

Abstract
Crystal structures of three members (BACOVA_00364 from Bacteroides ovatus, BACUNI_03039 from Bacteroides uniformis and BACEGG_00036 from Bacteroides eggerthii) of the Pfam domain of unknown function (DUF4488) were determined to 1.95, 1.66, and 1.81 Å resolutions, respectively. The protein structures adopt an eight-stranded, calycin-like, β-barrel fold and bind an endogenous unknown ligand at one end of the β-barrel. The amino acids interacting with the ligand are not conserved in any other protein of known structure with this particular fold. The size and chemical environment of the bound ligand suggest binding or transport of a small polar molecule(s) as a potential function for these proteins. These are the first structural representatives of a newly defined PF14869 (DUF4488) Pfam family.
Keywords: Bacteroides, DUF4488, sugar binding, calycins, unknown ligand, crystal structure
Introduction
Bacteroides are anaerobic, bile-resistant, non-spore forming, Gram-negative bacteria that inhabit the mammalian gastrointestinal tract. Bacteroides comprise nearly 25% of the 1011 organisms per gram of content typically found in the human gut.1 These bacteria maintain a commensal or mutualistic2 relationship with the host, playing a fundamental role in the processing of complex nutrients into simpler ones that can be readily processed by the host.3–5 However, some species can cause disease, such as sepsis, abscess formation in multiple organs, and bacteremia, when they escape the host intestine.4 Genomic and subsequent proteomic analyses of two Bacteroides species, B. thetaiotaomicron, and B. fragilis, reveal that a significant proportion of their genome is dedicated to nutrient-sensing and nutrient-metabolizing machinery, mainly carbohydrate degradation/acquisition/utilization systems.6–8 For example, the B. thetaiotaomicron genome encodes 172 glycosyl hydrolases and 163 starch-binding proteins (SusC and SusD homologs), which are involved in the breakdown of complex polysaccharides.3,6 As part of our efforts to explore and complement genomic studies of over-represented protein families in the human gut microbiome and expand the structural coverage of these proteins, the Joint Center for Structural Genomics (JCSG) has to date determined structures of 239 of a total of 544 Bacteroides protein structures in the PDB as of May 2014. Here, we report crystal structures of three homologous proteins of unknown function, BACOVA_00364 (ZP_02063416.1) from Bacteroides ovatus (B. ovatus), BACUNI_03039 (ZP_02071597.1) from Bacteroides uniformis (B. uniformis) and BACEGG_00036 (ZP_03457270.1) from Bacteroides eggerthii (B. eggerthii). These proteins share a sequence identity of 80% and are conserved in at least 22 of the 33 known Bacteroides species (http://www.ncbi.nlm.nih.gov/genome). The structures reveal an eight-stranded, β-barrel fold with a putative ligand binding site located at one end of the barrel, reminiscent of a group of proteins known as calycins. Interestingly, an unknown ligand (UNL) is bound at this site in each of the three structures. The nature of the ligand and its interactions with the proteins suggest that these proteins bind small polar molecules, such as carbohydrates, thereby indicating a possible function in nutrient binding/acquisition/transport. Further analysis of these structures indicated that they belong to a separate protein family and resulted in the creation of a new Pfam family, PF14869 (DUF4488).
Results
Overall structure
The crystal structure of BACOVA_00364 contains eight protein molecules (residues 25–163 in chain A, 31–163 in chain B, 28–163 in chain C, 31–163 in chain D, 23–163 in chain E, 28–163 in chain F, 28–163 in chain G, and 28–133, 140–163 in chain H), one sodium ion, one acetate ion, four glycerol molecules, five UNLs and 555 water molecules in the crystallographic asymmetric unit (asu). The BACUNI_03039 structure contains four protein molecules (residues 23–163 in chain A, 28–163 in chain B, 28–163 in chain C and 31–163 in chain D), one glycerol molecule, five polyethylene glycol fragments, four UNLs and 628 water molecules in the asu. The BACEGG_00036 structure contains two protein molecules (residues 27–163 in chain A and 27–163 in chain B), two UNLs and 293 water molecules in the asu. (n.b. possible oligomeric assemblies relevant for biological function in vivo are discussed below). A few residues at the N-terminus of most chains in BACOVA_00364 and BACUNI_03039 were not modeled in the structures due to lack of interpretable electron density. The Matthews’ coefficient (VM)9 and the estimated solvent content are 2.36 Å3/Da and 47.8% for BACOVA_00364, 2.73 Å3/Da and 54.9% for BACUNI_03039, and 2.69 Å3/Da and 54.3% for BACEGG_00036, respectively. The Ramachandran plots produced by MolProbity10 show more than 98.0% of the residues are in favored regions with no outliers for all three structures.
All structures adopt a β-barrel fold, comprised of eight anti-parallel β-strands and one 310-helix (residue 105–109) (Fig. 1). BACOVA_00364 contains an additional 310-helix at the N-terminus in one of its chains. A UNL is bound at the more open end of the β-barrel in all three structures.
Figure 1.
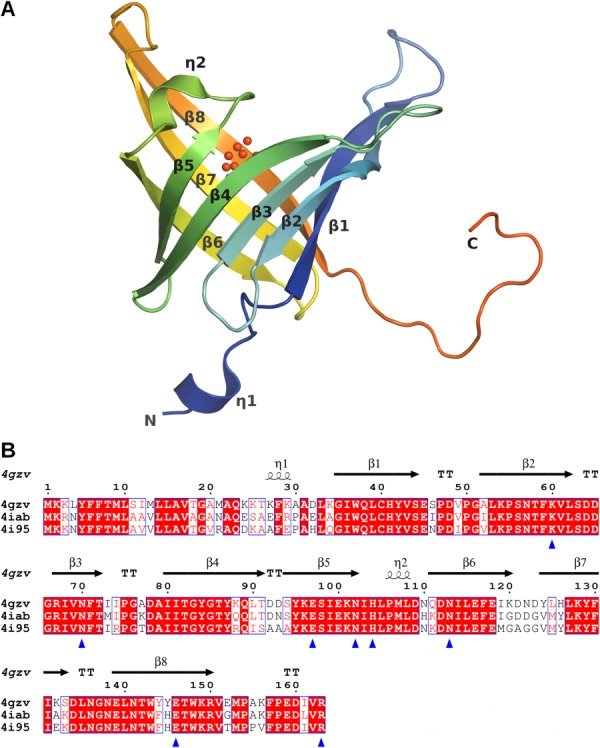
Overall structure and sequence alignment of the PF14869 monomer. (A) The eight-stranded β-barrel fold of the BACOVA_00364 protomer (pdb code 4gzv, chain A) is shown in a rainbow cartoon representation from blue to red with the secondary structure elements labeled. The N- and C-termini are also labeled. BACOVA_00364 has an additional 310-helix near the N-terminus that is absent in BACUNI_03039 (pdb code 4iab) and BACEGG_00036 (pdb code 4i95). The UNL is shown as red spheres. This and other figures were prepared with PyMOL.11 (B) The sequences were aligned using ClustalW12 and rendered using EsPript.13 The secondary structural elements corresponding to the 4gzv structure are identified at the top of the sequence. Identical residues are in bold white font on a red background while similar residues are in red font against a white background. The residues interacting with the ligand are indicated by blue triangles.
Similarity among the three structures
As expected, the structures of the three proteins are essentially identical [Fig. 2(A)]. A multiple structure alignment of the proteins by EBI-SSM server14 returns an overall Cα atom RMSD of 0.84 Å, overall Q-score of 0.91, and sequence identity of 80% using 135 equivalent residues in the alignment [Fig. 1(B)]. The presence of an additional 310-helix at the N-terminus and a different orientation of this region in BACOVA_00364 in one chain (likely due to crystal packing interactions that influence the local structure of this region in this chain but not the others) and some variations in loop orientations constitute the only structural variability among these three proteins [Fig. 2(B)].
Figure 2.
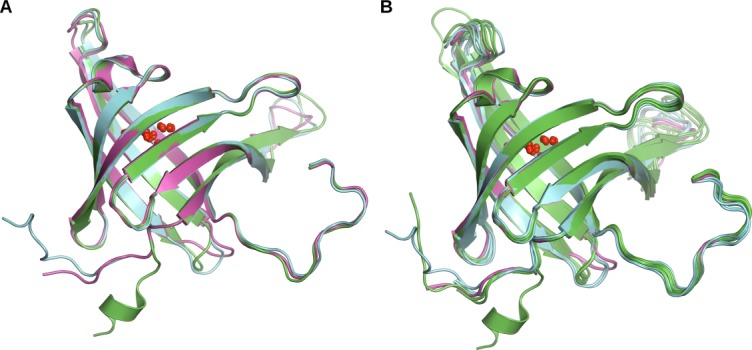
Structural comparison of BACOVA_00364, BACUNI_03039, and BACEGG_00036. (A) Cartoon representation of the superposition of the three structures (chain A) showing the high structural similarity with only minor differences at the N-terminus and a couple of loops. BACOVA_00364 is in green, BACUNI_03039 in cyan and BACEGG_00036 in magenta. (B) The structural similarities and differences are highlighted by a superposition of all chains in the asu in all three structures.
Ligand binding site
A putative ligand binding site consisting of a small cavity lined by conserved polar residues (Lys60, Asn70, Glu97, Asn102, His104, Asn113, and Glu146) was initially identified based on the presence of unaccounted for electron density, as illustrated in Figures 1(B) and 3(A). The volume of this cavity calculated with the CASTp server15 ranges from 235 to 250 Å3, depending on the specific protein and chain. A virtual library screen containing 144,110 small molecules of less than 20 non-hydrogen atoms identified many potential ligands, with the highest scoring candidate (binding affinity of −6.9 kcal/mol) 4,8-dihydro-6H-1,2,5-oxadiazolo[3,4-e]1,2,3-triazolo[4,5-b] pyrazine. However, closer inspection of the top 100 hits did not identify any ligand with a good fit to the electron density. Thus, a UNL was modeled at corresponding sites when warranted by the density (five out of eights chains in BACOVA_00364 and all chains in BACUNI_03039 and BACEGG_00036); a glycerol molecule was modeled at this site in the other three chains of BACOVA_00364 as this gave the best fit to the electron density.
Figure 3.
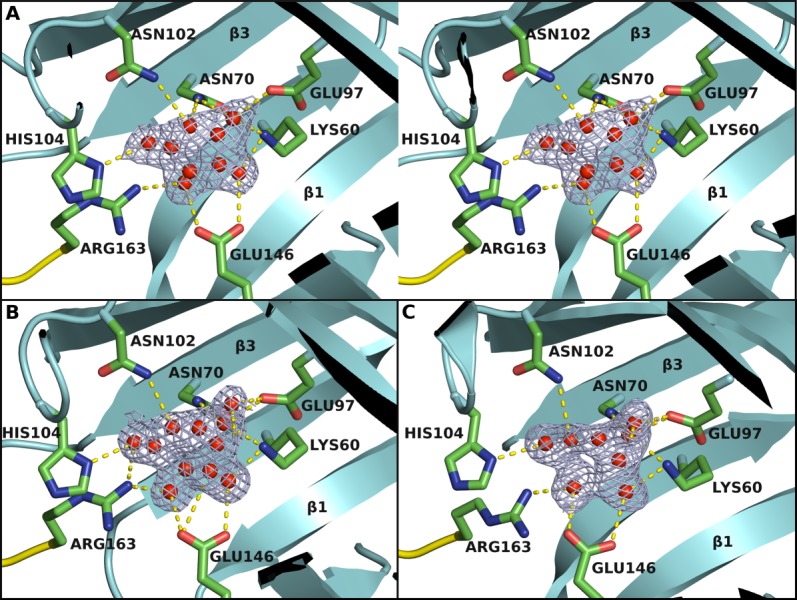
The ligand binding site. The binding site is located at one end of the β-barrel as identified from binding of a UNL (red spheres). (A) Stereo view of the UNL and its interaction with the protein in BACOVA_00364 (pdb code 4gzv). Omit map is contoured at 1.25 σ level above the mean density. The residues interacting with the UNL are shown in stick representation. Arg163 that interacts with the UNL comes from an adjacent protomer in the biological tetramer. (B) and (C) are the corresponding UNL binding sites for BACUNI_03039 (pdb code 4iab) and BACEGG_00036 (pdb code 4i95), respectively, where the same or similar UNL is bound.
Interestingly, the C-terminus (Arg150–Arg163) reaches over and inserts its tail into the putative ligand binding site pocket of the adjacent molecule in the tetramer and the terminal Arg163 residue interacts with the UNL (Fig. 3). The shape of the UNL and the residues interacting with it suggest the same small molecule is bound in all three proteins. As nothing similar was present in the purification or crystallization reagents, this compound likely originated from the bacterial growth medium used during protein expression. Despite the possibility that this may not be the natural substrate, the ligand shape, interatomic distances, and chemical environment suggest a ribose/erythrofuranose derivative, thus hinting at a sugar binding/transport/uptake-like function for these proteins. However, we cannot exclude the possibility that the Arg163 side chain could be acting as a partial surrogate for a larger substrate and could be displaced in the presence of the natural ligand.
Oligomeric state
Analytical size exclusion chromatography indicates BACEGG_00036 is a tetramer in solution while BACOVA_00364 and BACUNI_03039 are mixtures of monomers and dimers. Crystal packing analysis of all three proteins using the PISA server16 suggests that the proteins most likely exist as a tetramer (dimer of dimers; Fig. 4), with dimeric arrangements predicted as well. For the tetrameric association, the total buried surface area and change in solvent free energy are 11,810 Å2 and −70.0 kcal/mol for BACOVA_00364, 14,680 Å2 and −66.0 kcal/mol for BACUNI_03039, and 13,110 Å2 and −60.0 kcal/mol for BACEGG_00036. The proteins’ biologically relevant oligomeric state must be at least dimeric since the C-terminus (Arg163) of one protomer forms an integral part of the active site of the neighboring protomer (Fig. 3). This dimeric interface buries a total surface area of 1967 Å2 in BACEGG_00036 (the other dimeric interface between two subunits buries a surface area of 322 Å2). However, the tetrameric association seems probable because of the conservation of residues at the oligomeric interface and the fact that this arrangement is conserved in the different space groups (P1, C2221, P6322) in which these proteins were crystallized.
Figure 4.
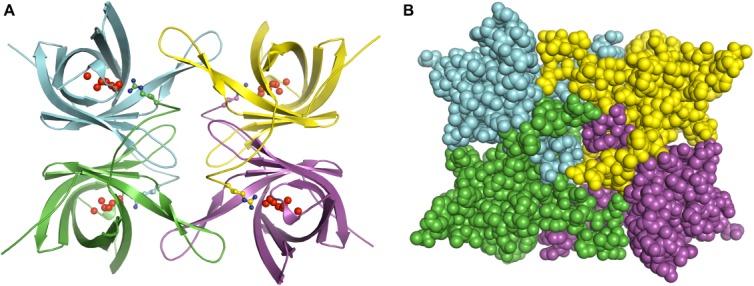
Putative biologically relevant oligomeric state. The proteins associate as a tetramer (dimer of dimers) shown in cartoon (A) and space-filling (B) representations and colored by chain in all three structures even when crystallized in different space groups. The UNL is shown as red spheres. Arginine 163 from a neighboring protomer that interact with the UNL is shown in ball and stick representation.
Sequence and structural similarities to proteins in UniProtKB and in the Protein Data Bank
BACOVA_00364, BACUNI_03039, and BACEGG_00036 are members of the Pfam family DUF4488 (PF14869).17 We ran the most recent version of the PF14869 family HMM (Supporting Information “PF14869-HMM.docx”) against the UniProtKB18 (March 2014 version) using the hmmer website (http://hmmer.janelia.org/) (hmmsearch with Pfam manually curated significance bit score thresholds of 27.0). This analysis identified 146 distinct, significant matches and uncovered an overlap with a region assigned by Pfam to the lipocalin_5-PF13924 family in the calycin clan (CL0116), indicating PF14869 may also belong to this clan (note: PF14869 [Pfam release 27.0 March 2013] contains only 63 sequences and no clan assignment, but this information will be updated in the next release). Except for four regions found in proteins from unclassified organisms (from metagenomics data), all other matches were with proteins from the phylum Bacteroidetes [Fig. 5(A)]. Most of these appear to be single-domain PF14869 proteins [Fig. 5(B)]. However, in 19 sequences, the PF14869 domain is found downstream of a domain that is a member of the TonB_C-PF03544 family. TonB_C is generally located at the C-terminus of TonB, a protein mostly involved in iron transport in Gram-negative bacteria. In one instance, F9YU00 (UniProt Id: F9YU00_CAPCC), the PF14869 domain is found upstream of a peptidase M60-like-PF13402 domain and, in another, R5PRP8 (UniProt Id: R5PRP8_9BACT), downstream of a FGE-sulfatase-PF03781 domain.
Figure 5.

Sequence alignment, family architecture and calycin signature residues in the DUF4488/PF14869 Pfam family. (A) Multiple sequence alignment of DUF4488/PF14869 Pfam family members using a redundancy cutoff of 80% sequence identity. For each sequence, we report the UniProtKB id, the position of first and last residue in the alignment, the percent sequence identity with respect to A7LRD6_BACOV (i.e., BACOVA_00364) and the amino acid sequence. Shades of gray in alignment columns reflect average similarity at a given position as calculated from the BLOSUM62 amino acid substitution matrix (black most conserved, white least conserved). Dashes (-) represent deletions. Lower case letters represent inserted residues and dots (.) in the same columns are fillers for sequences lacking the inserted residues. The red boxes mark the position of residues involved in the calcyin motif (GxW/R). Alignment visualized with Belvu (http://sonnhammer.sbc.su.se/Belvu.html). (B) General architecture of DUF4488/PF14869 family members. Out of 146 members, 125 contain a single DUF4488 domain; 19 also contain a TonB_C domain, one contains a M60-like domain, and one a FGE-sulfatase domain. ‘SP’ indicates a predicted signal peptide (according to PHOBIUS19). (C) Superposition of strand 1 and strand 8 of the β-barrel of 4gzv (green), 4iab (cyan), and 4i95 (magenta) in cartoon representation, highlighting the calycin motif residues tryptophan and arginine packing against each other.
Indeed, structural comparisons revealed further similarities between the three structures presented here and members of PF14869 family, and a group of proteins known as calycins,20 which include lipocalins, streptavidins/avidins, triabins, fatty acid-binding proteins and metallopeptidase inhibitors (part of the IK MEROPS clan21; referred to simply as IK inhibitors from now on). These are all characterized by a calyx-like β-barrel constituted from eight strands (10 strands in fatty acid-binding proteins) with an up-and-down +1 topology (except for triabin, which features a strand swap). The shear number, a measure of the barrel stagger,22 is 12 for lipocalins, triabin and fatty acid-binding proteins, whereas streptavidins, avidins and IK inhibitors have a shear number of 10, reflecting a generally straighter β-barrel. Accordingly, the structural classification database SCOP23 puts streptavidins, avidins, and IK inhibitors in separate folds (streptavidin-like fold versus lipocalins fold). Pfam groups the lipocalins, fatty acid-binding proteins and triabin into the calycin-CL0116 clan, IK inhibitors into the bBprotInhib-CL0354 clan, whereas avidins and streptavidins are in a family that is not assigned to any clan (Avidin-PF01382).
Although calycins are generally very diverse at the sequence level, most feature a conserved signature motif, typically Gx[WY]/[RK]. The first part of this motif (Gx[WY]) is located on the first strand of the β-barrel, whereas the second ([RK]) is located on the last strand. Typically, the positively charged residue located on the last strand of the β-barrel interacts with an aromatic residue on the first strand engaging in a cation-π interaction.24,25 The importance of this motif in β-barrel stability has been demonstrated.26,27 Conservation of this motif and other structural elements are often key to identifying calycins. All members of Pfam family DUF4488-PF14869 feature the calycin signature motif (GxW/R), except for a few proteins that appear to lack the N-terminal portion of the domain altogether (F9YU00 and A5ZKH0) and, thus, only have the arginine residue [Fig. 5(A)]; one protein (S7VPS0) lacks the final arginine. Additionally, four members in the family have the arginine residue substituted with lysine. Other conserved residues in the Pfam alignment [Fig. 5(A)] are identified with important structural/functional roles by mapping the sequence to the structure. For example, Lys60, Asn70, Glu97, Glu146 interact with the UNL, Gln38, Ile81, Val151 are involved in oligomerization, while Tyr95, Tyr129, Trp148 line the binding cavity (without making direct interaction with UNL). Interaction between the tryptophan and the arginine is observed in all three of our structures [Fig. 5(C)].
A structural similarity search using DALI28 provides several significant hits. All annotated proteins at the top of the list are calycins. These include, among others, bilin-binding protein 1bbp (Z-score 8.3), apolipoprotein D 2hzq (Z-score 7.6) and retinol-binding protein 1fem (Z-score 7.1) (all lipocalins), avidin 1avd (Z-score 7.1) and fatty acid-binding protein 1o8v (Z-score 7.0). Although the location of the UNL in our structures of PF14869 members coincides with that of the ligand in the bilin-binding protein structure (1bbp), the residues interacting with the UNL in our structures are not conserved in any of these other proteins (Fig. 6). The eight-stranded β-barrel in the PF14869 structures has a shear value of 12 (based on the 4gzv structure), suggesting a shape more reminiscent of structures in the lipocalins SCOP fold than those in the streptavidin-like SCOP fold. Lipocalins feature an additional helix+strand structural motif at the C-terminus [Fig. 6(B)] that is not generally found in other calycins or in the PF14869 proteins.
Figure 6.
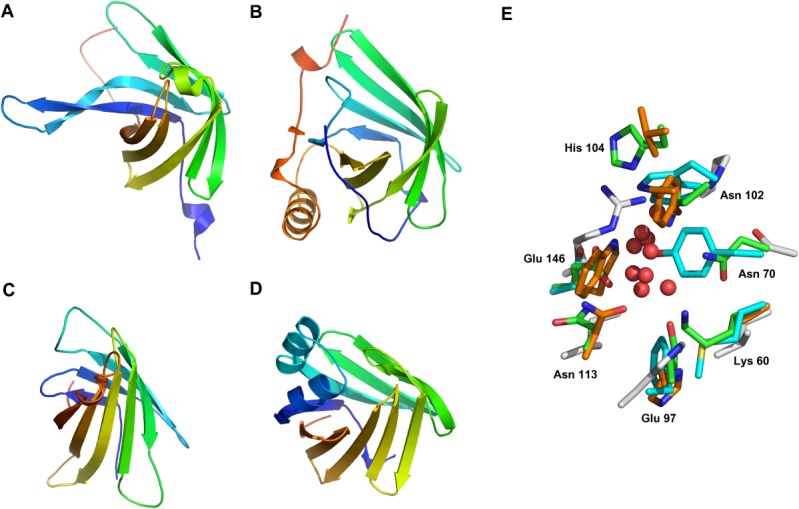
Comparison of the overall architecture and binding site of representative members of the calycin family. (A) BACOVA_00364 (pdb code 4gzv, chain A) in rainbow representation (N-terminus blue and C-terminus red). The structures of (B) Lipocalin (pdb code 1bpp), (C) Avidin (pdb code 1avd), and (D) fatty acid-binding protein (pdb code 1o8v) are shown is same orientation to illustrate the similarity of their overall structures. Lipocalins feature an additional helix+strand structure near the C-terminus. (E) The residues in the binding site are not conserved among members of the calycin family. The residues (green sticks) interacting with UNL (red spheres) in BACOVA_00364 are labeled and the corresponding residues in Lipocalin (orange), 1avd (cyan) and 1o8v (gray) are shown.
Discussion
The BACOVA_00364, BACUNI_03039, and BACEGG_00036 structures adopt an eight-stranded, β-barrel fold similar to calycins. Neither sequence nor structure searches reveal any clear function for these proteins despite overall structural similarity to proteins of known function. Likewise, genome context and potential protein–protein interactions analysis (using STRING29 and SEED30) did not provide any clear insight into the potential functions of these Bacteroides proteins. Calycin function is generally heavily influenced by loop conformation at the end of the β-barrel and conservation of specific binding residues.31 As a consequence, the overall β-barrel structural similarity alone is not sufficient to infer function, as confirmed by the diversity of functions represented within a very small range of Z-scores for the proteins returned by a DALI search. However, the presence of a UNL in the crystal structures helps identify the putative ligand binding site. The size and composition of the binding pocket suggests that the ligand could be a small polar, cyclical molecule, such as a sugar. The proteins likely assemble as a tetramer as indicated by crystal packing analysis in three independent space groups. These proteins provide the first representative structures of a newly defined Pfam family PF14869, (Pfam 27.0, March 2013).
Materials and Methods
Cloning, expression, purification and crystallization
The B. ovatus genomic DNA was extracted from cells (ATCC Number 8483) obtained from the American Type Culture Collection (ATCC), that for B. uniformis extracted from cells (ATCC 8492) provided by The Human Microbiome Project, and genomic B. eggerthii DNA was extracted from cells (DSM 20697) obtained from the DSMZ (The German Collection of Microorganisms and Cell Cultures). Clones were generated using the Polymerase Incomplete Primer Extension (PIPE) cloning method.32 The genes encoding the three proteins were amplified by polymerase chain reaction (PCR) from genomic DNA using PfuTurbo DNA polymerase (Stratagene) and I-PIPE (Insert) primers that included sequences for the predicted 5′ and 3′ ends. The primers used were forward primer, 5′-ctgtacttccagggcCAGAAGAAAACC AAATTCAAAGCGGCCG-3′; reverse primer, 5′-aatt aagtcgcgttaTCTCACAATATCTTCCGGAAATTTAGCC- 3′ for BACOVA_00364 from B. ovatus, forward primer, 5′-ctgtacttccagggcCAGGAGAGCGCAGAGTTT AGGCCTGCGG-3′; reverse primer, 5′-aattaagtcgcgt taACGGACAAGGTCCTCGGGGAATTTCGCG-3′ for BACUNI_03039 from B. uniformis, and forward primer, 5′-ctgtacttccagggcCAGGATAAGGCCGCTTTT GAGCCTGCGC-3′; reverse primer, 5′-aattaagtcgcg ttaCCGGACAATGTCTTCCGGAAACACCGGC-3′ for BACEGG_00036 from B. eggerthii; the target sequences are in upper case. The expression vector, pSpeedET, which encodes an amino-terminal tobacco etch virus (TEV) protease-cleavable expression and purification tag (MGSDKIHHHHHHENLYFQ/G), was PCR amplified with V-PIPE (Vector) primers (forward primer: 5′-taacgcgacttaattaactcgtttaaacggtctccagc-3′, reverse primer: 5′-gccctggaagtacaggttttcgtgatgatgatgatgatg-3′). V-PIPE and I-PIPE PCR products were mixed to anneal the amplified DNA fragments together. Escherichia coli GeneHogs (Invitrogen) competent cells were transformed with the I-PIPE/V-PIPE mixture and dispensed on selective LB-agar plates. The cloning junctions were confirmed by DNA sequencing. Using the PIPE method, the gene segment encoding residues M1-A22 were deleted from expression construct to produce soluble protein since it is predicted to contain either a signal peptide using SignalP33 or transmembrane helices using TMHMM-2.0.34
Expression was performed in a selenomethionine-containing medium at 37°C and selenomethionine was incorporated via inhibition of methionine biosynthesis.35 At the end of fermentation, lysozyme was added to the culture to a final concentration of 250 μg/mL, and the cells were harvested and frozen. After one freeze/thaw cycle, the cells were homogenized and sonicated in lysis buffer [40 mM Tris-HCl, 300 mM NaCl, 10 mM imidazole, 1 mM Tris (2-carboxyethyl) phosphine-HCl (TCEP)-HCl, pH 8.0]. Any remaining nucleic acids were digested with the addition of 0.4 mM MgSO4 and 1 µL of 250 U/µL benzonase (Sigma) to the lysate. The lysate was clarified by centrifugation at 32,500g for 25 min. The soluble fraction was passed over nickel-chelating resin (GE Healthcare) pre-equilibrated with lysis buffer, the resin was washed with wash buffer [40 mM Tris-HCl, 300 mM NaCl, 40 mM imidazole, 10% (v/v) glycerol and 1 mM TCEP-HCl, pH 8.0], and the protein was eluted with elution buffer [20 mM Tris, 300 mM imidazole, 10% (v/v) glycerol, 150 mM NaCl and 1 mM TCEP-HCl, pH 8.0]. The eluate buffer was exchanged with TEV buffer [20 mM Tris-HCl, 150 mM NaCl, 30 mM imidazole, 1 mM TCEP-HCl, pH 8.0] using a PD-10 column (GE Healthcare), and incubated with 1 mg of TEV protease per 15 mg of eluted protein for 2 h at ambient temperature followed by overnight at 4°C. The protease-treated eluate was passed over nickel-chelating resin (GE Healthcare) pre-equilibrated with crystallization buffer [20 mM Tris, 150 mM NaCl, 30 mM imidazole, and 1 mM TCEP-HCl, pH 8.0] and the resin was washed with the same buffer. The flow-through and wash fractions were combined and concentrated to ∼20 mg/mL by centrifugal ultrafiltration (Millipore) for crystallization trials.
The proteins were crystallized using the nanodroplet vapor diffusion method36 with standard JCSG crystallization protocols.37 Sitting drops composed of 100 nL protein solution mixed with 100 nL crystallization solution (0.1M Tris-HCl pH 8.5, 15% glycerol, 25.5% polyethylene glycol 4000, 0.17M sodium acetate for BACOVA_00364, and 1.0M lithium chloride, 20% polyethylene glycol 6000, 0.1M MES, pH 6.0 for BACUNI_03039 and BACEGG_00036) were equilibrated against a 35–50 μL reservoir at 277 K for 27–43 days prior to harvest. 20% (v/v) glycerol was added to the crystals of BACUNI_03039 and BACEGG_00036 as a cryoprotectant while no additional cryoprotectant was added to the crystal of BACOVA_00364. Initial screening for diffraction was carried out using the Stanford Automated Mounting system38 at the Stanford Synchrotron Radiation Lightsource (SSRL, Menlo Park, CA).
Analytical size exclusion chromatography
The oligomeric state of the proteins in solution was determined using a 0.8 cm × 30 cm Shodex Protein KW-803 size exclusion column (Thomson Instruments)32 pre-calibrated with gel filtration standards (Bio-Rad). The mobile phase consisted of 20 mM Tris-HCl pH 8.0, 150 mM NaCl, and 0.02% (w/v) sodium azide.
Data collection, structure solution, refinement
Multi-wavelength anomalous diffraction (MAD) data were collected to 1.95 Å resolution at wavelengths corresponding to inflection, peak, and high energy remote of the selenium edge on beamline BL9-2 at SSRL for BACOVA_00364. A similar MAD dataset for BACUNI_03039 was collected to 1.70 Å resolution at beamline BL8.2.2 at Advanced Light Source (ALS, Berkeley, CA) and a SAD dataset for BACEGG_00036 was collected to 1.81 Å resolution at beamline BL12-2 at SSRL. The data sets were collected at 100 K using a MAR325 CCD detector (BL9-2) or a Dectris Pilatus 6M pixel detector (BL12-2) with the BLU-ICE data collection environment39 at SSRL and an ADSC Q315 CCD detector at ALS. The data were processed with MOSFLM40 and scaled with SCALA41 for BACOVA_00364 while they were processed with XDS42 and scaled with XSCALE43 for BACUNI_03039 and BACEGG_00036. Phasing was performed with SHELXD44 and autoSHARP45 with a mean figure of merit of 0.27 for BACOVA_00364 (with two selenium sites per protein chain), 0.52 for BACUNI_03039 (with six sites per chain), and 0.33 for BACEGG_00036 (with four sites per chain). Automatic model building was performed with BUCCANEER.46 Model completion and refinement were performed with COOT47 and REFMAC.48 Experimental phase restraints in the form of Hendrickson-Lattman coefficients from SHARP, NCS restraints (except for BACUNI_03039), and TLS parameters were used during refinement. Data collection and refinement statistics are summarized in Table 1.
Table 1.
Crystallographic data and refinement statistics for BACOVA_00364 (4gzv), BACUNI_03039 (4iab), and BACEGG_00036 (4i95)
| Protein (PDB ID) | BACOVA_00364 (4gzv) | BACUNI_03039 (4iab) | BACEGG_00036 (4i95) | ||||
|---|---|---|---|---|---|---|---|
| Data Set | λ1 (remote) | λ2 (inflection) | λ3 (peak) | λ1 (inflection) | λ2 (remote) | λ3 (peak) | λ1 (peak) |
| Data collection | |||||||
| Space group | P1 | C2221 | P6322 | ||||
| Unit cell parameters (Å) | a = 44.78, b = 66.32, c = 109.74, 〈α = 88.2°, β = 82.3°, γ = 74.8° | a = 43.73, b = 213.88, c = 153.64 | a = 103.07, b = 103.07, c = 114.98 | ||||
| Wavelength (Å) | 0.9116 | 0.9792 | 0.9791 | 0.9795 | 0.9184 | 0.9793 | 0.9795 |
| Resolution range (Å) | 29.85–1.95 (2.00–1.95) | 29.85–1.95 (2.00–1.95) | 29.83–1.95 (2.00–1.95) | 46.19–1.66 (1.72–1.66) | 46.19–1.70 (1.76–1.70) | 46.22–1.73 (1.79–1.73) | 48.33–1.81 (1.87–1.81) |
| No. of observations | 250,141 | 242,093 | 243,550 | 306,941 | 286,554 | 271,057 | 1,307,232 |
| No. of unique reflections | 86,141 | 85,819 | 85,941 | 84,998 | 79,136 | 75,282 | 33,477 |
| Completeness (%) | 97.8 (96.7) | 97.4 (95.7) | 97.6 (96.4) | 99.1 (98.9) | 99.0 (98.7) | 99.0 (100.0) | 100.0 (100.0) |
| Mean I/σ (I) | 10.5 (1.8) | 8.1 (1.7) | 9.4 (1.6) | 15.5 (2.2) | 15.0 (2.3) | 12.3 (2.1) | 15.7 (2.9) |
| Rmerge on Ia (%) | 7.1 (44.5) | 7.4 (45.7) | 6.6 (50.7) | 4.6 (56.0) | 5.0 (53.6) | 5.9 (53.8) | 19.3 (156.2) |
| Rmeas on Ib (%) | 8.7 (62.8) | 9.2 (64.7) | 8.1 (71.7) | 5.3 (66.0) | 5.9 (63.0) | 7.0 (63.3) | 19.6 (158.2) |
| Rpim on Ic (%) | 4.9 (44.2) | 5.3 (45.7) | 4.7 (50.7) | 2.7 (35.8) | 3.0 (34.3) | 3.6 (34.4) | 3.1 (25.8) |
| CC1/2d | 0.997 (0.572) | 0.997 (–)i | 0.998 (–)i | 0.999 (0.732) | 0.999 (0.780) | 0.998 (0.761) | 0.992 (0.907) |
| Wilson B (Å2) | 27.5 | 27.6 | 29.0 | 21.7 | 20.6 | 20.8 | 21.9 |
| Model and refinement statistics | |||||||
| Resolution range (Å) | 29.85–1.95 | 46.19–1.66 | 48.33–1.81 | ||||
| No. of reflections (total)e | 86,030 | 84,954 | 33,450 | ||||
| No. of reflections (test) | 4305 | 4249 | 1693 | ||||
| Completeness (%) | 97.7 | 99.1 | 100.0 | ||||
| Data set used in refinement | λ1 | λ1 | λ1 | ||||
| Cutoff criteria | |F|>0 | |F|>0 | |F|>0 | ||||
| Rcrystf | 0.189 | 0.163 | 0.174 | ||||
| Rfreef | 0.220 | 0.184 | 0.204 | ||||
| Ramachandran stats | |||||||
| Favored (%) | 98.0 | 98.5 | 98.9 | ||||
| Outliers | 0 | 0 | 0 | ||||
| Restraints (r.m.s.d. observed) | |||||||
| Bond angles (°) | 1.80 | 1.57 | 1.76 | ||||
| Bond lengths (Å) | 0.012 | 0.012 | 0.016 | ||||
| Mean isotropic B valueg (Å2) | |||||||
| All | 48.3 | 27.6 | 27.0 | ||||
| Protein | 48.4 | 25.8 | 26.0 | ||||
| ESUh based on Rfree (Å) | 0.15 | 0.08 | 0.11 | ||||
| Protein residues/atoms | 1084/8763 | 547/4627 | 274/2273 | ||||
| Waters/solvent/UNLs | 555/6/5 | 628/6/4 | 293/0/2 | ||||
Values in parentheses are for the highest resolution shell.
Rmerge = ΣhklΣi|Ii(hkl) − (I(hkl))|/Σhkl Σi(hkl).
Rmeas = Σhkl[N/(N − 1)]1/2Σi|Ii(hkl) − (I(hkl))|/ΣhklΣiIi(hkl).49
CC1/2 is the correlation of an observed dataset with the underlying true signal.52
Typically, the number of unique reflections used in refinement is slightly less than the total number that were integrated and scaled. Reflections are excluded owing to negative intensities and rounding errors in the resolution limits and unit-cell parameters.
Rcryst = Σhkl||Fobs| − |Fcalc||/Σhkl|Fobs|, where Fcalc and Fobs are the calculated and observed structure-factor amplitudes, respectively. Rfree is the same as Rcryst but for 5% of the total reflections chosen at random and omitted from refinement.
This value represents the total B that includes TLS and residual B components.
The number of replicates for λ2 and λ3 in the high resolution shell was too small for scala to compute the CC1/2 value, but that the CC1/2 value in the 2.06–2.12 shell was 0.864, 0.860, 0.847 for λ1, λ2 and λ3 respectively.
Virtual library screen
A virtual ligand screen was performed at the putative ligand binding site in chain A of BACOVA_00364 (pdb 4gzv) with AutoDock Vina55 against the “ChemBridge_FullLibrary2011” from ZINC.12 Hydrogens and partial charges were added using MGLTools55 and the search limited to a 10 × 10 × 10 Å3 box around the ligand site. Based on the size of the binding site, the docking was limited to library entries ranging from 5 to 20 non-hydrogen atoms. The docking results (docked poses) were visually analyzed in COOT to identify candidates with acceptable fit to the electron density.
Validation and deposition
The quality of the crystal structures was analyzed using the JCSG Quality Control server (http://smb.slac.stanford.edu/jcsg/QC/). This server reports the stereochemical quality of the model using AutoDepInputTool,56 MolProbity,10 and Phenix,57 the agreement between the atomic model and the data using RESOLVE,21 the protein sequence using CLUSTALW,58 the ADP distribution using Phenix, and differences in Rcryst/Rfree, expected Rfree/Rcryst and various other items including nomenclature, atom occupancies, consistency of NCS pairs, ligand interactions, special positions, and so forth, using in-house scripts to analyze refinement log file and PDB header. Protein quaternary structure analysis was carried out using the PISA server.16 Atomic coordinates and experimental structure factors have been deposited in the PDB and are accessible under the codes 4gzv (BACOVA_00364), 4iab (BACUNI_03039), and 4i95 (BACEGG_00036).
Acknowledgments
The authors thank the members of the JCSG high-throughput structural biology pipeline for their contribution to this work. Portions of this research were carried out at SSRL and ALS. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Glossary
- ALS
Advanced Light Source
- JCSG
Joint Center for Structural Genomics
- LB
Luria-Bertani broth
- MAD
multi-wavelength anomalous diffraction
- MES
2-(N-morpholino)ethanesulfonic acid
- MgSO4
magnesium sulfate
- NCS
non-crystallographic symmetry
- NaCl
sodium chloride
- PCR
polymerase chain reaction
- PIPE
polymerase incomplete primer extension
- SAD
single-wavelength anomalous diffraction
- SSRL
Stanford Synchrotron Radiation Lightsource
- TCEP
tris(2-carboxyethyl)phosphine
- TEV
tobacco etch virus
- TLS
translation, libration, screw
- Tris
tris(hydroxymethyl)aminomethane
- UNL
unknown ligand.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
References
- Salyers AA. Bacteroides of the human lower intestinal tract. Annu Rev Microbiol. 1984;38:293–313. doi: 10.1146/annurev.mi.38.100184.001453. [DOI] [PubMed] [Google Scholar]
- Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- Xu J, Gordon JI. Honor thy symbionts. 2003;100:10452–10459. doi: 10.1073/pnas.1734063100. Proc Natl Acad Sci USA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexler HM. Bacteroides: the good, the bad, and the nitty-gritty. Clin Microbiol Rev. 2007;20:593–621. doi: 10.1128/CMR.00008-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Mahowald MA, Ley RE, Lozupone CA, Hamady M, Martens EC, Henrissat B, Coutinho PM, Minx P, Latreille P, Cordum H, Van Brunt A, Kim K, Fulton RS, Fulton LA, Clifton SW, Wilson RK, Knight RD, Gordon JI. Evolution of symbiotic bacteria in the distal human intestine. PLoS Biol. 2007;5:e156. doi: 10.1371/journal.pbio.0050156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, Hooper LV, Gordon JI. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science. 2003;299:2074–2076. doi: 10.1126/science.1080029. [DOI] [PubMed] [Google Scholar]
- Kuwahara T, Yamashita A, Hirakawa H, Nakayama H, Toh H, Okada N, Kuhara S, Hattori M, Hayashi T, Ohnishi Y. Genomic analysis of bacteroides fragilis reveals extensive DNA inversions regulating cell surface adaptation. 2004;101:14919–14924. doi: 10.1073/pnas.0404172101. Proc Natl Acad Sci USA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerdeno-Tarraga AM, Patrick S, Crossman LC, Blakely G, Abratt V, Lennard N, Poxton I, Duerden B, Harris B, Quail MA, Barron A, Clark L, Corton C, Doggett J, Holden MT, Larke N, Line A, Lord A, Norbertczak H, Ormond D, Price C, Rabbinowitsch E, Woodward J, Barrell B, Parkhill J. Extensive DNA inversions in the B. fragilis genome control variable gene expression. Science. 2005;307:1463–1465. doi: 10.1126/science.1107008. [DOI] [PubMed] [Google Scholar]
- Matthews BW. Solvent content of protein crystals. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodinger LLC. 2010. The PyMOL molecular graphics system, version 1.3r1.
- Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: a free tool to discover chemistry for biology. J Chem Inf Model. 52:1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouet P, Robert X, Courcelle E. ESPript/ENDscript: extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 2003;31:3320–3323. doi: 10.1093/nar/gkg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34:W116–W118. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer EL, Eddy SR, Bateman A, Finn RD. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40:D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kall L, Krogh A, Sonnhammer EL. A combined transmembrane topology and signal peptide prediction method. J Mol Biol. 2004;338:1027–1036. doi: 10.1016/j.jmb.2004.03.016. [DOI] [PubMed] [Google Scholar]
- Flower DR, North AC, Sansom CE. The lipocalin protein family: structural and sequence overview. Biochim Biophys Acta. 2000;1482:9–24. doi: 10.1016/s0167-4838(00)00148-5. [DOI] [PubMed] [Google Scholar]
- Terwilliger TC. Statistical density modification using local pattern matching. Acta Crystallogr D Biol Crstallogr. 2003;59:1688–1701. doi: 10.1107/S0907444903015142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin AG, Lesk AM, Chothia C. Principles determining the structure of beta-sheet barrels in proteins. I. A theoretical analysis. J Mol Biol. 1994;236:1369–1381. doi: 10.1016/0022-2836(94)90064-7. [DOI] [PubMed] [Google Scholar]
- Andreeva A, Howorth D, Chandonia JM, Brenner SE, Hubbard TJ, Chothia C, Murzin AG. Data growth and its impact on the SCOP database: new developments. Nucleic Acids Res. 2008;36:D419–D425. doi: 10.1093/nar/gkm993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallivan JP, Dougherty DA. Cation-pi interactions in structural biology. 1999;96:9459–9464. doi: 10.1073/pnas.96.17.9459. Proc Natl Acad Sci USA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasymov OK, Abduragimov AR, Glasgow BJ. Cation-pi interactions in lipocalins: structural and functional implications. Biochemistry (Mosc) 2012;51:2991–3002. doi: 10.1021/bi3002902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katakura Y, Totsuka M, Ametani A, Kaminogawa S. Tryptophan-19 of beta-lactoglobulin, the only residue completely conserved in the lipocalin superfamily, is not essential for binding retinol, but relevant to stabilizing bound retinol and maintaining its structure. Biochim Biophys Acta. 1994;1207:58–67. doi: 10.1016/0167-4838(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Greene LH, Chrysina ED, Irons LI, Papageorgiou AC, Acharya KR, Brew K. Role of conserved residues in structure and stability: tryptophans of human serum retinol-binding protein, a model for the lipocalin superfamily. Protein Sci. 2001;10:2301–2316. doi: 10.1110/ps.22901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Sander C. Dali: a network tool for protein structure comparison. Trends Biochem Sci. 1995;20:478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, Jensen LJ. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang HY, Cohoon M, de Crecy-Lagard V, Diaz N, Disz T, Edwards R, Fonstein M, Frank ED, Gerdes S, Glass EM, Goesmann A, Hanson A, Iwata-Reuyl D, Jensen R, Jamshidi N, Krause L, Kubal M, Larsen N, Linke B, McHardy AC, Meyer F, Neuweger H, Olsen G, Olson R, Osterman A, Portnoy V, Pusch GD, Rodionov DA, Ruckert C, Steiner J, Stevens R, Thiele I, Vassieva O, Ye Y, Zagnitko O, Vonstein V. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005;33:5691–5702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flower DR. Multiple molecular recognition properties of the lipocalin protein family. J Mol Recognit. 1995;8:185–195. doi: 10.1002/jmr.300080304. [DOI] [PubMed] [Google Scholar]
- Klock HE, Koesema EJ, Knuth MW, Lesley SA. Combining the polymerase incomplete primer extension method for cloning and mutagenesis with microscreening to accelerate structural genomics efforts. Proteins. 2008;71:982–994. doi: 10.1002/prot.21786. [DOI] [PubMed] [Google Scholar]
- Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol. 1993;229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- Santarsiero BD, Yegian DT, Lee CC, Spraggon G, Gu J, Scheibe D, Uber DC, Cornell EW, Nordmeyer RA, Kolbe WF, Jin J, Jones AL, Jaklevic JM, Schultz PG, Stevens RC. An approach to rapid protein crystallization using nanodroplets. J Appl Cryst. 2002;35:278–281. [Google Scholar]
- Lesley SA, Kuhn P, Godzik A, Deacon AM, Mathews I, Kreusch A, Spraggon G, Klock HE, McMullan D, Shin T, Vincent J, Robb A, Brinen LS, Miller MD, McPhillips TM, Miller MA, Scheibe D, Canaves JM, Guda C, Jaroszewski L, Selby TL, Elsliger MA, Wooley J, Taylor SS, Hodgson KO, Wilson IA, Schultz PG, Stevens RC. ) Structural genomics of the Thermotoga maritima proteome implemented in a high-throughput structure determination pipeline. 2002;99:11664–11669. doi: 10.1073/pnas.142413399. Proc Natl Acad Sci USA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AE, Ellis PJ, Miller MD, Deacon AM, Phizackerley RP. An automated system to mount cryo-cooled protein crystals on a synchrotron beamline, using compact sample cassettes and a small-scale robot. J Appl Cryst. 2002;35:720–726. doi: 10.1107/s0021889802016709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. Blu-ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. J Synchrotron Radiat. 2002;9:401–406. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- Leslie AGW. 1992. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4+ESF-EAMCB Newsletter on Protein Crystallography 26.
- Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr D Biol Crystallogr. 2010;66:133–144. doi: 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Vonrhein C, Blanc E, Roversi P, Bricogne G. Automated structure solution with autoSHARP. Methods Mol Biol. 2007;364:215–230. doi: 10.1385/1-59745-266-1:215. [DOI] [PubMed] [Google Scholar]
- Cowtan K. Fitting molecular fragments into electron density. Acta Crystallogr D Biol Crystallogr. 2008;64:83–89. doi: 10.1107/S0907444907033938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Murshudov GN, Papiz MZ. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003;374:300–321. doi: 10.1016/S0076-6879(03)74014-2. [DOI] [PubMed] [Google Scholar]
- Diederichs K, Karplus PA. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat Struct Biol. 1997;4:269–275. doi: 10.1038/nsb0497-269. [DOI] [PubMed] [Google Scholar]
- Weiss MS, Hilgenfeld R. On the use of the merging R factor as a quality indicator for X-ray data. J Appl Crystallogr. 1997;30:203–205. [Google Scholar]
- Weiss M. Global indicators of X-ray data quality. J Appl Crystallogr. 2001;34:130–135. [Google Scholar]
- Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Cruickshank DW. Remarks about protein structure precision. Acta Crystallogr D Biol Crystallogr. 1999;55:583–601. doi: 10.1107/s0907444998012645. [DOI] [PubMed] [Google Scholar]
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Guranovic V, Dutta S, Feng Z, Berman HM, Westbrook JD. Automated and accurate deposition of structures solved by X-ray diffraction to the Protein Data Bank. Acta Crystallogr D Biol Crystallogr. 2004;60:1833–1839. doi: 10.1107/S0907444904019419. [DOI] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crsystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.