

Abstract
HIV patients on combination oral drug therapy experience insufficient drug levels in lymph nodes, which is linked to viral persistence. Following success in enhancing lymph node drug levels and extending plasma residence time of indinavir formulated in lipid nanoparticles, we developed multidrug anti-HIV lipid nanoparticles (anti-HIV LNPs) containing lopinavir (LPV), ritonavir (RTV), and tenofovir (PMPA). These anti-HIV LNPs were prepared, characterized, scaled up, and evaluated in primates with a focus on plasma time course and intracellular drug exposure in blood and lymph nodes. Four macaques were subcutaneously administered anti-HIV LNPs and free drug suspension in a crossover study. The time course of the plasma drug concentration as well as intracellular drug concentrations in blood and inguinal lymph nodes were analyzed to compare the effects of LNP formulation. Anti-HIV LNPs incorporated LPV and RTV with high efficiency and entrapped a reproducible fraction of hydrophilic PMPA. In primates, anti-HIV LNPs produced over 50-fold higher intracellular concentrations of LPV and RTV in lymph nodes compared to free drug. Plasma and intracellular drug levels in blood were enhanced and sustained up to 7 days, beyond that achievable by their free drug counterpart. Thus, multiple antiretroviral agents can be simultaneously incorporated into anti-HIV lipid nanoparticles to enhance intracellular drug concentrations in blood and lymph nodes, where viral replication persists. As these anti-HIV lipid nanoparticles also prolonged plasma drug exposure, they hold promise as a long-acting dosage form for HIV patients in addressing residual virus in cells and tissue.
Introduction
Highly active antiretroviral therapy (HAART), a combination of antiretroviral drugs with different viral targets, can clear virus from the blood and maintain aviremia for several years. However, if daily oral therapy is interrupted, plasma viremia rapidly rebounds.1,2 Virus persists in lymph nodes and lymphoid tissues,3 where viral DNA and RNA remain detectable in patients on HAART even with plasma aviremia.4,5 HIV+ patients on HAART have lower drug concentrations in lymphoid tissues relative to concurrent plasma concentrations,6–8 which has been linked to persistent lymphatic viral replication.6,9,10 Enhancing and extending drug exposure in lymphoid tissues are essential for clearing residual virus to find a cure for HIV.
We have previously reported that lipid nanoparticles (LNPs) containing a protease inhibitor, indinavir (IDV), produced elevated drug levels in lymph nodes throughout the body and extended plasma residence time.7,11 In HIV-infected primates, treatment with these IDV-LNPs reversed CD4+ T cell decline and reduced viral RNA in plasma and lymph nodes.11 These LNPS have been documented to incorporate other protease inhibitors as well as a hydrophilic drug tenofovir (PMPA), a nucleotide analog reverse transcriptase inhibitor (NRTI) and the active drug in Viread.12 Since monodrug regimens may promote resistance,13 and combination drug therapies targeted to multiple HIV proteins reduce mortality,14,15 multiple drugs formulated in a single particle may enhance therapeutic potency and tissue viral clearance.
Therefore, we developed and evaluated in primates a three-drug combination lipid nanoparticle (anti-HIV LNP) containing lopinavir (LPV), ritonavir (RTV), and PMPA. LPV and RTV were selected due to their acid stability and hydrophobicity, which promotes lipid association.16 Ritonavir, a metabolic and transport inhibitor, is used clinically to enhance the efficacy of coadministered drugs.17,18 PMPA, being an NRTI, provides an additional site of antiviral action, and its phosphorylated form is retained intracellularly, thus prolonging antiviral activity.19 This combination is clinically relevant and favorable to extending and enhancing intracellular drug exposure. We found that a subcutaneous injection of anti-HIV LNPs in primates produced enhanced intracellular drug concentrations in mononuclear cells of lymph nodes (LNMCs) and peripheral blood (PBMCs) and prolonged residence time in PBMCs and plasma compared to free drug in suspension. Long-acting anti-HIV lipid nanoparticles have the potential to overcome drug insufficiency and the associated viral persistence in lymphoid tissues.
Materials and Methods
Materials and animals
1,2-Distearoyl-sn-glycero-3-phosphocholine (DSPC) and N-(carbonylmethoxypolyethyleneglycol-2000)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine, sodium salt (MPEG-2000-DSPE) were purchased from Corden Pharma (Liestal, Switzerland). LPV, RTV, and PMPA ([(2R)-1-(6-aminopurin-9-yl)propan-2-yl]oxymethylphosphonic acid) were purchased from Waterstone Technology (Carmel, IN). Other reagents were analytical grade or higher.
Four young adult male macaques (Macaca nemestrina, 2.9–4.0 kg) were housed and cared for by the Washington National Primate Research Center (WaNPRC) under an approved Institutional Animal Care and Use Committee protocol. One animal developed an unrelated illness that necessitated discarding that individual's second round PBMC data.
Lipid nanoparticle preparation and in vitro characterization
LNPs composed of DSPC: MPEG-2000-DSPE (8:2 or 9:1 molar ratio), LPV, RTV, and PMPA (115:10:5:15 lipid:LPV:RTV:PMPA molar ratio) were prepared aseptically according to previously established methods.7 Briefly, lipids and protease inhibitors were dissolved in chloroform:ethanol (3:1 v/v) and PMPA was added from a stock solution of 30 mg/ml in aqueous 150 mM NaHCO3. Solvent was removed by rotary evaporation and vacuum desiccation. The dry film was rehydrated to 200 mM lipid in 0.4% NaCl with 20 mM NaHCO3 buffer at 60°C. Particle size was reduced at 60°C by bath sonication (laboratory scale) or high-pressure homogenization using an Emulsiflex-C5 (Avestin, Ottawa, Canada) (clinical scale). LNPs were maintained at 60°C to anneal for 30 min prior to cooling and stored at 4°C. Particle size was determined by photon correlation spectroscopy using a NICOMP 380 ZLS (Particle Sizing Systems, Santa Barbara, CA). Osmolality (Vapro 5520 osmometer; Wescor, Logan, UT) and pH (Hydrion paper) were assessed.
The anti-HIV LNP formulations contain both unbound drug and drugs bound to lipid nanoparticles. To determine drug incorporation/encapsulation efficiencies, anti-HIV LNPs were dialyzed (MWCO 6,000–8,000) at room temperature against 1,000× volume bicarbonate-buffered saline for 4 h to remove unbound drug. Drugs were quantified by LC-MS/MS using acetonitrile precipitation. Encapsulation efficiency (EE) was calculated as follows:
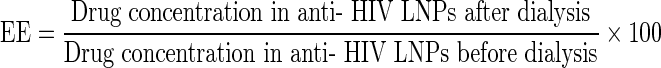 |
The antiviral activity of the three-drug combination in soluble or LNP form was evaluated in HIV-infected CEM-174 cells as described previously.7 After 4 days of incubation with drug, cells were observed for syncytia, and viral infection was confirmed by ELISA detection of HIV-2 p27. The ratio of LPV, RTV, and PMPA was fixed at 1:1:0.5 (m/m/m) for antiviral activity evaluation. The dose of drugs added to cells represents the sum of bound and unbound drugs in this anti-HIV LNP formulations.
Preparation of free drug suspension for primate study
Two free drug suspensions were prepared at the same molar ratios as for anti-HIV LNPs (LPV:RTV:PMPA 2:1:3, m/m/m). The vehicle for the first suspension was bicarbonate-buffered saline with 3% ethanol and 0.2% bovine serum albumin. The vehicle for the second suspension was bicarbonate-buffered saline with 8% DMSO and 0.1% Tween 20.
Sterility of preparations for injection
All preparations for injection were prepared using injection-grade aqueous solutions and an aseptic technique. Nonsterile aqueous components were sterilized by passage through a 0.22-μm cellulose acetate filter. Sterility was verified by a blood agar culture test for 7 days at 37°C.
Subcutaneous administration of free and lipid-associated drug combination to macaques
Four macaques were divided into two treatment groups (two animals per group). Each animal received both free drug and anti-HIV LNPs (bound and free drug mixture) in a cross-over study with a washout period of over 12 weeks between experiments. Anti-HIV LNPs were dosed at 25.0, 14.3, and 17.1 mg/kg of LPV, RTV, and PMPA, respectively, in 20 ml delivered subcutaneously on the back. Free drug suspension was dosed slightly lower (20.0, 11.5, and 13.7 mg/kg LPV, RTV, and PMPA, respectively) in the same volume due to limited solubility. Data were normalized to the anti-HIV LNP dose for comparative analysis.
Blood and tissue sample collection and processing
Blood samples were collected in EDTA by femoral venous puncture at 0, 0.5, 1, 3, 5, 8, 24, and 168 h, plus 48 h (both groups) and 120 h (anti-HIV LNP group) in the second round of the study. Plasma was removed, PBMCs were isolated by the density gradient method,20 and cells were aliquoted into pellets of approximately 2 million PBMCs each. An inguinal lymph node was surgically excised 24 h after drug administration (n=2 per treatment), LNMCs were isolated by passage through a 100-μm nylon cell strainer (Corning, Tewksbury, MA), and cells were aliquoted into pellets of approximately 1–2 million LNMCs each. All samples were stored at −80°C prior to drug analysis.
Determination of drug concentrations in plasma and cells
Plasma concentrations of all three drugs were analyzed simultaneously by liquid chromatography–tandem mass spectrometry using a recently published method.16 PBMC and LNMC pellets were lysed using 200 μl water/methanol (1:1 v/v), sonicated for 10 min, and then extracted and analyzed using the same method as cited above. Intracellular concentrations were converted to ng/ml with the assumption that each cell has a volume of 4×10−9 ml.21
Determination of plasma drug exposure
Area under the curve (AUC) was calculated for the total evaluated time period (0–168 h) as well as from 0 to 24 and 24 to 168 h using the trapezoidal rule. Data were analyzed by paired two-tailed Student's t-test with p<0.05 considered statistically significant.
Assessment of immunological and inflammatory response
Blood was collected at least 1 week prior to and exactly 7 days after drug administration for analysis of complete blood count, serum chemistry, C-reactive protein, and total complement. Reference values for Macaca nemestrina were provided by the WaNPRC. Human reference values were used for C-reactive protein and total complement due to limited primate data. Animals were observed daily for physical or behavioral changes.
Results
Physicochemical properties and antiviral activity of anti-HIV nanoparticles composed of lopinavir, ritonavir, and tenofovir
In preparation for primate studies, anti-HIV LNPs containing LPV, RTV, and PMPA were optimized on a laboratory scale (0.1–0.4 ml) and scaled up to clinical scale (14–40 ml) using equipment capable of multiliter capacity. Protease inhibitor incorporation efficiency was reproducibly high, with clinical scale batches showing over 90% LPV and RTV incorporation (Table 1). PMPA association was consistent and reproducible (Table 1). High-pressure homogenization resulted in slightly smaller particles (∼50 nm diameter) compared to those produced by sonication (∼70 nm diameter). The pH and osmolality of the anti-HIV LNP preparations were physiologically compatible, and a culture test verified sterility suitable for primate studies.
Table 1.
Physicochemical Characteristics of Anti-HIV Lipid Nanoparticles Produced on a Laboratory or Clinical Scale
| Degree of incorporation (% total) | ||||||
|---|---|---|---|---|---|---|
| Lopinavir | Ritonavir | Tenofovir | Particle diameter (nm)a | pH | Osmolality | |
| Laboratory scaleb (0.1–0.4 ml) | 74.2±7.3 | 74.2±8.4 | 11.8±5.1 | 68.3±14.0 | 7.0±0.3 | 334±39 |
| Clinical scalec (15–40 ml) | 93.6±5.5 | 90.7±7.4 | 11.9±3.0 | 52.4±9.1 | 7.0±0.5 | 266±31 |
Particle size was determined by Nicomp intensity-weighted analysis.
Anti-HIV LNPs were reproduced in 0.1–0.4 ml batches, with values expressed as mean±standard deviation of five batches.
Anti-HIV LNPs were reproduced in 15–40 ml batches, with values expressed as mean±standard deviation of three batches.
Anti-HIV LNPs were prepared at a lipid:LPV:RTV:PMPA molar ratio of 115:10:5:15 and a final lipid concentration of 200 mM. The degree of drug incorporation in the three drugs—LPV:RTV:PMPA (2:1:3 mole ratio)—was determined as described in Materials and Methods for reproducibility and other characteristics. The anti-HIV LNPs contain both free and bound drugs in a well-defined fraction.
LNP, lipid nanoparticle; LPV, lopinavir; RTV, ritonavir; PMPA, tenofovir.
The antiviral potency of LNP-associated LPV, RTV, and PMPA was evaluated in HIV-infected CEM-174 cells and compared to soluble free drugs combined in equivalent ratios or used individually. At a 1:1:0.5 (m/m/m) drug ratio, respectively, anti-HIV LNPs exhibited a 30-fold increase in potency over the drugs combined in soluble form (LPV, RTV, and PMPA EC50 3.0±0.8, 3.0±0.8, and 1.5±0.1 nM in LNP form vs. 98±0.3, 98±0.3, and 49±0.2 nM in free form). Single agent EC50 values for LPV, RTV, and PMPA were 600±0.04, 530±0.2, and 1150±0.7 nM, respectively. Control lipid nanoparticles exhibited no antiviral effects.
Effects of LNP association on plasma time course and total drug exposure
Primates received a single subcutaneous dose of LPV, RTV, and PMPA in either anti-HIV LNPs or free suspension form. Following free drug administration, plasma concentrations peaked within 8 h and dropped to low or undetectable levels by 24 h (Fig. 1). In contrast, macaques treated with anti-HIV LNPs demonstrated elevated plasma concentrations of LPV and RTV at all time points evaluated and higher peak plasma concentrations of LPV and RTV (p<0.05); all three drugs remained detectable in plasma at 7 days (168 h) postadministration. LNP-associated PMPA demonstrated two peaks: the first peak was similar to that seen after free drug administration, while the second peak was not seen with free drug (Fig. 1E and F).
FIG. 1.

Plasma concentration of lopinavir (LPV), ritonavir (RTV), and tenofovir (PMPA) over time after subcutaneous administration in either free (A, C, E) or lipid nanoparticle (B, D, F) formulation at a normalized dose of 25 mg/kg LPV, 14.3 mg/kg RTV, and 17.1 mg/kg PMPA. Data points represent individual animals (circle, M10066; triangle, M10068; square, R10142; diamond, Z11084; open, free drug; closed, LNP). Time points with a mean value of less than 10 ng/ml are annotated. *Outlier omitted: 3285.76 ng/ml at 8 h for animal M10068.
Total drug exposure was assessed by calculating the area under the plasma drug concentration curve (AUC). As shown in Table 2, anti-HIV LNPs provided a 7-fold or greater increase in total drug exposure for all three drugs when compared to free drug suspension (≤0.05, 0.07, and 0.173 for LPV, RTV, and PMPA, respectively). Additionally, the percentage of total drug exposure that occurred after the first 24 h was increased for all three drugs with anti-HIV LNPs. This shift was most dramatic for hydrophilic PMPA (0.1% with free drug, 51.2% with anti-HIV LNPs) (Table 2). These data indicate that the LNP formulation provides enhanced and extended plasma drug exposure for all three drugs, including water-soluble PMPA with only 12% drug encapsulation.
Table 2.
Effects of Anti-HIV Lipid Nanoparticles on Plasma Drug Exposure in Primates Treated with Lopinavir, Ritonavir, and Tenofovir in Combination
| AUC (μg·h·ml−1)a | AUCfraction (% of total)b | ||||||
|---|---|---|---|---|---|---|---|
| Free drug | Anti-HIV LNP | Free drug | Anti-HIV LNP | ||||
| Drug | 0–168 h | 0–168 h | LNP/free ratio | 0–24 h | 24–168 h | 0–24 h | 24–168 h |
| Lopinavir (LPV) | 3.83±4.04 | 69.6±10.7 | 18.2 | 56.6±9.4 | 43.5±9.4 | 37.2±9.9 | 62.8±9.9 |
| Ritonavir (RTV) | 1.39±1.18 | 19.4±12.2 | 14.0 | 84.1±18.9 | 15.9±18.9 | 43.0±12.5 | 57.0±12.5 |
| Tenofovir (PMPA) | 56.6±17.04 | 395.0±344.5 | 7.0 | 99.9±0.2 | 0.1±0.2 | 48.8±33.7 | 51.2±33.7 |
Four animals were treated with lopinavir (25 mg/kg), ritonavir (14.3 mg/kg), and tenofovir (17.1 mg/kg) administered subcutaneously in either free or LNP form. Plasma drug concentrations were measured and area under the curve (AUC) was calculated using the trapezoidal rule. Data are expressed as mean±standard deviation for four animals over a 168-h period.
AUC was analyzed from 0 to 24 h, which represents the standard dosing period for current HAART, and from 24 to 168 h to demonstrate the extended plasma exposure provided by anti-HIV LNPs. Values are expressed as a percent of the total AUC, mean±standard deviation.
Effect of LNP association on intracellular LPV, RTV, and PMPA concentrations in blood and lymph node mononuclear cells
While plasma drug levels indicate sustained drug exposure, intracellular levels are essential for inhibition of viral replication. Thus, mononuclear cells in blood (PBMCs) were isolated and intracellular concentrations of LPV, RTV, and PMPA were determined. As illustrated in Fig. 2, macaques treated with anti-HIV LNPs had detectable intracellular drug levels at 7 days postadministration that exceeded the experimental EC50 reported above. In contrast, intracellular drug levels after free drug administration generally fell below detection by 48 h (Fig. 2). LPV and RTV concentrations after anti-HIV LNP administration were comparable to free drug at early time points and up to 20-fold greater at later time points. Water-soluble PMPA exhibited lower intracellular drug levels for the first five time points with anti-HIV LNPs; however, by 8 h postadministration, intracellular PMPA concentrations were over 50-fold higher in LNP-treated animals (Fig. 2).
FIG. 2.

Mean intracellular concentration of (A) lopinavir (LPV), (B) ritonavir (RTV), and (C) tenofovir (PMPA) in peripheral blood mononuclear cells (PBMCs) over time after subcutaneous administration in either free (open symbols) or lipid nanoparticle (closed symbols) formulation at a normalized dose of 25 mg/kg LPV, 14.3 mg/kg RTV, and 17.1 mg/kg PMPA. Concentrations are expressed as ng/ml (left y axis) and nmol/liter (right y axis). Error bars show standard error of the mean (SEM). For free drug, n=4 except at 48 h and 120 h n=2. For anti-HIV LNP, n=3, except at 48 h and 120 h n=1. *Cannot calculate SEM for anti-HIV LNPs at 48 h and 120 h time points due to n of 1.
To determine the effect of LNP association on intracellular drug concentrations in LNMCs, an inguinal lymph node was collected 24 h after drug administration. LNMCs were isolated and intracellular drug concentrations were compared between test groups (LNP-to-free ratio) and to concurrent plasma drug concentrations (Table 3). LPV and RTV concentrations were low or undetectable in LNMCs of animals treated with free drug combination, and comparable to plasma concentrations. In contrast, concentrations of LPV and RTV in LNMCs after anti-HIV LNP administration were 2-fold and 10-fold higher, respectively, than concurrent plasma concentrations and over 50-fold higher than LNMC concentrations after free drug administration (p<0.05). Additionally, plasma concentrations of LPV and RTV at 24 h were approximately 25-fold higher with anti-HIV LNPs compared to free drug (Table 3). PMPA, while consistently detectable in LNMCs, showed no significant difference in intracellular lymph node concentrations between the two test groups (p=1.0). Intracellular drug levels in PBMCs and LNMCs at 24 h were comparable. Taken together, anti-HIV LNPs containing LPV, RTV, and PMPA enhanced and prolonged intracellular levels of all three drugs in blood as well as in lymph nodes, and produced greater concentrations of LPV and RTV in LNMCs than in plasma at 24 h postadministration.
Table 3.
The Effect of Anti-HIV Lipid Nanoparticles on the Intracellular Concentrations of Lopinavir, Ritonavir, and Tenofovir in Lymph Nodes Compared to Plasma Concentrations at 24 h
| Mean drug concentration (ng/ml) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Free drug treatmenta | Anti-HIV LNP treatmenta | Ratio comparison between treatments (LNP/free)b | ||||||
| Drug | Lymph node | Plasma | LN/plasma ratioc | Lymph node | Plasma | LN/plasma ratioc | Lymph node | Plasma |
| Lopinavir | 0.0±0.0 | 24.5±26.3 | <0.1 | 1212.3±9.8 | 596.7±122.7 | 2.0 | >1212.3 | 24.4 |
| Ritonavir | 32.6±32.6 | 5.9±9.3 | 5.5 | 1641.8±19.0 | 163.6±115.6 | 10.0 | 50.3 | 27.9 |
| Tenofovir | 255.7±31.5 | 0.9±1.1 | 284.1 | 189.9±22.8 | 3598.9±3508.5 | 0.1 | 0.7 | 3992.8 |
Animals were administered anti-HIV LNPs or free drug at a normalized dose of 25 mg/kg LPV, 14.3 mg/kg RTV, and 17.1 mg/kg PMPA subcutaneously. A blood sample and inguinal lymph node were collected 24 h after drug administration and lymph node mononuclear cells were isolated for drug analysis. Intracellular drug concentrations were calculated as described in Materials and Methods. Drug concentrations are reported as mean±standard deviation (n=2).
Mean anti-HIV LNP drug concentration divided by mean free drug concentration. In cases where the drug level was below detectable limits, the number was taken as “1” to calculate the ratio.
Mean lymph node intracellular drug concentration divided by mean plasma drug concentration. In cases where the drug level was below detectable limits, the number was taken as “1” to calculate the ratio.
Safety evaluation of immunological and inflammatory response
To evaluate safety, primate blood samples taken before and after drug administration were analyzed for complete blood count, serum chemistry panel, C-reactive protein, and total complement. Relevant values are summarized in Table 4. Indicators of renal and hepatic function (blood urea nitrogen, creatinine, liver enzymes) stayed within normal limits. No animals demonstrated increased C-reactive protein or white blood cell count to a value outside of the reference range. Total complement, while highly variable, showed no significant changes. A slight cholesterol level variation with anti-HIV LNP treatment was not statistically significant (p=0.07). Naive animals receiving free drug demonstrated a local reaction consisting of firm, nonerythematous swellings that resolved over the following weeks along with mildly increased platelet counts. Naive animals that received anti-HIV LNPs demonstrated no local reaction and their platelet counts remained within normal limits.
Table 4.
Changes in Selected Inflammatory Indicators and Cholesterol After Subcutaneous Administration of Lopinavir, Ritonavir, and Tenofovir in Free or Anti-HIV Lipid Nanoparticle Form
| Baselinea | After free drugb | p-valuec | After LNPb | p-valuec | |
|---|---|---|---|---|---|
| WBC (thousands/μl) | 10.1±4.3 | 8.6±1.9 | 0.92 | 11.7±3.6 | 0.93 |
| Platelet (thousands/μl) | 496±102 | 576±125 | 0.04 | 636±117 | 0.10 |
| Total complement (U/ml) | 129±73 | 146±76 | 0.31 | 139±59 | 0.76 |
| C-Reactive protein (mg/liter) | 0.2±0.0 | <0.2±0.0 | 1.00 | <0.2±0.0 | 0.39 |
| Cholesterol (mg/dl) | 162±16 | 162±24 | 0.80 | 191±15 | 0.07 |
To establish a baseline, blood was collected at least 7 days prior to drug administration.
Blood was collected 7 days after drug administration.
p-value was determined by paired two-tailed Student's t-test.
Venous blood samples were analyzed for complete blood count, serum chemistry, total complement, and C-reactive protein. Values are expressed as mean±standard deviation. No parameters excluded from this table showed a postadministration increase to a value outside of the reference range in any primates (n=4).
Discussion
Capitalizing on the ability of lipid nanoparticles to efficiently incorporate two lipophilic protease inhibitors, LPV and RTV, and simultaneously encapsulate a significant fraction of PMPA, we constructed a drug combination anti-HIV LNP and analyzed intracellular drug concentrations and plasma kinetics in macaques (M. nemestrina). Primates dosed subcutaneously with anti-HIV LNPs exhibited extended plasma drug levels (Fig. 1), had higher total drug exposure (Table 2), and sustained intracellular concentrations of LPV, RTV, and PMPA in blood (Fig. 2). Importantly, drugs in anti-HIV LNPs provided much higher intracellular levels of LPV and RTV in lymph nodes, beyond those achieved with free drug (Table 3). These findings indicate that lipid nanoparticles may overcome the drug insufficiency reported with HIV-infected patients on oral HAART.6–8
Employing a simple, reproducible, and scalable production method, these nanoparticles stably incorporate LPV and RTV with >90% efficiency while simultaneously encapsulating a hydrophilic reverse transcriptase inhibitor, PMPA (Table 1). The PMPA association was consistent and well-defined, and unassociated PMPA was left in the solution as free drug, reducing drug wastage and the risk of contamination as well as eliminating the need for costly and time-consuming purification. Due to space limitations, additional details on optimization and in vitro characterization of anti-HIV LNPs are beyond the scope of this report and will be reported separately.
A subcutaneously administered three-drug combination of anti-HIV LNPs increased peak plasma concentrations, prolonged plasma residence time, and increased plasma drug exposure as evidenced by an increase in AUC (Fig. 1 and Table 2). Considering the 12% encapsulation efficiency of PMPA, these findings indicate that limited encapsulation of hydrophilic PMPA still makes a marked impact on drug distribution and/or metabolism. Enhanced stability and reduced renal and hepatic drug clearance provided by the LNPs may contribute to these findings, although the behavior and form of anti-HIV LNPs in vivo remain to be explored.
Intracellular drug exposure is a vital consideration if a drug is to maximally suppress viral replication in cells. For all three drugs, a lipid-drug association enhanced intracellular drug concentrations in PBMCs and LNMCs, suggesting that LNPs are internalized and retained in mononuclear cells (Fig. 2 and Table 3). While this nonterminal study allowed analysis of only a single peripheral lymph node, previous work with indinavir-containing LNPs has shown elevated indinavir concentrations in lymph nodes throughout the body (including mesenteric, tonsillar, bronchial, and others).11 Thus, a widespread lymphatic distribution of drug-containing LNPs is achievable. Therefore, anti-HIV LNPs hold promise as a vehicle for simultaneous widespread lymphatic distribution of multiple anti-HIV drugs, overcoming lymphatic drug insufficiency in sites of viral persistence.
Presenting multiple drugs with different viral targets within a single particle is important for improving antiviral efficacy and reducing the risk of viral resistance. While a number of investigators have developed single-drug nanoparticles to be combined at the time of administration,22,23 this approach may result in heterogeneity of intracellular drug concentrations and potentially harbor drug-resistant virus. Solid polymeric particles 100–600 nm in diameter containing multiple crystalline drugs have been synthesized,24 but these are much larger than anti-HIV LNPs and may not be suitable for lymphatic uptake.25 While the mechanisms of drug action in anti-HIV LNPs remain to be elucidated, cellular uptake of intact nanoparticles followed by pH- or phospholipase-dependent release of active drug likely leads to enhanced intracellular levels. This mechanism, using acid-stable drugs in combination,16 provides a concerted antiviral effect compared to drugs in free form or separate nanoparticles. Further studies are planned to evaluate the therapeutic efficacy of multidrug anti-HIV LNPs in an HIV-infected macaque model.
With daily oral HAART therapy, treatment interruption is often correlated with adverse side effects or drug abuse. Gastrointestinal side effects cause patients to skip doses,26 and patients with a history of drug abuse are three times more likely to discontinue treatment,27 leading to rapid viral rebound.1,2 The elevated and sustained plasma and intracellular drug exposures achieved with a single dose of anti-HIV LNPs indicate the feasibility of less frequent (e.g., once weekly) dosing, reducing the likelihood of treatment interruption. While dose-ranging and multiple-dose studies are needed to optimize dose and dosing frequency, our predictive model indicates that once weekly dosing with anti-HIV LNPs is feasible. Studies evaluating intracellular drug levels in lymph nodes throughout the body and lipid-particle distribution using gadolinium tracers28 are planned, but are beyond the scope of this report.
In addition to presenting multiple antiviral compounds, the anti-HIV LNP platform offers the possibility of cell and organelle targeting with pH-dependent drug release,12,29 expression of a CD4-binding peptide,30 or other surface modifications (for review, see Gunaseelan et al.31). Eliciting little or no local or systemic inflammatory reaction, these anti-HIV lipid nanoparticles hold promise for delivery of HIV drugs such as LPV, RTV, and PMPA to the sites of viral persistence in blood and lymphoid tissues.
Acknowledgments
We thank the staff of the WaNPRC for animal care and observation, as well as Michael Gough, Jason Ogle, and Chris English of the WaNPRC Research Support Group for their contributions in conducting the primate study. We thank Jake Kraft and Jenny Zhang for critical review of the manuscript. This work was supported by NIH grants AI 077390 (S1, S2, S3), RR00166, RR025014, and UL1TR000423. R.J.Y.H. is also supported by the Milo Gibaldi endowment.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Chun TW, Davey RTJ, Engel D, et al. : Re-emergence of HIV after stopping therapy. Nature 1999;401(6756):874–875 [DOI] [PubMed] [Google Scholar]
- 2.Wong JK, Gunthard HF, Havlir DV, et al. : Reduction of HIV-1 in blood and lymph nodes following potent antiretroviral therapy and the virologic correlates of treatment failure. Proc Natl Acad Sci USA 1997;94(23):12574–12579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horiike M, Iwami S, Kodama M, et al. : Lymph nodes harbor viral reservoirs that cause rebound of plasma viremia in SIV-infected macaques upon cessation of combined antiretroviral therapy. Virology 2011;423(2):107–118 [DOI] [PubMed] [Google Scholar]
- 4.Lafeuillade A, Chollet L, Hittinger G, et al. : Residual human immunodeficiency virus type 1 RNA in lymphoid tissue of patients with sustained plasma RNA of <200 copies/mL. J Infect Dis 1998;177(1):235–238 [DOI] [PubMed] [Google Scholar]
- 5.Siliciano JD, Kajdas J, Finzi D, et al. : Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med 2003;9(6):727–728 [DOI] [PubMed] [Google Scholar]
- 6.Fletcher CV, Staskus K, Wietgrefe SW, et al. : Persistent HIV-1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proc Natl Acad Sci USA 2014;111(6):2307–2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kinman L, Brodie SJ, Tsai CC, et al. : Lipid-drug association enhanced HIV-1 protease inhibitor indinavir localization in lymphoid tissues and viral load reduction: A proof of concept study in HIV-2287-infected macaques. J Acquir Immune Defic Syndr 2003;34(4):387–397 [DOI] [PubMed] [Google Scholar]
- 8.Solas C, Lafeuillade A, Halfon P, et al. : Discrepancies between protease inhibitor concentrations and viral load in reservoirs and sanctuary sites in human immunodeficiency virus-infected patients. Antimicrob Agents Chemother 2003;47(1):238–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cavert W, Notermans DW, Staskus K, et al. : Kinetics of response in lymphoid tissues to antiretroviral therapy of HIV-1 infection. Science 1997;276(5314):960–964 [DOI] [PubMed] [Google Scholar]
- 10.Pantaleo G, Graziosi C, Demarest JF, et al. : HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nature 1993;362(6418):355–358 [DOI] [PubMed] [Google Scholar]
- 11.Kinman L, Bui T, Larsen K, et al. : Optimization of lipid-indinavir complexes for localization in lymphoid tissues of HIV-infected macaques. J Acquir Immune Defic Syndr 2006;42(2):155–161 [DOI] [PubMed] [Google Scholar]
- 12.Choi SU, Bui T, and Ho RJ: pH-dependent interactions of indinavir and lipids in nanoparticles and their ability to entrap a solute. J Pharm Sci 2008;97(2):931–943 [DOI] [PubMed] [Google Scholar]
- 13.Notermans DW, Jurriaans S, de Wolf F, et al. : Decrease of HIV-1 RNA levels in lymphoid tissue and peripheral blood during treatment with ritonavir, lamivudine and zidovudine. Ritonavir/3TC/ZDV Study Group. AIDS 1998;12(2):167–173 [DOI] [PubMed] [Google Scholar]
- 14.Martinez E, Arnedo M, Giner V, et al. : Lymphoid tissue viral burden and duration of viral suppression in plasma. AIDS 2001;15(12):1477–1482 [DOI] [PubMed] [Google Scholar]
- 15.Palella FJ, Jr., Delaney KM, Moorman AC, et al. : Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med 1998;338(13):853–860 [DOI] [PubMed] [Google Scholar]
- 16.Koehn J. and Ho RJ: Novel liquid chromatography-tandem mass spectrometry method for simultaneous detection of anti-HIV drugs lopinavir, ritonavir, and tenofovir in plasma. Antimicrob Agents Chemother 2014;58(5):2675–2680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu A, Granneman GR, and Bertz RJ: Ritonavir. Clinical pharmacokinetics and interactions with other anti-HIV agents. Clin Pharmacokinet 1998;35(4):275–291 [DOI] [PubMed] [Google Scholar]
- 18.Storch CH, Theile D, Lindenmaier H, et al. : Comparison of the inhibitory activity of anti-HIV drugs on P-glycoprotein. Biochem Pharmacol 2007;73(10):1573–1581 [DOI] [PubMed] [Google Scholar]
- 19.Piliero PJ: Pharmacokinetic properties of nucleoside/nucleotide reverse transcriptase inhibitors. J Acquir Immune Defic Syndr 2004;37(Suppl 1):S2–S12 [DOI] [PubMed] [Google Scholar]
- 20.Boyum A: Isolation of mononuclear cells and granulocytes from human blood. Isolation of mononuclear cells by one centrifugation, and of granulocytes by combining centrifugation and sedimentation at 1 g. Scand J Clin Lab Invest Suppl 1968;97:77–89 [PubMed] [Google Scholar]
- 21.Alberts B, Johnson A, Lewis J, et al. : Molecular Biology of the Cell, 4 ed. Garland Science, New York, 2002 [Google Scholar]
- 22.Kuo YC. and Chung CY: Solid lipid nanoparticles comprising internal Compritol 888 ATO, tripalmitin and cacao butter for encapsulating and releasing stavudine, delavirdine and saquinavir. Colloids Surf B Biointerfaces 2011;88(2):682–690 [DOI] [PubMed] [Google Scholar]
- 23.Nowacek AS, McMillan J, Miller R, et al. : Nanoformulated antiretroviral drug combinations extend drug release and antiretroviral responses in HIV-1-infected macrophages: Implications for neuroAIDS therapeutics. J Neuroimmune Pharmacol 2010;5(4):592–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shibata A, McMullen E, Pham A, et al. : Polymeric nanoparticles containing combination antiretroviral drugs for HIV type 1 treatment. AIDS Res Hum Retroviruses 2013;29(5):746–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oussoren C, Zuidema J, Crommelin DJ, and Storm G: Lymphatic uptake and biodistribution of liposomes after subcutaneous injection. II. Influence of liposomal size, lipid composition and lipid dose. Biochim Biophys Acta 1997;1328(2):261–272 [DOI] [PubMed] [Google Scholar]
- 26.Robison LS, Westfall AO, Mugavero MJ, et al. : Short-term discontinuation of HAART regimens more common in vulnerable patient populations. AIDS Res Hum Retroviruses 2008;24(11):1347–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cicconi P, Cozzi-Lepri A, Castagna A, et al. : Insights into reasons for discontinuation according to year of starting first regimen of highly active antiretroviral therapy in a cohort of antiretroviral-naive patients. HIV Med 2010;11(2):104–113 [DOI] [PubMed] [Google Scholar]
- 28.Bui T, Stevenson J, Hoekman J, et al. : Novel Gd nanoparticles enhance vascular contrast for high-resolution magnetic resonance imaging. PLoS One 2010;5(9):e13082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Straubinger RM, Duzgunes N, and Papahadjopoulos D: pH-sensitive liposomes mediate cytoplasmic delivery of encapsulated macromolecules. FEBS Lett 1985;179(1):148–154 [DOI] [PubMed] [Google Scholar]
- 30.Endsley AN. and Ho RJ: Enhanced anti-HIV efficacy of indinavir after inclusion in CD4-targeted lipid nanoparticles. J Acquir Immune Defic Syndr 2012;61(4):417–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gunaseelan S, Gunaseelan K, Deshmukh M, et al. : Surface modifications of nanocarriers for effective intracellular delivery of anti-HIV drugs. Adv Drug Deliv Rev 2010;62(4–5):518–531 [DOI] [PMC free article] [PubMed] [Google Scholar]