

Abstract
In enterohemorrhagic Escherichia coli (EHEC), sigma factor N (σN) regulates glutamate-dependent acid resistance (GDAR) and the locus of enterocyte effacement (LEE); discrete genetic systems that are required for transmission and virulence of this intestinal pathogen. Regulation of these systems requires nitrogen regulatory protein C, NtrC, and is a consequence of NtrC-σN-dependent reduction in the activity of sigma factor S (σS). This study elucidates pathway components and stimuli for σN-directed regulation of GDAR and the LEE in EHEC. Deletion of fliZ, the product of which reduces σS activity, phenocopied rpoN (σN) and ntrC null strains for GDAR and LEE control, acid resistance, and adherence. Upregulation of fliZ by NtrC-σN was shown to be indirect and required an intact flagellar regulator flhDC. Activation of flhDC by NtrC-σN and FlhDC-dependent regulation of GDAR and the LEE was dependent on σN-promoter flhDP2, and a newly described NtrC upstream activator sequence. Addition of ammonium chloride significantly altered expression of GDAR and LEE, acid resistance, and adherence, independently of rpoN, ntrC, and the NtrC sensor kinase, ntrB. Altering the availability of NtrC phosphodonor acetyl phosphate by growth without glucose, with acetate addition, or by deletion of acetate kinase ackA, abrogated NtrC-σN-dependent control of flhDC, fliZ, GDAR, and the LEE.
Keywords: acid resistance, EHEC O157, H7, LEE, NtrC, rpoN, sigma factor N
Introduction
Alternative sigma factor N (σN) when bound to RNA polymerase directs the transcription of genes for carbon and nitrogen metabolism, stress fitness, and regulation (Reitzer and Schneider 2001). In an increasing number of bacterial pathogens, σN also regulates genes for virulence and transmission, and is required for complete in vivo disease progression (Okada et al. 2008; Barchiesi et al. 2009; Albert-Weissenberger et al. 2010; Damron et al. 2012; Iyer and Hancock 2012; Mills et al. 2012; Sheng et al. 2012; Wang et al. 2012). For most pathogens, the mechanism underlying σN-dependent regulation of pathogenesis remains unknown; two exceptions to this include Borrelia burgdorferi, and to a lesser extent, enterohemorrhagic Escherichia coli (EHEC). In B. burgdorferi, the causative agent of Lyme borreliosis, σN activates the expression of genes encoding outer surface lipoproteins (OspA and OspC) essential for transmission from the tick vector to a mammalian host, and for establishment of infection (Pal et al. 2000; Hubner et al. 2001; Grimm et al. 2004). This Osp activation pathway requires another sigma factor, σS, the transcription of which is directly activated from a σN-promoter in what has been dubbed a σN-σS regulatory cascade (Smith et al. 2007; He et al. 2008). In EHEC serotype O157:H7, a food-borne pathogen attributed to outbreaks and sporadic cases of bloody diarrhea (hemorrhagic colitis) (Rangel et al. 2005), σN (encoded by rpoN) represses transcription of glutamate-dependent acid resistance (GDAR) genes, while activating the locus of enterocyte effacement (LEE) pathogenicity island (Riordan et al. 2010). The GDAR system allows for low oral infectious dose during gastric passage (Chart 2000; Teunis et al. 2004), while the LEE encodes a type III secretion (T3S) apparatus that translocates virulence factors into host intestinal cells mediating intimate adherence and immune subversion (McDaniel and Kaper 1997; Elliott et al. 1998; Perna et al. 1998). Thus, σN in EHEC regulates major determinants of fecal–oral transmission and colonization.
Like B. burgdorferi, a σN-σS regulatory pathway has been described for EHEC, and has been further implicated in the control of GDAR and LEE genes in this pathogen (Riordan et al. 2010). However for EHEC, the underlying mechanism by which σS is regulated is not completely understood. σS controls the expression of hundreds of genes in E. coli (Hengge-Aronis 2002); it is an activator of GDAR system genes (gad genes) (Ma et al. 2003), and can both activate and repress the LEE (Iyoda and Watanabe 2005; Tomoyasu et al. 2005; Laaberki et al. 2006). Strains null for rpoN are characterized by a phenotype of increased GDAR and decreased LEE expression that is dependent on an intact rpoS (encoding σS), and while deletion of rpoN in either EHEC or laboratory E. coli strain K-12 MG1655 has no impact on rpoS transcription, both the stability and activity of σS have been shown to increase (Riordan et al. 2010; Dong et al. 2011; Mitra et al. 2012). Mitra et al. (2012) demonstrated that this effect of σN on σS stability/activity is indirect and dependent on transcription from a σN promoter, and not competition of these sigma factors for core RNA polymerase (RNAP). What additional regulatory component(s) is required downstream of σN for control of σS, GDAR, and the LEE is not yet known. Unlike other E. coli sigma factors, the initiation of transcription by σN requires activation by enhancer-binding proteins (EBP) that communicate various environmental signals to the RNAP-σN holoenzyme complex (EσN) (Shingler 1996). Of the 11 EBPs encoded within the EHEC O157:H7 background, only deletion of ntrC (encoding NtrC) phenotypically reproduces the rpoN null background for control of σS, GDAR, and the LEE (Mitra et al. 2012). Nitrogen regulatory protein C, NtrC (also NRI), is the response regulator of a two-component system that activates σN-dependent transcription of genes for the assimilation and utilization of nitrogen, relieving slowed growth under nitrogen-limiting conditions (Zimmer et al. 2000). It is thus plausible that nitrogen availability plays a fundamental role in activation of the σN-σS regulatory pathway in EHEC. The objective of this study was to identify additional regulatory factors required for σN-dependent control of σS, acid resistance, and the LEE, and to examine the role for nitrogen availability in the stimulation of this pathway. The study identifies flhD and fliZ as new genetic determinants of this pathway and provides evidence that NtrC-σN-FlhDC-dependent activation of fliZ, the product of which modulates σS activity, is needed for regulation of GDAR and the LEE. Furthermore, the availability of acetyl phosphate, not ammonia, is shown to be an important factor for pathway activation.
Experimental Procedures
Bacterial strains and growth conditions
All strains and plasmids used in this study are listed in Table1. Luria-Bertani (LB) starter cultures were inoculated with a single colony of each strain and grown at 37°C with shaking (200 rpm) to an optical density at 600 nm (OD600) of 0.5. Unless otherwise indicated, these cultures were used to inoculate either Dulbecco's Modified Eagle's Medium (DMEM) (Sigma-Aldrich, St. Louis, MO) buffered with 50 mmol/L 3-morpholinopropane-1-sulfonic acid (MOPS) and containing 0.4% (w/v) glucose, or MOPS minimal medium. MOPS medium was prepared as described in Neidhardt et al.'s (1974) study, and contained 0.4% (w/v) glucose, 0.1% (w/v) NH4Cl, and 0.1% (w/v) l-glutamine. Cultures were grown for 18–20 h before inoculating into fresh DMEM or MOPS to a final OD600 = 0.05, respectively, using a 1:10 ratio of media-to-flask volume and grown at 37°C, 200 rpm. Appropriate antibiotics were added to cultures as required.
Table 1.
Strains and plasmids used in this study.
| Strain/Plasmid | Relevant characteristics | Source/Reference |
|---|---|---|
| Strain name | ||
| DH5α | Vector propagation, recA1 endA1 | |
| BL-21 | Miroux and Walker (1996) | |
| TW14359 | WT O157:H7 2006 outbreak, western US (NC_013008.1) | Manning et al. (2008) |
| EcRPF-6 | TW14359ΔrpoN | Mitra et al. (2012) |
| EcRAM-26 | TW14359ΔntrC | Mitra et al. (2012) |
| EcRAM-43 | TW14359ΔrpoNGln+, suppressor mutant for Gln auxotrophy | This study |
| EcRAM-45 | EcRAM-43ΔglnA | This study |
| EcRAM-47 | TW14359crl::kan KanR | This study |
| EcRAM-49 | TW14359ΔfliZ | This study |
| EcRAM-51 | EcRFP-6 pRAM-3 AmpR | This study |
| EcRAM-52 | EcRAM 26 pRAM-3 AmpR | This study |
| EcRAM-53 | EcRAM 49 pRAM-3 AmpR | This study |
| EcRAM-58 | TW14359ΔflhDC | This study |
| EcRAM-59 | EcRAM 58 pRAM-4 AmpR | This study |
| EcRAM-60 | EcRAM 58 pRAM-5 AmpR | This study |
| EcRAM-61 | EcRAM 58 pRAM-6 AmpR | This study |
| EcRAM-63 | TW14359ΔackA | This study |
| EcRAM-66 | TW14359ΔfliZpRAM-8 | This study |
| EcRAM-68 | TW14359ΔackApRAM-9 | This study |
| Plasmid name | ||
| pACYC177 | Low-copy cloning vector, AmpR KanR P15A | Chang and Cohen (1978) |
| pET-24d | IPTG-inducible His-tagging vector, KanR | Novagen |
| pBAD22 | Mid-copy arabinose-inducible cloning vector, AmpR | Guzman et al. (1995) |
| pSC-B | High-copy cloning vector, AmpR KanR | StrataClone |
| pBAD-TA | Mid-copy arabinose-inducible cloning vector, AmpR | Invitrogen |
| pRAM-1 | rpoN::pACYC177, AmpR KanS | Mitra et al. (2012) |
| pRAM-3 | flhDC::pBAD22, AmpR | This study |
| pRAM-4 | flhDC::pACYC177 positions +948 to −1994 relative to start codon | This study |
| pRAM-5 | flhDC::pACYC177 positions +948 to −825 relative to start codon | This study |
| pRAM-6 | flhDC::pACYC177 positions +948 to −728 relative to start codon | This study |
| pRAM-7 | ntrC::pET-24d containing ORF, KanR | This study |
| pRAM-8 | fliZ::pBAD, AmpR | This study |
| pRAM-9 | ackA::pSC-B, AmpR KanR | This study |
Procedures for genetic manipulation
Nonpolar gene deletion mutants were constructed using the λ Red recombinase-assisted approach (Datsenko and Wanner 2000; Murphy and Campellone 2003) and as described previously (Riordan et al. 2010). Primers used for the construction of deletion mutants are listed in Table S1. For overexpression of flhDC, a 932-bp polymerase chain reaction (PCR) fragment containing flhDC of strain TW14359 (nucleotide positions 2485400–2484469) was generated using primers flhDC-F/EcoRI and flhDC-R/XbaI. An EcoRI/XbaI digested fragment of the product was cloned into similarly digested arabinose-inducible expression vector pBAD22 (Guzman et al. 1995) to produce pRAM-3. pRAM-3 purified from DH5α transformants was then used to transform TW14359 and derivative strains producing EcRAM-51 through EcRAM-53. For flhDC promoter expression studies, a 2942-bp XhoI/BamHI digested PCR fragment (nucleotide positions 2487394–2484453) was generated using primers flhD-1994/XhoI and flhC+595/BamHI. This fragment contained the flhDC open reading frames (ORFs) and 1994 bp of DNA upstream of the flhD start codon including a σN promoter (2486152–2486138), a σ70 promoter (2485633–2485604), and a predicted NtrC box (2487152–2487132). This was ligated into XhoI/BamHI digested pACYC177 to produce pRAM-4. The same approach was used for pRAM-5 and pRAM-6 construction, however, the cloned fragment in pRAM-5, generated using primers flhD-825/XhoI and flhD+595/BamHI (positions 2486228-2484453), did not include the predicted NtrC box. For the fragment in pRAM-6, generated using primers flhD-728/XhoI and flhD+595/BamHI (positions 2486128–2484453), both the NtrC box and the σN promoter were excluded. Plasmids were purified from DH5α transformants and used to transform TW14359ΔflhDC producing strains EcRAM-59 to EcRAM-61. For fliZ complementation, a 552-bp PCR fragment containing the fliZ ORF was created using primers fliZ-Clone/F and fliZ-Clone/R and cloned into the arabinose-inducible pBAD-TA vector (Invitrogen, Grand Island, NY) to yield pRAM-8, which was then used to transform EcRAM-49 to produce EcRAM-66. For ackA complementation, the ackA ORF was amplified using primers ackA-Clone/F and ackA-Clone/R and cloned into the high copy pSC-B vector (Agilent, Santa Clara, CA) to create pRAM-9, which was then used to transform EcRAM-63 to produce EcRAM-68. The rpoN complement strain EcRAM-36 was constructed previously (Mitra et al. 2012). All genetic constructs were validated using a combination of restriction mapping, DNA sequencing, and quantitative real-time PCR (qRT-PCR).
Quantitative real-time PCR
RNA purification, cDNA synthesis, qRT-PCR cycling conditions, and data analysis for relative quantitation of gene expression followed previously described protocols (Riordan et al. 2010; Mitra et al. 2012; Morgan et al. 2013). Analysis was performed using a Realplex2 Mastercycler (Eppendorf, Hauppauge, NY). Cycle threshold (Ct) data were normalized to rrsA (16S rRNA gene) and normalized Ct values (ΔCt) were transformed to arbitrary gene expression units using 2−ΔCt/10−6 as described by Livak and Schmittgen (2001). A previous method was used for the quantitation of flhD mRNA copy number (Bustin 2000). Briefly, a 154-bp PCR product containing flhD was generated using flhD+63 and flhD+216, column purified (Qiagen, Valencia, CA) and serially diluted in molecular grade water. Ct was measured for each dilution to generate a standard curve plotting Ct as a linear function of DNA concentration (ng/μL). The strength of linearity was estimated by the correlation coefficient (r2), which exceeded 0.90 for all curves. DNA concentration was extrapolated from a standard curve using experimental Ct values and then converted to flhD copy number based on the estimated weight of a single 154-bp flhD dsDNA fragment of 47-kDa. Gene expression levels and flhD copy number were compared between samples using the appropriate t-test or by analysis of variance (ANOVA) and Tukey's HSD (n ≥ 3, α = 0.05) using R v. 2.13.0.
Protein extraction, sodium dodecyl sulfate polyacrylamide gel electrophoresis, and western blots
Protein extraction, purification, and procedures for western blots followed a previously described protocol (Mitra et al. 2012; Morgan et al. 2013). Monoclonal antibodies for σS and GroEL were acquired from Neoclone (Madison, WI) and Bio-Rad (Carlsbad, CA), respectively. Densitometry was used to estimate differences in protein levels using a ChemiDoc XRS+ Imaging System and Image Lab 3.0 (Bio-Rad, Hercules, CA). Western blots were repeated a minimum of three times in independent trials.
Purification of NtrC
A 1425-bp NcoI/XhoI-digested PCR fragment generated using primers ntrC+F/NcoI and ntrC-R/XhoI was cloned into similarly digested pET-24d producing pRAM7 and replacing the ntrC stop codon with a C-terminal 6xHis tag. pRAM7 was transformed into propagating E. coli strain BL-21, which was grown in LB containing ampicillin (100 μg/mL) to OD600 = 0.4 before induction of 6xHis-tagged ntrC with 1 mmol/L Isopropyl β-D-1-thiogalactopyranoside for 16 h at 20°C (200 rpm). Cultures were harvested by centrifugation (5000g, 20 min) and 6xHis-NtrC was purified using a nickel Ni-NTA Protein Purification Kit (Qiagen) according to the manufacturer's instruction.
Electrophoretic mobility shift assay
Electrophoretic mobility shift assay (EMSA) was performed using the LightShift Chemiluminescence EMSA Kit (Pierce, Rockford, IL) according to the manufacturer's instruction. Biotin end-labeled DNA probes were generated by PCR using flhD-1842/Biotin and flhD-1634/Biotin for the flhDP promoter probe, and glnA-311/Biotin and glnA-112/Biotin for the glnAP2 promoter probe; biotin end-labeled Epstein–Barr nuclear antigen (EBNA) DNA was supplied with the kit. The flhDP promoter probe (strain TW14359 nucleotide position 2487034–2487242) contained a putative NtrC-binding site flanked by 0.1 kb. For the glnAP2 promoter probe, a confirmed NtrC box (nucleotide position 4913213–4913228) was flanked by 0.1 kb. Binding reactions (20 μL per reaction) contained 20 fmol of biotin end-labeled DNA probe, 50 mmol/L KCl, 5 mmol/L MgCl2, 1% (v/v) glycerol, 0.05% (v/v) NP-40, 50 ng/μL poly(dI-dC) copolymer competitor, 10x molar excess Bovine Serum Albumin (10 mg/mL), and 0, 2, 4, 8, or 16 μmol/L purified C-terminally labeled 6xHis-NtrC. Reactions were incubated for 40 min at 4°C, and were then separated by electrophoresis using 8% nondenaturing acrylamide gels prepared in 0.5x Tris-borate-EDTA buffer at 4°C for 80 min at 160 V, and DNA/protein complexes transferred to a nylon membrane (Fisher, Pittsburgh, PA). Membranes were UV cross-linked at 120,000 mJ/cm2 for 1 min and detected by chemiluminescence using the Biotin Detection System (Pierce) and a ChemiDoc XRS+ Imaging System including Image Lab 3.0 (Bio-Rad, Hercules, CA).
Selection of suppressor mutants for glutamine auxotrophy
Spontaneous suppressor mutants for glutamine auxotrophy were selected in the TW14359ΔrpoN background by growth in MOPS minimal media without the addition of glutamine. Briefly, overnight cultures of TW14359ΔrpoN grown in MOPS media were inoculated into fresh MOPS containing 0.4% glucose and 0.1% NH4Cl and grown at 37°C (200 rpm). The outgrowth of suppressor mutants (TW14359ΔrpoN Gln+) consistently occurred following 48-h incubation. Single colonies of suppressor mutants were obtained by subculture from MOPS media to LB with 1.5% agar, and confirmed by growth in MOPS containing 0.2% glucose and 0.1% (w/v) l-histidine as described by Reitzer et al. (1987) and by qRT-PCR analysis of glutamine synthetase glnA expression. Three independent suppressor mutants were selected and validated by this approach. The mutation leading to suppression was determined using a combination of PCR and Sanger sequencing of amplified DNA fragments (MWG Operon, Huntsville, AL) and next-generation whole genome sequencing.
Whole genome next-generation DNA sequencing and analysis
Genomic DNA was extracted from TW14359ΔrpoN and a single suppressor mutant of TW14359ΔrpoN (TW14359ΔrpoN Gln+) using Puregene® Kits (Gentra, Minneapolis, MN). One microgram of DNA from each strain was enzymatically sheared into libraries of ∼200-bp fragments using the Ion Xpress™ Plus Fragment Library Kit (Life Technologies, Grand Island, NY). Each DNA library was purified using the E-Gel® SizeSelect™ 2% Agarose system (Invitrogen), and the integrity and quantity of each was determined using a Bioanalyzer high-sensitivity DNA chip (Agilent). Libraries were diluted and template-positive Ion Sphere Particles (ISPs) prepared using the Ion OneTouch 200 Template Kit (Life Technologies). ISPs were sequenced using an IonTorrent™ Personal Genome Machine and the Ion PGM 200 Sequencing Kit (Life Technologies) following the manufacturer's instructions. Whole genome sequencing data were exported from the Ion Torrent Server and analyzed using the Genomics Suite software package (CLC Bio, Boston, MA). Genomes were assembled using the TW14359 genome (NC_013008, NCBI) as a reference, followed by quality-based variant detection to identify polymorphisms with a minimum coverage of 10x and 100% detection frequency. Polymorphisms common to both strains (relative to the reference TW14359 genome), and those in homopolymeric nucleotide tracts, were excluded resulting in the identification of specific genetic variations between TW14359ΔrpoN and TW14359ΔrpoN Gln+.
Adherence assay
Adherence to epithelial cells was determined following a previously described protocol (Morgan et al. 2013). Briefly, human HT-29 colonic epithelial cells were grown to confluence on polylysine-treated glass coverslips placed within the wells of 24-well culture plates at 37°C with 5% CO2. Overnight DMEM cultures were diluted 1:40 (v/v) in fresh DMEM and 0.05 mL of this dilution was used to inoculate each well which already contained 0.45 mL of sterile DMEM. After 3 h, plate wells were washed five times with PBS (137 mmol/L NaCl, 2.7 mmol/L KCl, 10 mmol/L Na2HPO4, pH 7) to remove nonadherent bacteria from the coverslips, and fresh DMEM was then added before incubating for an additional 3 h. Plate wells were subsequently washed three times in PBS, and then fixed with ice cold (−20°C) 100% methanol for 10 min before staining with Giemsa diluted in PBS 1:20 (v/v) for 20 min. Giemsa-stained coverslips were examined at 1000× magnification by oil immersion, and microcolonies were scored as discrete clusters of five or more bacterial cells as previously defined (McKee and O'Brien 1995; Abe et al. 2002; Iyoda and Watanabe 2004). For each sample, a minimum of 10 viewing frames were observed and the average number of microcolonies were reported per 50 HT-29 cells. Microcolony counts were compared between strains by Tukey's HSD following a significant F-test (n ≥ 3, α = 0.05) (R v. 2.13.0).
Tests for acid resistance
Acid resistance by the glutamate-dependent system was measured for exponential phase cultures grown in DMEM as previously described (Riordan et al. 2010; Mitra et al. 2012) with slight adaptations. Strains were grown in DMEM to OD600 = 0.5 before inoculating (106 CFU/mL final cell density) into E minimal glucose (EG) media containing 5.7 mmol/L l-glutamate adjusted with HCl to pH 7 (control) or pH 2. Cultures were sampled for counts (CFU/mL) after 1 h incubation at 37°C (200 rpm) by plating serial dilutions to LB with 1.5% agar and incubating overnight. Experiments were repeated a minimum of three times in independent trials.
Results
NtrC-σN require fliZ for control of σS activity, GDAR, and the LEE
Previous studies have revealed NtrC and σN negatively regulates GDAR and positively regulates the LEE by reducing the activity of alternative sigma factor S (σS) (Riordan et al. 2010; Mitra et al. 2012). For this to occur, NtrC-σN must increase or decrease the expression of a gene(s) whose product, in-turn, alters σS-dependent transcription. One of two proteins were predicted to fulfill this role: Crl or FliZ. Crl enhances RNAP-σS holoenzyme formation, thus increasing transcription from σS promoters (Pratt and Silhavy 1998; Typas et al. 2007), whereas FliZ interferes with σS promoter-binding and transcription initiation, thus reducing σS-dependent transcription (Pesavento et al. 2008; Pesavento and Hengge 2012). During growth in DMEM (OD600 = 0.5), both crl and fliZ expression were shown to be reduced in TW14359ΔntrC and TW14359ΔrpoN when compared to TW14359 (P < 0.05) (Fig.1A), however, only TW14359ΔfliZ phenocopied TW14359ΔntrC and TW14359ΔrpoN for the control of GDAR and LEE genes (Fig.1B). In TW14359Δcrl, both gadE and gadB were increased in expression compared to TW14359 (P < 0.05), but less than for TW14359ΔrpoN, TW14359ΔntrC, and TW14359ΔfliZ, in which expression levels for all genes were nearly identical (P < 0.01) (Fig.1B). The expression of LEE genes ler, tir, espA, and cesT did not differ between TW14359 and TW14359Δcrl, but were uniformly reduced in TW14359ΔntrC, TW14359ΔrpoN, and TW14359ΔfliZ backgrounds (P < 0.05) (Fig.1B). Both gadE and ler expressions were restored to near wild-type levels in fliZ complement strain TW14359ΔfliZpRAM-8 or by the deletion of rpoS in TW14359ΔfliZ (Fig.1B). Consistent with the effect of fliZ deletion on gadE and gadB expression, CFU/mL of TW14359ΔfliZ recovered following exposure to acidified (pH 2) EG media for 1 h increased by 10- to 100-fold compared to TW14359, TW14359ΔfliZpRAM-8, and TW14359ΔfliZΔrpoS, yet remained ∼10-fold less than that observed for TW14359ΔntrC and TW14359ΔrpoN (Fig.1C). Furthermore, the ability to form microcolonies on HT-29 intestinal cells was decreased in TW14359ΔfliZ compared to TW14359 (P = 002), and matched that observed for TW14359ΔntrC and TW14359ΔrpoN (Fig.1D). Thus, NtrC-σN positively regulate fliZ during exponential growth, the product of which is predicted, downregulates GDAR and upregulates the LEE by reducing the activity of extant σS.
Figure 1.
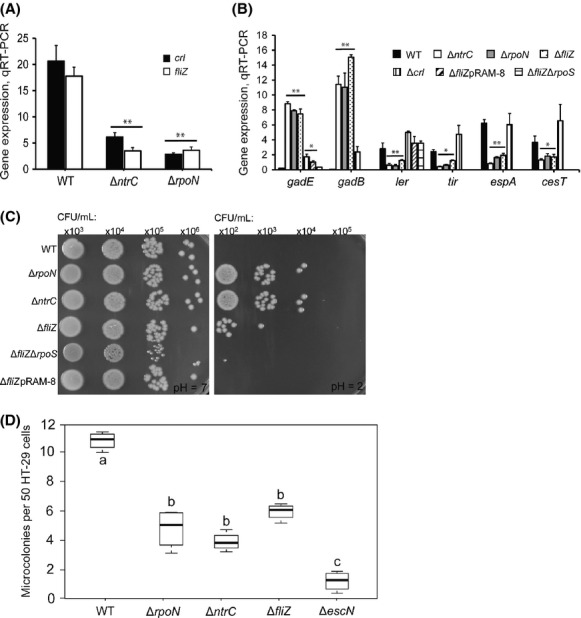
Effect of fliZ deletion on GDAR and LEE expression, acid resistance, and adherence. (A) Expression of crl (black) and fliZ (white) plotted for wild-type and derivative strains. (B) Expression of GDAR (gadE, gadB) and LEE (ler, tir, espA, cesT) genes plotted for wild type (black), ΔntrC (white), ΔrpoN (gray), ΔfliZ (stippled), and Δcrl (vertical lines); in strains ΔfliZpRAM-8 (diagonal) and ΔfliZΔrpoS (dashed), only gadE and ler were measured. (C) Representative colony-forming units (CFU/mL) on LBA for wild-type and derivative strains following 1-h challenge in EG media (pH 7 vs. pH 2). (D) Counts for microcolonies on HT-29 cells plotted for wild-type and mutant derivative strains. For A and B, asterisks denote significant differences between wild-type and derivative strains by t-test (*P < 0.05, **P < 0.01, n ≥ 3). Error bars denote standard deviation. For D, boxplot boundaries represent the 25th and 75th percentiles, whiskers represent the maximum and minimum values, and the median is given by the horizontal line. Plots that differ in lowercase letter differ significantly by Tukey's HSD following a significant F-test (n ≥ 3, P < 0.05). GDAR, glutamate-dependent acid resistance; LEE, locus of enterocyte effacement; EG, E minimal glucose.
Requirement for flhDC in the activation of fliZ by NtrC-σN
fliZ is encoded as the second gene of a three gene operon (fliAZY), the transcription of which is directed from at least two promoters, fliAP1 and fliAP2. Neither of these promoters are σN-dependent, however, fliAP1 is activated by the regulator of flagellar biosynthesis and motility FlhDC, for which there is a predicted σN-dependent promoter, flhDP2 (Zhao et al. 2010). In addition, a putative activator sequence (UAS) for NtrC is present ∼1-kb upstream of flhDP2. It was thus hypothesized that the control of fliZ by NtrC-σN is a consequence of direct activation of flhDC transcription from this promoter.
In agreement with this, flhDC expression was similarly reduced in both TW14359ΔntrC and TW14359ΔrpoN backgrounds compared to TW14359 during growth in DMEM (OD600 = 0.5) (P < 0.05) (Fig.2A). Also, flhDC significantly decreased gadE levels and increased ler levels when overexpressed in TW14359ΔntrC and TW14359ΔrpoN (P < 0.05), but not in TW14359ΔfliZ (Fig.2B). To define cis-elements of the flhDC promoter region important for NtrC-σN-dependent regulation, flhDC mRNA copy number was measured from three promoter fragments (Fig.2C) cloned into arabinose-inducible vector pBAD22 and transformed into TW14359ΔflhDC. As anticipated, flhDC copy number was reduced when expressed from a fragment in which the putative NtrC UAS was removed (Frag. II) compared to the wild-type flhDC promoter fragment (Frag. I) (P = 0.004) (Fig.2C). flhDC copy number was further reduced when expressed from a fragment in which both the NtrC UAS and putative σN promoter flhDCP2 were removed (Frag. III), but not significantly less than for Frag. II. Correspondingly, gadE expression increased (P < 0.01) and ler expression decreased (P < 0.05) in TW14359ΔflhDC expressing either Frag. II or Frag. III when compared to Frag. I (Fig.2C). Thus, the putative NtrC UAS site and σN promoter flhDCP2 are required for full expression of flhDC and for regulation of gadE and ler. Purified 6xHis-NtrC was observed to retard the mobility by EMSA of a 200-bp flhD promoter probe containing the putative NtrC UAS in a manner similar to the NtrC–dependent glutamine synthetase promoter, glnAP2 (Fig.2D). No shift was observed for flhD or glnA promoter probes in the absence of 6xHis-NtrC, or for the negative control EBNA DNA probe (Fig.2D).
Figure 2.
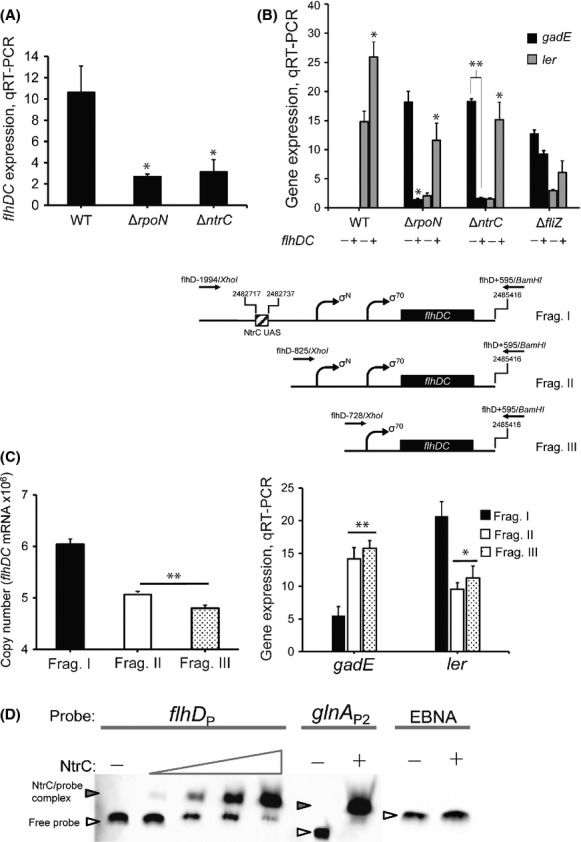
Regulation of flhDC by NtrC-σN and effect on gadE and ler expression. (A) Expression levels for flhDC plotted for wild-type and derivative strains. The asterisk denotes a significant difference between wild-type and mutated strains by t-test (P < 0.05, n ≥ 3). (B) Expression levels for gadE (black) and ler (gray) plotted for wild-type and derivative strains containing pRAM-3 (flhDC::pBAD22); expression of pRAM-3 is either uninduced (−) or induced (+) with arabinose. Asterisks indicate significant differences between uninduced and induced treatments by t-test (*P < 0.05, **P < 0.01, n ≥ 3). (C) Absolute flhDC mRNA copy number and expression levels for gadE and ler measured in the ΔflhDC background expressing cloned flhDC fragments, Frag. I (black), Frag. II (white), and Frag. III (stippled); topology of flhDC promoter fragments are included, top right (C). See text for details. (D) EMSA for NtrC binding to the flhDCP promoter and glnAP2 promoter; EBNA is EBNA DNA. Inset arrows indicate the location of the NtrC/probe complex (filled arrow) or free probe (empty arrow). See text for details. Error bars denote standard deviation for all panels. EMSA, electrophoretic mobility shift assay.
Glutamine is not essential for NtrC-σN-dependent regulation of GDAR and the LEE
The preceding experiments reveal NtrC-σN to directly activate flhDC transcription, the product of which upregulates fliZ. FliZ, in-turn, reduces the activity of σS and consequently, σS-dependent control of GDAR and LEE expression. While much is understood as to how σS regulates GDAR and the LEE (Sperandio et al. 1999; Foster 2004; Iyoda and Watanabe 2005; Laaberki et al. 2006), the mechanistic basis for activation of NtrC-σN-dependent control of these discrete genetic systems is as yet unknown. NtrC-σN direct the transcription of nitrogen-regulated (Ntr) response genes, the primary function of which is to assimilate nitrogen through induction of transport/scavenging systems and nitrogen degradation pathways (reviewed in Reitzer and Schneider 2001). Under these conditions, glutamine synthetase (GS) catalyzes the synthesis of l-glutamine from ammonia and l-glutamate. The gene for GS (glnA) is maximally expressed from the σN promoter glnAP2 in a manner dependent on NtrC. As such, strains that are null for rpoN or ntrC cannot initiate transcription from glnAP2 and are auxotrophic for glutamine when nitrogen is limiting. The significance of glutamine metabolism to NtrC-σN-dependent control of GDAR and the LEE was thus examined by selecting a suppressor mutant of glutamine auxotrophy in TW14359ΔrpoN and observing its effect on GDAR and LEE gene expression, acid resistance, and adherence. Growth of TW14359ΔrpoN in MOPS media containing 0.2% glucose and 0.1% l-histidine (i.e., high energy but nitrogen limiting) is impaired due to auxotrophy for glutamine (Gln−) (Fig.3A). However, after 48 h the outgrowth of a prototrophic (Gln+) suppressor mutant (TW14359ΔrpoNGln+) was repeatedly observed in which wild-type growth in MOPS media was restored (Fig.3A), and in which the expression of glnA was significantly increased compared to TW14359ΔrpoN during growth in DMEM (OD600 = 0.5) (P = 0.013) (Fig.3B); glnA expression was still, however, slightly but significantly lower in TW14359ΔrpoNGln+ when compared to TW14359 (P = 0.02). Mutations which suppress Gln− in E. coli have been mapped to ntrC, and to cis-elements controlling glnA transcription. glnA can be transcribed from three promoters: glnAP1 and glnAP3 are σ70 promoters that are repressed by NtrC during nitrogen-limitation, whereas glnAP2 is a σN promoter that is activated by NtrC under the same conditions. Mutations in the DNA-binding domain of NtrC (amino acid residues 400–470) at the C-terminus result in the derepression of glnAP1 and/or glnAP3, while mutations in the promoter(s) enhance transcription from glnAP1 or result in formation of a de novo σ70 consensus at glnAP2 (Reitzer et al. 1987). DNA sequencing of ntrC and the glnA promoter region did not reveal any of these described mutations in TW14359ΔrpoNGln+. Sequencing of the TW14359ΔrpoNGln+ genome, however, revealed a single adenine deletion in the ntrC ORF at nucleotide position 4,910,080 (accession NC_013008, NCBI), resulting in a frameshift mutation. This mutation occurs early in the ORF at +285 relative to the start codon and results in a premature stop codon or opal (UGA) mutation at amino acid position 106. It was thus suspected that increased expression of glnA, and growth in the absence of glutamine for TW14359ΔrpoNGln+ (Fig.3B), reflects derepression at the glnAP1 and glnAP3 promoters due to NtrC inactivation.
Figure 3.
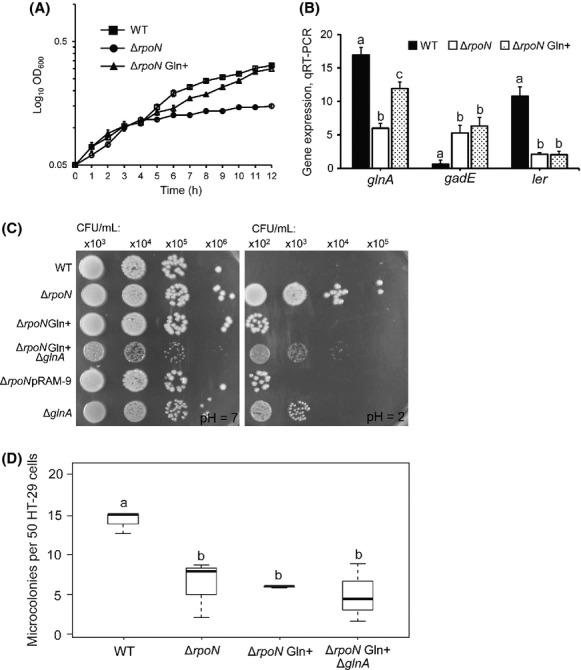
Impact of glutamine metabolism on the GDAR and LEE expression phenotype of TW14359ΔrpoN. (A) Mean (n = 3) log10 transformed optical density at 600 nm (log10 OD600) plotted for wild type (square), ΔrpoN (circles), and suppressor mutant ΔrpoNGln+ (triangles) as a function of time during growth in nitrogen-limiting MOPS media (2 g/L glucose, 1 g/L l-histidine, pH 7). (B) Expression levels for glnA, gadE, and ler plotted for wild type (black), ΔrpoN (white), and ΔrpoNGln+ (stippled). Error bars denote standard deviation for A and B. (C) Representative colony-forming units (CFU/mL) on LBA for wild-type and derivative strains following 1-h challenge in EG media (pH 7 vs. pH 2). (D) Counts for microcolonies on HT-29 cells plotted for wild-type and mutant derivative strains. Boxplots are as described for Figure1D. For B and D, plots that differ in lowercase letter for each gene (B) or strain (D) differ significantly by Tukey's HSD following a significant F-test (n ≥ 3, P < 0.05). GDAR, glutamate-dependent acid resistance; LEE, locus of enterocyte effacement.
Expression levels for gadE and ler did not differ between TW14359ΔrpoN and TW14359ΔrpoNGln+ during growth in DMEM (OD600 = 0.5), indicating that glutamine availability has no impact on GDAR and LEE gene regulation. Interestingly, however, CFU/mL recovered from acidified EG media were decreased by ∼1000-fold for TW14359ΔrpoNGln+ when compared to TW14359ΔrpoN (Fig.3C). Deletion of glnA in TW14359ΔrpoNGln+ again restored survival in acid comparable to that of TW14359ΔrpoN (Fig.3C), suggesting that glutamine synthetase production plays an indirect role in EHEC acid resistance. Overexpression of glnA in TW14359ΔrpoN (strain TW14359ΔrpoNpRAM-9) similarly mitigated the acid resistance phenotype of TW14359ΔrpoN (Fig.3C), clearly demonstrating a role for glnA in the complete acid resistance phenotype of TW14359ΔrpoN. Adding to this, CFU/mL recovered from acidified EG increased by ≥100-fold in TW14359ΔglnA compared to TW14359. Consistent with qRT-PCR data on ler (Fig.3A), the number of microcolonies formed on HT-29 cells in TW14359ΔrpoNGln+ was significantly reduced when compared to TW14359 (P < 0.05), but did not differ from TW14359ΔrpoN or TW14359ΔrpoNΔglnAGln+, collectively suggesting that changes in glutamine availability has no effect on σN-dependent LEE expression and adherence to intestinal cells (Fig.3D).
Acetyl phosphate stimulates the NtrC-σN-pathway controlling GDAR and LEE expression
When E. coli is cultivated in media without ammonia, intracellular levels of glutamine are low, culminating in the phosphorylation and activation of NtrC by sensor kinase NtrB and NtrC-σN-dependent transcription. It was thus suspected that the absence of ammonia in DMEM may prompt NtrC-σN-dependent transcription of flhDC, activating the pathway for GDAR and LEE regulation, and that supplementation of DMEM with ammonia would offset this effect. If so, ammonia would be expected to stimulate gad gene expression and repress the LEE in TW14359, but to have no effect in the TW14359ΔrpoN and TW14359ΔntrC backgrounds.
While the addition of ammonium chloride (2 g/L NH4Cl) was observed to slightly but not significantly increase GDAR gene (gadE and gadB) expression in TW14359, expression in TW14359ΔntrC and TW14359ΔrpoN uniformly decreased (P < 0.05) (Fig.4A). Correspondingly, ammonium addition reduced CFU/mL recovered for TW14359ΔntrC and TW14359ΔrpoN by ∼100- to 1000-fold but had no observable effect on CFU/mL recovered for TW14359 (Fig.4B). For the LEE, ammonium addition increased ler, tir, espA, and cesT expression in all backgrounds (Fig.4C) and correspondingly increased the number of microcolonies formed on HT-29 cells for all strains (P < 0.05). The same observations were made when substituting equimolar ammonium sulfate for ammonium chloride (data not shown). These results reveal that ammonium does in fact influence GDAR and LEE gene expression, but by a mechanism that is independent of ntrC and rpoN. In support of these data, the expression of pathway components (gadE, ler, flhDC, and fliZ) for control of GDAR and the LEE were not altered in a strain deleted for the NtrC cognate sensor kinase, ntrB (Fig. S1). Interestingly, growth in DMEM containing ammonium was observed to significantly reduce rpoS expression in TW14359, TW14359ΔntrC, and TW14359ΔrpoN (P < 0.01), while having no impact on flhDC or fliZ expression in these backgrounds (Fig.4E). This reduction in rpoS transcript levels correlated with a reduction in σS levels in all backgrounds with ammonium, however, σS levels were not as strongly reduced in TW14359ΔrpoN when compared to TW14359 or TW14359ΔntrC (Fig.4F).
Figure 4.

Role for ammonium in the NtrC-σN-dependent pathway controlling GDAR and the LEE. (A) Expression levels for gadE (filled) and gadB (empty) without (−) and with (+) the addition of NH4Cl plotted for wild-type and derivative strains; asterisks denote significant difference between treatments by t-test (*P < 0.05, **P < 0.01, n≥3). (B) Representative colony-forming units (CFU/mL) on LBA for wild-type and derivative strains grown without (−NH4) or with (+NH4) NH4Cl added to DMEM and following 1-h challenge in EG media (pH 7 vs. pH 2). (C) Expression levels for ler (black), tir (white), espA (stippled), and cesT (gray) for wild-type and derivative strains grown without (−NH3) or with (+NH3) NH4Cl added to DMEM. (D) Counts for microcolonies on HT-29 cells plotted for wild-type and mutant derivative strains grown without (−NH4) or with (+NH4) NH4Cl. Boxplots are as described for Figure1D. (E) Expression levels of flhDC, fliZ, and rpoS plotted for wild type (black), ΔntrC (white), and ΔrpoN (stippled). (F) Representative western blot for σS and GroEL (control) in wild type, ΔrpoN, and ΔntrC grown without (−) or with (+) NH4Cl added to DMEM. For A, C, and E, asterisks denote significant differences between treatments by t-test (*P < 0.05, **P < 0.01, n ≥ 3). For D, plots that differ in lowercase letter differ significantly by Tukey's HSD following a significant F-test (n ≥ 3, P < 0.05). Error bars denote standard deviation. GDAR, glutamate-dependent acid resistance; LEE, locus of enterocyte effacement.
Feng et al. (1992) demonstrated phosphotransfer to, and activation of, NtrC in E. coli by the small molecule phosphodonor acetyl phosphate (acetyl∼P). Acetyl∼P readily accumulates during growth on glucose or in the presence of excess acetate, but not during growth on glycerol (McCleary and Stock 1994; Wolfe 2005). It was thus of interest to determine the effect of glucose and acetyl∼P availability on NtrC-σN-dependent control of pathway components for the regulation of GDAR and the LEE. During growth in MOPS media containing glucose (2 g/L) and NH4Cl (1 g/L) (OD600 = 0.5), the expression of flhDC, fliZ, and ler was decreased and gadB increased in TW14359ΔntrC and TW14359ΔrpoN when compared to TW14359 (P < 0.05) (Fig.5A), similar to that observed during growth in DMEM media (Figs.1A, B and 2A). Substituting 0.2% (v/v) glycerol for glucose as the sole carbon source reduced flhDC, fliZ, and ler expression in TW14359 and rpoN complement strain TW14359ΔrpoNpRAM-1 (P < 0.05), but not in TW14359ΔntrC and TW14359ΔrpoN (Fig.5A). Likewise, glycerol substitution increased gadB expression in TW14359 and TW14359ΔrpoNpRAM-1 (P < 0.05), but not in TW14359ΔntrC and TW14359ΔrpoN. The addition of sodium acetate (2 g/L) to glycerol treatments restored flhDC, fliZ, and ler expression to levels observed for glucose in TW14359, however, gadB expression was slightly but not significantly increased when compared to glycerol treatments (Fig.5A). In TW14359ΔntrC and TW14359ΔrpoN, acetate was still observed to generally increase fliZ, flhDC, and ler expression, yet had no impact on gadB expression in these backgrounds, which may reflect a more generalized, ntrC- and rpoN-independent effect of acetate on the expression of these genes. To further examine the effect of acetate and acetyl∼P availability on this regulatory pathway, gadB and ler expressions were measured in a strain null for acetate kinase (ackA), the product of which catalyzes the interconversion of acetate to acetyl∼P (Rose et al. 1954). In TW14359ΔntrC, TW14359ΔrpoN, and TW14359ΔackA, gadB expression was significantly and uniformly increased when compared to TW14359 (P < 0.01) (Fig.5B). Complementation with ackA (strain TW14359ΔackApRAM-8) restored gadB expression to wild-type levels. For ler, expression was similarly reduced in TW14359ΔackA, TW14359ΔntrC, and TW14359ΔrpoN when compared to TW14359 and TW14359ΔackApRAM-8 (P < 0.05). Together, these data provide evidence that regulation of GDAR and the LEE by NtrC-σN is insensitive to changes in nitrogen availability (i.e., glutamine/ammonium), but instead is influenced by the availability of acetyl∼P.
Figure 5.
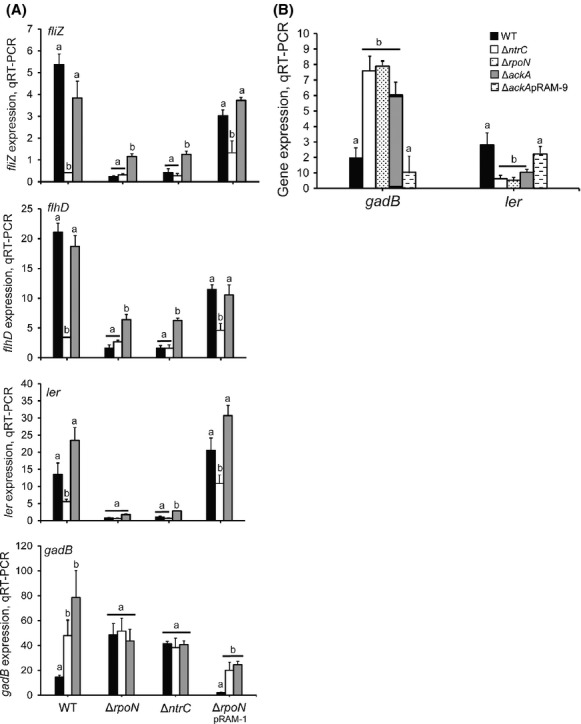
The effect of acetyl∼P availability on the expression of essential components for σN-dependent regulation of GDAR and the LEE. (A) Expression levels of genes in order from bottom to top: gadE, ler, flhD, and fliZ plotted for wild-type and derivative strains grown in MOPS with glucose (black), glycerol (white), or glycerol and acetate (gray). (B) gadE and ler expression levels plotted for wild-type (black), ΔntrC (white), ΔrpoN (stippled), ΔackA (gray), and ackA complement strain ΔackApRAM-8 (hatched). Plots that differ in lowercase letter for each strain (A) or gene (B) differ significantly by Tukey's HSD following a significant F-test (n ≥ 3, P < 0.05). Error bars denote standard deviation. GDAR, glutamate-dependent acid resistance; LEE, locus of enterocyte effacement.
Discussion
In the present study, NtrC and σN have been shown to positively regulate the expression of crl and fliZ, the products of which control the activity of σS. It is predicted that of the two, only FliZ is a required component of the σN pathway controlling σS, GDAR, and the LEE. What impact crl upregulation in TW14359ΔrpoN has on σS, if any, is as yet unclear. Crl and FliZ play antagonistic roles in the regulation of σS. Crl directly binds σS facilitating interaction with RNA polymerase and holoenzyme (EσS) formation (Bougdour et al. 2004), whereas FliZ acts downstream of EσS formation, binding to the -10 box of σS promoters (Pesavento and Hengge 2012) precluding promoter recognition by EσS. Thus, FliZ may be dominant to Crl in σN-directed control of σS activity. Alternatively, Crl reduces σS stability in an RssB-dependent manner during all stages of growth (Typas et al. 2007). It is therefore plausible that the increased stability of σS in rpoN null backgrounds (Dong et al. 2011; Mitra et al. 2012) results from reduced crl expression. This is consistent with the observation that in TW14359ΔrpoN the GDAR and LEE expression phenotype cannot be reproduced by increasing σS stability alone (Mitra et al. 2012).
The transcription of fliZ is largely determined by FlhDC, a global regulator of motility genes (Francez-Charlot et al. 2003). FlhD forms a heterodimer with FlhC, directly activating transcription of the fliAZY operon from the σ70-dependent promoter fliAP. This study determined that flhDC was required for σN-directed regulation of GDAR and LEE genes in a manner that was dependent on an intact fliZ. Based on our results, it is predicted that NtrC and σN directly activate transcription of flhDC during exponential growth in DMEM (4 g/L glucose, with no NH4) requiring the putative σN-promoter flhDP2, and a newly identified NtrC box at positions 2481732–2487152 (Fig.6). This NtrC box is nearly identical to the predicted NtrC consensus (Ferro-Luzzi Ames and Nikaido 1985), differing by a single nucleotide in the dyad repeat region. Upregulation of FlhDC leads to increased transcription of fliZ (Francez-Charlot et al. 2003), the product of which decreases the activity of σS (Pesavento and Hengge 2012) (Fig.6). This suggests that during exponential growth NtrC-σN keep the activity of extant σS in check by increasing FlhDC-dependent transcription of fliZ. One consequence of this reduced σS activity in EHEC is an increase in LEE expression (Riordan et al. 2010) and correspondingly, increased in vitro microcolony formation. This could occur by at least two discrete mechanisms: by upregulation of pchA or through the downregulation of gadE (Fig.6). PchA is a LEE activator that is negatively regulated by σS (Iyoda and Watanabe 2005), whereas GadE represses the LEE and is activated by σS through upregulation of gadX (Ma et al. 2003). While the involvement of PchA in this pathway cannot be ruled out, only gadE and gadX expressions are significantly altered in the rpoN null background (Riordan et al. 2010). Even though FlhDC has been shown to effect adherence in E. coli, until now, the association has been negative. Leatham et al. (2005) reported that the deletion of flhDC in E. coli K-12 increased colonization of a mouse, while constitutive expression of flhDC in another study, reduced adherence of EHEC to HeLa cells (Iyoda et al. 2006). As the former study is in the K-12 MG1655 background, the effect of flhDC on colonization is clearly LEE independent. For EHEC, however, flhDC and the LEE are known to be inversely regulated; expression of LEE-encoded GrlA downregulates flhDC and motility in a manner dependent on RcsB, a response regulator of the Rcs phosphorelay system (Iyoda et al. 2006; Morgan et al. 2013). Perhaps FlhDC is used by σN to initiate transcription of the LEE, and then is repressed as GrlA accumulates as part of a GrlA-RcsB feedback loop initiating intimate adherence. This would be consistent with the transience and growth-phase dependence of σN-dependent regulation of the LEE (Mitra et al. 2012).
Figure 6.
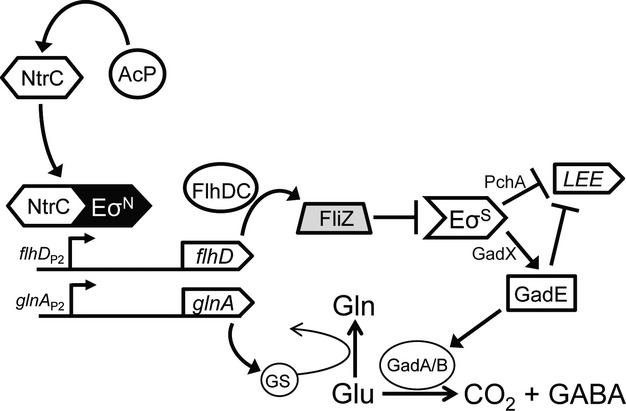
Model predicting NtrC-σN-dependent regulation of GDAR and the LEE. During exponential growth in DMEM (a nitrogen-limiting media), NtrC activates transcription from σN-dependent promoters for flhD and glnA. FlhDC (regulator of flagellar biosynthesis) directly activates fliZ, the product of which reduces the activity of σS-RNAP (EσS) holoenzyme. σS indirectly downregulates LEE expression by repressing the LEE activator pchA by an unknown mechanism, while upregulating the GDAR activator gadE through increased transcription of gadX. GadE has also been shown to directly repress transcription of ler. The upregulation of glnA (encoding glutamine synthetase, GS) increases the conversion of extant glutamate (Glu) to glutamine (Gln), thus depleting the substrate for GDAR system decarboxylases (GadA/GadB) and the potential for proton scavenging and acid detoxification. Acetyl∼P (AcP) is a noncognate phosphodonor that can activate NtrC-dependent transcription from σN promoters for flhD and glnA. The model is an amalgam of experimental observations inferred from this and previous studies (Reitzer et al. 1989; Feng et al. 1992; Tomoyasu et al. 2005; Kailasan Vanaja et al. 2009; Zhao et al. 2010; Lee et al. 2011; Pesavento and Hengge 2012; Branchu et al. 2014). See the text for further details. GDAR, glutamate-dependent acid resistance; LEE, locus of enterocyte effacement.
By reducing the activity of σS, σN also helps to maintain a low level of GDAR gene expression during exponential growth (Fig.6). However, unlike σN-dependent LEE regulation, it is predicted that full expression of the GDAR phenotype in rpoN null strains is a consequence of two discrete but concurrent mechanisms. One requires σS for the activation of GDAR system genes (gad genes), the products of which confer acid resistance by a proton-scavenging mechanism involving the decarboxylation (GadA/GadB decarboxylases) and subsequent protonation of glutamate to yield γ-amino butyric acid (GABA) (Fig.6). In the absence of glutamate, GDAR is defective in protecting E. coli from acid stress (reviewed in Foster 2004). It is this cellular glutamate that is the source of the corresponding mechanism. Specifically, under nitrogen-limiting conditions (ex. growth in DMEM), NtrC-σN activate transcription of glutamine synthetase (glnA), which catalyzes the conversion of glutamate (Glu) to glutamine (Gln) (Fig.6). Strains null for rpoN or ntrC are therefore unable to activate glnA in response to reduced nitrogen availability, leading to glutamate accumulation and auxotrophy for glutamine. These strains are thus characterized by elevated levels of both the components (i.e., gadE, gadA/B, gadC) and substrate (glutamate) for GDAR. This mechanistic duality is reflected in the observation that neither fliZ nor glnA deletion can fully recapitulate the GDAR phenotype of an rpoN null background. Since as many as 60% of σN-regulated genes have been shown to be antagonistically controlled by σS in E. coli (Dong et al. 2011), the interplay of these sigma factors likely has a more global impact on virulence, fitness, and metabolism than simply control of GDAR and the LEE.
The precise activating signal for NtrC-σN-dependent regulation of GDAR and the LEE is as yet unknown. Phosphorylation and activation of NtrC is sensitive to changes in the intracellular levels of glutamine. When E. coli is grown in the absence of ammonium, glutamine levels are low, signaling the phosphorylation of NtrC by its cognate sensor kinase NtrB, and NtrC-dependent activation of σN promoters for nitrogen assimilation (Reitzer 2003). Although the addition of ammonium to DMEM did have a significant impact on GDAR and LEE expression, it did so independently of ntrC and rpoN. This effect of ammonium on the expression of E. coli colonizing factors has been formerly observed in EPEC, as well as for enterotoxigenic E. coli (ETEC). In EPEC, ammonium reduces expression of the bundle-forming pilus genes bfpA and bfpT, and reduces T3S-secretion of the EspA, EspB, and EspC translocon proteins (Puente et al. 1996; Kenny et al. 1997; Martinez-Laguna et al. 1999). For ETEC, ammonium increased expression of the 987P fimbria genes fasH and fasA (Edwards and Schifferli 1997). Changes in EPEC and ETEC colonizing factor expression in response to ammonium correlate with differences in tissue tropism and reflect the availability of ammonium in the intestine; its concentration gradually increases toward the distal small intestine (Toskes 1993; Edwards and Schifferli 1997; Martinez-Laguna et al. 1999). This natural gradient of intestinal ammonium may have a significant influence on the decision for colonization in all E. coli. However for EPEC, repression of bfp was shown to require a trans-acting factor that was absent, or present, but not functional in E. coli K-12 (Martinez-Laguna et al. 1999). How the ammonium signal is communicated to GDAR in EHEC and to the LEE in EHEC and EPEC requires further study.
Based on the findings of this study, it is proposed that NtrC is autophosphorylated by a noncognate phosphodonor in the σN pathway controlling GDAR and the LEE. Acetyl∼P is a plausible candidate (Fig.6), as it is a known NtrC phosphodonor (Feng et al. 1992; Atkinson and Ninfa 1998), and experimental alteration of acetyl∼P levels by substituting either glycerol or glycerol and acetate for glucose, or by the deletion of acetate kinase (ackA), altered the expression of pathway components for regulation of GDAR and the LEE in a manner dependent on rpoN and ntrC. Requirement for acetyl∼P is consistent with the growth-phase dependency of σN for GDAR and LEE regulation. The cellular pool of acetyl∼P during growth with glucose peaks during exponential phase, and drops off precipitously during transition into stationary phase (Takamura and Nomura 1988; Pruss and Wolfe 1994). Correspondingly, control of gad and LEE genes by NtrC and σN is restricted to the mid-exponential phase of growth (Riordan et al. 2010; Mitra et al. 2012). Remarkably, acetyl∼P also serves as a phosphodonor for Rrp2, a σN EBP found in B. burgdorferi and required for activation of the σN-σS pathway regulating virulence expression in this pathogen (Xu et al. 2010). Thus, the use of acetyl∼P for autophosphorylation of σN EBPs may be a phenomenon that is conserved across different species of bacteria. Why acetyl∼P would be used in place of the cognate sensor kinase NtrB in E. coli is not yet known. It has been formerly proposed that the phosphorylation of NtrC by acetyl∼P may be used to initiate transcription of Ntr genes during transition to a nitrogen poor environment, as cellular NtrB levels are very low when nitrogen is abundant (Feng et al. 1992). Yet, in this study ntrB was clearly dispensable for GDAR and LEE regulation when grown in nitrogen-limiting media containing glucose, suggesting that acetyl∼P alone is sufficient to activate this pathway. It remains to be determined if ntrB is required for GDAR and LEE regulation by NtrC-σN in nitrogen-limiting media lacking glucose. The broader significance of this finding is that acetyl∼P levels in E. coli are sensitive to many factors, including nutrients, temperature, anaerobiosis, and pH (Wolfe 2005), suggesting that it may be used by NtrC to communicate various environmental cues to σN.
Conflict of Interest
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Effect of ntrB deletion on the expression of genes for GDAR and LEE control. Gene expression levels plotted for wild type (black), ΔrpoN (white), ΔntrC (hatched), and ΔntrB (gray). Asterisks denote significant difference between wild-type and respective mutants by t-test (*P < 0.05, **P < 0.01, n ≥ 3). Error bars denote standard deviation.
Growth curve for TW14359 and mutant derivative strains in DMEM. Optical density (OD600) is plotted for each strain as a function of time. Samples were taken every hour for 12 h. OD600 measurements differed by less than 5% for each time point and strain.
References
- Abe H, Tatsuno I, Tobe T, Okutani A. Sasakawa C. Bicarbonate ion stimulates the expression of locus of enterocyte effacement-encoded genes in enterohemorrhagic Escherichia coli O157:H7. Infect. Immun. 2002;70:3500–3509. doi: 10.1128/IAI.70.7.3500-3509.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert-Weissenberger C, Sahr T, Sismeiro O, Hacker J, Heuner K. Buchrieser C. Control of flagellar gene regulation in Legionella pneumophila and its relation to growth phase. J. Bacteriol. 2010;192:446–455. doi: 10.1128/JB.00610-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson MR. Ninfa AJ. Role of the GlnK signal transduction protein in the regulation of nitrogen assimilation in Escherichia coli. Mol. Microbiol. 1998;29:431–447. doi: 10.1046/j.1365-2958.1998.00932.x. [DOI] [PubMed] [Google Scholar]
- Barchiesi J, Espariz M, Checa SK. Soncini FC. Downregulation of RpoN-controlled genes protects Salmonella cells from killing by the cationic antimicrobial peptide polymyxin B. FEMS Microbiol. Lett. 2009;291:73–79. doi: 10.1111/j.1574-6968.2008.01437.x. [DOI] [PubMed] [Google Scholar]
- Bougdour A, Lelong C. Geiselmann J. Crl, a low temperature-induced protein in Escherichia coli that binds directly to the stationary phase sigma subunit of RNA polymerase. J. Biol. Chem. 2004;279:19540–19550. doi: 10.1074/jbc.M314145200. [DOI] [PubMed] [Google Scholar]
- Branchu P, Matrat S, Vareille M, Garrivier A, Durand A, Crepin S, et al. NsrR, GadE, and GadX interplay in repressing expression of the Escherichia coli O157:H7 LEE pathogenicity island in response to nitric oxide. PLoS Pathog. 2014;10:e1003874. doi: 10.1371/journal.ppat.1003874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin SA. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 2000;25:169–193. doi: 10.1677/jme.0.0250169. [DOI] [PubMed] [Google Scholar]
- Chang AC. Cohen SN. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J. Bacteriol. 1978;134:1141–1156. doi: 10.1128/jb.134.3.1141-1156.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chart H. VTEC enteropathogenicity. Symp. Ser. Soc. Appl. Microbiol. 2000;88:12S–23S. doi: 10.1111/j.1365-2672.2000.tb05328.x. [DOI] [PubMed] [Google Scholar]
- Damron FH, Owings JP, Okkotsu Y, Varga JJ, Schurr JR, Goldberg JB, et al. Analysis of the Pseudomonas aeruginosa regulon controlled by the sensor kinase KinB and sigma factor RpoN. J. Bacteriol. 2012;194:1317–1330. doi: 10.1128/JB.06105-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA. Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong T, Yu R. Schellhorn H. Antagonistic regulation of motility and transcriptome expression by RpoN and RpoS in Escherichia coli. Mol. Microbiol. 2011;79:375–386. doi: 10.1111/j.1365-2958.2010.07449.x. [DOI] [PubMed] [Google Scholar]
- Edwards RA. Schifferli DM. Differential regulation of fasA and fasH expression of Escherichia coli 987P fimbriae by environmental cues. Mol. Microbiol. 1997;25:797–809. doi: 10.1046/j.1365-2958.1997.5161875.x. [DOI] [PubMed] [Google Scholar]
- Elliott SJ, Wainwright LA, McDaniel TK, Jarvis KG, Deng YK, Lai LC, et al. The complete sequence of the locus of enterocyte effacement (LEE) from enteropathogenic Escherichia coli E2348/69. Mol. Microbiol. 1998;28:1–4. doi: 10.1046/j.1365-2958.1998.00783.x. [DOI] [PubMed] [Google Scholar]
- Feng J, Atkinson MR, McCleary W, Stock JB, Wanner BL. Ninfa AJ. Role of phosphorylated metabolic intermediates in the regulation of glutamine synthetase synthesis in Escherichia coli. J. Bacteriol. 1992;174:6061–6070. doi: 10.1128/jb.174.19.6061-6070.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferro-Luzzi Ames G. Nikaido K. Nitrogen regulation in Salmonella typhimurium. Identification of an ntrC protein-binding site and definition of a consensus binding sequence. EMBO J. 1985;4:539–547. doi: 10.1002/j.1460-2075.1985.tb03662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster JW. Escherichia coli acid resistance: tales of an amateur acidophile. Nat. Rev. Microbiol. 2004;2:898–907. doi: 10.1038/nrmicro1021. [DOI] [PubMed] [Google Scholar]
- Francez-Charlot A, Laugel B, Van Gemert A, Dubarry N, Wiorowski F, Castanie-Cornet MP, et al. RcsCDB His-Asp phosphorelay system negatively regulates the flhDC operon in Escherichia coli. Mol. Microbiol. 2003;49:823–832. doi: 10.1046/j.1365-2958.2003.03601.x. [DOI] [PubMed] [Google Scholar]
- Grimm D, Tilly K, Byram R, Stewart PE, Krum JG, Bueschel DM, et al. Outer-surface protein C of the Lyme disease spirochete: a protein induced in ticks for infection of mammals. Proc. Natl. Acad. Sci. USA. 2004;101:3142–3147. doi: 10.1073/pnas.0306845101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman LM, Belin D, Carson MJ. Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Oman T, Xu H, Blevins J, Norgard MV. Yang XF. Abrogation of ospAB constitutively activates the Rrp2-RpoN-RpoS pathway (sigmaN-sigmaS cascade) in Borrelia burgdorferi. Mol. Microbiol. 2008;70:1453–1464. doi: 10.1111/j.1365-2958.2008.06491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengge-Aronis R. Signal transduction and regulatory mechanisms involved in control of the sigma(S) (RpoS) subunit of RNA polymerase. Microbiol. Mol. Biol. Rev. 2002;66:373–395. doi: 10.1128/MMBR.66.3.373-395.2002. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner A, Yang X, Nolen DM, Popova TG, Cabello FC. Norgard MV. Expression of Borrelia burgdorferi OspC and DbpA is controlled by a RpoN-RpoS regulatory pathway. Proc. Natl. Acad. Sci. USA. 2001;98:12724–12729. doi: 10.1073/pnas.231442498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer VS. Hancock LE. Deletion of sigma(54) (rpoN) alters the rate of autolysis and biofilm formation in Enterococcus faecalis. J. Bacteriol. 2012;194:368–375. doi: 10.1128/JB.06046-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyoda S. Watanabe H. Positive effects of multiple pch genes on expression of the locus of enterocyte effacement genes and adherence of enterohaemorrhagic Escherichia coli O157: H7 to HEp-2 cells. Microbiology. 2004;150:2357–2571. doi: 10.1099/mic.0.27100-0. [DOI] [PubMed] [Google Scholar]
- Iyoda S. Watanabe H. ClpXP protease controls expression of the type III protein secretion system through regulation of RpoS and GrlR levels in enterohemorrhagic Escherichia coli. J. Bacteriol. 2005;187:4086–4094. doi: 10.1128/JB.187.12.4086-4094.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyoda S, Koizumi N, Satou H, Lu Y, Saitoh T, Ohnishi M, et al. The GrlR-GrlA regulatory system coordinately controls the expression of flagellar and LEE-encoded type III protein secretion systems in enterohemorrhagic Escherichia coli. J. Bacteriol. 2006;188:5682–5692. doi: 10.1128/JB.00352-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kailasan Vanaja S, Bergholz TM. Whittam TS. Characterization of the Escherichia coli O157:H7 Sakai GadE regulon. J. Bacteriol. 2009;191:1868–1877. doi: 10.1128/JB.01481-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny B, Abe A, Stein M. Finlay BB. Enteropathogenic Escherichia coli protein secretion is induced in response to conditions similar to those in the gastrointestinal tract. Infect. Immun. 1997;65:2606–2612. doi: 10.1128/iai.65.7.2606-2612.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laaberki MH, Janabi N, Oswald E. Repoila F. Concert of regulators to switch on LEE expression in enterohemorrhagic Escherichia coli O157:H7: interplay between Ler, GrlA, HNS and RpoS. Int. J. Med. Microbiol. 2006;296:197–210. doi: 10.1016/j.ijmm.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Leatham MP, Stevenson SJ, Gauger EJ, Krogfelt KA, Lins JJ, Haddock TL, et al. Mouse intestine selects nonmotile flhDC mutants of Escherichia coli MG1655 with increased colonizing ability and better utilization of carbon sources. Infect. Immun. 2005;73:8039–8049. doi: 10.1128/IAI.73.12.8039-8049.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YY, Barker CS, Matsumura P. Belas R. Refining the binding of the Escherichia coli flagellar master regulator, FlhD4C2, on a base-specific level. J. Bacteriol. 2011;193:4057–4068. doi: 10.1128/JB.00442-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ. Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Ma Z, Richard H. Foster JW. pH-Dependent modulation of cyclic AMP levels and GadW-dependent repression of RpoS affect synthesis of the GadX regulator and Escherichia coli acid resistance. J. Bacteriol. 2003;185:6852–6859. doi: 10.1128/JB.185.23.6852-6859.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning SD, Motiwala AS, Springman AC, Qi W, Lacher DW, Ouellette LM, et al. Variation in virulence among clades of Escherichia coli O157:H7 associated with disease outbreaks. Proc. Natl. Acad. Sci. USA. 2008;105:4868–4873. doi: 10.1073/pnas.0710834105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Laguna Y, Calva E. Puente JL. Autoactivation and environmental regulation of bfpT expression, the gene coding for the transcriptional activator of bfpA in enteropathogenic Escherichia coli. Mol. Microbiol. 1999;33:153–166. doi: 10.1046/j.1365-2958.1999.01460.x. [DOI] [PubMed] [Google Scholar]
- McCleary WR. Stock JB. Acetyl phosphate and the activation of two-component response regulators. J. Biol. Chem. 1994;269:31567–31572. [PubMed] [Google Scholar]
- McDaniel TK. Kaper JB. A cloned pathogenicity island from enteropathogenic Escherichia coli confers the attaching and effacing phenotype on E. coli K-12. Mol. Microbiol. 1997;23:399–407. doi: 10.1046/j.1365-2958.1997.2311591.x. [DOI] [PubMed] [Google Scholar]
- McKee ML. O'Brien AD. Investigation of enterohemorrhagic Escherichia coli O157:H7 adherence characteristics and invasion potential reveals a new attachment pattern shared by intestinal E. coli. Infect. Immun. 1995;63:2070–2074. doi: 10.1128/iai.63.5.2070-2074.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills DC, Gundogdu O, Elmi A, Bajaj-Elliott M, Taylor PW, Wren BW, et al. Increase in Campylobacter jejuni invasion of intestinal epithelial cells under low-oxygen coculture conditions that reflect the in vivo environment. Infect. Immun. 2012;80:1690–1698. doi: 10.1128/IAI.06176-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miroux B. Walker JE. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 1996;260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- Mitra A, Fay PA, Morgan JK, Vendura KW, Versaggi SL. Riordan JT. Sigma factor N, liaison to an ntrC and rpoS dependent regulatory pathway controlling acid resistance and the LEE in enterohemorrhagic Escherichia coli. PLoS One. 2012;7:e46288. doi: 10.1371/journal.pone.0046288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JK, Vendura KW, Stevens SM. Riordan JT. RcsB determines the locus of enterocyte effacement (LEE) expression and adherence phenotype of Escherichia coli O157:H7 spinach outbreak strain TW14359 and coordinates bicarbonate-dependent LEE activation with repression of motility. Microbiology. 2013;159:2342–2353. doi: 10.1099/mic.0.070201-0. [DOI] [PubMed] [Google Scholar]
- Murphy KC. Campellone KG. Lambda Red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol. Biol. 2003;4:11. doi: 10.1186/1471-2199-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neidhardt FC, Bloch PL. Smith DF. Culture medium for enterobacteria. J. Bacteriol. 1974;119:736–747. doi: 10.1128/jb.119.3.736-747.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Makino S, Okada N, Asakura H, Yamamoto S. Igimi S. Identification and analysis of the osmotolerance associated genes in Listeria monocytogenes. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk. Assess. 2008;25:1089–1094. doi: 10.1080/02652030802056634. [DOI] [PubMed] [Google Scholar]
- Pal U, de Silva AM, Montgomery RR, Fish D, Anguita J, Anderson JF, et al. Attachment of Borrelia burgdorferi within Ixodes scapularis mediated by outer surface protein A. J. Clin. Invest. 2000;106:561–569. doi: 10.1172/JCI9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perna NT, Mayhew GF, Posfai G, Elliott S, Donnenberg MS, Kaper JB, et al. Molecular evolution of a pathogenicity island from enterohemorrhagic Escherichia coli O157:H7. Infect. Immun. 1998;66:3810–3817. doi: 10.1128/iai.66.8.3810-3817.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesavento C. Hengge R. The global repressor FliZ antagonizes gene expression by sigmaS-containing RNA polymerase due to overlapping DNA binding specificity. Nucleic Acids Res. 2012;40:4783–4793. doi: 10.1093/nar/gks055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesavento C, Becker G, Sommerfeldt N, Possling A, Tschowri N, Mehlis A, et al. Inverse regulatory coordination of motility and curli-mediated adhesion in Escherichia coli. Genes Dev. 2008;22:2434–2446. doi: 10.1101/gad.475808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt LA. Silhavy TJ. Crl stimulates RpoS activity during stationary phase. Mol. Microbiol. 1998;29:1225–1236. doi: 10.1046/j.1365-2958.1998.01007.x. [DOI] [PubMed] [Google Scholar]
- Pruss BM. Wolfe AJ. Regulation of acetyl phosphate synthesis and degradation, and the control of flagellar expression in Escherichia coli. Mol. Microbiol. 1994;12:973–984. doi: 10.1111/j.1365-2958.1994.tb01085.x. [DOI] [PubMed] [Google Scholar]
- Puente JL, Bieber D, Ramer SW, Murray W. Schoolnik GK. The bundle-forming pili of enteropathogenic Escherichia coli: transcriptional regulation by environmental signals. Mol. Microbiol. 1996;20:87–100. doi: 10.1111/j.1365-2958.1996.tb02491.x. [DOI] [PubMed] [Google Scholar]
- Rangel JM, Sparling PH, Crowe C, Griffin PM. Swerdlow DL. Epidemiology of Escherichia coli O157:H7 outbreaks, United States, 1982-2002. Emerg. Infect. Dis. 2005;11:603–609. doi: 10.3201/eid1104.040739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitzer L. Nitrogen assimilation and global regulation in Escherichia coli. Annu. Rev. Microbiol. 2003;57:155–176. doi: 10.1146/annurev.micro.57.030502.090820. [DOI] [PubMed] [Google Scholar]
- Reitzer L. Schneider BL. Metabolic context and possible physiological themes of sigma(54)-dependent genes in Escherichia coli. Microbiol. Mol. Biol. Rev. 2001;65:422–444. doi: 10.1128/MMBR.65.3.422-444.2001. and, table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitzer LJ, Bueno R, Cheng WD, Abrams SA, Rothstein DM, Hunt TP, et al. Mutations that create new promoters suppress the sigma 54 dependence of glnA transcription in Escherichia coli. J. Bacteriol. 1987;169:4279–4284. doi: 10.1128/jb.169.9.4279-4284.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitzer LJ, Movsas B. Magasanik B. Activation of glnA transcription by nitrogen regulator I (NRI)-phosphate in Escherichia coli: evidence for a long-range physical interaction between NRI-phosphate and RNA polymerase. J. Bacteriol. 1989;171:5512–5522. doi: 10.1128/jb.171.10.5512-5522.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan JT, Tietjen JA, Walsh CW, Gustafson JE. Whittam TS. Inactivation of alternative sigma factor 54 (RpoN) leads to increased acid resistance, and alters locus of enterocyte effacement (LEE) expression in Escherichia coli O157: H7. Microbiology. 2010;156:719–730. doi: 10.1099/mic.0.032631-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose IA, Grunberg-Manago M, Korey SR. Ochoa S. Enzymatic phosphorylation of acetate. J. Biol. Chem. 1954;211:737–756. [PubMed] [Google Scholar]
- Sheng L, Gu D, Wang Q, Liu Q. Zhang Y. Quorum sensing and alternative sigma factor RpoN regulate type VI secretion system I (T6SSVA1) in fish pathogen Vibrio alginolyticus. Arch. Microbiol. 2012;194:379–390. doi: 10.1007/s00203-011-0780-z. [DOI] [PubMed] [Google Scholar]
- Shingler V. Signal sensing by sigma 54-dependent regulators: derepression as a control mechanism. Mol. Microbiol. 1996;19:409–416. doi: 10.1046/j.1365-2958.1996.388920.x. [DOI] [PubMed] [Google Scholar]
- Smith AH, Blevins JS, Bachlani GN, Yang XF. Norgard MV. Evidence that RpoS (sigmaS) in Borrelia burgdorferi is controlled directly by RpoN (sigma54/sigmaN) J. Bacteriol. 2007;189:2139–2144. doi: 10.1128/JB.01653-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperandio V, Mellies JL, Nguyen W, Shin S. Kaper JB. Quorum sensing controls expression of the type III secretion gene transcription and protein secretion in enterohemorrhagic and enteropathogenic Escherichia coli. Proc. Natl. Acad. Sci. USA. 1999;96:15196–15201. doi: 10.1073/pnas.96.26.15196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamura Y. Nomura G. Changes in the intracellular concentration of acetyl-CoA and malonyl-CoA in relation to the carbon and energy metabolism of Escherichia coli K12. J. Gen. Microbiol. 1988;134:2249–2253. doi: 10.1099/00221287-134-8-2249. [DOI] [PubMed] [Google Scholar]
- Teunis P, Takumi K. Shinagawa K. Dose response for infection by Escherichia coli O157:H7 from outbreak data. Risk Anal. 2004;24:401–407. doi: 10.1111/j.0272-4332.2004.00441.x. [DOI] [PubMed] [Google Scholar]
- Tomoyasu T, Takaya A, Handa Y, Karata K. Yamamoto T. ClpXP controls the expression of LEE genes in enterohaemorrhagic Escherichia coli. FEMS Microbiol. Lett. 2005;253:59–66. doi: 10.1016/j.femsle.2005.09.020. [DOI] [PubMed] [Google Scholar]
- Toskes PP. Bacterial overgrowth of the gastrointestinal tract. Adv. Intern. Med. 1993;38:387–407. [PubMed] [Google Scholar]
- Typas A, Barembruch C, Possling A. Hengge R. Stationary phase reorganisation of the Escherichia coli transcription machinery by Crl protein, a fine-tuner of sigmas activity and levels. EMBO J. 2007;26:1569–1578. doi: 10.1038/sj.emboj.7601629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Liu E, Song S, Wang X, Zhu Y, Ye J, et al. Characterization of Edwardsiella tarda rpoN: roles in sigma(70) family regulation, growth, stress adaption and virulence toward fish. Arch. Microbiol. 2012;194:493–504. doi: 10.1007/s00203-011-0786-6. [DOI] [PubMed] [Google Scholar]
- Wolfe AJ. The acetate switch. Microbiol. Mol. Biol. Rev. 2005;69:12–50. doi: 10.1128/MMBR.69.1.12-50.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Caimano MJ, Lin T, He M, Radolf JD, Norris SJ, et al. Role of acetyl-phosphate in activation of the Rrp2-RpoN-RpoS pathway in Borrelia burgdorferi. PLoS Pathog. 2010;6:e1001104. doi: 10.1371/journal.ppat.1001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Liu M. Burgess RR. Promoter and regulon analysis of nitrogen assimilation factor, sigma54, reveal alternative strategy for E. coli MG1655 flagellar biosynthesis. Nucleic Acids Res. 2010;38:1273–1283. doi: 10.1093/nar/gkp1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer DP, Soupene E, Lee HL, Wendisch VF, Khodursky AB, Peter BJ, et al. Nitrogen regulatory protein C-controlled genes of Escherichia coli: scavenging as a defense against nitrogen limitation. Proc. Natl. Acad. Sci. USA. 2000;97:14674–14679. doi: 10.1073/pnas.97.26.14674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect of ntrB deletion on the expression of genes for GDAR and LEE control. Gene expression levels plotted for wild type (black), ΔrpoN (white), ΔntrC (hatched), and ΔntrB (gray). Asterisks denote significant difference between wild-type and respective mutants by t-test (*P < 0.05, **P < 0.01, n ≥ 3). Error bars denote standard deviation.
Growth curve for TW14359 and mutant derivative strains in DMEM. Optical density (OD600) is plotted for each strain as a function of time. Samples were taken every hour for 12 h. OD600 measurements differed by less than 5% for each time point and strain.