

Abstract
Understanding commonalities and differences in glutamatergic synaptic signaling is essential for understanding cortical functional diversity, especially in the highly complex primate brain. Previously, we have shown that spontaneous EPSCs differed markedly in layer 3 pyramidal neurons of two specialized cortical areas in the rhesus monkey, the high-order lateral prefrontal cortex (LPFC) and the primary visual cortex (V1). Here, we used patch-clamp recordings and confocal and electron microscopy to determine whether these distinct synaptic responses are due to differences in firing rates of presynaptic neurons and/or in the features of presynaptic or postsynaptic entities. As with spontaneous EPSCs, TTX-insensitive (action potential-independent) miniature EPSCs exhibited significantly higher frequency, greater amplitude, and slower kinetics in LPFC compared with V1 neurons. Consistent with these physiological differences, LPFC neurons possessed higher densities of spines, and the mean width of large spines was greater compared with those on V1 neurons. Axospinous synapses in layers 2–3 of LPFC had larger postsynaptic density surface areas and a higher proportion of large perforated synapses compared with V1. Axonal boutons in LPFC were also larger in volume and contained ∼1.6× more vesicles than did those in V1. Further, LPFC had a higher density of AMPA GluR2 receptor labeling than V1. The properties of spines and synaptic currents of individual layer 3 pyramidal neurons measured here were significantly correlated, consistent with the idea that significantly more frequent and larger synaptic currents are likely due to more numerous, larger, and more powerful synapses in LPFC compared with V1.
Keywords: electron microscopy, GluR2, glutamate, layers 2/3 pyramidal neurons, Macaca mulatta, whole-cell patch clamp
Introduction
The primate cerebral cortex has evolved into a highly complex multifaceted structure comprised of specialized anatomically and functionally distinct areas that communicate with each other through excitatory glutamatergic synaptic pathways. These pathways emanate principally from layers 2 and 3 corticocortical pyramidal neurons that have local projections within a given area and long distance projections that target other cortical areas in a distinctive layer-specific manner (Pandya and Kuypers, 1969; for review, see Somogyi et al., 1998; Callaway, 2002; DeFelipe et al., 2002; Douglas and Martin, 2004; Barbas et al., 2013). A detailed understanding of the commonalities and differences in glutamatergic synaptic signaling is an essential prerequisite for an understanding of cortical functional diversity and the selective vulnerability of different cortical areas in disease states.
We have recently demonstrated marked differences in the functional and structural properties of layer 3 (L3) pyramidal neurons in two cortical areas in the rhesus monkey: the high-order cognitive area 46 in the lateral prefrontal cortex (LPFC) and the primary sensory area 17 in the visual cortex (V1) (Amatrudo et al., 2012). One of the most notable differences is in the properties of spontaneous glutamatergic EPSCs, which exhibit higher frequencies and amplitudes and slower decay times in LPFC than in V1 neurons. The present study was designed to unveil the mechanism(s) underlying these functionally important differences by assessing TTX-insensitive (action potential [AP]-independent) miniature EPSCs and the structural and neurochemical features of glutamatergic synapses in V1 and LPFC. Glutamatergic synaptic efficacy is determined by a variety of morphological and physiological features of the presynaptic (the axonal bouton and its vesicles) and postsynaptic (the postsynaptic density [PSD], receptors, and sites) elements. The properties of the presynaptic terminal, such as bouton size and number of synaptic vesicles, are correlated with the probability of neurotransmitter release (for review, see Stevens, 2004; Denker and Rizzoli, 2010). Postsynaptic features, such as the surface area of the PSD, the presence of a perforation on the PSD, and the volume of the targeted spine, are all correlated with the number of AMPA receptors activated during an EPSC (Desmond and Weinberg, 1998; Matsuzaki et al., 2001; Ganeshina et al., 2004; for review, see Bourne and Harris, 2008).
We characterized the detailed morphological features of glutamatergic synaptic elements in LPFC and V1 to determine whether differences in these structures could plausibly account for the observed differences in synaptic efficacy. The distribution and morphology of dendritic spines, the sites of excitatory input, were assessed on individual L3 pyramidal neurons from LPFC and V1 using high-resolution confocal microscopy, and the fine structure of excitatory synapses in layers 2–3 neuropil in these areas was compared using serial electron microscopy and 3D reconstruction. Because glutamate receptor subunit composition is another key determinant of synaptic efficacy (for review, see Isaac et al., 2007), we assessed the distribution of the GluR2 AMPA receptor subunit in the two areas. We present evidence of marked structural and functional diversity in the basic components of glutamatergic synaptic signaling between these functionally specialized and distinctive cortical areas.
Materials and Methods
Experimental subjects
A total of 13 adult rhesus monkeys (Macaca mulatta; 11.9 ± 1.5 years old; range = 5–20 years old; 5 male, 8 female) were used for the electrophysiological and morphological studies. A second cohort of three adult monkeys (ages 6–8 years old; all female) was used for ultrastructural studies. All rhesus monkeys were part of a larger program of studies examining the impact of normal aging on the brain. Monkeys were initially obtained from the Yerkes National Primate Research Center at Emory University (Atlanta). Animals were housed individually in the Laboratory Animal Science Center at Boston University School of Medicine and kept under a 12 h light/dark cycle. Both the Yerkes National Primate Research Center and the Boston University School of Medicine Laboratory Animal Science Center are fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, with animal research and maintenance conducted in strict accordance with the guidelines established by the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Electrophysiological analyses of spontaneous and miniature EPSCs of layer 3 pyramidal neurons
Monkeys were sedated with ketamine hydrochloride (10 mg/ml), then deeply anesthetized with sodium pentobarbital (to effect, 15 mg/kg, i.v.), and a craniotomy was then performed, as described previously (Amatrudo et al., 2012; Luebke and Amatrudo, 2012; Luebke et al., 2013). After a thoracotomy, animals were perfused through the ascending aorta with ice-cold Krebs-Henseleit buffer (concentrations, in mm as follows: 6.4 Na2HPO4, 1.4 Na2PO4, 137 NaCl, 2.7 KCl, 5 glucose, 0.3 CaCl2, 1 MgCl2, pH 7.4; Sigma-Aldrich). Blocks of tissue (10 mm3) from lateral prefrontal area 46 (LPFC, from the mid-caudal half of the principal sulcus) and visual cortex area 17 (V1, from the operculum of area 17) were removed from the left hemisphere, and within <3 min, were sectioned into 300-μm-thick coronal slices in ice-cold Ringer's solution (concentrations, in mm as follows: 26 NaHCO3, 124 NaCl, 2 KCl, 3 KH2PO4, 10 glucose, 1.3 MgCl2, pH 7.4; Sigma-Aldrich) with a vibrating microtome. Slices were then immediately placed into room temperature, oxygenated (95% O2, 5% CO2) Ringer's solution.
After a 1 h equilibration period, individual slices were placed into submersion-type recording chambers (Harvard Apparatus) mounted on the stages of Nikon E600 infrared-differential interference contrast microscopes (Micro Video Instruments). Slices were continuously superfused with room temperature, oxygenated Ringer's solution at a rate of 2–2.5 ml/min. Although it is known that temperature influences a number of physiological parameters recorded in vitro, it is very unlikely that identically treated LPFC and V1 slices are differentially sensitive to temperature. Thus, we conducted all physiological experiments at room temperature, which improves the viability and duration of recordings from monkey cortical slices.
Standard tight-seal, whole-cell patch-clamp recordings with simultaneous biocytin filling were obtained from L3 pyramidal cells as described previously (Chang et al., 2005; Amatrudo et al., 2012; Luebke and Amatrudo, 2012; Luebke et al., 2013). Cells were visualized under infrared-differential interference contrast optics, and electrodes were fabricated on a horizontal Flaming and Brown micropipette puller (model P-87, Sutter Instruments). Potassium methane sulfonate-based solution (concentrations, in mm as follows: 122 KCH3SO3, 2 MgCl2, 5 EGTA, 10 NaHEPES, with 1% biocytin, pH 7.4; Sigma-Aldrich) was used as internal solution in electrodes with resistances of 3–6 mΩ in the external Ringer's solution. Data were acquired using EPC-9 or EPC-10 patch-clamp amplifiers (HEKA Elektronik), using PatchMaster acquisition software (HEKA Elektronik). Access resistance was monitored throughout the duration of each experiment, and signals were low-pass filtered at 10 kHz. For cell inclusion in electrophysiological analyses, cells were required to have a resting membrane potential ≤−55 mV, stable access resistance, an AP overshoot, and the ability to fire repetitive APs in response to prolonged depolarizing current steps, as described previously (Chang et al., 2005; Amatrudo et al., 2012; Luebke and Amatrudo, 2012; Luebke et al., 2013).
Spontaneous EPSCs (sEPSCs) were recorded for 2 min at a holding potential of −80 mV. In a subset of cells, sEPSC recording was followed by recordings of miniature EPSCs (mEPSCs) for a minimum of 2 min at a holding potential of −80 mV in the continuous presence of TTX (300 nm). mEPSCs were fully blocked by the AMPA receptor antagonist CNQX (10 μm) and unaffected by bicuculline methiodide (10 μm). Analyses were performed using MiniAnalysis software (Synaptosoft), with event detection threshold set at maximum RMS noise level (5 pA). Recordings of synaptic events were assessed for frequency (Hz), amplitude (pA), and integral (area under the curve, pA/ms). The kinetics (rise and decay time constants, and half-width, ms) of EPSCs was determined from exponential fits to averaged EPSC waveforms (all events in a given cell).
Labeling and confocal imaging of individual layer 3 pyramidal neurons
Following recording with simultaneous cell filling with biocytin, slices were fixed for 2 d at 4°C in 4% PFA/0.1 m PBS, pH 7.4. Following a PBS rinse, cells were placed in 1% Triton X-100 in PBS for 2 h at room temperature and then for 2 d at 4°C in streptavidin-Alexa-488 (1:500; Invitrogen). Slices were mounted on slides and coverslipped using Prolong Gold mounting medium (Invitrogen).
For cell inclusion in morphological analyses, the criteria were as follows: an intact soma, completely filled dendritic arbors, and no cut dendrites in the proximal third of the apical dendritic arbor. Image stacks were acquired using a Zeiss 510 confocal laser-scanning microscope. Neurons labeled with Alexa-488 probe were imaged using an Argon excitation laser. For imaging entire neurons to assess whole-cell dendritic and spine topology, stacks were acquired using a 40×/1.3 NA oil-immersion objective (210 μm working distance; Plan-Apochromat, Zeiss) at a resolution of 0.1 × 0.1 × 0.2 μm per voxel. For determination of spine subtype distributions and measurement of the maximum spine head width and assessment of spines immunolabeled for GluR2, a second series of image stacks were acquired using a 100×/1.3 NA oil-immersion objective (UPlan-FL, Olympus or Plan-Apochromat, Zeiss). These 100× scans were done either at a resolution of 0.022 × 0.022 × 0.1 μm per voxel or 0.044 × 0.044 × 0.2 μm per voxel. Images were acquired from one complete basilar dendritic branch (∼100 μm in length), the middle third of the main apical trunk (∼45 μm in length), and one distal apical dendritic branch (∼140 μm in length) of each neuron. To reduce signal blurring in the z-plane, each acquired stack of images was deconvolved using AutoQuant software (Media Cybernetics). Adjacent deconvolved image stacks were then imported into Volume Integration and Alignment System software (Rodriguez et al., 2003), aligned in 3D, and integrated into a single volumetric dataset.
Assessment of spine size and distribution on individual layer 3 pyramidal neurons
Image stacks of dendritic segments obtained at 100× were used to measure the maximum widths of spine heads (widest part of spine head perpendicular to the neck). Dendritic segments were reconstructed in 3D using the 64-bit version of NeuronStudio (Rodriguez et al., 2003, 2006; Wearne et al., 2005). Dendritic spines were then manually marked on the reconstructed dendrites and automatically measured for the maximum spine head width perpendicular to the neck and maximum distance from the shaft, using the Rayburst-based algorithm (Rodriguez et al., 2008). Spines were manually classified into subtypes according to previous criteria (for review, see Luebke et al., 2010b; Amatrudo et al., 2012): spines with a head width of <0.6 μm were classified as either thin or filopodia, with thin spines having a neck length ≤3 μm and filopodia having a neck length >3 μm. Spines with a head width of >0.6 μm were classified as mushroom. Spines lacking a neck were classified as stubby. Densities of total spines and of each subtype were calculated as the number of spines per micron of dendritic length. The resulting measured spine head widths for each cell were then used to assess the size of spines. K-means cluster analysis was used to obtain a cutoff point (0.52 μm) that separates the population of spines into two size clusters: “large” and “small” spines.
To assess spine number and distribution by size across entire layer 3 pyramidal neurons, volumetric datasets of entire neurons from image stacks scanned at 40× were imported into NeuronStudio and reconstructed in 3D, as described previously (Luebke et al., 2013). Spines were marked, measured for maximum spine head width as above, and classified as either large or small. Sholl analysis using concentric spheres placed at 20 μm increments and originating at the soma (Sholl, 1953) was used to determine the number and mean maximum spine width of total, small, and large spines across different distances from the soma.
Serial electron microscopy and 3D reconstruction of synapses
The morphology and distribution of asymmetric excitatory synapses in the neuropil of layers 2–3 in V1 and LPFC were assessed using electron microscopy (EM). Monkeys used only for ultrastructural studies were perfused with 1% PFA and 1.25% glutaraldehyde in 0.1 m cacodylate or phosphate buffer, pH 7.4, as described previously (Peters et al., 2000, 2001, 2008). Upon removal, one-half of the brain was immersed in a stronger aldehyde solution, containing 2% PFA and 2.5% glutaraldehyde in the same buffer used for the perfusion for at least 1 week at 4°C. Small blocks of tissue taken from V1 and LPFC were osmicated and embedded in Araldite resin, as described previously (Peters et al., 2000, 2001, 2008).
Semithick (1 μm) sections were cut from the Araldite-embedded blocks and oriented so that the plane of section was parallel to the vertical axis of the apical dendrites, and included the entire depth of cortex from the pia to the white matter. These semithick sections were mounted on glass slides and stained with toluidine blue for light microscopic examination. Using a light microscope with a camera lucida, layers 2–3 were demarcated from ∼100 μm deep to the pia, extending to ∼300 μm for V1 and 500 μm for area 46 toward the white matter. The block was trimmed using a diamond trim tool (Diatome) and an ultramicrotome (Ultracut; Leica) to include the entire depth of layers 2–3. The average block face area for V1 was ∼150 × 300 μm, and for area 46 was 150 × 500 μm.
Approximately 80–100 serial ultrathin sections (50 nm) were then cut from each block using a diamond knife (Diatome). The ribbons of sections were collected on single slot pioloform-coated grids and counterstained with 3% uranyl acetate in water and Reynolds lead citrate (Ted Pella). Serial ultrathin sections were examined at 60 kV using a JEOL 100S transmission electron microscope with a film camera, or at 80 kV using a JEOL JEM 1011 (JEOL) with a digital camera (Gatan). From each block, 1–2 fields in the center of the sampling area in layers 2–3 and devoid of cell bodies of neurons and neuroglial cells were photographed throughout 30–60 serial sections at 6000× magnification for film or at 25,000× magnification for digital camera. For micrographs captured using film, EM negatives were developed and digitally scanned (Epson America). Serial images were aligned and analyzed using Reconstruct software, as described previously (Fiala, 2005; Medalla et al., 2007). The thickness of the imaged sections was estimated using the method of cylindrical diameters (Fiala and Harris, 2001b).
Synapses in the series were counted using stereologic methods described previously (Fiala and Harris, 2001a). Excitatory asymmetric synapses were identified based on three classic criteria (Peters et al., 1991): the presence of a dense PSD, round vesicles, and wide synaptic clefts. Asymmetric synapses were then characterized based on postsynaptic targets (axospinous or axodendritic) and the presence or absence of perforations (perforated vs nonperforated). For a subset of asymmetric synapses completed in the series, object contours of PSDs, postsynaptic spines, and presynaptic boutons were manually traced section by section and used to calculate the surface area of the PSD and the volume of spines and boutons. For serial sections photographed at high magnification, vesicles with a visible clear core were counted within every other contour (to avoid repeated counting) of each fully reconstructed axospinous bouton. The total number of vesicles per bouton was estimated as the number of vesicles counted per volume sampling fraction (ratio of the sampled volume/total bouton volume). In randomly selected boutons, the outer diameter of vesicles with a visible clear center was measured, as described previously (Schikorski and Stevens, 1997, 2001). Reconstruct was used to generate a 3D model (Virtual Reality Modeling Language), which was imported in to 3D Studio Max (2015, Autodesk) for additional rendering.
Immunofluorescence labeling of the GluR2 receptor subunit
To assess the distribution of the AMPA receptor subunit GluR2 on spines and dendrites of filled neurons, slices containing biocytin-filled neurons labeled with streptavidin-Alexa-488 were subsequently processed for GluR2 immunohistochemistry using a complementary red or far-red fluorescent probe. To assess the overall distribution of GluR2 in the layer 1 and layers 2–3 neuropil, thin 50-μm sections cut from blocks directly adjacent to blocks used for in vitro slices were processed for GluR2 single immunohistochemistry. These adjacent blocks were extracted during perfusion with cold oxygenated Krebs (see Electrophysiological methods), postfixed overnight in 4% PFA in 0.1 m PBS at 4°C, and cut into 50 μm sections using a vibrating microtome. Sections were first treated with 10 mm sodium citrate buffer (pH 8.4 at 50°C–60°C) for antigen retrieval using a variable wattage microwave (150 W for 15 min; Biowave, Ted Pella) and then incubated for 2 h in 50 mm glycine (at room temperature) to further unmask binding sites. After rinsing, sections were then preblocked in 5% normal goat serum and 5% BSA with 0.1% Triton-X (2 h for 300 μm sections and 1 h for 50 μm sections at room temperature). Sections were then incubated in either a monoclonal primary antibody against the N-terminal extracellular domain of the GluR2 subunit (1:500; anti-GluR2 mouse monoclonal, catalog # MAB397, Millipore) or a polyclonal antibody against the carboxyl terminals of the GluR2 and, to a lesser extent, the GluR3 subunits (1:500, anti-GluR2/3 rabbit polyclonal, catalog # 07-598, Millipore). Primary antibody incubation was first done using the microwave (150 W for 10 min at room temperature) to assist in tissue penetration, followed by 4 d (for 300 μm sections) or 1–2 d (for 50 μm sections) at 4°C with mild agitation. After multiple rinses over 30 min to 1 h, 300 μm slices were incubated in a goat anti-rabbit secondary F(ab′)2 IgG conjugated either to Alexa-546 (red) or Alexa-633 (far-red; 1:400; Invitrogen) using a microwave (150 W for 10 min at room temperature), followed by 3 d at 4°C. Thin 50 μm sections were processed using a tyramide signal amplification kit (PerkinElmer). These sections were first incubated in biotinylated goat anti-rabbit secondary F(ab) IgG (The Jackson Laboratory) for 3–4 h at 4°C, followed by Avidin-Biotin-HRP complex (Vector Laboratories) for 1 h at 4°C, then for 30 min in biotinylated tyramide (0.025% in PBS with 0.002% H2O2 at room temperature; PerkinElmer), followed by overnight in streptavidin-Alexa-633 (1:500, at 4°C; Invitrogen). All antibodies were diluted in 1% normal goat serum, 0.2% acetylated BSA (BSA-c, Aurion), and 0.1% Triton-X. Sections were then mounted and coverslipped with Prolong anti-fade medium (Invitrogen). Control experiments wherein the primary antibody was omitted or preabsorbed with the control peptide were conducted, and no immunolabeling was detected.
Confocal imaging and assessment of GluR2+ spines on filled layer 3 neurons
Laser scanning confocal images of filled dendritic segments together with GluR2 receptors were acquired at 100× (1.3 NA, oil-immersion), using a Zeiss-510 confocal microscope (0.044 × 0.044 × 0.2 μm per voxel resolution). Under the multitrack mode, filled dendrites were imaged using an Argon laser, and GluR2 receptors were imaged with either a Helium-Neon 543 nm laser (red) or 633 nm laser (far-red). Images from two channels (green and red or green and far-red) were deconvolved separately using AutoQuant software. The channels were merged as an RGB stack in ImageJ (http://imagej.nih.gov/ij/; Rasband, WS, 1997–2014, National Institutes of Health, Bethesda, Maryland), and colocalization of immunolabel with spines was assessed using a manually set threshold for each channel, as described previously (Medalla and Barbas, 2012). Spines with colocalized points were marked and counted using either NeuronStudio or Neurolucida (Microbrightfield).
Confocal imaging and assessment of GluR2 receptors in layers 1–3 neuropil
In addition to assessing GluR2 localization on spines of filled neurons, the overall distribution GluR2 subunits was assessed in layers 1 and 2–3 neuropil. For this analysis, either the same 300 μm slices containing filled neurons or thin 50 μm sections cut from blocks directly adjacent to recording sites were immunohistochemally processed for GluR2 label with either red or far-red fluorescent probes, as described above. Confocal stacks were captured at z-depths between 2 and 10 μm in each slice using either a Zeiss-510 at 100× for 300 μm slices (as described above) or using a Zeiss-710 confocal microscope at 63× (1.3 NA, oil-immersion, Plan-Apochromat, Zeiss; 0.07 × 0.07 × 0.2 μm per voxel resolution) for 50 μm sections. Image stacks were then deconvolved. In each section of the image stack, the total area with fluorescence label above a set intensity threshold was quantified using the particle analysis function of ImageJ. The average area of fluorescence label across all sections in each stack was used as a density measure of GluR2 label. One to two sites each in layers 1, 2, and 3 were captured from each area per case (n = 5).
GluR2 pre-embedding immunohistochemistry and assessment for electron microscopy
To study the distribution of GluR2 at the synaptic level, thin 50 μm sections were processed for pre-embedding GluR2 immunohistochemistry using gold-conjugated secondary antibodies, as described previously (Medalla and Barbas, 2012). Briefly, sections were incubated in 10 mm sodium citrate, pH 8.5, buffer for antigen retrieval using the microwave (45°C–50°C, 150 W for 15 min), and then in 50 mm glycine for 1 h. Sections were preblocked and incubated in primary antibody against GluR2 as described above, except with 0.025% Triton-X for all incubation steps. Sections were then incubated in gold-conjugated secondary goat anti-mouse or goat anti-rabbit F(ab′)2 IgG (1:100, Aurion), first in the microwave (10 min at 150 W) followed by overnight at 4°C. Sections were then silver enhanced for ∼45 min (at 20°C; R-Gent SE-EM Silver Enhancement Reagent, Aurion). Tissue was then postfixed in 4% PFA and 6% glutaraldehyde in 0.1 m PB using the microwave until sample temperature reached >30°C (2–5 min at 150 or 250 W). After rinsing in PB, sections were osmicated, dehydrated in ascending series of alcohols with en bloc uranyl acetate staining in 70% ethanol, infiltrated with propylene oxide, and finally flat-embedded in Araldite resin using Aclar plastic (Ted Pella), as described previously (Medalla et al., 2007; Medalla and Barbas, 2009, 2010).
Small blocks of tissue from layers 2–3, extending 300 μm for V1 and 500 μm for LPFC from the top of layer 2, were cut from Aclar flat-embedded sections and then re-embedded in blocks for tissue sectioning as described above. A series of 10 sections per block was cut ∼2 μm from the block surface. Two to three random fields were imaged throughout the 10-section series and GluR2+ asymmetric synapses were quantified stereologically as described above.
Statistics
Differences between groups were assessed statistically using Student's t test or repeated-measures ANOVA and Tukey's or Fisher's LSD post hoc tests. Analyses of absolute, relative (n in each bin/total n), and cumulative frequency distribution histograms of mEPSC and synaptic properties were conducted. The coefficient of variation (CV) and skew were calculated for frequency distribution histograms of mEPSC amplitudes and integrals and for synapse size measurements (PSD area, spine volume, bouton volume). The CV was defined as σ/μ where σ is the sample SD and μ the sample mean. Skew is defined as Σ ((xi − μ)/σ)3/N, where the sum is over the N data points xi. To test for normality, the distributions were fitted to a Gaussian distribution, and a Kolmogorov–Smirnov (K-S) test was used to compare two cumulative distribution histograms. Relationships between variables were examined with linear regression analyses using Pearson's correlation. All values were reported as mean ± SEM from multiple animals.
Results
Significantly more frequent and larger EPSCs in L3 pyramidal neurons in LPFC compared with V1
Recordings of sEPSCs and mEPSCs were obtained from LPFC and V1 L3 pyramidal neurons to compare the properties of quantal glutamatergic synaptic neurotransmission (Fig. 1). The AP-independent mEPSCs recorded from V1 and LPFC pyramidal neurons differed significantly with regard to frequency and size, consistent in direction with differences in sEPSCs (AP-dependent plus AP-independent) shown here (Fig. 1A,C) and in our previous study (Amatrudo et al., 2012). Spontaneous EPSCs in LPFC neurons (n = 8, from 5 cases) exhibited significantly higher frequencies (t test, p = 0.0003), larger amplitudes (p = 0.002), slower rise times (p = 0.029), and longer half-widths (p = 0.018) than in V1 neurons (n = 15, from 8 cases; Fig. 1C). Application of TTX to block AP firing significantly reduced the frequency of synaptic currents in both LPFC (t test, p = 0.04) and V1 neurons (p = 0.015) but did not significantly alter their size and kinetics (p > 0.05). Consistent with findings for sEPSCs, the mean frequency of mEPSCs was 5.3 times higher in LPFC neurons (n = 9, from 2 cases) than in V1 neurons (n = 11, from 4 cases; 1.54 ± 0.4 vs 0.3 ± 0.04 Hz; t test, p = 0.001; Fig. 1A,C). The mean amplitude of mEPSCs did not differ significantly between the two areas (t test, p = 0.07; Fig. 1C). Although the mean rise time of mEPSCs did not differ (t test, p = 0.13), their decay time was significantly slower (p = 0.001) and half-widths significantly greater in LPFC neurons (p = 0.00009; Fig. 1B,C). The mean integral of mEPSCs, a measure that correlates with both amplitude and decay time, was more than twice as high in LPFC than in V1 neurons (51 ± 9 vs 23.5 ± 2.1 pA/ms; t test, p = 0.003; Fig. 1B,C).
Figure 1.
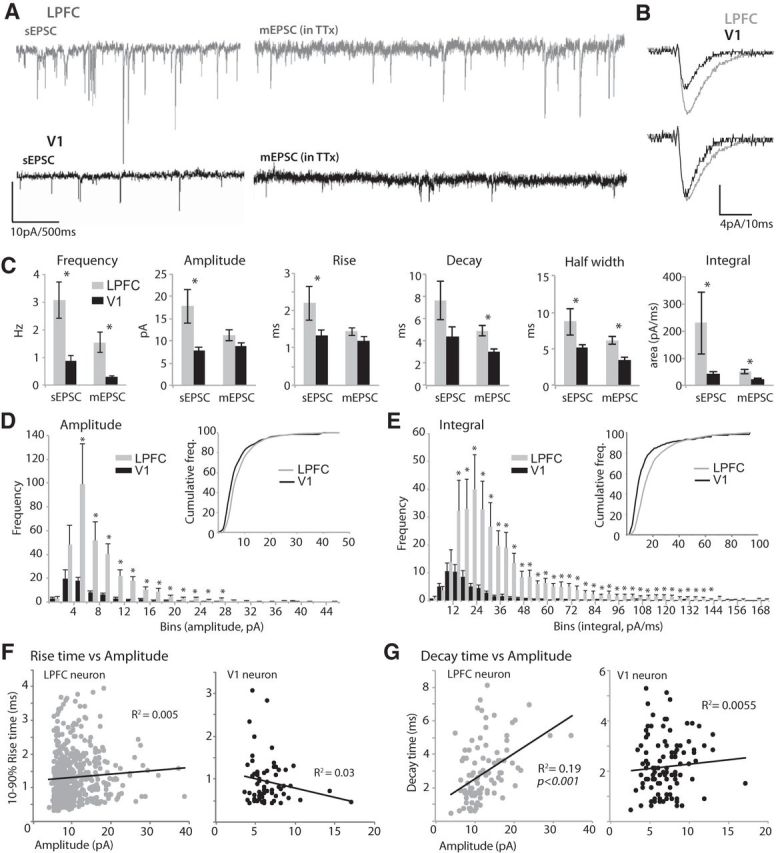
Distinct properties of EPSCs in LPFC and V1 neurons. A, Representative traces of sEPSCs (left) and mEPSCs (in TTX, right) recorded from LPFC and V1 L3 neurons held at −80 mV. B, Top, Averaged waveforms of mEPSCs from representative LPFC and V1 neurons. Bottom, Superimposed mEPSC waveforms after normalizing to peak amplitude. Note the longer decay time of mEPSCs of LPFC neurons (light gray) compared with V1 neurons (black). C, Bar graphs comparing mean sEPSC and mEPSC frequency, amplitude, kinetics (rise time, decay time, half-width), and integral (area under the curve) in LPFC versus V1 neurons. *p < 0.02. D, Frequency distribution histogram of mEPSC amplitudes. *p < 0.04. Inset, Cumulative distribution. E, Frequency distribution histogram of mEPSC integrals. *p < 0.05. Inset, Cumulative distribution. F, Plots of 10%–90% rise time versus amplitude of mEPSCs for representative LPFC and V1 neurons. Note the lack of relationship between the variables. G, Plots of decay time versus amplitude of mEPSCs for representative LPFC and V1 neurons. Note the positive correlation between decay time and amplitude in the LPFC neuron, but no significant relationship in the V1 neuron. Error bars indicate SEM.
Amplitude (Fig. 1D) and integral (Fig. 1E) distribution histograms of mEPSCs show that, whereas events with small amplitudes (<4 pA) and integrals (<12 pA/ms) did not differ in frequency, those with amplitudes >4 pA and integrals ≥12 pA/ms were significantly higher in frequency in LPFC compared with V1 L3 neurons (t test for each comparison, p < 0.02). Cumulative frequency distribution histograms of mEPSC amplitudes and integrals showed a significant rightward shift of the LPFC distribution relative to V1 (Fig. 1D,E, insets; K-S test, p < 0.0001).
For mEPSC amplitude and integral distribution histograms, we analyzed the skewness to assess the symmetry of the distribution, and the CV, a measure of spread or variation from the mean (Bekkers and Clements, 1999). The distribution of mEPSC amplitudes and integrals in both LPFC and V1 did not fit a Gaussian function and had large positive skews. However, LPFC neurons had a significantly larger average positive skew, with a longer right hand tail (amplitude skew = 2.4 ± 0.3, integral skew = 5.8 ± 1.6) compared with that for mEPSCs in V1 neurons (amplitude skew = 1.1 ± 0.3, integral skew = 2.1 ± 0.3; Fig. 1D,E, t test, p < 0.04). Amplitude and integral CV of mEPSC distributions were large for both LPFC (amplitude CV = 0.46 ± 0.03; integral CV = 1.3 ± 0.3) and V1 (amplitude CV = 0.38 ± 0.04; integral CV = 0.83 ± 0.1), consistent with the high variability in presumed quantal events in these two cortical areas (see Bekkers and Clements, 1999).
Because LPFC neurons have much larger and more complex dendritic arbors than V1 neurons (Amatrudo et al., 2012), we sought to determine whether dendritic filtering of synaptic events differed in the two populations. Previous studies have shown that, if substantial dendritic filtering is present, there is an inverse relationship between rise time and amplitude (Bekkers and Stevens, 1996; Bekkers and Clements, 1999). We compared these quantities in our data (Fig. 1F). No such relationship was observed in either LPFC or V1 neurons (p > 0.1), likely because the distribution of mEPSC amplitudes and rise times were highly variable, as has been shown in rodent dentate granule and CA1 pyramidal neurons (Bekkers and Stevens, 1996; Bekkers and Clements, 1999). However, consistent with the difference in mean mEPSC integral between the two areas, a significant linear correlation between mEPSC decay time and amplitude was observed in all LPFC neurons (R2 = 0.32–0.57, p < 0.001) but none of the V1 neurons (p > 0.1; Fig. 1G).
Significantly more numerous and larger dendritic spines on LPFC compared to V1 L3 pyramidal neurons
The distribution and sizes of dendritic spines, the major sites of excitatory synaptic inputs, were assessed in LPFC (n = 10, from 4 cases) and V1 (n = 8, from 3 cases) L3 neurons using high-resolution confocal microscopy (Fig. 2A). LPFC neurons had a higher density of spines in both apical and basal dendrites compared to V1 neurons, confirming our previous report (Amatrudo et al., 2012). This difference was principally due to a significantly higher density of thin spines in LPFC than in V1 neurons in both apical and basal arbors (Fig. 2B). In both LPFC and V1 neurons, thin spines comprised the majority (∼60%) of the total spine population, mushroom spines comprised ∼22%, stubby comprised ∼17%, and filopodia spines were rare (∼2%).
Figure 2.
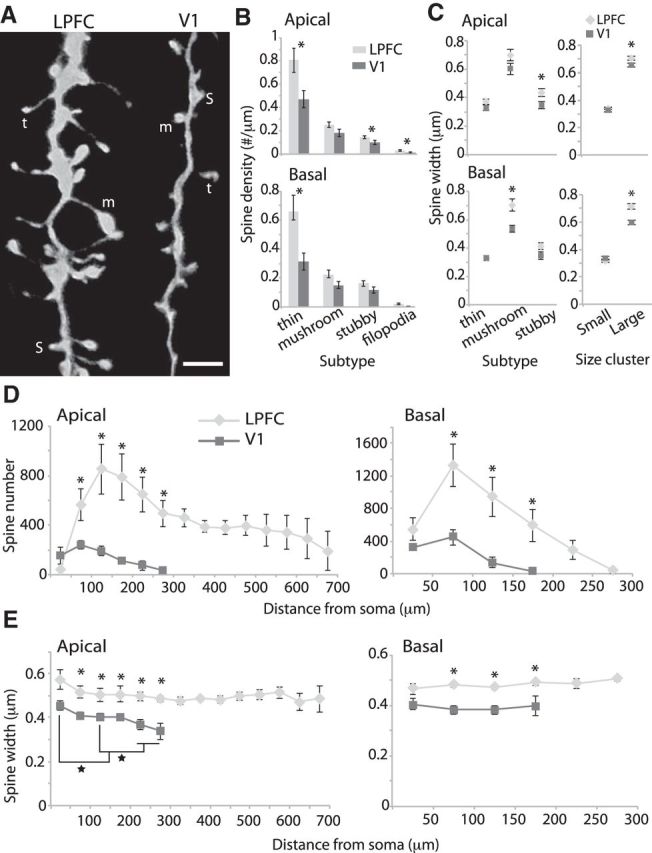
Spine distribution and size in LPFC and V1 pyramidal neurons. A, Maximum xy-projection of 100× confocal image stacks of distal apical branches of LPFC and V1 neurons showing examples of spine subtypes: t, thin; m, mushroom; s, stubby. Scale bar, 2 μm B, Comparison of spine density by subtype. *p < 0.03. C, Maximum widths of spine heads in apical (top) and basal (bottom) dendrites of LPFC versus V1 neurons. Left, Mean spine head widths of distinct spine subtypes. *p < 0.03. Right, Mean spine head widths of small (<0.52 μm in width) and large (≥0.52 μm; *p < 0.02) spines. D, Sholl analysis showing mean spine number (*p < 0.04) and; E, mean maximum spine head widths (*p < 0.04) as a function of distance from the soma across the entire apical and basal dendritic arbors of LPFC and V1 neurons. Brackets represent comparisons between bins (★p ≪ 0.0001). Error bars indicate SEM.
To assess the relative sizes of spines, we measured the maximum widths of spine heads, perpendicular to the neck, in dendritic segments sampled from apical and basal dendritic arbors of LPFC and V1 neurons, imaged at high magnification (100×). The two populations of neurons did not differ in thin spine head widths, but LPFC neurons had larger spine widths than V1 neurons for stubby spines in apical arbors (p = 0.028) and mushroom spines in basal arbors (p = 0.007; Fig. 2C, left). Further, K-means cluster analysis of spine head widths was performed to determine a numerical cutoff that separates the total population into small (<0.52 μm) and large (≥0.52 μm) spines. This method simplifies the dataset to objectively assess how spine size differed between the two areas independent of spine subtype. The mean head width of large spines was significantly greater in LPFC compared to V1 neurons, in both apical (p = 0.017) and basal arbors (p = 0.0006; Fig. 2C, right).
The spatial distribution of spines and the head width of spines across dendrites of entire neurons (scanned at 40×) are shown in Figure 2D, E. The overall spine distribution pattern was similar for LPFC and V1 neurons, peaking in number at ∼100–150 μm from the soma and gradually declining distally (Fig. 2D). However, the mean spine number was higher in LPFC than in V1 L3 pyramidal neurons at all distances from the soma in both apical and basal arbors (p = 0.007 for apical and p = 0.01 for basal arbors, repeated-measures ANOVA; Fig. 2D, p < 0.04, t test for each Sholl ring), with the exception of the first 50 μm, which is sparsely spiny. Moreover, mean spine widths were significantly greater in LPFC than in V1 neurons at all distances >50 μm from the soma in both apical and basal arbors (p = 0.01 for apical, p = 0.002 for basal, repeated-measures ANOVA; Fig. 2E, p < 0.05, t test for each Sholl ring).
Figure 3 shows the relative distribution of large versus small spines as a function of distance from the soma, across entire LPFC and V1 L3 pyramidal neurons. The population of large and small spines had similar distribution profiles to the total spine population (Fig. 2D): the numbers of both large and small spines peaked at ∼100–150 μm from the soma and gradually declined distally (Fig. 3B,C, top). Large and small spines were both significantly higher in number in LPFC than V1 neurons across the entire apical and basal arbors (p < 0.02 for apical, p < 0.01 for basal, repeated-measures ANOVA; Fig. 3B,C, top, p < 0.04, t test for each Sholl ring). This was due to LPFC neurons having an overall higher density of total spines and greater dendritic extent (Figs. 2B and 3A). Thus, to assess the relative distribution of large and small spines in the two neuronal populations, we plotted the percentage of large spines in each Sholl ring (Fig. 3B,C, bottom). LPFC neurons displayed a significantly higher proportion of large spines (and thus concomitantly lower proportion of small spines) than V1 neurons at all distances from the soma (p = 0.03 apical, p = 0.003 basal, repeated-measures ANOVA; Fig. 3B,C, bottom, p < 0.05, t test for each Sholl ring), with the exception of the sparsely spinous proximal 50 μm of the apical and basal arbors.
Figure 3.

Distribution of large and small spines across entire LPFC and V1 neurons. A, Maximum xy-projections of tiled 40× confocal image stacks showing small (light blue) and large (dark blue) spines marked across entire dendritic arbors of LPFC and V1 neurons. Scale bar, 100 μm. B, C, Sholl analysis comparing mean number of small and large spines (top: *p < 0.04) and the relative proportion of large spines (bottom: *p < 0.05) across apical (B) and basal (C) arbors of LPFC versus V1 neurons. Brackets represent comparisons between bins (★p < 0.05). Error bars indicate SEM.
Interestingly, in the apical arbors, the mean spine widths and the proportion of large spines at distinct distances from the soma changed differentially in LPFC and V1 L3 neurons. In LPFC apical arbors, mean spine width (p = 0.95, one-way ANOVA; Fig. 2E, light gray) and proportion of large spines (p = 0.99, Fig. 3B, bottom) did not change with distance from the soma. In contrast, in V1 apical arbors, there was a significant progressive decline in spine width (Fig. 2E, left, dark gray, p ≪ 0.0001) and in proportion of large spines (Fig. 3B, bottom, solid line, p < 0.05) from proximal to distal distances from the soma. Pairwise comparisons (ANOVA, post hoc Fisher's LSD, Figs. 2E and 3B, brackets) showed that adjacent Sholl distances in V1 apical arbors had similar spine size (p > 0.05), whereas distant Sholl distances were significantly different such that spine size became progressively smaller from proximal (>50 μm from the soma), to middle (100–200 μm), to distal (250–300 μm) Sholl distances from the soma (all significant differences, p < 0.05; Figs. 2E, left, dark gray and 3B, bottom, solid line).
Correlations between physiological and morphological synaptic properties in L3 pyramidal neurons
To assess potential structure–function relationships, we performed correlation analyses between physiological and morphological synaptic features in a subset of L3 neurons for which stringent criteria for both types of data were met. In these LPFC and V1 L3 neurons, sEPSC frequency strongly correlated with mean spine density (Pearson's r = 0.75, p < 0.01; Fig. 4A, left), but not with mean spine width and the proportion of large spines (Fig. 4A, middle, right). Spontaneous ESPC amplitude (Fig. 4B), decay (Fig. 4C), and integral (data not shown) each significantly correlated (p < 0.05) with spine density (vs amplitude, r = 0.73; vs decay, r = 0.61; vs integral, r = 0.72), spine width (vs amplitude, r = 0.56; vs decay, r = 0.61; vs integral, r = 0.61), and proportion of large spines (vs amplitude, r = 0.61; vs decay, r = 0.64; vs integral, r = 0.62). Because of the lower n of neurons for which both mEPSC and spine data are available, we do not have the statistical power (degrees of freedom) to make meaningful conclusions about correlations between structural properties and mEPSC properties. Nevertheless, we found that morphological estimates of spine density and size strongly correlated with physiological measurements of frequency and size of spontaneous EPSCs in L3 pyramidal neurons.
Figure 4.

Correlation between properties of spines and synaptic events in LPFC and V1 neurons. Correlation analyses show linear relationships between structural features of spines (x-axes) and physiological features of spontaneous EPSCs (y-axes) in individual LPFC (n = 5) and V1 (n = 8) L3 neurons. A, Spontaneous EPSC frequency was significantly correlated with mean spine density (left: p < 0.01), but not with mean spine width (middle) and the proportion of large spines (right) in individual L3 pyramidal neurons. Spontaneous EPSC amplitude (B) and decay (C) were both significantly correlated with mean spine density (left), mean spine width (middle), and the proportion of large spines (right: p < 0.05) in individual L3 pyramidal neurons.
Density and location of asymmetric synapses in layers 2–3 neuropil of LPFC versus V1
Serial EM demonstrated that asymmetric (excitatory) synapses comprised the majority of synapses in both areas (88 ± 3% for LPFC, 85 ± 5% for V1), with the remaining being symmetric (inhibitory) synapses (Fig. 5A–C). The relative proportions and numerical density of asymmetric and symmetric synapses did not differ between the two cortical areas (Fig. 5C; Table 1). We further classified asymmetric synapses based on their postsynaptic targets, with the majority (81 ± 2% for LPFC; 75 ± 3% for V1) being axospinous, and the rest axodendritic, and the relative proportions of these did not differ between LPFC and V1 (Fig. 5C). Axospinous synapses in LPFC possessed significantly larger PSD surface areas (0.12 ± 0.007 μm2) compared to V1 (0.08 ± 0.01 μm2; t test, p = 0.03; Fig. 5D). This size difference is consistent with the finding of larger spines in LPFC versus V1 L3 pyramidal neurons shown using confocal microscopy.
Figure 5.
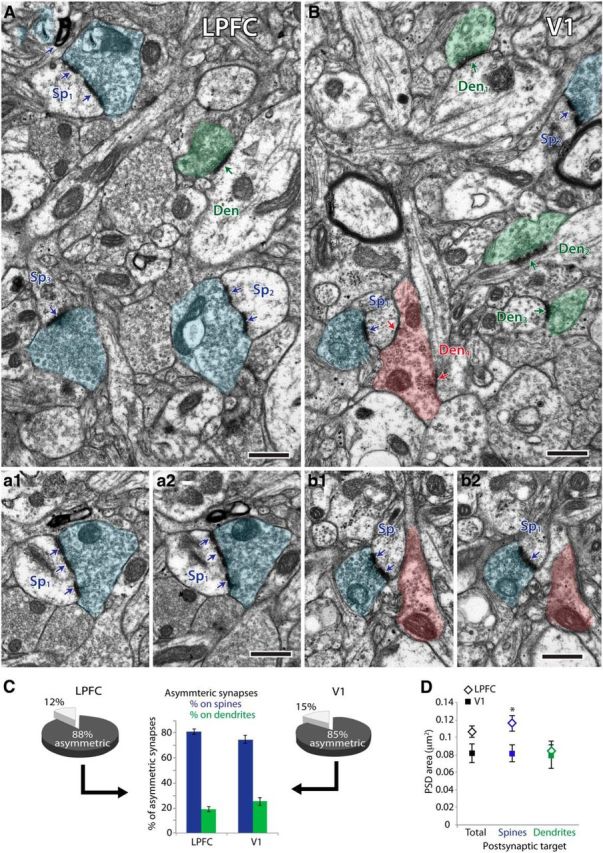
Excitatory synapses in layer 2–3 neuropil of LPFC and V1. A, B, Electron micrographs of layers 2–3 neuropil showing examples of synapses (arrows) and their presynaptic boutons (shaded structures) and postsynaptic spines (Sp) and dendrites (Den). A, Electron micrograph of LPFC neuropil showing three boutons (blue) each forming an asymmetric (excitatory) synapse with a spine (Sp1, Sp2, Sp3) and one bouton (green) forming an asymmetric synapse on a dendrite (Den). a1, a2, Serial images through one spine (Sp1) receiving one perforated asymmetric synapse. B, Electron micrograph of V1 neuropil showing two boutons (blue) each forming an asymmetric synapse with a spine (Sp1, Sp2), three boutons (shaded green) each forming an asymmetric synapse on a dendrite (Den1, Den2, Den3), and one bouton (shaded red) forming two symmetric (inhibitory) synapses: one on a spine (Sp1) and one on a dendrite (Den4). b1, b2, Serial images through one spine (Sp1) receiving one perforated asymmetric synapse and one symmetric synapse. Scale bar, 0.5 μm. C, Pie charts show proportions of asymmetric and symmetric synapses in layers 2–3 neuropil of LPFC and V1, stereologically counted using 3D serial electron microscopy. Middle bar graph shows proportions of asymmetric synapses formed on spines (blue) and dendrites (green). D, Surface area of PSDs of total asymmetric synapses, and the subpopulation formed on spines and dendrites. Asymmetric synapses formed on spines had significantly larger PSD areas in LPFC than in V1. *p = 0.03. Error bars indicate SEM.
Table 1.
Synapse density in layers 2–3 neuropil of LPFC and V1
| LPFC (Nv × 106 /mm3) | V1 (Nv × 106 /mm3) | p | |
|---|---|---|---|
| Asymmetric | 447.51 ± 46 | 484.58 ± 56 | 0.58 |
| Axospinous | 364.68 ± 46 | 365.27 ± 54 | 0.99 |
| Axodendritic | 83.52 ± 2 | 119.31 ± 10 | 0.02* |
| Symmetric | 63.03 ± 19 | 95.08 ± 29 | 0.34 |
| Axospinous | 11.67 ± 7 | 32.77 ± 7 | 0.039* |
| Axodendritic | 51.35 ± 13 | 62.31 ± 24 | 0.66 |
* Significant.
Significantly larger and more abundant perforated synapses in LPFC versus V1 neuropil
We reconstructed in 3D a subset of asymmetric axospinous synapses (including the postsynaptic spines) in layers 2–3 neuropil of LPFC (n = 431 synapses, 347 spines) and V1 (n = 331 synapses, 310 spines) from 4 subjects (Figs. 6 and 7). In addition to quantifying the surface area of the PSD, we assessed the presence of a perforation on the PSD and the volume of the targeted spine, both of which have previously been positively correlated with receptor density and synaptic efficacy (for review, see Bourne and Harris, 2008). Consistent both with the larger head diameter of spines found with confocal data (Fig. 2) and with the larger mean PSD surface area of asymmetric axospinous synapses in LPFC (Fig. 5), the mean volume of reconstructed spines was significantly larger in layers 2–3 of LPFC (0.102 ± 0.004 μm3) compared with V1 (0.066 ± 0.008 μm3; t test, p = 0.008). This was due specifically to differences in the subset of axospinous synapses that were perforated (Fig. 6, dark blue). There was a significantly higher proportion of perforated axospinous synapses in LPFC (35 ± 5%) than in V1 (20 ± 7%, p = 0.04, Fig. 6A,C). Moreover, these perforated axospinous synapses were ∼2 times larger in LPFC compared with V1, with significantly greater PSD surface areas (p = 0.005, Fig. 6B, dark blue, D, left) and larger spine volumes (p = 0.0004, Fig. 6D, right). In contrast, the nonperforated synapses (p = 0.92) and their targeted spines (p = 0.12) did not differ in size between the two areas (Fig. 6, light blue).
Figure 6.
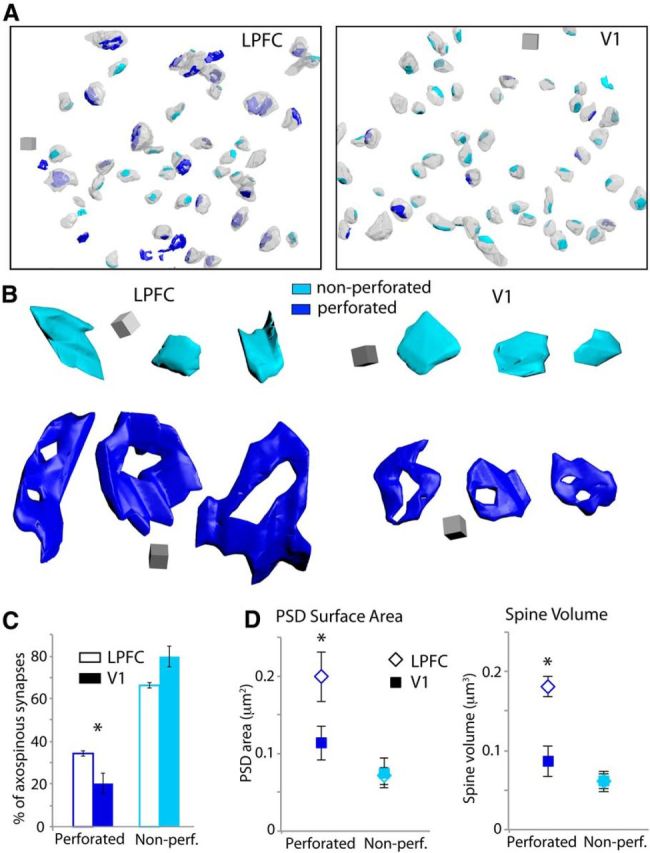
Perforated and nonperforated asymmetric axospinous synapses in LPFC and V1. A, 3D reconstructions of perforated (dark blue) and nonperforated (light blue) synapses and their associated spines (translucent light gray) in a volume of layers 2–3 neuropil in LPFC (left) and V1 (right). Scale cube, 0.5 μm3. B, 3D-reconstructed synapses in LPFC (left) and V1 (right). Nonperforated synapses (light blue) were similar in size between the two areas, but perforated synapses (dark blue) were significantly larger in surface area in LPFC than in V1. Scale cube, 0.1 μm3. C, Proportion of perforated and nonperforated axospinous synapses in LPFC and V1. *p = 0.04. D, Mean PSD surface area (left: *p = 0.005) and spine volume (right: *p = 0.0004) of perforated and nonperforated axospinous synapses in LPFC and V1. Error bars indicate SEM.
Figure 7.
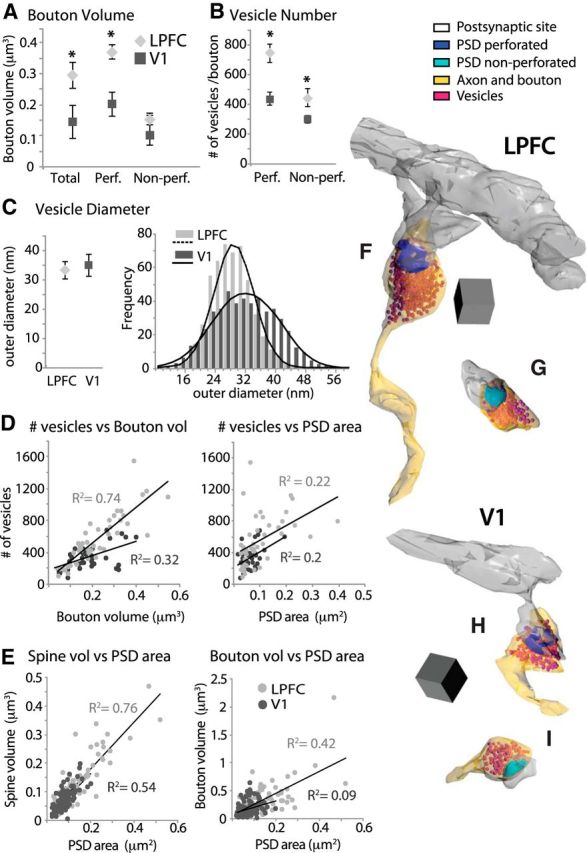
Relationship between presynaptic and postsynaptic features of synapses in LPFC and V1. A, Mean volume of boutons forming total, perforated, and nonperforated asymmetric axospinous synapses in layers 2–3 neuropil in LPFC and V1. *p < 0.05. B, Mean number of vesicles in boutons forming perforated and nonperforated synapses. *p < 0.02. C, Left, Mean outer diameters of vesicles measured in a subset of boutons in LPFC and V1 (n ∼ 500 vesicles per case). Right, Frequency distribution of vesicle diameters fitted to a Gaussian distribution. D, Linear correlation of number of synaptic vesicles with both bouton volume (p < 0.001) and PSD area (p < 0.01). E, Linear correlation of PSD surface area with both spine volume and bouton volume (p < 0.01). F–I, 3D reconstructions of presynaptic (boutons with vesicles) and postsynaptic (PSD and dendritic spines) elements of asymmetric synapses. Examples of boutons in LPFC (F, G) and in V1 (H, I) forming perforated (dark blue) and nonperforated (light blue) axospinous synapses. Scale cube, 0.05 μm3.
Significantly larger boutons containing more abundant vesicles in LPFC versus V1 neuropil
The size and morphology of the presynaptic elements of asymmetric axospinous synapses in LPFC and V1 were then studied from a subset of axon terminals (boutons) reconstructed in 3D from the same subjects (n = 87 boutons in LPFC, n = 117 in V1, from 3 animals; Fig. 7). Consistent with the differences in synapse and spine size shown above, the mean volume of axonal boutons in layers 2–3 of LPFC was significantly larger than those of V1 (t test, p = 0.05), specifically due to the population of boutons forming perforated synapses (p = 0.01; Fig. 7A). The population of boutons forming nonperforated synapses had comparable volumes across the two cortical areas (p = 0.55).
The volume of axonal boutons has been previously shown to be correlated with the number of synaptic vesicles (e.g. Germuska et al., 2006; Zikopoulos and Barbas, 2007), which is in turn correlated with the probability of neurotransmitter release during synaptic events (Tong and Jahr, 1994; Murthy et al., 1997, 2001; Schikorski and Stevens, 2001; Li et al., 2005). Here we measured the two-dimensional outer diameters of vesicles and counted the mean number of vesicles per bouton from a series of high-magnification EM images of LPFC versus V1 neuropil. The two areas were markedly different in the average number of vesicles per bouton. In the LPFC, there were ∼1.6× more vesicles per bouton than there were in V1 boutons (p = 0.0002; 555 ± 48 vs 337 ± 23 vesicles/bouton). This difference was significant for both the population of boutons forming perforated (p = 0.0008) and nonperforated synapses (p = 0.02) but was more prominent for boutons forming perforated synapses (Fig. 7B,F–I). The number of synaptic vesicles in boutons forming perforated synapses in LPFC (746 ± 62 vesicles/bouton) was almost twice as high than in V1 boutons (438 ± 44 vesicles/bouton; p = 0.0008). The mean outer diameter of synaptic vesicles was similar in the two areas (p > 0.05, Fig. 7C, left; V1 = 35 ± 4 nm; LPFC = 33 ± 3 nm) and was comparable to previous measurements in hippocampal and medial prefrontal synapses in the rat (Harris and Sultan, 1995; Schikorski and Stevens, 1997; Nava et al., 2014). The population frequency distribution histograms of vesicle diameters in the two areas both fit a Gaussian function (Fig. 7C, right; p < 0.01), consistent with those seen in glutamatergic synapses in other brain areas (Schikorski and Stevens, 1997). However, V1 exhibited a broader distribution with a significant shift toward higher frequencies of large vesicles than LPFC (K-S test, p < 0.0001).
Correlation between presynaptic and postsynaptic ultrastructural features in LPFC versus V1
There was a linear relationship between synapse size and the size of presynaptic and postsynaptic elements in LPFC and V1, consistent with findings in other areas of the monkey (e.g., Medalla et al., 2007; Medalla and Barbas, 2009, 2010; Timbie and Barbas, 2014) and rodent cortex (e.g., Harris et al., 1992; Rollenhagen et al., 2014; for review, see Rollenhagen and Lübke, 2006; Bourne and Harris, 2008). Linear regression analysis showed a positive correlation between bouton volume and number of synaptic vesicles in layers 2–3 of both cortical areas (Fig. 7D, left), but this relationship was stronger in LPFC (Pearson's r = 0.86, p < 0.001) than in V1 (r = 0.55, p < 0.001) where we noted some large boutons that contained relatively sparse vesicles (data not shown). The number of synaptic vesicles was also significantly correlated with PSD surface area of the synapse in LPFC (r = 0.47, p < 0.01) and V1 (r = 0.45, p < 0.01; Fig. 7D, right), indicating a relationship between vesicle pool and synapse size of axospinous asymmetric synapses. Further, a strong linear correlation of PSD surface area with the volume of the postsynaptic spine was found in both LPFC (r = 0.87, p < 0.001) and V1 (r = 0.73, p < 0.001; Fig. 7E, left). The synapse PSD surface area was also correlated with the volume of the presynaptic bouton, a relationship that was stronger in LPFC (r = 0.81, p < 0.001) than in V1 (r = 0.3, p < 0.001; Fig. 7E, right).
Figure 8 shows mean population distribution histograms of presynaptic and postsynaptic features of axospinous synapses, wherein relative frequency (n in each bin/total n) was calculated to normalize for the differences in total number of synapses counted for each case. V1 showed higher relative frequencies of small synapses (PSD area 0.06–0.08 μm2; Fig. 8A; p < 0.05) and spines (volumes between 0.04 and 0.1 μm3), which was evident in the significant leftward shift of cumulative frequency histograms, compared with LPFC (Fig. 8A,B, insets; K-S test, p < 0.0001). The population distribution of bouton volumes did not differ significantly between LPFC and V1 (Fig. 8C; K-S test, p = 0.32), but large boutons >0.7 μm3 were evident in LPFC but not in V1.
Figure 8.
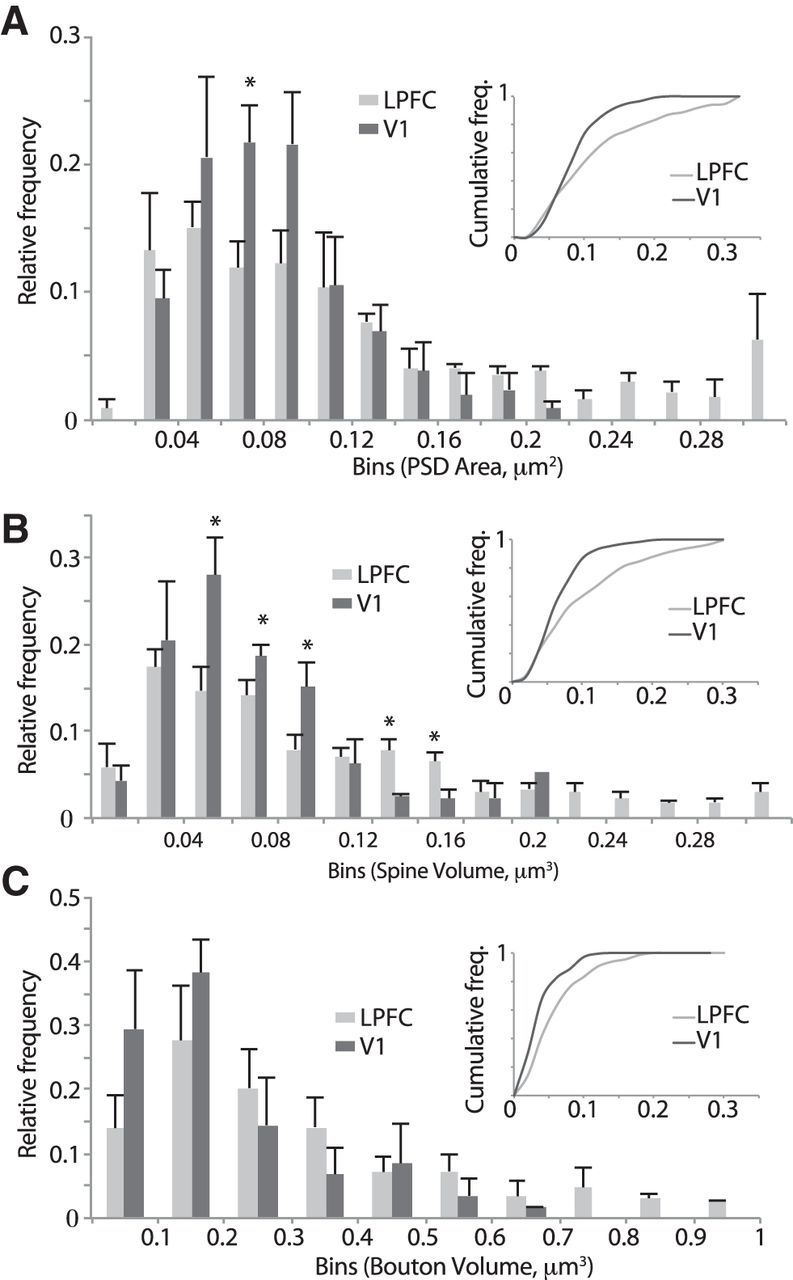
Population distribution histograms of presynaptic and postsynaptic features of synapses in LPFC and V1. Relative and cumulative frequency distribution histograms of the following: A, PSD areas. B, Spine volumes. C, Bouton volumes of asymmetric axospinous synapses in layers 2–3 neuropil of LPFC and V1. *p < 0.05. Note the more positively skewed frequency distribution and rightward shift of the cumulative distribution in LPFC versus V1. Error bars indicate SEM.
Distribution of GluR2 subunit containing AMPA receptors in LPFC versus V1
The presence or absence of the GluR2 subunit specifically leads to distinctive conductance states of the AMPA receptor ionophore complex (for review, see Isaac et al., 2007). Thus, we sought to determine whether differences in synaptic response properties in the two brain areas could potentially be accounted for by differences in the expression of postsynaptic GluR2 subunit-containing glutamate receptors. This was done using immunohistochemical GluR2 labeling and localization at the single-cell and areal levels (confocal) as well as at the ultrastructural (electron microscopic) level.
The distribution of spines with detectable levels of GluR2+ receptors was quantified along the dendrites of individual LPFC and V1 neurons filled during recordings, using high-resolution confocal microscopy (Fig. 9A). As shown in Figure 9B, in both areas the majority of the GluR2+ spines were thin, the most abundant spine subtype, although a significant proportion were mushroom and stubby. In V1 basal arbors, however, there were proportionally more GluR2+ thin spines than in LPFC (Fig. 9B, right; p = 0.036). Approximately 20% of the spines on apical and basal arbors of L3 neurons were GluR2+ (Fig. 9C, total). In both apical and basal arbors, the percentage of spines of each subtype that were GluR2+ did not differ significantly between LPFC and V1 neurons (Fig. 9C). However, in the population of mushroom spines on LPFC basal arbors, there was a higher overall proportion expressing GluR2+ receptors compared with thin spines (Fig. 9C; p = 0.035).
Figure 9.

Distribution of GluR2-containing AMPA receptors on spines of LPFC and V1 layer 3 pyramidal neurons. A, Confocal image stacks of filled dendritic segments (green, top) from LPFC and V1 neurons double labeled with GluR2 (red, middle), showing colocalization of label (merged, bottom; white pixels indicate red and green overlap) on dendrites and a subset of spines (white arrows). Scale bar, 2 μm. B, Relative distribution of GluR2+ spines by subtype in apical and basal arbors of L3 pyramidal neurons in LPFC and V1. Among GluR2+ spines in V1 basal arbors, there was a relatively higher proportion of thin spines than in LPFC. *p = 0.036. C, Proportion of GluR2+ spines in the total spine population and in distinct subtype subpopulations. In LPFC basal arbors, a higher proportion of mushroom spines were GluR2+ compared with thin spines. *p = 0.035. Error bars indicate SEM.
The overall distribution of GluR2+ AMPA receptors across layers 1 and 2–3 in LPFC and V1 was also assessed at the areal level using confocal microscopy (Fig. 10A,B). The mean fraction of the sampled area with label was measured and revealed a significantly higher density of GluR2+ label in layers 2–3 of LPFC compared with V1 (Fig. 10A,B; p = 0.05). Finally, the distribution of GluR2 receptors associated with asymmetric synapses in layers 2–3 neuropil was assessed using electron microscopy (Fig. 10C–I). The numerical density of GluR2+ axospinous synapses did not differ significantly in LPFC and V1 (Fig. 10C). A large majority of these receptors were localized on the membranes and in the cytoplasm of dendrites and spines in both LPFC (75%) and V1 (80%), and the rest were localized in the cytoplasm of presynaptic boutons (Fig. 10D). In both areas, the majority (53% in LPFC, 43% in V1) of label was localized to spines on perisynaptic sites (Fig. 10D, black, G–I). There was also no significant difference in the relative proportion of GluR2+ versus GluR2− axospinous synapses (Fig. 10E) in LPFC compared with V1. However, a significantly higher proportion of GluR2+ synapses were perforated in LPFC compared with V1 (Fig. 10F; p = 0.01).
Figure 10.

Distribution of GluR2-containing AMPA receptors in layers 2–3 neuropil of LPFC and V1. A, Maximum projections of confocal image stacks (each with 10 optical slices) of GluR2 label in fields sampled in layer 1, layers 2–3, and layer 3 of LPFC and V1. Bottom, Insets, Preabsorption control sections with no visible label. Scale bars, 20 μm. B, Density of GluR2 label quantified as the fractional area labeled with GluR2 in fields sampled from layers 1 and 2–3 of LPFC and V1. Error bars indicate SEM (n = 5 animals). *p = 0.05. C, Numerical density of GluR2+ asymmetric synapses in layers 2–3 of LPFC and V1. D, GluR2+ asymmetric synapses and their relative localization on presynaptic boutons and postsynaptic spines and dendrites. E, Proportion of GluR2+ and GluR2− asymmetric axospinous synapses. F, Proportion of GluR2+ axospinous synapses, which are perforated versus nonperforated. Error bars indicate SEM (n = 3 animals). *p = 0.01. G–I, Electron micrographs showing examples of axon terminals (At) forming asymmetric synapses (black arrowheads) on spines (Sp) with GluR2+ immunogold label (black arrows). G, Asymmetric axospinous synapse in LPFC (black arrowheads) with GluR2+ label (black arrows) localized on the perisynaptic membrane. Note the nearby unlabeled axospinous synapse (white arrowhead). H1, H2, Serial sections showing a perforated axospinous synapse in LPFC with GluR2 receptors localized on the perisynaptic membrane. I, Asymmetric axospinous synapse in V1 (black arrowheads) with GluR2+ label (black arrows) and a nearby unlabeled axospinous synapse (white arrowhead). Scale bar, 0.5 μm.
Discussion
Detailed information about excitatory synaptic signaling by pyramidal neurons in different cortical areas is required to understand how each area accomplishes its unique function. Here we report fundamental differences in glutamatergic synaptic physiology and structure between the high-order LPFC and the primary sensory area V1 in the rhesus monkey. Physiologically, AMPA receptor-mediated spontaneous and miniature EPSCs in LPFC L3 pyramidal neurons occurred at significantly higher frequencies, with a higher proportion of large synaptic events, compared with V1 neurons. Structurally, individual LPFC neurons had significantly more numerous and larger spines than did V1 neurons. Within layers 2–3 neuropil, asymmetric synapses and axonal boutons were larger in LPFC and the proportion of perforated synapses, number of vesicles per bouton, and density of GluR2 AMPA receptor subunits were significantly higher than in V1. Importantly, spine density and spine size correlated positively with the frequency and size of excitatory synaptic responses in individual L3 neurons, indicating that distinctive structural features underlie functionally important differences in glutamatergic signaling in the two brain areas.
A principal difference in excitatory synaptic physiology was the 5.3-fold higher frequency of mEPSCs in LPFC compared with V1 neurons. The frequency of synaptic events recorded with somatic voltage clamp is determined by complex interacting variables, including the number of activated synapses, degree of dendritic filtering, and the probability of neurotransmitter release (Bekkers and Clements, 1999). LPFC neurons possess ∼9 times more spines than do V1 neurons; and given that there is at least one glutamatergic synapse on each spine, the higher frequency of synaptic events is likely due, at least in part, to a higher number of activated synapses. Indeed, we found that spine density is strongly correlated with the frequency of spontaneous EPSCs in individual L3 neurons. We have previously shown that LPFC L3 pyramidal neurons possess larger and more complex dendritic arbors than V1 neurons (Amatrudo et al., 2012). Given this marked difference, synaptic attenuation due to dendritic filtering would be expected to be greater in LPFC than in V1 neurons and to reduce the frequency of events observed at the soma to a greater degree. Whether this is the case remains an open question, however, as dendritic filtering of mEPSCs was not observed in either LPFC or V1 neurons. This lack of empirically measurable dendritic filtering with whole-cell patch-clamp methods is consistent with previous studies and is not unexpected because of the stochastic nature of mEPSC amplitudes, the spatial limitations of somatic voltage clamp, and the likely presence of counterbalancing boosting mechanisms in dendritic processes (for review, see Magee, 2000; London and Häusser, 2005; Spruston, 2008; Poleg-Polsky and Diamond, 2011). Dendritic recordings will provide further insight into the effects dendritic filtering on synaptic events in future studies.
It is plausible that an increased probability of glutamate release from presynaptic boutons in LPFC compared with V1 contributes to a higher frequency of mEPSCs. Although direct physiological evidence for this idea is currently lacking, here we ruled out an effect of differences in AP-dependent release in the two areas by recording TTX-insensitive mEPSCs and presented morphological findings consistent with this idea. For example, axonal boutons in layers 2–3 of LPFC were significantly larger and contained ∼1.6× more synaptic vesicles than those in V1. The number of presynaptic vesicles is correlated with neurotransmitter release probability (for review, see Rizzoli and Betz, 2005; Schweizer and Ryan, 2006; Ostroff et al., 2012; Rollenhagen et al., 2014). Moreover, large boutons with more vesicles have been associated with an increased probability of multivesicular release (Tong and Jahr, 1994; Murthy et al., 1997; Prange and Murphy, 1999), which, if asynchronous, could contribute to the higher frequency of mEPSCs seen in LPFC.
In addition to occurring at higher frequencies, there was also a significantly higher proportion of large mEPSCs in LPFC than in V1 neurons. As a consequence of higher amplitudes and slower decay times, the mean integral of mEPSCs was twofold higher in LPFC than in V1 neurons. As with frequency, many factors could lead to larger synaptic events, including higher quantal content and/or increased number and conductance of AMPA receptors (for review, see Rollenhagen and Lübke, 2006; Spruston, 2008). The mean size of vesicles did not differ in LPFC versus V1, ruling out one mechanism of increased quantal content: increased vesicle size. The present study provided evidence for differences in presynaptic and postsynaptic structures in two distinct cortices that are consistent with the differences in the size of postsynaptic events. For example, spine widths and proportion of large spines were both positively correlated with the size of synaptic events in individual L3 neurons. Thus, larger EPSCs in LPFC than in V1 could result from larger spines with synapses that possess a higher number of functional AMPA receptors (Harris et al., 1992; Baude et al., 1995; Nusser et al., 1998; Takumi et al., 1999) and hence have a higher sensitivity to glutamate. Glutamate uncaging experiments have demonstrated that large mushroom spines have the largest EPSC responses of any spine subtype (Matsuzaki et al., 2001).
Compared with V1, LPFC had larger and more prevalent axospinous perforated synapses, which have been shown to express more AMPA receptors than do simple synapses (Desmond and Weinberg, 1998; Ganeshina et al., 2004) and likely produce larger synaptic events when activated. Previous studies have estimated that, during an EPSC, the areal spread of glutamate from a single vesicle covers ∼0.04–0.05 μm2 of the PSD area (Clements et al., 1992; Diamond and Jahr, 1997; Franks et al., 2003; Raghavachari and Lisman, 2004), which can saturate AMPA receptors on small simple synapses but not on large perforated synapses (Matsuzaki et al., 2001). Further, simulations of EPSCs have shown that multiquantal release of vesicles located 25–200 nm apart at single synapses can best estimate the amplitude and time course of large EPSCs recorded empirically (Raghavachari and Lisman, 2004). Thus, our finding of larger perforated synapses formed by larger boutons with more synaptic vesicles in LPFC than in V1 supports the idea that the probability of multivesicular glutamate release is greater in LPFC, leading to larger synaptic responses (Li et al., 2009).
In addition to the structural components of glutamatergic synapses, the subunit composition of AMPA receptors also influences the properties of EPSCs. The GluR2 subunit prevents calcium permeability, lowers single-channel conductance, prolongs channel closing time of AMPA channels, and is important for channel trafficking and membrane insertion (for review, see Malinow and Malenka, 2002; Isaac et al., 2007). Here we found a higher density of GluR2 label in layers 2–3 of LPFC, where large perforated synapses are more abundant than in V1. In light of this finding, it is interesting that GluR2 knock-out results in a decrease in number of large perforated synapses and an increase in small nonperforated synapses (Medvedev et al., 2008). Moreover, neurons lacking GluR2-containing AMPA receptors exhibit EPSCs with rapid kinetics (for review, see Isaac et al., 2007). Thus, the increased prevalence of GluR2-containing AMPA receptors in LPFC may play a role in the formation of more perforated synapses and the generation of longer lasting synaptic events in this area.
Glutamate signaling mechanisms are complex, and the frequency and size of synaptic events are regulated by a number of other presynaptic and postsynaptic factors that were not directly examined here, such as degree of saturation of postsynaptic receptors, vesicle opening and transmitter emission kinetics, and calcium dependence (for review, see Atwood and Karunanithi, 2002). That said, the significant relationships between the properties of spines and synaptic currents in L3 neurons and between presynaptic and postsynaptic ultrastructural features in the neuropil of LPFC and V1 provide important insight into the mechanisms of distinctive glutamatergic signaling in these two areas. Together, our results are consistent with the predictions from our computational models that the phenomenological “maximal conductance” parameter of each synaptic event (i.e., the parameter that determines postsynaptic conductance amplitude) is larger in LPFC than in V1 (Amatrudo et al., 2012). Future studies will explore the implications of the present findings for dendritic signaling and network function in these functionally distinct cortices.
Our data predict that synaptic integration at the cellular and network levels differs between V1, a primary sensory area for unimodal representation, and LPFC, a high-order area for complex multimodal processing (for review, see Fuster, 2001; Schummers et al., 2004). L3 pyramidal neurons in these two areas are distinctive with regard to dendritic topology and basic membrane physiology (Amatrudo et al., 2012). The highly excitable and electrically compact neurons in V1 are perhaps adapted to respond optimally to less numerous, small, and fast synapses, building a network with a limited dynamic range but well suited for signal transformations with relatively high input–output fidelity (for review, see Olshausen and Field, 2004; Vogels et al., 2005; Panzeri et al., 2010). A primary sensory area, such as V1, rapidly integrates and propagates precisely mapped unimodal data from the thalamus to higher-order cortices (for review, see Bullier, 2001; Guillery and Sherman, 2002; Lund et al., 2003). In contrast, perhaps more powerful and persistent synapses are needed to activate the less excitable neurons in LPFC with more complex dendritic arbors. Large and long-lasting synaptic currents are well suited for sustained activation, coincidence detection, and spike-timing-dependent plasticity, processes that have been associated with memory and integrative functions of LPFC (for review, see Constantinidis and Wang, 2004; Sjöström et al., 2008). The LPFC integrates and sustains multimodal information from a wide array of cortical and subcortical afferents to perform sophisticated executive tasks (for review, see Miller and Cohen, 2001; Luebke et al., 2010a). Indeed, a relatively larger dynamic range of integration of information conferred by larger and more numerous synapses would be a distinct advantage in a high-order area, such as LPFC, whereas it would be disadvantageous in V1 where more rapid signal transformations are required.
In conclusion, we propose that significantly more frequent and larger EPSCs in LPFC compared with V1 neurons are due, at least in part, to structurally more numerous, larger, and more powerful synapses in LPFC. These data are critical for understanding how excitatory signaling within neuronal networks differs between two functionally distinct cortical areas in the rhesus monkey. Excitatory transmission, however, is just one piece, albeit a large one, of the cortical circuitry puzzle. Assessment of inhibitory synaptic properties will be an important next step in the process of understanding differences in network signaling in V1 versus LPFC. Excitatory/inhibitory synaptic balance is a pivotal determinant of cortical activity (for review, see Vogels and Abbott, 2009) and likely plays a major role in conferring functional specificity in the intact and pathological cortical network.
Footnotes
This work was supported by NIH Grants P01 AG00001, R01 AG025062, R01 AG035071, and K99 MH101234. We thank Josh Gilman, Jingyi Wang, and Andy Gorden for help with data acquisition; Dr. Alan Peters, Dr. Helen Barbas, Claire Folger, and Marcia Feinberg for help with electron microscopy and their generosity in sharing space and equipment; Dr. Douglas Rosene for providing monkey tissue for slice preparation; and Dr. Christina Weaver for thoughtful comments and discussions.
The authors declare no competing financial interests.
References
- Amatrudo JM, Weaver CM, Crimins JL, Hof PR, Rosene DL, Luebke JI. Influence of highly distinctive structural properties on the excitability of pyramidal neurons in monkey visual and prefrontal cortices. J Neurosci. 2012;32:13644–13660. doi: 10.1523/JNEUROSCI.2581-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood HL, Karunanithi S. Diversification of synaptic strength: presynaptic elements. Nat Rev Neurosci. 2002;3:497–516. doi: 10.1038/nrn876. [DOI] [PubMed] [Google Scholar]
- Barbas H, Bunce JG, Medalla M. Prefrontal pathways that control attention. In: Stuss DT, Knight R, editors. Principles of frontal lobe functions. Ed 2. New York: Oxford UP; 2013. pp. 31–48. [Google Scholar]
- Baude A, Nusser Z, Molnár E, McIlhinney RA, Somogyi P. High-resolution immunogold localization of AMPA type glutamate receptor subunits at synaptic and non-synaptic sites in rat hippocampus. Neuroscience. 1995;69:1031–1055. doi: 10.1016/0306-4522(95)00350-R. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Clements JD. Quantal amplitude and quantal variance of strontium-induced asynchronous EPSCs in rat dentate granule neurons. J Physiol. 1999;516:227–248. doi: 10.1111/j.1469-7793.1999.227aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Cable properties of cultured hippocampal neurons determined from sucrose-evoked miniature EPSCs. J Neurophysiol. 1996;75:1250–1255. doi: 10.1152/jn.1996.75.3.1250. [DOI] [PubMed] [Google Scholar]
- Bourne JN, Harris KM. Balancing structure and function at hippocampal dendritic spines. Annu Rev Neurosci. 2008;31:47–67. doi: 10.1146/annurev.neuro.31.060407.125646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullier J. Integrated model of visual processing. Brain Res Brain Res Rev. 2001;36:96–107. doi: 10.1016/S0165-0173(01)00085-6. [DOI] [PubMed] [Google Scholar]
- Callaway EM. Cell type specificity of local cortical connections. J Neurocytol. 2002;31:231–237. doi: 10.1023/A:1024165824469. [DOI] [PubMed] [Google Scholar]
- Chang YM, Rosene DL, Killiany RJ, Mangiamele LA, Luebke JI. Increased action potential firing rates of layer 2/3 pyramidal cells in the prefrontal cortex are significantly related to cognitive performance in aged monkeys. Cereb Cortex. 2005;15:409–418. doi: 10.1093/cercor/bhh144. [DOI] [PubMed] [Google Scholar]
- Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL. The time course of glutamate in the synaptic cleft. Science. 1992;258:1498–1501. doi: 10.1126/science.1359647. [DOI] [PubMed] [Google Scholar]
- Constantinidis C, Wang XJ. A neural circuit basis for spatial working memory. Neuroscientist. 2004;10:553–565. doi: 10.1177/1073858404268742. [DOI] [PubMed] [Google Scholar]
- DeFelipe J, Alonso-Nanclares L, Arellano JI. Microstructure of the neocortex: comparative aspects. J Neurocytol. 2002;31:299–316. doi: 10.1023/A:1024130211265. [DOI] [PubMed] [Google Scholar]
- Denker A, Rizzoli SO. Synaptic vesicle pools: an update. Front Synaptic Neurosci. 2010;2:135. doi: 10.3389/fnsyn.2010.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmond NL, Weinberg RJ. Enhanced expression of AMPA receptor protein at perforated axospinous synapses. Neuroreport. 1998;9:857–860. doi: 10.1097/00001756-199803300-00017. [DOI] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE. Transporters buffer synaptically released glutamate on a submillisecond time scale. J Neurosci. 1997;17:4672–4687. doi: 10.1523/JNEUROSCI.17-12-04672.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas RJ, Martin KA. Neuronal circuits of the neocortex. Annu Rev Neurosci. 2004;27:419–451. doi: 10.1146/annurev.neuro.27.070203.144152. [DOI] [PubMed] [Google Scholar]
- Fiala JC. Reconstruct: a free editor for serial section microscopy. J Microsc. 2005;218:52–61. doi: 10.1111/j.1365-2818.2005.01466.x. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Harris KM. Extending unbiased stereology of brain ultrastructure to three-dimensional volumes. J Am Med Inform Assoc. 2001a;8:1–16. doi: 10.1136/jamia.2001.0080001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala JC, Harris KM. Cylindrical diameters method for calibrating section thickness in serial electron microscopy. J Microsc. 2001b;202:468–472. doi: 10.1046/j.1365-2818.2001.00926.x. [DOI] [PubMed] [Google Scholar]
- Franks KM, Stevens CF, Sejnowski TJ. Independent sources of quantal variability at single glutamatergic synapses. J Neurosci. 2003;23:3186–3195. doi: 10.1523/JNEUROSCI.23-08-03186.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster JM. The prefrontal cortex—an update: time is of the essence. Neuron. 2001;30:319–333. doi: 10.1016/S0896-6273(01)00285-9. [DOI] [PubMed] [Google Scholar]
- Ganeshina O, Berry RW, Petralia RS, Nicholson DA, Geinisman Y. Differences in the expression of AMPA and NMDA receptors between axospinous perforated and nonperforated synapses are related to the configuration and size of postsynaptic densities. J Comp Neurol. 2004;468:86–95. doi: 10.1002/cne.10950. [DOI] [PubMed] [Google Scholar]
- Germuska M, Saha S, Fiala J, Barbas H. Synaptic distinction of laminar-specific prefrontal-temporal pathways in primates. Cereb Cortex. 2006;16:865–875. doi: 10.1093/cercor/bhj030. [DOI] [PubMed] [Google Scholar]
- Guillery RW, Sherman SM. Thalamic relay functions and their role in corticocortical communication: generalizations from the visual system. Neuron. 2002;33:163–175. doi: 10.1016/S0896-6273(01)00582-7. [DOI] [PubMed] [Google Scholar]
- Harris KM, Sultan P. Variation in the number, location and size of synaptic vesicles provides an anatomical basis for the nonuniform probability of release at hippocampal CA1 synapses. Neuropharmacology. 1995;34:1387–1395. doi: 10.1016/0028-3908(95)00142-S. [DOI] [PubMed] [Google Scholar]
- Harris KM, Jensen FE, Tsao B. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J Neurosci. 1992;12:2685–2705. doi: 10.1523/JNEUROSCI.12-07-02685.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac JT, Ashby MC, McBain CJ. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 2007;54:859–871. doi: 10.1016/j.neuron.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Li GL, Keen E, Andor-Ardó D, Hudspeth AJ, von Gersdorff H. The unitary event underlying multiquantal EPSCs at a hair cell's ribbon synapse. J Neurosci. 2009;29:7558–7568. doi: 10.1523/JNEUROSCI.0514-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Burrone J, Tyler WJ, Hartman KN, Albeanu DF, Murthy VN. Synaptic vesicle recycling studied in transgenic mice expressing synaptopHluorin. Proc Natl Acad Sci U S A. 2005;102:6131–6136. doi: 10.1073/pnas.0501145102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London M, Häusser M. Dendritic computation. Annu Rev Neurosci. 2005;28:503–532. doi: 10.1146/annurev.neuro.28.061604.135703. [DOI] [PubMed] [Google Scholar]
- Luebke JI, Amatrudo JM. Age-related increase of sI(AHP) in prefrontal pyramidal cells of monkeys: relationship to cognition. Neurobiol Aging. 2012;33:1085–1095. doi: 10.1016/j.neurobiolaging.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luebke J, Barbas H, Peters A. Effects of normal aging on prefrontal area 46 in the rhesus monkey. Brain Res Rev. 2010a;62:212–232. doi: 10.1016/j.brainresrev.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luebke JI, Weaver CM, Rocher AB, Rodriguez A, Crimins JL, Dickstein DL, Wearne SL, Hof PR. Dendritic vulnerability in neurodegenerative disease: insights from analyses of cortical pyramidal neurons in transgenic mouse models. Brain Struct Funct. 2010b;214:181–199. doi: 10.1007/s00429-010-0244-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luebke JI, Medalla M, Amatrudo JM, Weaver CM, Crimins JL, Hunt B, Hof PR, Peters A. Age-related changes to layer 3 pyramidal cells in the Rhesus monkey visual cortex. Cereb Cortex. 2013 doi: 10.1093/cercor/bht336. doi: 10.1093/cercor/bht336. Advanced online publication. Retrieved Dec. 8, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund JS, Angelucci A, Bressloff PC. Anatomical substrates for functional columns in macaque monkey primary visual cortex. Cereb Cortex. 2003;13:15–24. doi: 10.1093/cercor/13.1.15. [DOI] [PubMed] [Google Scholar]
- Magee JC. Dendritic integration of excitatory synaptic input. Nat Rev Neurosci. 2000;1:181–190. doi: 10.1038/35044552. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci. 2001;4:1086–1092. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medalla M, Barbas H. Synapses with inhibitory neurons differentiate anterior cingulate from dorsolateral prefrontal pathways associated with cognitive control. Neuron. 2009;61:609–620. doi: 10.1016/j.neuron.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medalla M, Barbas H. Anterior cingulate synapses in prefrontal areas 10 and 46 suggest differential influence in cognitive control. J Neurosci. 2010;30:16068–16081. doi: 10.1523/JNEUROSCI.1773-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medalla M, Barbas H. The anterior cingulate cortex may enhance inhibition of lateral prefrontal cortex via m2 cholinergic receptors at dual synaptic sites. J Neurosci. 2012;32:15611–15625. doi: 10.1523/JNEUROSCI.2339-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medalla M, Lera P, Feinberg M, Barbas H. Specificity in inhibitory systems associated with prefrontal pathways to temporal cortex in primates. Cereb Cortex. 2007;17(Suppl 1):i136–i150. doi: 10.1093/cercor/bhm068. [DOI] [PubMed] [Google Scholar]
- Medvedev NI, Rodríguez-Arellano JJ, Popov VI, Davies HA, Tigaret CM, Schoepfer R, Stewart MG. The glutamate receptor 2 subunit controls post-synaptic density complexity and spine shape in the dentate gyrus. Eur J Neurosci. 2008;27:315–325. doi: 10.1111/j.1460-9568.2007.06005.x. [DOI] [PubMed] [Google Scholar]
- Miller EK, Cohen JD. An integrative theory of prefrontal cortex function. Annu Rev Neurosci. 2001;24:167–202. doi: 10.1146/annurev.neuro.24.1.167. [DOI] [PubMed] [Google Scholar]
- Murthy VN, Sejnowski TJ, Stevens CF. Heterogeneous release properties of visualized individual hippocampal synapses. Neuron. 1997;18:599–612. doi: 10.1016/S0896-6273(00)80301-3. [DOI] [PubMed] [Google Scholar]
- Murthy VN, Schikorski T, Stevens CF, Zhu Y. Inactivity produces increases in neurotransmitter release and synapse size. Neuron. 2001;32:673–682. doi: 10.1016/S0896-6273(01)00500-1. [DOI] [PubMed] [Google Scholar]
- Nava N, Chen F, Wegener G, Popoli M, Nyengaard JR. A new efficient method for synaptic vesicle quantification reveals differences between medial prefrontal cortex perforated and nonperforated synapses. J Comp Neurol. 2014;522:284–297. doi: 10.1002/cne.23482. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Lujan R, Laube G, Roberts JD, Molnar E, Somogyi P. Cell type and pathway dependence of synaptic AMPA receptor number and variability in the hippocampus. Neuron. 1998;21:545–559. doi: 10.1016/S0896-6273(00)80565-6. [DOI] [PubMed] [Google Scholar]
- Olshausen BA, Field DJ. Sparse coding of sensory inputs. Curr Opin Neurobiol. 2004;14:481–487. doi: 10.1016/j.conb.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Ostroff LE, Cain CK, Jindal N, Dar N, Ledoux JE. Stability of presynaptic vesicle pools and changes in synapse morphology in the amygdala following fear learning in adult rats. J Comp Neurol. 2012;520:295–314. doi: 10.1002/cne.22691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya DN, Kuypers HG. Cortico-cortical connections in the rhesus monkey. Brain Res. 1969;13:13–36. doi: 10.1016/0006-8993(69)90141-3. [DOI] [PubMed] [Google Scholar]
- Panzeri S, Brunel N, Logothetis NK, Kayser C. Sensory neural codes using multiplexed temporal scales. Trends Neurosci. 2010;33:111–120. doi: 10.1016/j.tins.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Peters A, Palay SL, Webster HD. The fine structure of the nervous system. New York: Oxford UP; 1991. [Google Scholar]
- Peters A, Moss MB, Sethares C. Effects of aging on myelinated nerve fibers in monkey primary visual cortex. J Comp Neurol. 2000;419:364–376. doi: 10.1002/(SICI)1096-9861(20000410)419:3<364::AID-CNE8>3.0.CO%3B2-R. [DOI] [PubMed] [Google Scholar]
- Peters A, Sethares C, Killiany RJ. Effects of age on the thickness of myelin sheaths in monkey primary visual cortex. J Comp Neurol. 2001;435:241–248. doi: 10.1002/cne.1205. [DOI] [PubMed] [Google Scholar]
- Peters A, Sethares C, Luebke JI. Synapses are lost during aging in the primate prefrontal cortex. Neuroscience. 2008;152:970–981. doi: 10.1016/j.neuroscience.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poleg-Polsky A, Diamond JS. Imperfect space clamp permits electrotonic interactions between inhibitory and excitatory synaptic conductances, distorting voltage clamp recordings. PLoS One. 2011;6:e19463. doi: 10.1371/journal.pone.0019463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prange O, Murphy TH. Analysis of multiquantal transmitter release from single cultured cortical neuron terminals. J Neurophysiol. 1999;81:1810–1817. doi: 10.1152/jn.1999.81.4.1810. [DOI] [PubMed] [Google Scholar]
- Raghavachari S, Lisman JE. Properties of quantal transmission at CA1 synapses. J Neurophysiol. 2004;92:2456–2467. doi: 10.1152/jn.00258.2004. [DOI] [PubMed] [Google Scholar]
- Rizzoli SO, Betz WJ. Synaptic vesicle pools. Nat Rev Neurosci. 2005;6:57–69. doi: 10.1038/nrn1583. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Ehlenberger D, Kelliher K, Einstein M, Henderson SC, Morrison JH, Hof PR, Wearne SL. Automated reconstruction of three-dimensional neuronal morphology from laser scanning microscopy images. Methods. 2003;30:94–105. doi: 10.1016/S1046-2023(03)00011-2. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Ehlenberger DB, Hof PR, Wearne SL. Rayburst sampling, an algorithm for automated three-dimensional shape analysis from laser scanning microscopy images. Nat Protoc. 2006;1:2152–2161. doi: 10.1038/nprot.2006.313. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Ehlenberger DB, Dickstein DL, Hof PR, Wearne SL. Automated three-dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS One. 2008;3:e1997. doi: 10.1371/journal.pone.0001997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollenhagen A, Lübke JH. The morphology of excitatory central synapses: from structure to function. Cell Tissue Res. 2006;326:221–237. doi: 10.1007/s00441-006-0288-z. [DOI] [PubMed] [Google Scholar]
- Rollenhagen A, Klook K, Satzler K, Qi G, Anstotz M, Feldmeyer D, Lubke JH. Structural determinants underlying the high efficacy of synaptic transmission and plasticity at synaptic boutons in layer 4 of the adult rat ‘barrel cortex.’. Brain Struct Funct. 2014 doi: 10.1007/s00429-014-0850-5. doi: 10.1007/s00429-014-0850-5. Advance online publication. Retrieved Aug. 2, 2014. [DOI] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J Neurosci. 1997;17:5858–5867. doi: 10.1523/JNEUROSCI.17-15-05858.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF. Morphological correlates of functionally defined synaptic vesicle populations. Nat Neurosci. 2001;4:391–395. doi: 10.1038/86042. [DOI] [PubMed] [Google Scholar]
- Schummers J, Mariño J, Sur M. Local networks in visual cortex and their influence on neuronal responses and dynamics. J Physiol Paris. 2004;98:429–441. doi: 10.1016/j.jphysparis.2005.09.017. [DOI] [PubMed] [Google Scholar]
- Schweizer FE, Ryan TA. The synaptic vesicle: cycle of exocytosis and endocytosis. Curr Opin Neurobiol. 2006;16:298–304. doi: 10.1016/j.conb.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- Sjöström PJ, Rancz EA, Roth A, Häusser M. Dendritic excitability and synaptic plasticity. Physiol Rev. 2008;88:769–840. doi: 10.1152/physrev.00016.2007. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Tamás G, Lujan R, Buhl EH. Salient features of synaptic organisation in the cerebral cortex. Brain Res Brain Res Rev. 1998;26:113–135. doi: 10.1016/S0165-0173(97)00061-1. [DOI] [PubMed] [Google Scholar]
- Spruston N. Pyramidal neurons: dendritic structure and synaptic integration. Nat Rev Neurosci. 2008;9:206–221. doi: 10.1038/nrn2286. [DOI] [PubMed] [Google Scholar]
- Stevens CF. Presynaptic function. Curr Opin Neurobiol. 2004;14:341–345. doi: 10.1016/j.conb.2004.04.004. [DOI] [PubMed] [Google Scholar]
- Takumi Y, Ramírez-León V, Laake P, Rinvik E, Ottersen OP. Different modes of expression of AMPA and NMDA receptors in hippocampal synapses. Nat Neurosci. 1999;2:618–624. doi: 10.1038/10172. [DOI] [PubMed] [Google Scholar]
- Timbie C, Barbas H. Specialized pathways from the primate amygdala to posterior orbitofrontal cortex. J Neurosci. 2014;34:8106–8118. doi: 10.1523/JNEUROSCI.5014-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong G, Jahr CE. Multivesicular release from excitatory synapses of cultured hippocampal neurons. Neuron. 1994;12:51–59. doi: 10.1016/0896-6273(94)90151-1. [DOI] [PubMed] [Google Scholar]
- Vogels TP, Abbott LF. Gating multiple signals through detailed balance of excitation and inhibition in spiking networks. Nat Neurosci. 2009;12:483–491. doi: 10.1038/nn.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogels TP, Rajan K, Abbott LF. Neural network dynamics. Annu Rev Neurosci. 2005;28:357–376. doi: 10.1146/annurev.neuro.28.061604.135637. [DOI] [PubMed] [Google Scholar]
- Wearne SL, Rodriguez A, Ehlenberger DB, Rocher AB, Henderson SC, Hof PR. New techniques for imaging, digitization and analysis of three-dimensional neural morphology on multiple scales. Neuroscience. 2005;136:661–680. doi: 10.1016/j.neuroscience.2005.05.053. [DOI] [PubMed] [Google Scholar]
- Zikopoulos B, Barbas H. Parallel driving and modulatory pathways link the prefrontal cortex and thalamus. PLoS One. 2007;2:e848. doi: 10.1371/journal.pone.0000848. [DOI] [PMC free article] [PubMed] [Google Scholar]