

Abstract
Recent studies on the pathogenic mechanisms of recessive hyperekplexia indicate disturbances in glycine receptor (GlyR) α1 biogenesis. Here, we examine the properties of a range of novel glycine receptor mutants identified in human hyperekplexia patients using expression in transfected cell lines and primary neurons. All of the novel mutants localized in the large extracellular domain of the GlyR α1 have reduced cell surface expression with a high proportion of receptors being retained in the ER, although there is forward trafficking of glycosylated subpopulations into the ER-Golgi intermediate compartment and cis-Golgi compartment. CD spectroscopy revealed that the mutant receptors have proportions of secondary structural elements similar to wild-type receptors. Two mutants in loop B (G160R, T162M) were functional, but none of those in loop D/β2–3 were. One nonfunctional truncated mutant (R316X) could be rescued by coexpression with the lacking C-terminal domain. We conclude that a proportion of GlyR α1 mutants can be transported to the plasma membrane but do not necessarily form functional ion channels. We suggest that loop D/β2–3 is an important determinant for GlyR trafficking and functionality, whereas alterations to loop B alter agonist potencies, indicating that residues here are critical elements in ligand binding.
Keywords: assembly, biogenesis, glycine receptor, human hyperekplexia, rescue of function, subcompartimentalization
Introduction
Glycine receptors (GlyRs), which mediate fast inhibitory neurotransmission in the adult spinal cord and brainstem, are the major underlying cause of the human neurological motor disorder hyperekplexia (Online Mendelian Inheritance in Man #149400). Most human mutations have been described in the GLRA1 gene and follow a dominant or a recessive trait with incomplete penetrance. GLRA1 encodes the GlyR α1 subunit. Mutations of the GLRB gene, which is translated in the GlyR β subunit, and the SCL5A6 gene, which results in the presynaptic glycine transporter 2, may also lead to similar neuromotor phenotypes (Rees et al., 2006; Chung et al., 2013; James et al., 2013). Symptoms in human hyperekplexia range from exaggerated startle reactions due to unexpected noise or tactile stimuli to muscle stiffness, apnea, and loss of postural control upon startle. Approximately 55 GLRA1 mutations have been detected in humans (Bode and Lynch, 2014). Most dominant mutations have been localized in the ion channel domain or adjacent loops, thereby likely influencing channel properties. Defects in gating, desensitization, and open channel probabilities of GlyRs have been described (Schaefer et al., 2013). In contrast, recessive mutations are distributed throughout the GlyR α1 sequence and have been associated with disturbed biogenesis. Cell surface receptor expression is decreased for recessive mutants compared with wild-type (WT). A rescue of protein function by the coexpression with unaffected α- or β subunits has been described for some single mutants (e.g., R392H and E375X; Chung et al., 2010; Bode et al., 2013). Recessive mutants generate instable proteins that are degraded via proteasomal pathways (Villmann et al., 2009a). There have, however, been no further studies on the subcellular trafficking of mutant GlyR α1 protein.
GlyRs belong to the superfamily of Cys-loop receptors (CLRs) and are postsynaptic pentameric receptor complexes of α1- and β subunits anchored via the scaffolding protein gephyrin (Grudzinska et al., 2005). The large extracellular domains (ECDs) of GlyRs form ligand-binding sites at the interface between adjacent subunits, which is constituted by loops A–C from one subunit and loops D–F from the neighboring subunit (Brejc et al., 2001; Brams et al., 2011; Hibbs and Gouaux, 2011).
Here, we describe the distinct attribution of novel patient mutations in GlyR loop structures to subcellular receptor distribution in cell lines and primary neurons and their malfunctions. We show the functional rescue of a truncated nonfunctional human α1 variant R316X by complementation with the lacking protein domain. We further elucidate subcellular trafficking routes in primary neurons and transfected cell lines of recessive GlyR α1 loop D/β2–3 mutants. A high proportion of affected receptors get stuck in the ER, but forward trafficking of subpopulations of glycosylated receptors into the ER-Golgi intermediate compartment (ERGIC) and cis-Golgi compartment was observed. Therefore, a proportion of recessive α1 variants are transported to the outer membrane but do not form functional ion channels. Loop D/β2–3 was identified as an important determinant for GlyR trafficking and functionality, whereas loop B mutants affect either taurine or glycine affinity, indicating that residues here are critical elements in ligand binding.
Materials and Methods
Direct sequencing from genomic DNA.
Genomic DNA from male and female index patients was prepared according to the manufacture's protocol (DNeasy Blood and Tissue Kit; Qiagen). Specific intronic primers for exons 1–9 of the GLRA1 gene were used for PCR amplification of the appropriate exons followed by direct sequencing (LGC Genomics).
Site-directed mutagenesis.
cDNAs encoding the human WT or mutated GlyR α1 (G160R, T162M, W68C, D70N, R72H, R316X, W407R, iD-TM4-C) were subcloned into the eukaryotic expression vector pRK5 (CMV promoter). Mutations were introduced using site-directed mutagenesis. Primers (Life Technologies) containing the appropriate single nucleotide exchanges also harbored a restriction site for quick identification of generated clones. PCRs were set up as follows: 100 ng of template DNA; 10 mm each dATP, dCTP, dGTP, and dTTP; 100 pmol of sense and antisense primer, and 2 units of high-fidelity Taq polymerase (Roche) in the supplied polymerase buffer. PCR conditions were as follows: 28 cycles with 1 min at 95°C, 2 min at 55°C, and 3 min at 72°C. The last cycle ended with a 10 min 72°C amplification step. The final fragments were ligated into WT GlyR α1. Double mutants (W68C/R316X and D70N/W407R) were generated with restriction and religation into the respective other mutant. All mutated clones were sequenced across the PCR-generated sequence to verify successful mutagenesis using the ABI Sequencer system. GlyR α1 WT and mutants were subcloned into a low-copy-vector FUVAL_IRES2_GFP (kindly provided by R. Blum) under the control of a ubiquitin promoter to exclude effects resulting from overexpression systems.
Cell lines and primary neurons.
Human embryonic kidney (HEK293) cells were grown in minimum essential medium; COS7 (African Green Monkey kidney cells) cells were grown in Dulbecco's modified eagle medium (high glucose; all cell culture media were purchased from Life Technologies), supplemented with 10% fetal calf serum, l-glutamine (200 mm), and 50 U/ml penicillin and streptomycin at 37°C and 5% CO2. HEK293 cells were transiently transfected using a modified calcium-phosphate precipitation method. All experiments concerning immunocytochemistry and protein biochemistry were performed 48 h after transfection. HEK293 cells for FlexStation experiments were transfected at 60–90% confluency using polyethyleneimine (PEI, 25 kDa, linear; Polysciences). Briefly, 5 μg of DNA and 30 μl of 1 mg/ml PEI were added to 1 ml of serum-free medium. The transfection solution was mixed thoroughly and left for 10 min before adding dropwise to cells in 10 cm dishes. Forty-eight hours after transfection, cells were transferred to poly-lysine-coated, black, clear-bottomed, 96-well plates and left overnight to allow adherence.
COS7 cells were transfected using a mixture of 1 μg of DNA, 30 μl of PBS, and 62 μl of DEAE-Dextran (10 mg/ml) for a 3 cm dish. The medium was aspirated after 30 minutes and fresh medium was added, along with 12 μl of chloroquin (10 mm). The medium was exchanged again after 3 h.
Hippocampal neurons were prepared from embryonic day 18 mouse embryos and grown in neurobasal medium plus 5 ml of l-glutamin (200 mm) plus B27 supplement (Life Technologies) with an exchange of 50% medium after 6 d in culture. Hippocampal neurons were transfected 3 d after plating using a modified calcium-phosphate precipitation method. Then, 2.5 μg of DNA (1 μg/μl), 2.5 μl of CaCl2 (2.5 m), and 25 μl of 2× BBS (50 mm BES, 280 mm NaCl, 1.5 mm Na2HPO4, pH 7.05) were mixed and incubated for 30 min. The neuronal medium was aspirated from the hippocampal culture and stored until the end of transfection. Neurons were transfected with the DNA solution plus 450 μl of neurobasal medium for 30 min. The transfection mixture was aspirated and the cells were washed twice with HBSS medium and floated with the original neuronal medium.
Immunocytochemical staining.
Transfected HEK293 cells were fixed in 4% paraformaldehyde in PBS for 10 min, washed twice with PBS, and blocked with 5% goat serum in PBS (permeabilized with 0.2% Triton X-100) for 30 min at 22°C. Primary GlyR antibody incubation was done for 1 h at 22°C. The GlyR α1-specific antibody MAb2b was used without permeabilization and recognizes an epitope in the far N-terminus (residues 1–10 of mature protein). In permeabilized cells, MAb4A a pan-α antibody recognizing residues 96–105 (both from SYSY) or anti-rabbit GlyR α1 affinity-purified (Millipore) were used. The GlyR β subunit was detected via an N-terminal attached myc-epitope by a c-myc antibody (1:250; Santa Cruz Biotechnology). Antibodies for the compartmental marker proteins were calnexin (polyclonal anti-calnexin antibody ab22595; Abcam) or MA3–027 (Thermo Fisher Scientific), ERGIC-53 (purified mouse antibody against ER-Golgi intermediate compartment; Enzo Life Sciences), and GM130 (purified anti-mouse antibody; BD Transduction Laboratories). Secondary antibodies gαmCy3, gαrCy5, and gαrALEXA488 (1:500; Dianova) were applied for 1 h. Slides were mounted with Mowiol.
Biotinylation of cell surface protein.
Biotinylation experiments were performed as described previously (Unterer et al., 2012).
Membrane preparation and ligand-binding assay.
For membrane protein analysis, crude cell membranes were prepared from transfected cells. Western blots of membrane preparations from transfected HEK293 cells were stained with the pan-α antibody, MAb4a (1:100). Radioligand binding was performed using filtration assays with triplicate 80 μg of membrane protein. Samples were incubated for 30 min in a range of concentrations (1, 10, 20, 50, 100, and 200 nm) of [3H]strychnine (specific activity 30 Ci/mmol; DuPont NEN); nonspecific binding was determined using 30 mm glycine (Kling et al., 1997). Binding data were analyzed by a nonlinear algorithm provided by the program Origin 6.0 (Microcal Software).
EndoH and PNGase treatment.
Deglycosylation experiments were performed using the endoglycosidases EndoH and PNGaseF according to the manufacture's protocol (New England Biolabs).
Coimmunoprecipitation.
Immunoprecipitation assays followed the protocol by Unterer et al. (2012). In brief, 4 Petri dishes (10 cm diameter) transfected with the cDNA coding for GlyR variants were harvested after 2 min of incubation on ice with 1400 μl of lysis buffer and tubes were rotated for 40 min at 4°C. Lysates were centrifuged for 10 min at 14 000 rpm. The supernatant was incubated with the precipitating antibodies for 4 h at 4°C by rotating the tubes. Then 50 μl of equilibrated protein A/G agarose was added and incubated overnight at 4°C while rotating. The antibodies used for immunoprecipitation were as follows: mouse monoclonal anti-calnexin: 3 μl of MA3027, mouse monoclonal MAb2b (1:500), and anti-GlyR α1. GlyRs were detected in Western blots with the mouse monoclonal MAb4a (1:500) and calnexin by the polyclonal anti-calnexin antibody ab22595 at a dilution of 1:500.
FlexStation experiments.
Transfected cells on 96-well plates were washed with Flex buffer (140 mm NaCl, 4.7 mm KCl, 2.5 mm CaCl2, 1.2 mm MgCl2, 11 mm HEPES, 10 mm D-glucose, pH 7.4). To each well, 100 μl of fluorescent membrane potential (blue) dye (FLIPR Membrane Potential Blue Kit; Molecular Devices) diluted in Flex buffer was added. Cells were incubated at 37°C for 30 min before assay. Fluorescence was measured every 2 s for 200 s and agonists (50 μl) were added at 20 s. Data were normalized to the maximum change in fluorescence. Concentration-response data were fitted to the four-parameter logistic equation, F = Fmin + (Fmax − Fmin)/(1 + 10 log(EC50−[A])nH)), where Fmax is the maximum response, Fmin is the baseline fluorescence, [A] is the log concentration of agonist, denoted as [agonist] log M in the graph, and nH is the Hill coefficient, using Prism version 5.0 software (GraphPad).
Electrophysiological recordings.
Maximal current amplitudes (Imax) were measured by patch-clamp recordings in the whole-cell configuration from transfected HEK293 cells. Current signals were amplified with an EPC-9 amplifier (HEKA). After transfection (24–48 h), whole-cell recordings were performed by application of ligand using a U-tube system bathing the suspended cell in a laminar flow of solution, giving a time resolution for equilibration of 10–30 ms. Glycine was used at concentrations between 0.5 and 3 mm. The lower-efficacy agonist taurine was used at concentrations from 3 μm to 10 mm. The external buffer consisted of the following (in mm): 137 NaCl, 5.4 KCl, 1.8 CaCl2, 1 MgCl2, 5 HEPES, pH adjusted to 7.2 with NaOH; the internal buffer consisted of the following (in mm): 120 CsCl, 20 N(Et)4Cl, 1 CaCl2, 2 MgCl2, 11 EGTA, 10 HEPES, pH adjusted to 7.2 with CsOH. Recording pipettes were fabricated from borosilicate capillaries with an open resistance of 4–6 MΩ. Current responses were measured at a holding potential of −60 mV. All experiments were performed at room temperature (∼22°C).
Expression and refolding of the ECDs.
GlyR α1 ECD variants were cloned into pET30a harboring an N-terminal S-tag and hexa-His-tag. BL21 DE3 pLys-E. coli cells were transformed with these constructs for expression experiments (Vogel et al., 2009). All ECD variants produced in inclusion bodies were isolated, washed, solubilized in 8 m urea, and refolded using stepwise dialysis. Proteins were dialyzed against 10 mm NaxHyPO4, pH 7.4.
CD spectroscopy.
Recombinant proteins were dialyzed against 10 mm NaxHyPO4, pH 7.4, and the sample concentration was determined by measuring the A280 using a molar extinction coefficient of 30 160 m−1 cm−1 (for W68C 24785 m−1 cm−1). CD spectra were measured using a Jasco J810 spectropolarimeter equipped with a Peltier element in a 0.1 cm quartz cuvette. Spectra were recorded at 20°C between 260 and 190 nm with a 0.2 nm step size and an integration time of 1 s. A total of four accumulations were averaged and the buffer spectrum, which was obtained under identical conditions, was subtracted. The obtained spectra were subsequently smoothed by a Savitzky–Golay filter with a window size of 15 data points. The content of secondary structure of the proteins was estimated using the concentration independent method developed by Raussens et al. (2003, 2006).
Sucrose density centrifugation.
Membranes from transfected HEK293 cells with GlyR α1 variants were solubilized according to a previously published protocol (Seeger et al., 2012). Total protein concentrations were determined using the BCA method. Equal amounts of solubilized proteins (750–1000 μg) were loaded on a 30 ml of continuous sucrose gradient from 10% to 40% in 50 mm Tris, pH 7.4, 1% Triton X-100 on top of a 4 ml 60% sucrose layer. Centrifugation was done in a Kontron ultra-centrifuge 26 000 rpm (121 569.8 × g) for 18 h at 4°C. Next, 100 μl of methanol was added to 400 μl of manually taken fractions used for dot blots. GlyR α1 protein was stained with the MAb4 antibody (SYSY). For sucrose gradient calibration, marker proteins were cytochrome C, 12.3 kDa, 1.83 S; ovalbumin, 43 kDa, 3.55 S; adult human hemoglobin (α-β tetramer), 64 kDa, 4.27 S; bovine serum albumin, 66.5 kDa, 4.6 S; malate dehydrogenase, 70 kDa, 4.32 S; gamma globulin, 150 kDa, 6.3 S; catalase 232 kDa, 11.3 S; and ferritin, 440 kDa, ≈18 S.
Computational methods.
The homology model of the GlyR α1 was generated by using the crystal structure of the glutamate-gated chloride channel (GluCl) at 3.35 Å resolution (pdb code: 3RIF) as a template (Hibbs and Gouaux, 2011). The sequences of human GlyR α1 and GluCl were aligned according to the ClustalW algorithm using the default settings of the input form found at http://www.ch.embnet.org/software/ClustalW.html (Thompson et al., 1994). The template structure was modified by removing the Fab molecule bound at each GluCl subunit interface, as well as by mutating the glutamate ligands into glycine. Molecular modeling was performed using MODELLER9.9 (Sánchez and Sali, 2000) with the ligand module. The disulfide bridges were checked in the resulting model. The modeled structure was improved by 200 steps of conjugated gradient energy minimization using the Powell algorithm in Sybyl7.3 (Sybyl 6.9, Release 7.0A; Tripos). The quality of the model was verified by WHAT_CHECK (Hooft et al., 1996). The visualization of the structures was performed with DS Viewer Pro6.0 (Accelrys).
Statistical analysis.
Concentration-response curves were constructed from the peak current amplitudes obtained with at least seven appropriately spaced concentrations in the range 3–10 000 μm taurine. Using a nonlinear algorithm (Microcal Origin), concentration-response data were first analyzed using the following Hill equation Itaurine/Isat = [taurine]nH/[taurine]nH + EC50nH, where Itaurine refers to the current amplitude at a given taurine concentration, Isat is the current amplitude at saturating concentrations of taurine, EC50 is the taurine concentration producing half-maximal current responses, and nH is the Hill coefficient.
Results
Description of patients
We identified four novel GLRA1 mutations, one dominant and three recessive, in four patients from three unrelated families with classical clinical features of hyperekplexia (Fig. 1). Molecular genetic testing for GlyR mutations revealed heterozygosity for the novel allele GLRA1 (G160R, Patient 1), which was present in both father and infant. Patient 2 was homozygous for the recessive mutation T162M inherited from his consanguineous heterozygous parents. The mutations D70N and W407R were identified in a compound heterozygous patient (Patient 3) with both parents carrying one of the described recessive mutations.
Figure 1.
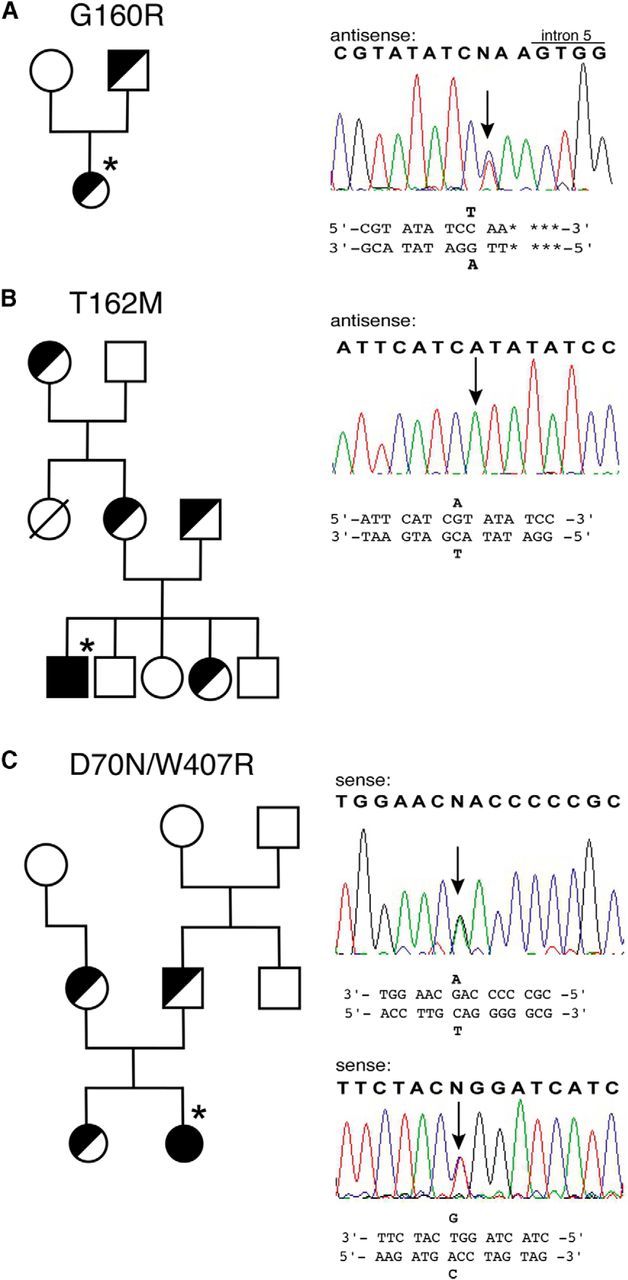
Pedigrees of hyperekplexia families. A–C, Affected individuals are indicated by filled symbols (index patients are marked with asterisks) and unaffected individuals are noted with open symbols. Half-closed symbols represent family members with only one mutant allele. Direct sequencing of affected exons is shown on the right. Heterozygosity or homozygosity (T162M) are marked with an arrow and listed in part of the GLRA1 sequence in either sense or antisense direction.
Clinical description of Patient 1 (GLRA1 G160R)
This female child was referred from a district hospital by the local pediatrician for assessment of generalized hypertonia. She was born at 41 weeks gestation by spontaneous vaginal delivery. Examination on the first day of life revealed an increased tone in a generalized distribution. Four days later, persisting hypertonia was observed. Family history demonstrated a similar history of hypertonia in infancy of the father. The patient had no dysmorphic features and an age-appropriate flexed posture of upper and lower limbs. Deep tendon reflexes were present throughout but were not brisk. When pulled from supine, there was sustained hypertonia and stiffness of the trunk and limbs. When awake, there was an exaggerated startle response to nose tap, which was persistent and did not habituate. At follow-up, hypertonia persisted and a delay in motor milestones was noted. At the age of 5 months, her tendency to excessive startle had improved.
Clinical description of Patient 2 (GLRA1 T162M)
This boy is the fifth child from healthy consanguineous parents of Turkish origin. An older brother has generalized epilepsy with onset at 6 years of age. The other siblings are healthy. The parents reported that a maternal aunt who was born ∼20 years ago had symptoms similar to what was observed in the patient. After uneventful pregnancy, the boy was born at term without perinatal complications. Directly after birth, continuous generalized stiffness was noted, which decreased during sleep. The newborn had an exaggerated startle response to tactile and auditory stimuli without habituation. Recurrent episodes of unprovoked generalized myoclonus lasting for a few minutes with apnea and cyanosis occurred during sleep. Phenobarbitone and later clobazame proved effective in ameliorating the nocturnal myoclonic attacks. Stiffness gradually decreased over the first year of life and excessive startle response slowly disappeared over a few years.
Clinical description of Patient 3 (GLRA1 D70N/W407R)
In the first days of life, the parents noted myocloni including flexion of forearms, clenched fists, and a tensed stomach. After 2 weeks, the young female patient showed tonic spasms, especially during sleep, in response to sudden and unexpected noise or light stimuli. Seizures could be interrupted by touching or holding for ∼10 s. The startle reaction observed during sleep led sometimes to cyanotic skin color. Hypertonic musculature, very vivid proprioceptive reflexes, and a persisting whole-body myoclonus to nose tapping, hand clapping, or light stimulus were observed. During sleep, the above-described stimuli provoked complex persisting seizures. At the age of 1 year and 3 months, the girl showed no developmental delay.
Conserved residues of the GlyR α1 affect loop structures
Novel identified mutations were localized in N-terminal loops B (G160R and T162M) and β2–3 (D70N) and in transmembrane domain 4 (TM4; W407R) of GlyR α1 (Fig. 2A–D). WT residues in loop B and D/β2–3 and W407 in TM4 are highly conserved among different GlyR subunits (e.g., α2, α3, and β; Fig. 2E). We screened for other mutations in these regions that have not yet been functionally investigated. Human GlyR mutations W68C and R72H are also localized close to D70N in loop D/β2–3, with W68C identified from a compound heterozygous patient together with R316X (Tsai et al., 2004; Coto et al., 2005). Similarly, WT loop D/β2–3 residues (W68, R72) are also conserved among all GlyR subunits and in GluCl, a protein that shares the highest homology (33%) with the GlyR family (Fig. 2D,E). The high degree of conservation suggests the importance of loop D/β2–3 for receptor function. Because W68C was identified in a compound heterozygous patient together with R316X, the truncated α1 mutant was also investigated (Fig. 2D,E).
Figure 2.
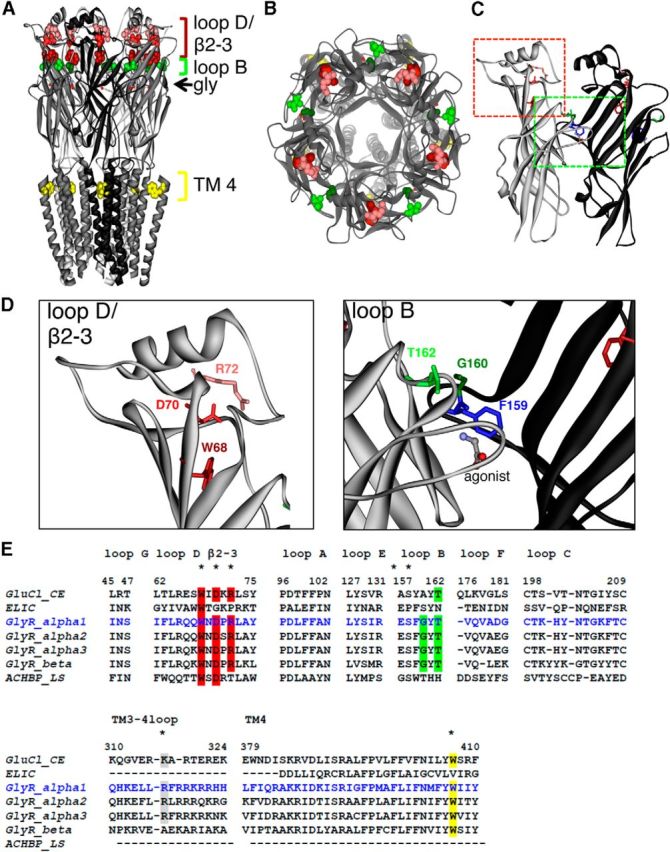
Structural model of GlyR based on GluCl structure. A, View parallel to lipid membrane. The glycine ligands, which are bound at each subunit, are represented in stick form. The amino acids are shown in spacefill representation: W68 (red), D70 (light red), and R72 (pink) are located in loop D/β2–3; G160 (dark green) and T162 (light green) are located in loop B. W407 (yellow) is located in TM4. B, View of the human GlyR looking down the pore axis toward cytosol. C, Dimer interface with colored rectangles pointing to loop B (green) and loop D/β2–3 (red). D, Enlarged view to loop B and loop D/β2–2; affected residues are marked. E, Alignment of various human GlyR sequences of subunits α1 (blue), α2, α3, and β compared with the AChBP from L. stagnalis, ELIC, a prokaryotic homolog of the CLR family, and GluCl from C. elegans. Loop sequences are shown from loops A–G, part of the ICD = TM3–4 loop covering residues 310–324, and TM4. Residues affected in patients with hyperekplexia are highlighted with different colors (red in loop D/β2–3, green in loop B, gray TM3–4 loop, yellow TM4). All mutated residues are highly conserved among sequences shown.
Influence of loop structure on cellular localization
The expression of mutant α1 variants was analyzed by subcellular immunostaining and compared with a biotinylation of surface proteins. We used LIVE cell staining compared with fixed and permeabilized transfected HEK293 cells 48 h after transfection. Loop B mutations G160R and T162M were expressed at the cell surface, T162M with less efficiency at the single-cell level (Fig. 3A, bottom right). Coexpression with the β subunit, which is important in the in vivo situation for receptor complex formation, did not enhance the expression of mutated α1T162M (Fig. 3A, bottom right). Using biotinylation to label surface receptors, the surface expression of T162M was reduced to ∼53% compared with WT α1, whereas no differences were observed for the loop B mutation G160R (Fig. 3D).
Figure 3.
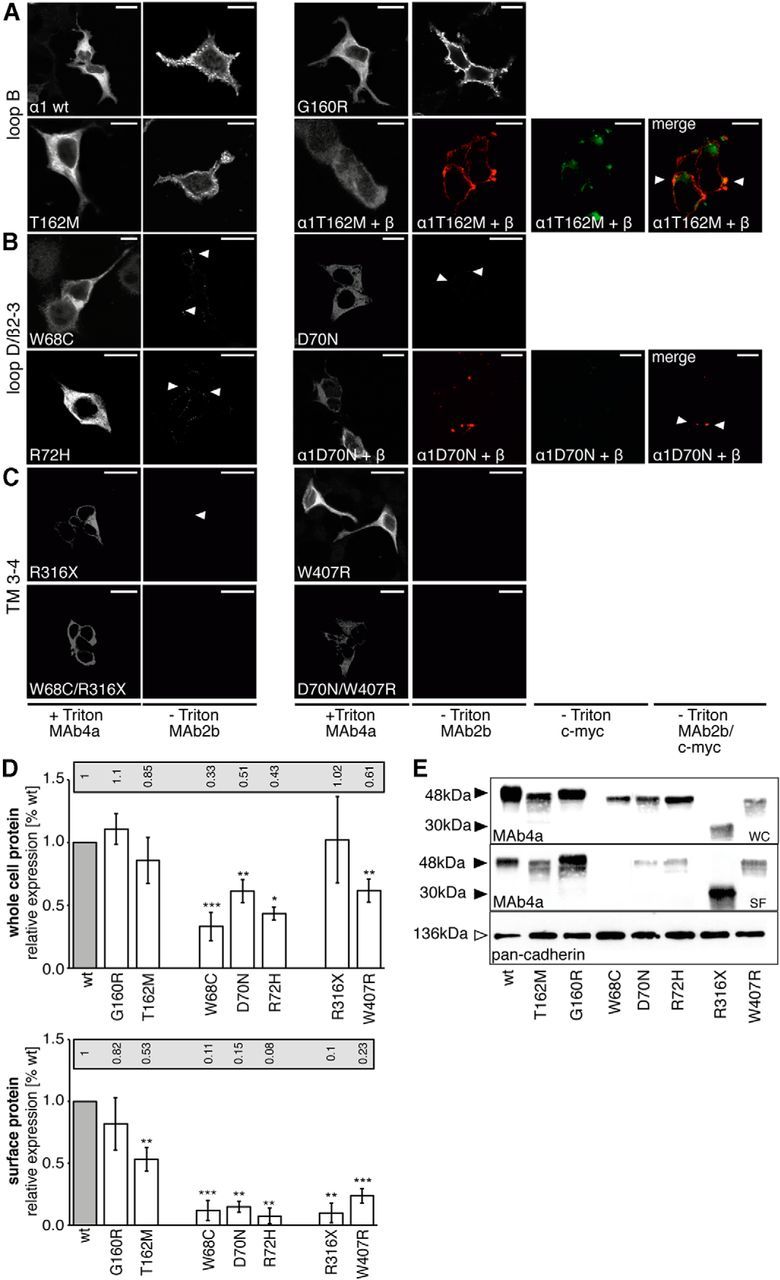
Loop D/β2–3 mutations interfere with trafficking of GlyR α1 polypeptides. A–C, Immunocytochemical staining of dominant and recessive GlyR α1 subunit mutants in transfected HEK293 cells. Variants are grouped according to their localization within the polypeptide sequence into loop B (WT, G160R, T162M), loop D/β2–3 (W68C, D70N, R72H), and TM3–4 (R316X, W407R, W68C/R316X, D70N/W407R) mutants. First and third column show the whole-cell expression of GlyR α1 variants in fixed and permeabilized cells stained with the monoclonal antibody 4a (MAb4a). Second and fourth columns represent live stainings of cell surface α1 protein with GlyR α1 with MAb2b. For T162M and D70N, coexpression with the GlyR β subunit is shown. GlyR β is detected via its N-terminal myc-epitope. Note that GlyR β is not able to enhance cell surface expression of affected α1 mutants. Scale bar, 40 μm. D, Biotinylation experiments to determine the amount of cell surface compared with whole-cell protein of dominant and recessive GlyR α1 variants from transfected HEK293 cells. Quantification was from at least six independent experiments; ratios compared with WT are shown above the blots. Top graph refers to the amount of whole-cell protein; bottom graph represents the biotinylated protein fraction reflecting surface GlyR α1. Note that R316X is stained at a lower molecular weight of 30 kDa compared with full-length GlyRα1 (48 kDa). Mutations in loop D/β2–3 result in a significant decrease of whole-cell and surface α1 receptors. *p < 0.05, **p < 0.01, ***p < 0.001. E, Representative Western blots of GlyR α1 mutants stained with MAb4a. Cadherin was used as a membrane marker protein for internal loading control (stained with a pan-cadherin antibody at 136 kDa).
For loop D/β2–3 GlyR α1 variants, only a few labeled clusters showed positive expression at the surface of transfected HEK293 cells, suggesting that the overall structure of a loop D/β2–3 subpopulation was not misfolded (Fig. 3B, second and fourth column). Forward trafficking of GlyR α1 mutants to the cellular surface was again not enhanced upon coexpression with the β subunit detected via an N-terminal myc-epitope (Fig. 3B, bottom right). Whole-cell expression of loop D/β2–3 variants revealed a significant reduction of GlyR α1 mutants compared with WT α1 protein (W68C 33%, D70N 51%, R72H 43%). Differences in expression levels were more pronounced at the cell surface with W68C 11%, D70N 15%, and R72H 8% of WT level (Fig. 3C,D).
The recessive mutant R316X, which is localized in the large intracellular TM3–4 loop, had relatively poor (10%) cell surface expression, although the level of whole-cell protein was comparable to WT levels (Fig. 3D). Truncated proteins are usually recognized by the ER control system and are not transported into the secretory pathway (Lecker et al., 2006). A similar GlyR α1 mutation in the TM3–4 loop present in the mouse mutant oscillator led to very low expression levels at the cell surface in vitro (Villmann et al., 2009b). A construct harboring both mutations present in the compound heterozygous patient W68C/R316X was almost absent from the plasma membrane (Fig. 3C, bottom left). There was reduced cell surface expression with W407R (23%) and the double mutant D70N/W407R behaved similarly (Fig. 3C, bottom right). The highly reduced number of receptors in the plasma membrane could explain the hyperekplexia symptoms observed in the patients.
Loop B mutants modify receptor function
To explore ion channel function, whole-cell patch-clamp recordings were performed after transfection of α1 variants into HEK293 cells. First, glycine was applied at two saturating concentrations (1 and 10 mm). Loop B variants did not show significant differences in maximal currents (Imax) compared with α1 WT (Fig. 4A,D). All other constructs carrying mutations in loop D/β2–3, the TM3–4 loop, or TM4 did not generate agonist-induced currents even at a very high concentration of glycine (10 mm; Fig. 4B,C). Expression of the double mutants W68C/R316X and D70N/W407R revealed similar results (Fig. 4C). For some cells expressing D70N alone (6 of 14 cells), very small currents were recorded (Fig. 4B, Table 1). This is consistent with the dotted expression pattern observed in LIVE cell images, which likely represents a small subpopulation of functional channels at the cell surface, at least for α1D70N (Fig. 4B, inset).
Figure 4.

Ion channel functionality of GlyR α1 variants. A–C, Whole-cell recordings from transfected HEK293 cells with GlyR α1 variants of loops D and B and TM3–4. Glycine-gated absolute current amplitudes were recorded using saturating glycine concentrations of 1 and 10 mm. Note that the small current amplitudes observed for D70N at 1 mm glycine reflects the small portion of receptor complexes at the cell surface of transfected cells (inset: enhanced contrast, white arrows point to receptor clusters). D, Rrepresentative traces of functional GlyR α1 loop B variants G160R and T162M from whole-cell recordings after application of 1 mm glycine for 1 s. E, Radioligand binding with the antagonist [3H] strychnine. KD and Bmax values were determined using the two-ligand binding model with the software Microcal Origin 6.0 (for values, see Table 2). F–H, EC50 values for loop D/β2–3 variants (W68C, D70N, R72H) and loop B GlyRα1 (G160R, T162M) determined using membrane-potential-sensitive dye. The normalized fluorescence values were plotted as a function of the agonist concentration (logarithmic scale). F, Loop D/β2–3 variants showing nonspecific increases in fluorescence indistinguishable from untransfected cells (dotted line). G, Concentration-response curves for loop B mutants (G160R black squares, T162M black triangles, WT filled circles) with taurine (left), β-alanine (right), or the full agonist glycine (middle). H, Graph plots from taurine, glycine, and β-alanine concentration-response curves for WT and G160R determined from whole-cell recordings.
Table 1.
Functional properties of GlyRα1 variants
| Localization | GlyR variant | Imax 1 mM gly (nA) | n | Imax 10 mM gly (nA) | n |
|---|---|---|---|---|---|
| Loop B | WT | 3.03 ± 0.5 | 25 | 2.2 ± 0.4 | 20 |
| G160R | 3.2 ± 0.7 | 10 | 5.3 ± 1.2 | 7 | |
| T162M | 4.8 ± 0.6 | 10 | 1.4 ± 0.2 | 11 | |
| Loop D/β2–3 | W68C | 0 ± 0 | 9 | 0 ± 0 | 10 |
| D70N | 0.02 ± 0.004* | 6/14* | 0 ± 0 | 10 | |
| R72H | 0 ± 0 | 10 | 0 ± 0 | 10 | |
| TM3–4 loop | R316X | 0 ± 0 | 10 | 0 ± 0 | 10 |
| TM3–4 loop, TM4, C-terminus | iD-TM4-C | 0 ± 0 | 10 | 0 ± 0 | 10 |
| TM3–4 loop | R316X + iD-TM4-C (R1) | 0.45 ± 0.12 | 16 | 0.7 ± 0.2 | 11 |
| Loop D/TM3–4 loop | W68C/R316X | 0 ± 0 | 10 | 0 ± 0 | 10 |
| Loop D/TM3–4 loop | W68C/R316X + iD-TM4-C (R2) | 0 ± 0 | 21 | 0 ± 0 | 10 |
| Loop D/TM3–4 loop | W68C + R316X + iD-TM4-C (R3) | 0.5 ± 0.13 | 13 | 0.12 ± 0.02 | 11 |
| TM4 | W407R | 0 ± 0 | 10 | 0 ± 0 | 10 |
| β2–3/TM4 | D70N/W407R | 0 ± 0 | 10 | 0 ± 0 | 16 |
| Localization | GlyR variant | FLEX station experiments |
|||||
|---|---|---|---|---|---|---|---|
| EC50 gly (μm)FLEX | n | EC50 tau (μm)FLEX | n | EC50 β-ala (μm)FLEX | n | ||
| Loop B | WT | 58 ± 2.5 | 24 | 443 ± 62 | 6 | 120 ± 11 | 6 |
| G160R | 43 ± 3 | 18 | 85 ± 5 | 6 | 43 ± 2 | 6 | |
| T162M | 409 ± 10 | 14 | >162000 | 3 | 480 ± 91 | 6 | |
| Loop D/β2–3 | W68C | NB | ND | ND | |||
| D70N | NB | ND | ND | ||||
| R72H | NB | ND | ND | ||||
| Localization | GlyR variant | Whole-cell recordings |
|||||
|---|---|---|---|---|---|---|---|
| EC50 gly (μm) | n | EC50 tau (μm) | n | EC50 β-ala (μm) | n | ||
| Loop B | WT | 26 ± 3 | 6 | 271 ± 22 | 6 | 63 ± 12 | 6 |
| G160R | 90 ± 8 | 6 | 55 ± 6 | 6 | 120 ± 25 | 6 | |
| T162M | ND | ND | ND | ||||
| Loop D/β2–3 | W68C | NF | NF | NF | |||
| D70N | NF | NF | NF | ||||
| R72H | NF | NF | NF | ||||
EC50 values represent mean values ± SEM. n, Number of measured cells out of different transfections; NB, nonbinding; ND, not determined; NF, nonfunctional.
* Calculated from responded cells only out of n cells recorded.
A radioligand-binding assay using the antagonist [3H]strychnine revealed that the binding affinity was not significantly changed for G160R compared with WT (Fig. 4E, Table 2). A reduced binding capacity (Bmax) for G160R is consistent with the slight decrease of cell surface receptors (Fig. 3D). The mutant protein T162M resulted in no specific binding (Fig. 4E). The levels of expressed loop D/β2–3 variants were either too low to be detected and/or the mutations had severely decreased the strychnine-binding affinity.
Table 2.
Ligand binding properties of GlyRα1 variants
| Localization | GlyR variant | Bmax (pmol × mg−1) | KD (nm) | n |
|---|---|---|---|---|
| Loop B | WT | 15367 ± 4025 | 115 ± 66 | 9 |
| G160R | 7650 ± 939 | 136 ± 12 | 3 | |
| T162M | NB | — | 4 | |
| Loop D/β2–3 | W68C | NB | — | 3 |
| D70N | NB | — | 3 | |
| R72H | NB | — | 3 | |
| TM3–4 loop | R316X | NB | — | 4 |
| TM3–4 loop, TM4, C-terminus | iD-TM4-C | NB | — | 3 |
| TM3–4 loop | R316X + iD-TM4-C | 533 ± 257 | 92 ± 12 | 4 |
| Loop D/TM3–4 loop | W68C/R316X | NB | — | 3 |
| Loop D/TM3–4 loop | W68C/R316X + iD-TM4-C | NB | — | 3 |
| TM4 | W407R | NB | — | 4 |
| β2–3/TM4 | D70N/W407R | NB | — | 3 |
n, Number of independent experiments; concentrations of 3(H)strychnine 0.1–200 nm were used. Bmax and KD values represent mean values ± SEM. NB, Nonbinding.
The FlexStation data revealed a glycine EC50 for G160R (43 ± 3 μm) mutant receptors similar to WT (58 ± 2.5 μm), but both β-alanine and taurine were more potent. A higher taurine affinity for G160R was also observed in whole-cell recording from transiently transfected HEK293 cells (Fig. 4H). The affinity of G160R for β-alanine was similar to WT (less than a factor of 2; Fig. 4H, Table 1). Compared with the WT, the mutant receptors G160R showed a slight decrease in glycine affinity (Fig. 4H, Table 1). Glycine activated T162M mutants (409 ± 10 μm), but was ∼10-fold less potent than at WT receptors. T162M mutants also had comparatively higher EC50 with β-alanine and did not respond to taurine. W68C, D70N, and R72H did not respond to glycine (Fig. 4F,G, Table 1).
Nonfunctional human variants can be restored in ion channel function
In addition to missense mutations, GlyR α1 mutations in human and mice associated with a neuromotor phenotype may also result from nonsense mutations (Fig. 5A). Here, coexpression of the truncated human mutant R316X together with a construct encoding the lacking C-terminal portion (tail) of GlyR α1 resulted in surface expression of R316X [R1 (Rescue1) = R316X + iD-TM4-C; cf. Figs. 5B, 3C]. A similar expression pattern was detected upon expression of the double mutant W68C/R316X together with the C-terminal tail (R2 = W68C/R316X + iD-TM4-C) or in coexpressed W68C and R316X with the tail construct (R3 = W68C + R316X + iD-TM4-C; Fig. 5B, middle and bottom). The quantification of surface protein revealed 10–17% expression of R316X at the plasma membrane and 86% at the whole-cell level compared with WT (Fig. 5D,E). Coexpressions of R316X with the C-terminal tail were most successful for functional rescue when the tail construct was expressed in excess. The surface expression levels of R316X (15–24%) in coexpressions with the C-terminal tail (iD-TM4-C) were similar compared with single expression (Fig. 5D). When three GlyRα1 domain variants were coexpressed, the surface expression of all three was enhanced (30–53%; Fig. 5E). Similarly, hippocampal neurons at in vitro day 3 were transfected with R316X, W68C/R316X, W68C, and R316X, together with the rescue construct iD-TM4-C (R1, R2, R3). The coexpressed C-terminal domain iD-TM4-C resulted in α1 polypeptides localized at the dendrites of primary hippocampal neurons for R1 and R3 (Fig. 5C), referring to similar trafficking routes of affected GlyR α1 mutants in neurons compared with transfected eukaryotic cell lines. In contrast, in R2, the construct iD-TM4-C was almost absent in dendrites of hippocampal neurons (Fig. 5C, middle), which is consistent with the lowest cell surface expression values quantified from membranes of transfected cell lines (Fig. 5E). Overall truncation of a GlyR α1 does not obligatorily prevent forward trafficking in transfected cells and primary neurons.
Figure 5.
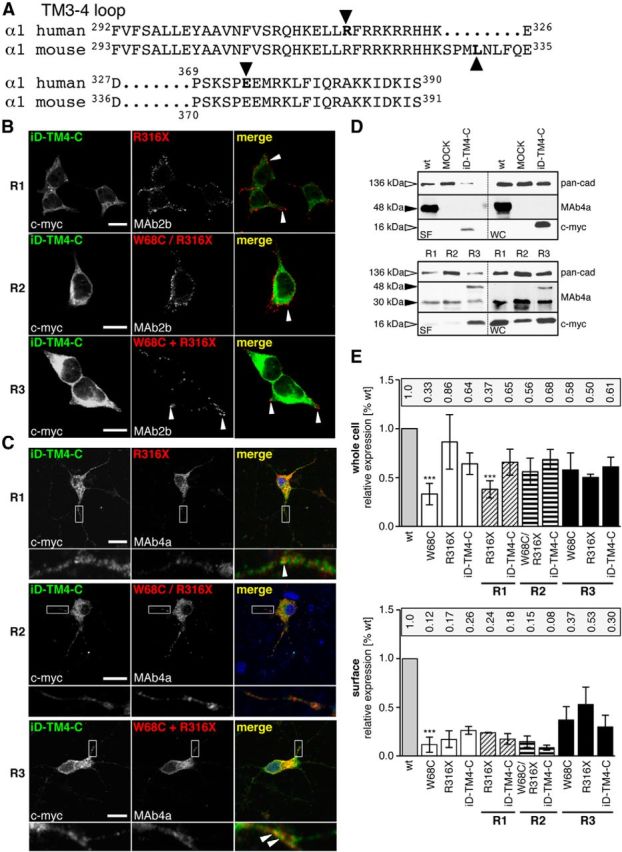
Rescue of truncated human GlyR α1 by domain coexpression. A, Alignment of the TM3–4 loop sequences from the human and mouse GlyR α1. Residues determined from human patients or in a mouse model for hyperekplexia (spdot = oscillator) leading to premature STOP codons are marked (bold letters and black arrows). B, The human GlyR α1 variants R316X, W68C/R316X, and W68C + R316X were coexpressed with the GlyR domain iD-TM4-C in HEK293 cells (refers to tail construct and represents the lacking protein sequence harboring residues of the TM3–4 loop starting with F317, TM4, and the C terminus); rescue 1 = R1 (R316X + iD-TM4-C), R2 (W68C/R316X + iD-TM4-C), R3 (W68C + R316X + iD-TM4-C). Cell surface expression of N-terminal GlyR α1 domains (W68C, R316X or W68C/R316X) was determined in LIVE stainings with the α1-specific antibody MAb2b recognizing a native epitope in the far N-terminal part of GlyR α1 (middle column). The C-terminal complementation tail domain was detected via its intracellularly located N-terminal myc-epitope (left column). Merged images show colocalization of both GlyR domains (right column). C, Constructs used as shown in B. Hippocampal neurons, which do not express endogenous GlyR α1, were transfected at in vitro day 3 and stained using MAb2b for GlyR α1 detection and anti-c-myc antibody for the tail constructs. Colocalization of coexpressed constructs along the dendrites is shown in merged images (enlarged insets show dendritic expressions). D, Representative Western blot of constructs used for rescues R1, R2, and R3 after surface labeling with biotin. Controls (e.g., WT, iD-TM4-C, MOCK) are shown at the top. Note that staining of both α1 variants W68C (48 kDa) and R316X (30 kDa) in R3 stained by MAb4a, iD-TM4-C was determined at 16 kDa. E, Quantification of whole-cell and surface GlyR α1 detected using the monoclonal antibody MAb4a from at least six independent experiments. Ratios compared with WT expression levels are labeled above the bar diagrams. Within R1, R2, and R3, the expression of all α1 domains is shown separately. *p < 0.05, **p < 0.01, ***p < 0.001.
On a similar theme, R316X coexpressed with iD-TM4-C was able to rescue strychnine binding, revealing a similar affinity to WT, although Bmax was reduced (30%), which is consistent with decreased levels of expressed receptors at the plasma membrane (Fig. 6A, Table 2). In contrast, the double mutant W68C/R316X was unable to restore strychnine binding, which is consistent with the lack of binding in the W68C mutant (Fig. 6A). When W68C was coexpressed with R316X and the C-terminal domain iD-TM4-C (R3), function was restored (Fig. 6B), presumably indicating that there were sufficient functional binding sites to activate the receptor. The mutant W68C itself shows enhanced transport to the cell surface when coexpressed with R316X and iD-TM4-C. Therefore, W68C did not hinder R316X from forward trafficking toward the cell surface to form functional GlyR ion channels together with the C-terminal domain GlyR α1 iD-TM4-C (Figs. 5E, 6B).
Figure 6.

Physiological characteristics of functionally restored GlyRs from independent domains. A, Ligand-binding experiments were performed using membrane preparations of transfected HEK293 with truncated GlyR α1 variants R316X and W68C/R316X together with the tail construct (transfected ratio was 1: 5 of truncated α1: C-terminal domain). B, Whole-cell recordings from transfected HEK293 cells with rescues R1, R2, and R3 coexpressing a truncated GlyR α1 variant together with the tail domain iD-TM4-C: R1 (R316X + iD-TM-C), R2 (W68C/R316X + iD-TM4-C), and R3 (W68C + R316X + iD-TM4-C). Glycine was applied at 1 mm. Representative traces are shown above the bars of the corresponding rescue condition. Imax values result from at least four independent experiments.
Folding status of the GlyR α1 variants
To investigate the impact of the amino acid exchanges in the ECD on the CLR fold, we subcloned the α1 mutations into appropriate vectors for expression in E. coli. The WT GlyR ECD has been shown to produce an unfolded protein deposited in inclusion bodies of E. coli cells that has to be refolded (Breitinger et al., 2004). Refolded α1 proteins used in this study are shown in the Coomassie-stained gel (Fig. 7A, bottom). We used circular dichroism spectroscopy to assess the contents of secondary structures. Similar fractions of α-helices and β-strands were observed for all GlyR ECDs analyzed (Fig. 7A, top). Quantitative evaluation of the secondary structure contents of the WT protein and all α1 variants using the method of Raussens et al. (2003, 2006) observed α-helical contents of 23–30% and 17–26% β-strand, which show good agreement within the accuracy of the method (Table 3). Therefore, it is concluded that all ECDs investigated exhibit very similar structural properties.
Figure 7.

Structural analysis of GlyRα1 variants. A, Circular dichroism spectroscopy of refolded GlyR ECDs of WT, loop B variants (G160R and T162M), and loop D/β2–3 variants. Colors are given in the figure legend next to the graph. Inset, Enlarged intensity maximum shown for W68C (orange) and D70N (light blue) compared with WT (black). Bottom, Coomassie-stained gel of the refolded WT and α1 ECD variants used for recording CD spectra. B, Sucrose density gradient centrifugation of the full-length GlyR WT and hyperekplexia variants. Peak distribution points to various receptor oligomerization states with monomers (left), over various assembly states to aggregates (far right) for loop B, D and TM3–4 loop variants. Arrows above the top graph point to the maximal peaks of the proteins used for calibration. Normalized AU, Arbitrary units determined from GlyR detection in various fractions using MAb4a. The line colors and symbols for each variant are given in the legends next to the graphs. The positions of the putative monomer and dimer obtained for Torpedo (Riganti et al., 2005) are marked by red arrowheads. Aggregrate formation is designated by gray boxes.
Table 3.
Estimation of secondary structure content from CD spectra of refolded GlyR ECDs
| Variant | α-Helix (%) | β-Strand (%) | Turns (%) | Unordered (%) | Sum (%) |
|---|---|---|---|---|---|
| WT | 22.5 | 23.9 | 12.9 | 35.1 | 94.0 |
| W68C | 30.4 | 17.4 | 12.5 | 33.8 | 94.1 |
| D70N | 23.8 | 22.9 | 12.5 | 34.5 | 93.7 |
| R72H | 23.5 | 23.1 | 12.5 | 34.4 | 93.6 |
| G160R | 23.1 | 25.5 | 12.5 | 34.7 | 95.8 |
| T162M | 22.5 | 25.0 | 12.5 | 35.0 | 95.0 |
| SD (%) | 11.9 | 11.1 | 4.15 | 10.3 |
Analysis was performed using the method of Raussens et al. (2003, 2006).
In W68C, the content of β-strands is reduced to 17% and the α-helical content slightly increased to 30% of that of WT according to the higher maximum intensity and the leftward shift of the intensity minimum to smaller wavelengths (Fig. 7A, insets).
To complement the results of the isolated refolded ECDs, we investigated the folding and assembly states of full-length GlyR variants expressed in HEK293 cells. A 10–40% sucrose gradient was used on top of a 60% sucrose layer to determine the oligomerization state and aggregation of the GlyR-ECD. The gradient was calibrated with selected globular proteins (arrows on top of WT; Fig. 7B). For the averaged WT receptor, dot blot signals gave five major peaks, the lowest corresponding to fraction 8 (low range peak) at ∼150 kDa and 6.3 S (Fig. 7B), most probably representing dimers or trimers. Although all five major peaks are present, the WT curve is rather flat due to the averaged values taken from all individual experiments. T162M and G160R showed a similar peak pattern as WT, with G160R having an additional peak at fraction 15, which could represent an intermediate assembly species. The lowest peak (fraction 3) for T162M matched the putative WT monomer peak, but was larger in amplitude. The high sucrose range peak of T162M gave a higher signal, which could point to an increased propensity for aggregation (Fig. 7B). The β2–3 variant D70N has a first peak shifted to lower sucrose densities compared with the first WT peak. The position between 43 kDa 3.55 S and 66.5 kDa 4.6 S fits to a monomer. Loop D/β2–3 variants differ much more from WT, pointing to pentamer assembly problems (Fig. 7B, middle). R316X and W407R have a low-density-range double peak. The rather broad peak between fractions 20 and 24 might result from aggregated proteins or GlyR oligomers associated with other proteins (Fig. 7B, bottom).
Disturbances in subcellular trafficking routes of GlyR α1 mutants
The human GlyR α1 variants of loop D/β2–3 have a higher tendency to form aggregates. Only a small subpopulation of GlyR α1 loop D/β2–3 mutants were observed at the cell surface. Therefore, we followed the loop D/β2–3 α1 proteins from the ER to the cell surface by cell compartment analysis using low copy vectors to exclude effects from overexpression. COS7 cells are suitable for compartment analysis due to their large cytoplasm (Fig. 8). Costaining of α1 with the ER chaperone calnexin revealed large accumulations of loop D/β2–3 variants and W407R in the ER (Fig. 8A, first column). In contrast, the α1 WT does not reside within the ER—here, forward trafficking toward the cell surface is favored (Fig. 8A). Mutants W68C and D70N were detected in ERGIC and cis-Golgi compartments in transfected COS7 cells (Fig. 8A,B). The α1 variant R72H showed colocalization with ERGIC, but was not detectable in cis-Golgi (Fig. 8A,B). Coimmunoprecipitation studies of GlyR α1 supported this finding. A low amount of α1 WT precipitated with calnexin, but increased protein–protein interactions with calnexin were observed for all loop D/β2–3 variants and W407R (Fig. 9A). Furthermore, these mutant GlyR α1 proteins showed protein instability seen by the large fraction of protein degradation compared with WT and loop B variants G160R and T162M.
Figure 8.
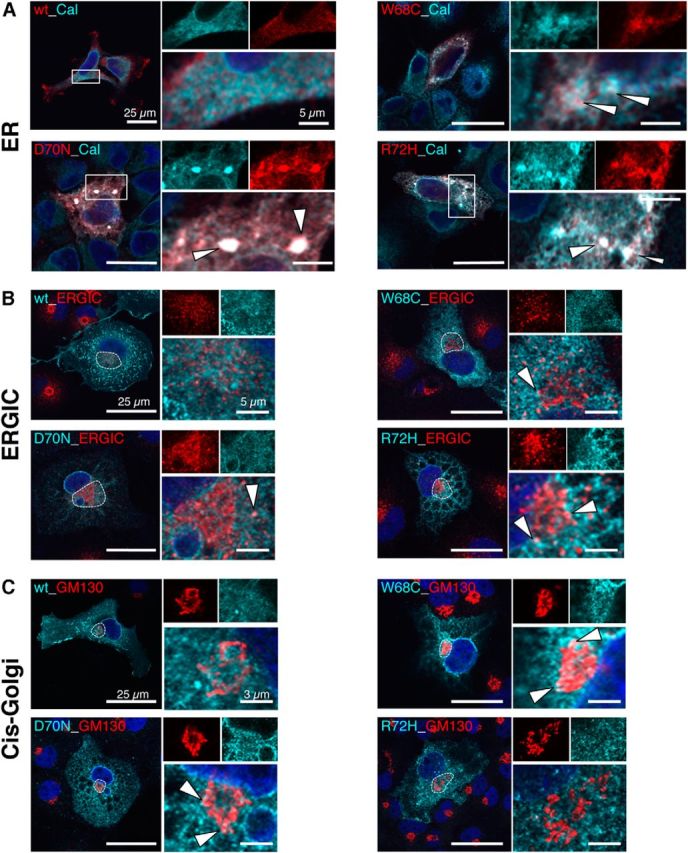
Subcellular trafficking routes of α1 loop D/β2–3 mutants. A–C, GlyR α1 variants W68C, D70N, and R72H were analyzed for their localization in cellular compartments ER, ERGIC, and cis-Golgi. To exclude effects from overexpression, α1 variants were subcloned into a low-expression vector under the control of the ubiquitin promoter. A, Merged images showing costaining of GlyR α1 (red) with the ER protein calnexin (cyan, rabbit anti-calnexin, 1:500). Right, Magnification of the white rectangle with white arrowheads pointing to accumulated GlyRα1. B, Colocalization of α1 (cyan) with the ERGIC protein 53 (red, mouse anti-ERGIC53, 1:500); the ERGIC compartment is marked with a white dotted line and shown in higher magnification at top right. C, Costaining with the cis-Golgi marker GM130 (red, mouse anti-GM130, 1:500). Color codes are indicated in labels within the left images. The nuclei are stained with DAPI (blue). White arrows point to colocalization of GlyR α1 variants with compartmental marker proteins. Scale bars: 25 μm in left images, 5 μm for ERGIC, and 3 μm for cis-Golgi in enlarged compartment images.
Figure 9.

GlyRα1 variants circumvent the ER quality control system. A, Immunoprecipiation of GlyR α1 loop D/β2–3 variants with anti-calnexin antibody stained for GlyRα1 with MAb4a (1:500). Input and IP are shown. Black arrows point to the appropriate molecular weight of GlyR α1 of 48 kDa. B, Analysis of the glycosylation pattern of loop D/β2–3 variants compared with WT. Endoglycosidases EndoH and PNGaseF were used for deglycosylation of α1 proteins and stained with MAb4a. Arrow points to the appropriate molecular weight of 48 kDa for glycosylated GlyR α1, 45 kDa of deglycosylated protein. Note the differences in the glycosylation pattern of R72H and W407R compared with WT and the other loop D/β2–3 mutants. C, Loop D/β2–3 variants transfected into hippocampal neurons. Images show GM130 costainings. White boxes mark the enlarged areas at higher magnification (bottom).
Forward trafficking from the ER requires protein glycosylation. The GlyR α1 is not a highly glycosylated protein, harboring only one glycosylation site in the ECD. A digest with endoglycosidases EndoH and PNGaseF showed differences in the glycosylation pattern, although all α1 variants were core glycosylated (Fig. 9B). Although PNGaseF digests mannose chains directly attached at asparagine, EndoH is only able to digest high-mannose-type glycans, leaving only one N-acetylglucosamine attached at the asparagine until the Golgi α-glucosidase II cleaves off two mannose molecules. Variants positive for forward cis-Golgi trafficking (W68C, D70N) possessed a higher sugar tree capable of being cut by PNGase F (Fig. 9B). GlyRs α1 R72H and W407R do only receive core glycosylation, enabling trafficking toward ERGIC but preventing transport to the Golgi secretory pathways. Similar protein transportation pathways were observed in transfected primary hippocampal neurons lacking endogenous GlyR α1 (Fig. 9C). These data argue that loop D/β2–3 mutations lead to trafficking ER-Golgi defects in neurons and transfected cell lines.
Discussion
The major causes of human hyperekplexia are defective GlyRs. So far, the focus on mutants has been to identify physiological changes in receptor function (Chung et al., 2010; Bode and Lynch, 2013; Bode et al., 2013), although recent studies have shown that recessive hyperekplexia may result from disturbed GlyR biogenesis (Vergouwe et al., 1999; Villmann et al., 2009a; Bode et al., 2013). Because GlyR trafficking has not yet been investigated at the subcellular level, we examined the underlying pathogenic mechanisms in terms of protein maturation and subcellular receptor trafficking. Due to the continuous expansion of genetic data, the number of identified compound heterozygous patients is currently increasing. If both alleles are affected by recessive mutations, it becomes important to resolve the impact of each single mutation on the assembly and trafficking of the pentameric GlyRs. We identified four novel mutations localized in extracellular loops B and D/β2–3 and one in TM4; all of these are residues conserved within the GlyR family. We have also considered other mutations (Tsai et al., 2004; Coto et al., 2005) identified in these domains to provide a general mechanism for mutant GlyR variants. GlyR α1 loop B variants had mutations of hydrophobic glycine to positively charged arginine or the polar threonine to nonpolar methionine, but expression levels demonstrated no differences for G160R and only a slight decrease for T162M. In contrast, mutants in TM4 and loop D/β2–3 were almost absent from the cell surface, suggesting that these positions are key determinants in protein biogenesis. Coexpression with GlyR β, which is present in vivo in the adult mammalian receptor complex that has been shown to promote surface accumulation for some recessive GlyR mutants (Chung et al., 2010), did not enhance mutant loop D/β2–3 populations at the plasma membrane. Loop D/β2–3 mutants are localized at the transition of the second β-sheet to a 310 helix in the GlyR α1 ECD. Alterations to residues F63 and R65, which are close to loop D/β2–3, have been shown to decrease in surface expression and strychnine potency (Brams et al., 2011; Bode et al., 2013). Here, we show that all loop D/β2–3 mutants W68C, D70N, and R72H were unable to bind ligand and result in nonfunctionality. The small subpopulation of receptors able to circumvent the quality control system and to traffic to the cell surface internalize very fast.
Receptors containing the TM4 mutant W407R remain intracellular, possibly due to a pentamerization defect. Aromatic residues present in TMs 1, 3, and 4 have been previously proposed to form an aromatic network required for pentamerization, with W407 being a key residue in this process (Haeger et al., 2010).
Interestingly, a truncation of the GlyR α1 protein at position 316 resulted in a subpopulation transported toward the cell surface. Nonsense mutations are usually coupled to nonsense-mediated decay mechanisms (Conti and Izaurralde, 2005). However, this general assumption seems not to apply for GlyR truncations. In a recent study, the incorporation of α1 E374X together with unaffected α subunits into functional receptor complexes were shown (Bode et al., 2013). A functional rescue of a truncated murine α1 variant was also demonstrated upon coexpression with the lacking C-terminal domain (Villmann et al., 2009b). The human α1 mutant R316X itself is functionally inactive, but here we show its functional rescue by domain coexpression. These data provide the first evidence that functional rescue by domain complementation is transferable to the human situation and may be a general mechanism for ion channel restoration of truncated receptors. The mutant binds ligand with no changes of glycine KD values compared with α1 WT. The observed lower Bmax within domain coexpression can be explained by the ratio of 1:5 of truncated α1 to the C-terminal domain. Due to early truncation, R316X lacks the basic motif 318RRKRR, which has been described as an important domain for surface transport (Sadtler et al., 2003) and can explain the decreased cell surface trafficking. Here, the presence of this motif in the C-terminal complementation domain is not sufficient to enable efficient cotransport of R316X. Coexpression of R316X and loop D variant W68C, together with the independent C-terminal domain, resulted into the formation of functional ion channels, whereas the presence of both mutations in one α1 subunit W68C/R316X led to nonfunction. Therefore, loop D/β2–3 residues are key players in receptor maturation. Amino acid exchanges in loop D/β2–3 can lead to misfolded protein, hindering forward trafficking of the main neuronal α1 receptor population, recognition by cellular control, and ER retention.
Loop B mutations are localized close to residue F159, which interacts via a cation-π interaction with the agonist glycine and the low-efficacy agonists β-alanine and taurine (Pless et al., 2008; Hibbs et al., 2009; Pless et al., 2011). Therefore, the neighboring residues might influence agonist binding. Glycine affinity was slightly decreased for G160R, but a gain of function for the low-efficacy agonist taurine was observed. A different effect was seen with T162M, where there was a decrease in glycine affinity by eightfold and no binding to taurine. Differences in side chains of loop B residues lead to changes in the overall conformation of the ligand-binding pocket, possibly by altering the positioning of F159. Alternatively, they could be affecting the ability of the receptor to undergo the gating transitions that translate ligand binding into pore opening.
The very low abundance of loop D/β2–3 GlyR variants at the cell surface and the inability to bind ligand suggest that these variants are actively retained by the cellular quality control system. One cause for this could be the abnormal folding of these loop D/β2–3 mutants, but our data from ECDs expressed in E. coli gave similar CD spectra of loop D/β2–3 variants and WT α1, with almost no obvious differences in the calculated content of secondary structures such as α-helices, β-strands, and turns. For W68C, increased contents of α-helices and decreased β-strand were observed using the method of Raussens et al. (2003, 2006). This could point to a basic folding problem due to a missing aromatic stacking core of the molecule. Therefore, the refolded ECDs adopt the common Ig-fold typical for all CLR ECDs.
In sucrose density gradient centrifugations, proteins are distributed according to their buoyancy, providing information about their quaternary and tertiary structure as local interactions within one protomer influence the compactness of the folded protein (Gotti et al., 1992; Mazzo et al., 2013). An improved solubilization protocol and a linear gradient of 10–40% achieved splitting of the GlyR signal into five different peaks, reflecting different assembly stages and aggregated protein. Loop D/β2–3 variants have a broader high peak range than the WT, which could be due to aggregation and/or increased interaction with chaperones of the protein quality machinery. Peaks for putative WT α1 dimers and pentamers were in a similar range (9 and 13.6 S) to previously published data for Torpedo californica nAChR (Riganti et al., 2005), but loop D/β2–3 variants lacked the peak that represents pentameric receptors, assembly of which may be the rate-limiting step for nAChR (Ross et al., 1991; Wanamaker and Green, 2007). The slowness of fine-tuned folding after the initial Ig-fold and the slow assembly explains why all assembly intermediates may have been detectable. Therefore, our data suggest that loop D/β2–3 and ICD variants are unable to achieve an ordered pentamerization.
Aggregation of nascent polypeptides does result from folding defects in the ER and are recognized by chaperones such as calnexin, which controls N-glycosylation of protein before forward trafficking into the secretory pathway. Large proportions of loop D/β2–3 variants interacted with calnexin leading to protein accumulations within the ER in primary neurons and cell lines. Glycosylation of the 45 kDa GlyR α1 protein plus 3 kDa N-glycan per protomer has been described as a prerequisite for ER exit and pentamerization (Griffon et al., 1999). Core glycosylation was observed for all GlyR α1 mutants. A distinct glycosylation pattern indicated differences among loop D/β2–3 variants, W407R, and WT in subcellular transport. Due to the presence of a subpopulation of EndoH-resistant α1 protein of mutants W68C and D70N, these GlyRs exited the ER and entered the Golgi apparatus. In contrast, R72H and W407R did not gain EndoH resistant carbohydrates and were retained in the ER. Some protein clusters of R72H were detected in ERGIC, but never in the cis-Golgi compartment. Therefore, loop D/β2–3 mutations affect trafficking at different compartmental levels. Although some subpopulations of loop D/β2–3 variants are able to leave the ER, they are presumably recognized by other control proteins localized within the ERGIC or Golgi that disable further transport toward the cell surface.
The GlyR α1 mutants described here are aberrant in respect to trafficking and assembly. The observed differences of α1 variants in forward and, presumably, reverse transport from ERGIC and cis-Golgi to the ER for further degradation seems to be highly dynamic and precisely controlled by as yet unknown proteins of the secretory pathway. Our data with compartmental subcellular localization and EndoH resistance as a measure for subcellular localization and trafficking of the GlyR α1 variants provide further insights into the pathogenic mechanism of the neuromotor disorder hyperekplexia.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (Grant DFG VI586 to C.V.) and the European Union (FP7 project Neurocypres to C.J.K., K.L.P., and S.C.R.L.). N. Schaefer and G.L. are supported by the GSLS Wuerzburg. S.C.R.L. is a Wellcome Trust Senior Research Fellow in Basic Biomedical Research. We thank Katrin Schiebel and Pamela Strissel for critical reading of the manuscript and helpful comments and Rosa Weber, Elke Spirk, and Marina Wenzel are for excellent technical assistance.
The authors declare no competing financial interests.
References
- Bode A, Wood SE, Mullins JG, Keramidas A, Cushion TD, Thomas RH, Pickrell WO, Drew CJ, Masri A, Jones EA, Vassallo G, Born AP, Alehan F, Aharoni S, Bannasch G, Bartsch M, Kara B, Krause A, Karam EG, Matta S, et al. New hyperekplexia mutations provide insight into glycine receptor assembly, trafficking, and activation mechanisms. J Biol Chem. 2013;288:33745–33759. doi: 10.1074/jbc.M113.509240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode A, Lynch JW. Analysis of hyperekplexia mutations identifies transmembrane domain rearrangements that mediate glycine receptor activation. J Biol Chem. 2013;288:33760–33771. doi: 10.1074/jbc.M113.513804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode A, Lynch JW. The impact of human hyperekplexia mutations on glycine receptor structure and function. Mol Brain. 2014;7:2. doi: 10.1186/1756-6606-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brams M, Pandya A, Kuzmin D, van Elk R, Krijnen L, Yakel JL, Tsetlin V, Smit AB, Ulens C. A structural and mutagenic blueprint for molecular recognition of strychnine and d-tubocurarine by different cys-loop receptors. PLoS Biol. 2011;9:e1001034. doi: 10.1371/journal.pbio.1001034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitinger U, Breitinger HG, Bauer F, Fahmy K, Glockenhammer D, Becker CM. Conserved high affinity ligand binding and membrane association in the native and refolded extracellular domain of the human glycine receptor alpha1-subunit. J Biol Chem. 2004;279:1627–1636. doi: 10.1074/jbc.M303811200. [DOI] [PubMed] [Google Scholar]
- Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB, Sixma TK. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 2001;411:269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- Chung SK, Vanbellinghen JF, Mullins JG, Robinson A, Hantke J, Hammond CL, Gilbert DF, Freilinger M, Ryan M, Kruer MC, Masri A, Gurses C, Ferrie C, Harvey K, Shiang R, Christodoulou J, Andermann F, Andermann E, Thomas RH, Harvey RJ, et al. Pathophysiological mechanisms of dominant and recessive GLRA1 mutations in hyperekplexia. J Neurosci. 2010;30:9612–9620. doi: 10.1523/JNEUROSCI.1763-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung SK, Bode A, Cushion TD, Thomas RH, Hunt C, Wood SE, Pickrell WO, Drew CJ, Yamashita S, Shiang R, Leiz S, Longardt AC, Raile V, Weschke B, Puri RD, Verma IC, Harvey RJ, Ratnasinghe DD, Parker M, Rittey C, et al. GLRB is the third major gene of effect in hyperekplexia. Hum Mol Genet. 2013;22:927–940. doi: 10.1093/hmg/dds498. [DOI] [PubMed] [Google Scholar]
- Conti E, Izaurralde E. Nonsense-mediated mRNA decay: molecular insights and mechanistic variations across species. Curr Opin Cell Biol. 2005;17:316–325. doi: 10.1016/j.ceb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Coto E, Armenta D, Espinosa R, Argente J, Castro MG, Alvarez V. Recessive hyperekplexia due to a new mutation (R100H) in the GLRA1 gene. Mov Disord. 2005;20:1626–1629. doi: 10.1002/mds.20637. [DOI] [PubMed] [Google Scholar]
- Gotti C, Hanke W, Schlue WR, Briscini L, Moretti M, Clementi F. A functional alpha-bungarotoxin receptor is present in chick cerebellum: purification and characterization. Neuroscience. 1992;50:117–127. doi: 10.1016/0306-4522(92)90386-G. [DOI] [PubMed] [Google Scholar]
- Griffon N, Büttner C, Nicke A, Kuhse J, Schmalzing G, Betz H. Molecular determinants of glycine receptor subunit assembly. EMBO J. 1999;18:4711–4721. doi: 10.1093/emboj/18.17.4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grudzinska J, Schemm R, Haeger S, Nicke A, Schmalzing G, Betz H, Laube B. The beta subunit determines the ligand binding properties of synaptic glycine receptors. Neuron. 2005;45:727–739. doi: 10.1016/j.neuron.2005.01.028. [DOI] [PubMed] [Google Scholar]
- Haeger S, Kuzmin D, Detro-Dassen S, Lang N, Kilb M, Tsetlin V, Betz H, Laube B, Schmalzing G. An intramembrane aromatic network determines pentameric assembly of Cys-loop receptors. Nat Struct Mol Biol. 2010;17:90–98. doi: 10.1038/nsmb.1721. [DOI] [PubMed] [Google Scholar]
- Hibbs RE, Gouaux E. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature. 2011;474:54–60. doi: 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbs RE, Sulzenbacher G, Shi J, Talley TT, Conrod S, Kem WR, Taylor P, Marchot P, Bourne Y. Structural determinants for interaction of partial agonists with acetylcholine binding protein and neuronal alpha7 nicotinic acetylcholine receptor. EMBO J. 2009;28:3040–3051. doi: 10.1038/emboj.2009.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooft RW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- James VM, Bode A, Chung SK, Gill JL, Nielsen M, Cowan FM, Vujic M, Thomas RH, Rees MI, Harvey K, Keramidas A, Topf M, Ginjaar I, Lynch JW, Harvey RJ. Novel missense mutations in the glycine receptor beta subunit gene (GLRB) in startle disease. Neurobiol Dis. 2013;52:137–149. doi: 10.1016/j.nbd.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kling C, Koch M, Saul B, Becker CM. The frameshift mutation oscillator (Glra1(spd-ot)) produces a complete loss of glycine receptor alpha1-polypeptide in mouse central nervous system. Neuroscience. 1997;78:411–417. doi: 10.1016/S0306-4522(96)00567-2. [DOI] [PubMed] [Google Scholar]
- Lecker SH, Goldberg AL, Mitch WE. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol. 2006;17:1807–1819. doi: 10.1681/ASN.2006010083. [DOI] [PubMed] [Google Scholar]
- Mazzo F, Pistillo F, Grazioso G, Clementi F, Borgese N, Gotti C, Colombo SF. Nicotine-modulated subunit stoichiometry affects stability and trafficking of alpha3beta4 nicotinic receptor. J Neurosci. 2013;33:12316–12328. doi: 10.1523/JNEUROSCI.2393-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pless SA, Millen KS, Hanek AP, Lynch JW, Lester HA, Lummis SC, Dougherty DA. A cation-pi interaction in the binding site of the glycine receptor is mediated by a phenylalanine residue. J Neurosci. 2008;28:10937–10942. doi: 10.1523/JNEUROSCI.2540-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pless SA, Hanek AP, Price KL, Lynch JW, Lester HA, Dougherty DA, Lummis SC. A cation-pi interaction at a phenylalanine residue in the glycine receptor binding site is conserved for different agonists. Mol Pharmacol. 2011;79:742–748. doi: 10.1124/mol.110.069583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raussens V, Ruysschaert JM, Goormaghtigh E. Protein concentration is not an absolute prerequisite for the determination of secondary structure from circular dichroism spectra: a new scaling method. Anal Biochem. 2003;319:114–121. doi: 10.1016/S0003-2697(03)00285-9. [DOI] [PubMed] [Google Scholar]
- Raussens V, Ruysschaert JM, Goormaghtigh E. Erratum to “Protein concentration is not an absolute prerequisite for the determination of secondary structure from circular dichroism spectra: A new scaling method”. Anal Biochem. 2006;359:150. doi: 10.1016/j.ab.2006.09.010. [DOI] [PubMed] [Google Scholar]
- Rees MI, Harvey K, Pearce BR, Chung SK, Duguid IC, Thomas P, Beatty S, Graham GE, Armstrong L, Shiang R, Abbott KJ, Zuberi SM, Stephenson JB, Owen MJ, Tijssen MA, van den Maagdenberg AM, Smart TG, Supplisson S, Harvey RJ. Mutations in the gene encoding GlyT2 (SLC6A5) define a presynaptic component of human startle disease. Nat Genet. 2006;38:801–806. doi: 10.1038/ng1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riganti L, Matteoni C, Di Angelantonio S, Nistri A, Gaimarri A, Sparatore F, Canu-Boido C, Clementi F, Gotti C. Long-term exposure to the new nicotinic antagonist 1,2-bisN-cytisinylethane upregulates nicotinic receptor subtypes of SH-SY5Y human neuroblastoma cells. Br J Pharmacol. 2005;146:1096–1109. doi: 10.1038/sj.bjp.0706434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross AF, Green WN, Hartman DS, Claudio T. Efficiency of acetylcholine receptor subunit assembly and its regulation by cAMP. J Cell Biol. 1991;113:623–636. doi: 10.1083/jcb.113.3.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadtler S, Laube B, Lashub A, Nicke A, Betz H, Schmalzing G. A basic cluster determines topology of the cytoplasmic M3–M4 loop of the glycine receptor alpha1 subunit. J Biol Chem. 2003;278:16782–16790. doi: 10.1074/jbc.M213077200. [DOI] [PubMed] [Google Scholar]
- Sánchez R, Sali A. Comparative protein structure modeling. Introduction and practical examples with modeller. Methods Mol Biol. 2000;143:97–129. doi: 10.1385/1-59259-368-2:97. [DOI] [PubMed] [Google Scholar]
- Schaefer N, Langlhofer G, Kluck CJ, Villmann C. Glycine receptor mouse mutants: model systems for human hyperekplexia. Br J Pharmacol. 2013;170:933–952. doi: 10.1111/bph.12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger C, Christopeit T, Fuchs K, Grote K, Sieghart W, Danielson UH. Histaminergic pharmacology of homo-oligomeric beta3 gamma-aminobutyric acid type A receptors characterized by surface plasmon resonance biosensor technology. Biochem Pharmacol. 2012;84:341–351. doi: 10.1016/j.bcp.2012.04.008. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CH, Chang FC, Su YC, Tsai FJ, Lu MK, Lee CC, Kuo CC, Yang YW, Lu CS. Two novel mutations of the glycine receptor gene in a Taiwanese hyperekplexia family. Neurology. 2004;63:893–896. doi: 10.1212/01.WNL.0000138566.65519.67. [DOI] [PubMed] [Google Scholar]
- Unterer B, Becker CM, Villmann C. The importance of TM3–4 loop subdomains for functional reconstitution of glycine receptors by independent domains. J Biol Chem. 2012;287:39205–39215. doi: 10.1074/jbc.M112.376053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergouwe MN, Tijssen MA, Peters AC, Wielaard R, Frants RR. Hyperekplexia phenotype due to compound heterozygosity for GLRA1 gene mutations. Ann Neurol. 1999;46:634–638. doi: 10.1002/1531-8249(199910)46:4<634::AID-ANA12>3.0.CO%3B2-9. [DOI] [PubMed] [Google Scholar]
- Villmann C, Oertel J, Melzer N, Becker CM. Recessive hyperekplexia mutations of the glycine receptor alpha1 subunit affect cell surface integration and stability. J Neurochem. 2009a;111:837–847. doi: 10.1111/j.1471-4159.2009.06372.x. [DOI] [PubMed] [Google Scholar]
- Villmann C, Oertel J, Ma-Högemeier ZL, Hollmann M, Sprengel R, Becker K, Breitinger HG, Becker CM. Functional complementation of Glra1(spd-ot), a glycine receptor subunit mutant, by independently expressed C-terminal domains. J Neurosci. 2009b;29:2440–2452. doi: 10.1523/JNEUROSCI.4400-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel N, Kluck CJ, Melzer N, Schwarzinger S, Breitinger U, Seeber S, Becker CM. Mapping of disulfide bonds within the amino-terminal extracellular domain of the inhibitory glycine receptor. J Biol Chem. 2009;284:36128–36136. doi: 10.1074/jbc.M109.043448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanamaker CP, Green WN. Endoplasmic reticulum chaperones stabilize nicotinic receptor subunits and regulate receptor assembly. J Biol Chem. 2007;282:31113–31123. doi: 10.1074/jbc.M705369200. [DOI] [PMC free article] [PubMed] [Google Scholar]