the determinants for lipid binding of PI3Kα by Williams and colleagues,10 we have designed a series of constructs lacking both the ABD and lipid binding site (LBS) to facilitate production of stable p110α in the absence of p85. Here we present the crystal structure of apo p110α (107-1048) at 2.65 Å and its complex with PF-04691502 at 2.85 Å, alongside with biochemical characterization and biophysical ligand binding studies of isolated p110α constructs with pan PI3Kα inhibitor.
Results
Generation of stable p110α, p110β and p110δ lacking ABD and lipid-binding motif and their biochemical analysis
We have designed a series of constructs with either a truncated LBS (Table 1) or mutated amino acids 1057–1059 to alanines, encoding for the WIF motif previously shown critical for lipid binding10 (Fig. 1). These modifications in putative helix 12 of the p110α subunit were previously described by Williams and colleagues as critical determinants for its lipid kinase activity.10 In the case of p110α, both modifications (mutation of the WIF motif or truncation of kα12) lead to an increased insect cell viability compared to wild type full length p110α subunit, in constructs with and without ABD. Furthermore, the truncated version of p110α (105-1048 and 105-1050) lacking the ABD and LBS, exhibited robust expression in Sf21 insect cells and were amendable to purification (Fig. 1; Table 1). When the ABD and LBS deletions were engineered in two other PI3K isoforms, we were able to successfully purify p110β and p110δ proteins (Table 1). The ΔABD-LBS (delta ABD and LBS) proteins for PI3Kα and PI3Kδ isoforms were purified to homogeneity and shown to be monodisperse, as judged by analytical size exclusion chromatography (aSEC), which generally aligns well with suitability of the protein construct for structural studies (Table 1).16 A PI3Kβ construct lacking ABD and LBS expressed at high level as well (Table 1) but showed some signs of degradation and aggregation upon removal of histidine tag.
Table 1.
PI3K α,β and δ Constructs
| Protein | Construct | Deletion/Mutation | Expression level (mg/L) | aSEC classification |
|---|---|---|---|---|
| PI3Kα | 2-1050 | ΔLBS | 1 | No Data |
| PI3Kα | 2-1068 | WT | <0.1 | No Data |
| PI3Kα | 2-1068 | WT; WIF to AAA | 1 | ++ |
| PI3Kα | 105-1048 | ΔABD-LBS | 5 | +++ |
| PI3Kα | 105-1050 | ΔABD-LBS | 5 | +++ |
| PI3Kα | 105-1068 | ΔABD | <0.5 | No Data |
| PI3Kα | 105-1068 | ΔABD; WIF to AAA | 5 | +++ |
| PI3Kβ | 117-1051 | ΔABD-LBS | 6 | ++ |
| PI3Kδ | 106-1034 | ΔABD-LBS | 3 | +++ |
Figure 1.
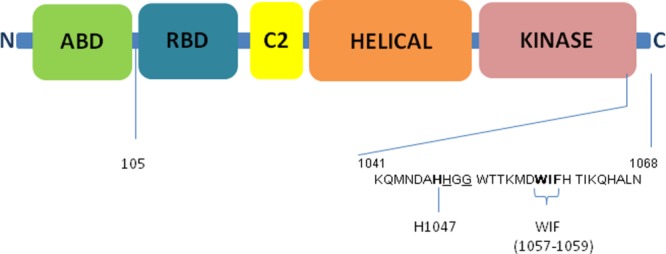
Domain structure of p110α catalytic subunit of PI3Kα. Key C-terminal residues and motifs are as labeled. Underlined residues are the sites of C-terminal truncations for p110α ΔABD-LBS described in the paper
Consistent with previous reports, when we examined the ATPase and lipid kinase activity of all purified ΔABD-LBS PI3K constructs,10 all PI3K constructs tested had detectable and robust ATPase activity, suggesting a functional ATP binding site. The p110 ΔABD-LBS constructs showed increased ATPase activity over their full-length counterparts, in agreement with the evidence supporting negative regulation of p110α activity by p85.17 ATPase activity of PI3Kβ and PI3Kδ lacking ABD and C-terminal lipid binding motif remained similar to their p110/p85 complex counterparts. PI3Kα, PI3Kβ, and PI3Kδ full-length p110/p85 complexes all showed robust lipid kinase activity as measured by phosphorylation of purified PIP2 in a competitive fluorescence polarization assay, while no lipid kinase activity was detected for constructs lacking the C-terminal lipid binding domain, at enzyme concentrations of up to 1 μM (Table 2). Interestingly, the PI3Kα construct lacking the ABD domain and containing the WIF motif mutation still retained lipid kinase activity, albeit significantly lower than full-length protein (Table II).
Table 2.
ATPase and Lipid Kinase Activity of PI3K α, β, and δ Constructs
| Protein | Construct | Mutation/Deletion | Specific activity (ATPase) μM ADP/s/μM enzyme | Specific activity (lipid kinase) μM PIP3/s/μM enzyme |
|---|---|---|---|---|
| PI3Kα | p110/p85 | None | 0.007 ± 0.004 | 0.392 ± 0.194 |
| PI3Kα | 105-1068 | ΔABD; WIF τ AAA | 0.03 ± 0.01 | 0.013 ± 0.009 |
| PI3Kα | 105-1048 | ΔABD-LBS | 0.03 ±0.01 | Not Detected |
| PI3Kα | 105-1050 | ΔABD-LBS | 0.08 ±0.05 | Not Detected |
| PI3Kβ | p110/p85 | None | 0.002 ± 0.001 | 0.058 ± 0.022 |
| PI3Kβ | 117-1051 | ΔABD-LBS | 0.003 ± 0.001 | Not Detected |
| PI3Kδ | p110/p85 | None | 0.002 ± 0.001 | 0.099 ± 0.014 |
| PI3Kδ | 106-1034 | ΔABD-LBS | 0.002 ± 0.001 | Not Detected |
Structural features of isolated p110α protein
The crystal structure of isolated p110α ΔABD-LBS has been determined at 2.65 Å resolution. It displays a conformation closely matching full-length p110α subunit as found in the p110α-p85 (niSH2) heterodimer (2RD0), denoted hetero-p110α (Fig. 2). A superposition of the two p110α proteins, using all common residues, gives a root mean square difference (RMSD) on Cα atoms of 1.76 Å. Most of the domains overlap well with the exception of C2, which has a slightly different orientation in the two structures. This is likely because of the absence of the interaction with p85 niSH2 in the p110α ΔABD-LBS structure, and to a lesser extent the absence of the ABD domain. When kinase domain residues are used for the alignment of the two p110α subunits from p110α ΔABD-LBS and hetero-p110α structures, the RMSD on Cα atoms is 1.14 Å. Comparison of the ATP binding sites of p110α ΔABD-LBS and hetero-p110 shows an even greater similarity, with RMSD of 0.6 Å for 33 Cα atoms within 6 Å of the ATP binding site. The integrity of the ATP site is also confirmed by direct binding studies with an ATP competitive inhibitors (see below).
Figure 2.
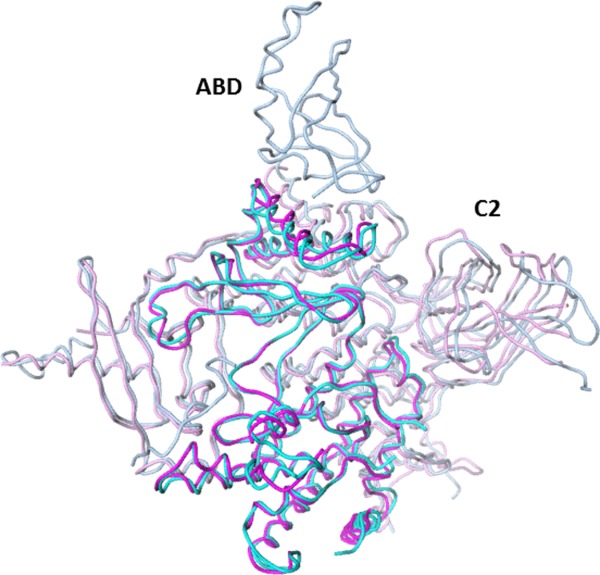
Superposition of the crystal structure of the p110α ΔABD-LBS (4TUU, magenta) and the equivalent residues (cyan) found in full length p110α-p85 niSH2 (2RD0). Kinase domains lie in front and are highlighted by darker magenta and cyan.
Structural and biophysical characterization of inhibitor binding to isolated p110α protein
The x-ray structure of p110α ΔABD-LBS in complex with PI3K/mTOR dual inhibitor PF-0469150218 was determined at 2.85 Å resolution. Electron density for the compound is well defined, and lies in the p110α kinase domain at the ATP-binding site, as expected (Fig. 3A). The observed protein–ligand interactions are very similar to those in the complex for the p110γ structure determined using a construct lacking ABD.18 This was previously used as a surrogate for p110α. In both cases, the inhibitor’s aminopyrimidine forms key hydrogen bonds with hinge residue Val882, the methoxypyridine is bound in the selectivity pocket, and the cyclopentyl side chain is positioned in the ribose-binding pocket (Fig. 3B).
Figure 3.
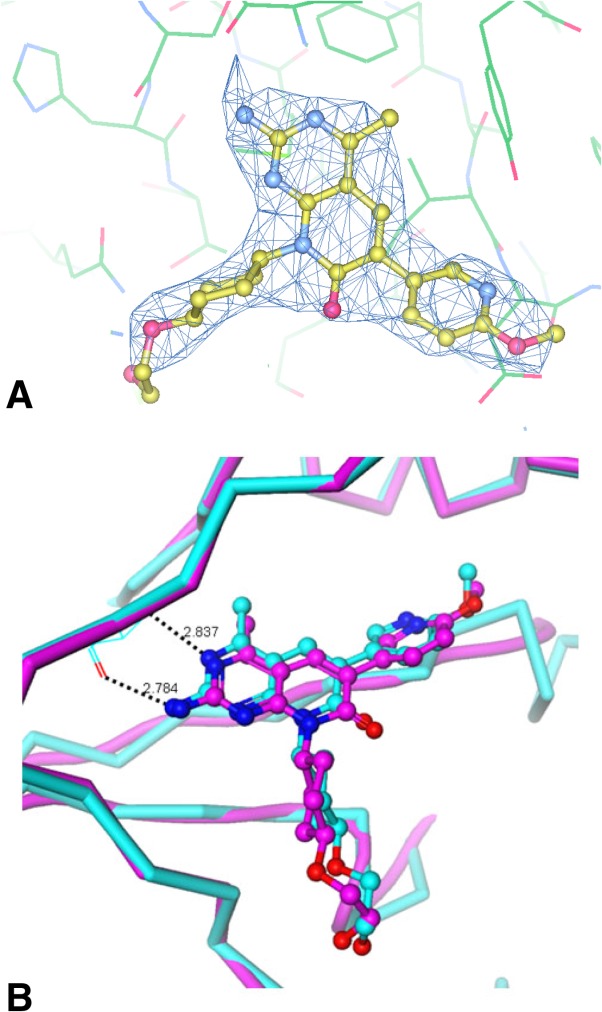
(A) The ligand molecule is shown superimposed with the refined 2Fo-Fc electron density map contoured at 1.0σ;and (B) Crystal structure of PF-04691502 (4TV3) bound to p110α ΔABD-LBS (magenta carbon atoms) compared with the structure of the same compound bound to PI3Kγ(cyan carbon atoms), a surrogate for PI3Kα. Protein–ligand interactions are similar in both complexes.
To further characterize ligand binding properties of the p110α ΔABD-LBS and its applicability for structural studies with the diverse set of PI3Kα inhibitors, we have conducted comparative direct binding studies of p110α ΔABD-LBS, p110α-p85 niSH2, and full-length p110α-p85 complexes by surface plasmon resonance (SPR) and isothermal titration calorimetry (ITC). These techniques complement structural information by providing a readout on the noncovalent interactions between the compound and the ATP binding pocket of the protein. Changes in the structural features and dynamics of the backbone residues in the ATP binding pocket and surrounding regions impact the kinetic and thermodynamic parameters for compound binding. Example sensograms for PF-04691502 binding to the full-length p110α-p85 and p110α ΔABD-LBS are shown in Figure 4 and kinetic parameters for PF-04691502 binding to the p110α ΔABD-LBS and p110α-p85 niSH2 are compared to full length p110α-p85 in Table 3. The PF-04691502 kinetic and equilibrium dissociation constant is almost insensitive to the change in PI3Kα protein construct. ITC studies provide further insight into the binding mode of pan-PI3Kα inhibitor providing a direct measure of the binding enthalpy (ΔH), KD, and entropy of binding (ΔS) (Table 4). Stoichiometric binding was observed for PF-04691502 to the p110α-p85 niSH2 complex allowing determination of ΔH but only an upper limit of KD (5 nM), which was consistent with SPR. The ΔH for PF-04691502 was not significantly different between p110α-p85 niSH2 and p110α ΔABD-LBS. Together the SPR and ITC data suggest that the binding mode of PF-04691502 is unaffected by the structural changes in the PI3Kα protein associated with the loss of p85 binding and deletions of C-terminal lipid binding motif.
Figure 4.
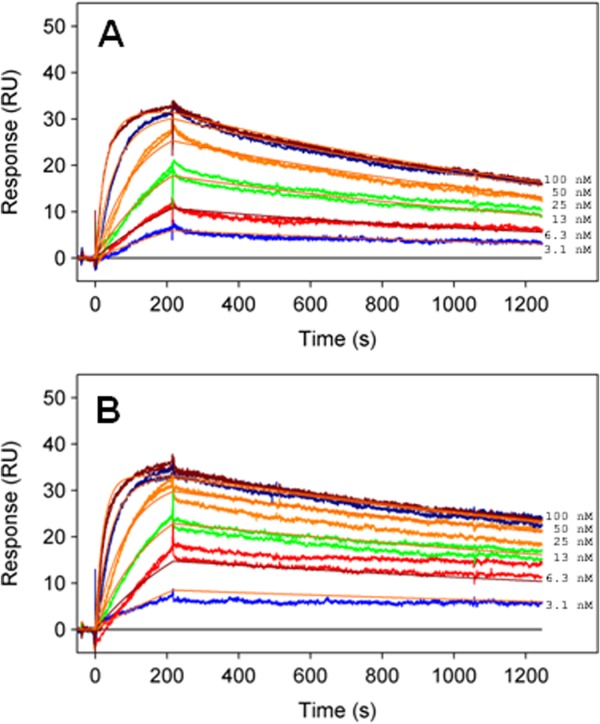
Sensograms for PF-04691502 binding to (A) p110α ΔABD-LBS and (B) full-length p110α-p85. Compound was injected in duplicate at 100, 50, 25, 12.5, 6.25, and 3.13 nM. No significant difference in the association and dissociation rate of the compound is observed between the full-length p110α-p85 and p110α ΔABD-LBS proteins.
Table 3.
Kinetic Parameters for PF-04691502 Binding to PI3Kα Constructs at 10°C
| Compound | kon (M−1 s−1) | koff (s−1) | T1/2 (min) | KD (nM) |
|---|---|---|---|---|
| p110α-p85 | 4.7 ± 0.5 | 4.5 ± 1.2 | 26 | 1.0 ± 0.4 |
| p110α-p85 niSH2 | 3.7 ± 0.2 | 5.9 ± 1.4 | 19.6 | 1.6 ± 0.5 |
| p110α ΔABD-LBS | 3.4 ± 0.6 | 6.3 ± 1.7 | 18.0 | 1.9 ± 0.1 |
Table 4.
Thermodynamic Parameters for PF-04691502 Binding to the PI3Kα Constructs
| Compound | KD (nM) | ΔH |
|---|---|---|
| p110a-p85 niSH2 | <5 | −12.1 |
| ΔABD-LBS | <5 | −11.5 ± 0.7 |
Discussion
Previous studies suggest that the key challenge for generation of stable p110α, β and δ subunits from Class IA PI3-kinases, is their dependence on the respective p85 binding partner for intrinsic stabilization.17 Conversely, the p110γ subunit lacking ABD can be produced as a stable protein enabling determination of its crystal structure.19 We and others have previously observed that a major limitation in production of stable PI3Kα H1047R complex in insect cells is related to its high intrinsic activity, leading to low cell viability and low protein recovery, which was successfully mitigated by generation of a kinase dead mutation of PI3Kα.10 This was also mitigated by co-expression with PI3Kα inhibitor to generate protein for structural studies of PI3Kα H1047R oncogenic mutant.8 Thus even in the presence of p85 subunit, the activation of the PI3Kα complex by the H1047R mutation is an impediment for protein production. Recent work by Williams and colleagues has established the key role of the C-terminal WIF motif (located in kα12) in maintaining lipid kinase activity of PI3Kα.10 This observation led us to hypothesize that the critical issue for production of isolated p110α subunit stems from an elevated lipid kinase activity caused by removal of p85 regulatory subunit. Significantly, the retention of ATPase activity by the LBS truncated PI3Kα complex suggests that the complex retains a functional ATP binding site.10
We have pursued this observation for the production of p110 α, β and δ subunits in insect cells by removing the ABD and C-terminal LBS, then evaluating the purified protein’s behavior and biochemical properties. p110α, β and δ subunits lacking the ABD and C-terminal LBS motif expressed at relatively high level in insect cells. Constructs with C-terminal truncations of the LBS behaved similar to the construct with the WIF motif mutated to AAA indicating that the lipid kinase activity of p110α ΔABD-LBS is the key limitation with regards to protein production. The biochemical characterization of the purified constructs indicates that the p110 ΔABD-LBS subunits have increased ATPase activity compared to the full length p110-p85 complexes. Interestingly, there is ∼3-fold increase in ATPase activity for the p110α construct 105-1050 compared to the 105-1048 suggesting that small changes in the protein construct translate in significant increase in ATPase activity (Table II). In the majority of the previously published PI3Kα complex structures there is a break in the electron density at residue 1048 in p110α subunit, suggesting a high degree of flexibility of the C-terminal LBS.8–10 The low ATPase activity for full length PI3Kα complexes suggests a critical role of p85 subunit in regulation of ATPase activity. While the biological significance of an increase in ATPase activity for p110α ΔABD-LBS is not clear, the activation of PI3Kα by phosphopeptides and helical domain oncogenic mutations that remove inhibitory contacts between p110 and p85 subunits, may lead to the increase in ATPase activity. Our study also provides insight into the differences in regulation of p110α versus p110β by their respective p85 subunits. The analysis of p110β/iSH2-cSH2 (icSH2) complex structure reveals weaker interactions along p110β-C2/p85β-iSH2 interface than corresponding interaction in PI3Kα while revealing novel regulatory contacts between cSH2 and p110β kinase domain.20 In a recent review,21 Vogt postulated that the additional inhibitory contacts between p110β kinase domains with cSH2 of corresponding p85 subunits is compensated for by loosening of C2/iSH2 interface resulting in overall constitutively active PI3Kβ complex. The lack of difference in ATPase activity between p110β and p110β/p85 complex may indirectly support the notion that p85 has no significant role in regulation of PI3Kβ activity (Table II). As expected, the p110 ΔABD-LBS subunits lacked lipid kinase activity, confirming that the C-terminal LBS is a critical determinant for lipid binding.
With the development of a structural biology platform for the p110α ΔABD-LBS in mind, we then compared direct binding properties of p110α ΔABD-LBS constructs with full length PI3Kα complexes and a p110α-niSH2 complex that was previously characterized structurally. We evaluated binding for a pan-PI3K inhibitor PF-04691502. No significant differences were observed in the binding characteristics of PF-04691502 to the PI3K constructs measured by SPR and ITC studies. Thus our biochemical and direct binding studies confirm the relevance of p110α ΔABD-LBS protein for further structural work.
An overall goal of our study was to enable development of a crystallization platform for p110α and improve overall resolution of PI3Kα inhibitor co-crystal structures, allowing co-crystallization with multiple PI3Kα inhibitor classes using novel protein constructs. In previously described PI3Kα crystal forms, all of which share the same space group and unit cell parameters, the ATP site is partially occluded by the RBD loop from a neighboring, symmetry-related molecule (Fig. 5). This steric hindrance precludes soaking of inhibitors into apo PI3Kα crystals, and also prevents crystallization of pre-formed inhibitor-complexes depending on compound chemotype. In the few cases where successful co-crystallization with PI3Kα has been reported, the neighboring RBD loop undergoes modest reconfiguration in order to accommodate a covalent ligand, wortmannin (PDB ID: 3HHM) or a relatively small noncovalent ligand (PDB ID: 4A55).8,10 We feel that the success of co-crystallization with PI3Kα specific inhibitors is dependent on inhibitor chemotype as our attempts to co-crystallize PF-04691502 or similar compounds with full length p110α-p85 niSH2 complex were unsuccessful. Conversely, in the p110α ΔABD-LBS crystal form, there are no intrusions of symmetry related residues into the active site. The p110α ΔABD-LBS crystal form is therefore compatible with a wide-range of bound inhibitors, and may also be available to ligands that are soaked into crystals. In fact, we were able to obtain additional co-crystal structures of PI3Kα inhibitors using the p110α ΔABD-LBS protein (Ya-Li Deng and Ping Chen, unpublished data).
Figure 5.
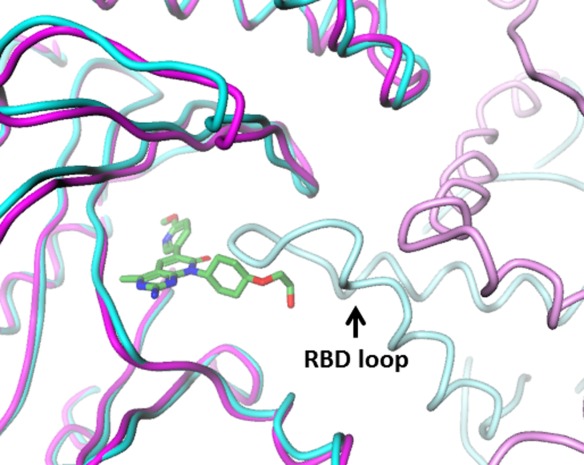
Crystal packing around the ATP site for full length p110α-p85 niSH2 (dark cyan), showing symmetry-related RBD loop (light cyan) that precludes occupancy of site by most ligands. In the orthorhombic crystal form of isolated P110a, the ATP site (dark magenta, with bound PF-04691502) is largely free of potential symmetry-related contacts (light magenta).
In summary, we have isolated and characterized p110 ΔABD-LBS subunits of PI3Kα, β and δ. These proteins retain a native ATP binding site based on biochemical analysis. Further studies with p110α ΔABD-LBS revealed that it is capable of binding pan inhibitor with characteristics comparable to intact PI3Kα heterodimer. We also determined crystal structures of p110α ΔABD-LBS subunit alone at 2.65 Å (PDB ID: 4TUU) and bound to PF-04691502 at 2.85 Å (PDB ID: 4TV3). Unlike previously published crystal forms of p110α-p85 niSH2, the p110α ΔABD-LBS crystal form can likely accommodate a wide range of PI3Kα inhibitor chemotypes and potentially serve as a platform for SBDD of novel PI3Kα therapeutic agents.
Material and Methods
Expression and purification of ΔABD-LBS constructs for p110α, p110β and p110δ
Genes encoding for p110α (105-1048), p110β (117-1051), p110δ (106-1034) were optimized using human codon table and synthesized at GeneScript (Piscataway, NJ) followed by subcloning into modified pFastBacHtB vector containing N-terminal histidine tag followed by Tobacco Etch Virus (TEV) cleavage site. Recombinant baculovirus was generated using Bac-to-Bac protocol (Life Technologies, Carlsbad, CA) and large scale expression was conducted in Sf21 cells at MOI = 1 for 72 hours. Cells were lyzed in 50 mM Tris pH 8.0, 250 mM NaCl, 0.25 mM TCEP, and 20 mM imidazole. The p110 subunits were purified from clarified supernatant using Immobilized Metalo Affinity Chromatography (IMAC). The protein was eluted from the column using 50 mM Tris pH 8.0, 200 mM NaCl, 0.25 mM TCEP, and 200 mM imidazole. After elution TEV protease was added to the protein and TEV cleavage was performed overnight concurrent with the dialysis against 50 mM Tris pH 8.0, 200 mM NaCl, 0.25 mM TCEP, and 40 mM imidazole. The flow through fractions containing p110 subunits were concentrated and loaded on Superdex 200 26/60 SEC column equilibrated in 50 mM Tris pH 8.0, 100 mM NaCl, 2% Ethylene glycol and 1 mM TCEP. After SEC peak fractions were pulled and concentrated to ∼5–6 mg/mL. Purity and integrity of the complex was confirmed using LCMS, analytical SEC and SDS-PAGE analysis.
Expression and purification of p110α/p85 and p110α/niSH2 complex for biochemical and biophysical studies
Genes encoding p110α and p85 subunits of PI3Kα complex were subcloned from human cDNA into pFASTBAC Dual vector. Gene encoding p110α subunit was subcloned into polyhedrine promoter while gene encoding p85 subunit (or niSH2 p85 322-600) was subcloned into p10 promoter. Additionally, sequence encoding for histidine tag and TEV cleavage site preceded p110α ORF. Recombinant baculovirus was generated using Bac-to-Bac protocol and large scale expression was conducted in Sf21 cells at MOI = 1 for 72 hours. Cells were lyzed in 50 mM Tris pH 8.0, 250 mM NaCl, 5% glycerol and 0.25 mM TCEP, and 20 mM imidazole. The p110a/p85 complex was purified from clarified supernatant using Immobilized Metalo Affinity Chromatography (IMAC). The protein was eluted from the column using 50 mM Tris pH 8.0, 200 mM NaCl, 5% glycerol, and 0.25 mM TCEP, 200 mM imidazole and further desalted into 50 mM Tris pH 8.0, 20 mM NaCl, 0.25 mM TCEP prior loading on MonoQ sepharose. PI3Kα complex was eluted from MonoQ sepharose over 20 column volumes using 0–30% gradient of buffer B (50 mM Tris pH 8.0, 1M NaCl, 0.25 mM TCEP). The peak fractions were pulled together and loaded on Superdex 200 26/60 SEC column equilibrated in 50 mM Tris pH 8.0, 200 mM NaCl, 0.5 mM TCEP. After SEC peak fractions were pulled and concentrated to ∼1–2 mg/mL. Purity and integrity of the complex was confirmed using LCMS, analytical SEC and SDS-PAGE analysis.
Crystallization of p110α (105-1048)
Crystallization conditions were found initially by sitting-drop vapor-diffusion, employing a Mosquito robot (TTPLabtech, Cambridge, MA), using an in-house crystallization screen derived from multiple commercial screens. Initial hits were obtained by mixing complex of protein in complex with inhibitor with 0.2% (w/v) BOG in 1:1 ratio with the reservoir solution (0.1 μL + 0.1 μL) at 13°C. The crystals (both apo and inhibitor) used for X-ray data collection grew in hanging drops with micro-seeding made by mixing 1.5 μL protein complex containing 1:5 molar ratio of human p110α (5.8 mg/mL) with and without compounds and 1.5 μL of reservoir solution consisting 8–10% (w/v) PEG 6K, 0.6M NaFormate, 0.1M CHES, pH 9.0–10 and 5 mM TCEP, pH 7.0 at a temperature of 20°C. These crystals belong to space group P212121 with unit cell dimensions a = 57.7, b = 136.5 and c = 143.1. There is one molecule per asymmetric unit. Crystals were flash-frozen in liquid nitrogen after transferring to 2 μL reservoir solution containing 25% (v/v) glycerol as a cryoprotectant and then stored in liquid nitrogen.
Data collection and structure determination
Diffraction data were collected using a Pilatus 6M detector on beamline 17-ID at the Advanced Photon Source (Argonne National Laboratory), and processed with auto-Proc. Data statistics for the p110α ΔABD-LBS (apo) includes R-merge of 0.05 (0.5 for high resolution shell 2.78-2.64); I/σ(I) of 29 (3.3); completeness of 100% (100%); and redundancy of 6.5 (6.3). Data statistics for the p110α ΔABD-LBS/PF-04691502 complex includes R-merge of 0.05 (0.49 for high resolution shell 3.0-2.85); I/σ(I) of 26 (3.5); completeness of 100% (99.5%); and redundancy of 6.5 (5.6). The structure was determined using coordinates of the p110 subunit from our in-house structure of PI3Kα heterodimer structure. The final p110α ΔABD-LBS (apo) with 31 water molecules was refined at 2.65 Å using BUSTER to R/Rfree values of 20.5/24.5, with RMSD bond and angle deviations of 0.01 Å and 1.1°, respectively. The final p110α ΔABD-LBS/PF-04691502 complex with four water molecules was refined at 2.85 Å using BUSTER to R/Rfree values of 21.2/24.5, with RMSD bond and angle deviations of 0.009 Å and 1.05°, respectively. The coordinates have been deposited into the PDB with code 4TUU and 4TV3 respectively.
Enzyme assays
Lipid kinase activity
PI3K lipid kinase activity was measured by a competitive fluorescence polarization assay, as described previously.22 Reaction buffer contained 50 mM Hepes, pH 7.5, 5 mM MgCl2, 150 mM NaCl, 0.05% CHAPS, 30 μM PIP2 (PtdIns-(4,5)-P2, 1,2 dioctanoyl, Cayman Chemical, Ann Arbor, MI) and 5 mM DTT. The PI3K reaction was initiated with 100 μM ATP and allowed to proceed at room temperature for 1 hr. The reaction was quenched with EDTA at a final concentration of 10 mM, and PIP3 product formation was detected by mixing a portion of the reaction mix with 240 nM GST-Grp1 PH domain (Univ. of Dundee) and 6 nM TAMRA-PIP3 (Echelon Biosciences, Salt Lake City, UT). After a 15-min incubation, fluorescence polarization was read on an LJL Analyst HT (Molecular Devices, Sunnyvale, CA) with excitation at 530 nm, emission at 580 nm, and beam splitter of 561 nm, with a G-factor of 0.85. PIP3 formed was calculated through the use of a PIP3 standard curve. All reactions were linear with respect to time and enzyme concentration.
ATPase activity
PI3K ATPase activity was measured using the Transcreener ADP2 Fluorescence Intensity assay (BellBrook Labs, Madison, WI) according to the manufacturer’s protocol. Reaction buffer contained 50 mM Hepes, pH 7.5, 5 mM MgCl2, 150 mM NaCl, 0.05% CHAPS, and 5 mM DTT, in the absence of PIP2. The reaction was initiated with 100 μM ATP, and allowed to proceed at room temperature for 1 hr. The reaction was terminated in the presence of 46.9 μg/mL ADP2 antibody and 4 nM ADP Tracer in 1× Stop Buffer containing EDTA. The detection step was incubated at room temperature with gentle shaking for 1 hr, and fluorescence intensity was read on a Tecan M1000 (Mannedorf, Switzerland) with excitation of 590 nm and emission of 617 nm. ADP formed was calculated through the use of an ADP standard curve.
Surface plasmon resonance binding studies
SPR binding studies were carried out on a Biacore 3000 instrument (GE Healthcare, Piscataway NJ) at 10°C in 150 mM NaCl, 25 mM HEPES, pH 8.0, 5 mM MgCl2, 10% glycerol, 1% DMSO, 0.5 mM TCEP, 0.005% P20. PI3K proteins were immobilized on a CM5 sensorchip (GE Healthcare, Piscataway, NJ) by standard amine coupling. Injections (100, 50, 25, 12.5, 6.25, 3.13 nM) were made, in duplicate, using the Kinject mode at 50 μM/min with a 1200 s compound dissociation time. Data analysis was performed using the Scrubber2 software (BioLogic Software, Pty., Australia). Compound injections were referenced to a blank surface and by a buffer blank. Data were fit to a 1:1 kinetic model, including bulk refractive index correction, in the Scrubber2 program. Standard deviation was calculated from 3 to 5 replicate binding experiments.
Isothermal titration calorimetry
ITC experiments were carried out on a VP ITC instrument (GE Healthcare, Piscataway NJ) at 20°C. Samples were extensively dialyzed into a buffer containing, 150 mM NaCl, 25 mM HEPES, pH 8.0, 5 mM MgCl2, 10% glycerol, 1 mM TCEP. Concentrations were determined spectrophotometrically using an A280 of 208450 for M−1cm−1 p110α-p85 niSH2 and 143130 M−1cm−1for ΔABD-LBS PI3Kα. In a typical experiment, nineteen 15 μL injection of 50 μM compound was made into a 5 μM PI3Kα. Data were analyzed using the ORIGIN software provided with the instrument and fit to a simple 1:1 binding model.
Acknowledgments
The authors would like to acknowledge John Kath, Shubha Bagrodia and Al Stewart for discussion and critical review of manuscript. We would like to acknowledge Nicole Garble for large scale protein expression in insect cells and small scale expression/purification screening. We are also thankful to Martin Edwards and Stephan Grant for their support and attention to this work.
References
- Vadas O, Burke JE, Zhang X, Berndt A, Williams RL. Structural basis for activation and inhibition of class I phosphoinositide 3-kinases. Sci Signal. 2011;4:re2. doi: 10.1126/scisignal.2002165. [DOI] [PubMed] [Google Scholar]
- Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr GA, Backer JM. Regulation of the p85/p110 phosphatidylinositol 3′-kinase: stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol Cell Biol. 1998;18:1379–1387. doi: 10.1128/mcb.18.3.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: The path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13:195–203. doi: 10.1038/nrm3290. [DOI] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol. 1999;9:125–128. doi: 10.1016/s0962-8924(99)01519-6. [DOI] [PubMed] [Google Scholar]
- Vogt PK, Kang S, Elsliger MA, Gymnopoulos M. Cancer-specific mutations in phosphatidylinositol 3-kinase. Trends Biochem Sci. 2007;32:342–349. doi: 10.1016/j.tibs.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Sun M, Hillmann P, Hofmann BT, Hart JR, Vogt PK. Cancer-derived mutations in the regulatory subunit p85alpha of phosphoinositide 3-kinase function through the catalytic subunit p110alpha. Proc Natl Acad Sci U S A. 2010;107:15547–15552. doi: 10.1073/pnas.1009652107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt PK, Hart JR, Gymnopoulos M, Jiang H, Kang S, Bader AG, Zhao L, Denley A. Phosphatidylinositol 3-kinase: the oncoprotein. Curr Top Microbiol Immunol. 2010;347:79–104. doi: 10.1007/82_2010_80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelker D, Gabelli SB, Schmidt-Kittler O, Zhu J, Cheong I, Huang CH, Kinzler KW, Vogelstein B, Amzel LM. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc Natl Acad Sci U S A. 2009;106:16996–17001. doi: 10.1073/pnas.0908444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CH, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu VE, Kinzler KW, Vogelstein B, Gabelli SB, Amzel LM. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007;318:1744–1748. doi: 10.1126/science.1150799. [DOI] [PubMed] [Google Scholar]
- Hon WC, Berndt A, Williams RL. Regulation of lipid binding underlies the activation mechanism of class IA PI3-kinases. Oncogene. 2012;31:3655–3666. doi: 10.1038/onc.2011.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabelli SB, Huang CH, Mandelker D, Schmidt-Kittler O, Vogelstein B, Amzel LM. Structural effects of oncogenic PI3Kalpha mutations. Curr Top Microbiol Immunol. 2010;347:43–53. doi: 10.1007/82_2010_53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JE, Williams RL. Dynamic steps in receptor tyrosine kinase mediated activation of class IA phosphoinositide 3-kinases (PI3K) captured by H/D exchange (HDX-MS) Adv Biol Regul. 2013;53:97–110. doi: 10.1016/j.jbior.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JE, Perisic O, Masson GR, Vadas O, Williams RL. Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110alpha (PIK3CA) Proc Natl Acad Sci U S A. 2012;109:15259–15264. doi: 10.1073/pnas.1205508109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacht M, Qiao L, Sheets MP, St Martin T, Labenski M, Mazdiyasni H, Karp R, Zhu Z, Chaturvedi P, Bhavsar D, Niu D, Westlin W, Petter RC, Medikonda AP, Singh J. Discovery of a potent and isoform-selective targeted covalent inhibitor of the lipid kinase PI3Kalpha. J Med Chem. 2013;56:712–721. doi: 10.1021/jm3008745. [DOI] [PubMed] [Google Scholar]
- Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, Balla T, Weiss WA, Williams RL, Shokat KM. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell. 2006;125:733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullan D, Canaves JM, Quijano K, Abdubek P, Nigoghossian E, Haugen J, Klock HE, Vincent J, Hale J, Paulsen J, Lesley SA. High-throughput protein production for X-ray crystallography and use of size exclusion chromatography to validate or refute computational biological unit predictions. J Struct Funct Genomics. 2005;6:135–141. doi: 10.1007/s10969-005-2898-1. [DOI] [PubMed] [Google Scholar]
- Wu H, Yan Y, Backer JM. Regulation of class IA PI3Ks. Biochem Soc Trans. 2007;35:242–244. doi: 10.1042/BST0350242. [DOI] [PubMed] [Google Scholar]
- Cheng H, Hoffman JE, Le PT, Pairish M, Kania R, Farrell W, Bagrodia S, Yuan J, Sun S, Zhang E, Xiang C, Dalvie D, Rahavendran SV. Structure-based design, SAR analysis and antitumor activity of PI3K/mTOR dual inhibitors from 4-methylpyridopyrimidinone series. Bioorg Med Chem Lett. 2012;23:2787–2792. doi: 10.1016/j.bmcl.2013.02.020. [DOI] [PubMed] [Google Scholar]
- Walker EH, Perisic O, Ried C, Stephens L, Williams RL. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature. 1999;402:313–320. doi: 10.1038/46319. [DOI] [PubMed] [Google Scholar]
- Zhang X, Vadas O, Perisic O, Anderson KE, Clark J, Hawkins PT, Stephens LR, Williams RL. Structure of lipid kinase p110beta/p85beta elucidates an unusual SH2-domain-mediated inhibitory mechanism. Mol Cell. 2011;41:567–578. doi: 10.1016/j.molcel.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt PK. PI3K p110beta: more tightly controlled or constitutively active? Mol Cell. 2011;41:499–501. doi: 10.1016/j.molcel.2011.02.017. [DOI] [PubMed] [Google Scholar]
- Yuan J, Mehta PP, Yin MJ, Sun S, Zou A, Chen J, Rafidi K, Feng Z, Nickel J, Engebretsen J, Hallin J, Blasina A, Zhang E, Nguyen L, Sun M, Vogt PK, McHarg A, Cheng H, Christensen JG, Kan JL, Bagrodia S. PF-04691502, a potent and selective oral inhibitor of PI3K and mTOR kinases with antitumor activity. Mol Cancer Ther. 2011;10:2189–2199. doi: 10.1158/1535-7163.MCT-11-0185. [DOI] [PubMed] [Google Scholar]