

Abstract
HIP/RPL29 is a heparan sulfate (HS) binding protein with diverse activities including modulation of heparanase (HPSE) activity. We examined HIP/RPL29’s ability to modulate actions of HS-binding growth factors (HBGFs) in angiogenesis. Between 1–2.5 μg/ml (ca. 60–150 nM), HIP/RPL29 inhibited HBGF-stimulated endothelial cell tube formation. Aortic explant outgrowth also was inhibited, but at higher concentrations (40 μg/ml). At this concentration, HIP/RPL29 had no effect on HBGF- stimulated MAPK phosphorylation or VEGF-stimulated receptor-2 phosphorylation at site Y-996. Partial inhibition occured at VEGF receptor-2 site Y951, associated with cell migration. HBGF displacement from HS-bearing perlecan domain I showed that HIP/RPL29 released 50% of bound HBGF at 20 μg/ml, a dose where endothelial tube formation is inhibited. Similar FGF2 release occurred at pH 5.0 and 7.0, conditions where HPSE is highly and residually active, respectively. We considered that HIP/RPL29 inhibits HPSE-dependent release of HS-bound HBGFs. At pH 5.0, release of soluble HS was inhibited by 64% at concentrations of 5 μg/ml and by 77% at 40 μg/ml, indicating that HIP/RPL29 antagonizes HPSE activity. At concentrations up to 40 μg/ml (ca 2.5 mM) where angiogenic processes are inhibited, release of FGF2 occurred in the presence of HPSE and HIP/RPL29. The majority of this FGF2 is not bound to soluble HS. Studies of HIP/RPL29 binding to HS indicated that many structural features of HS are important in modulation of HBGF activities. Our findings suggest that inhibition of angiogenic processes by HIP/RPL29 involves attenuation of the formation of soluble, biologically active HBGF:HS complexes that activate HBGF receptors.
Keywords: HIP/RPL29, heparan sulfate, VEGF, FGF2, heparanase
Introduction
Heparan sulfate (HS) binding growth factors (HBGFs) modulate a variety of biological responses including angiogenesis, morphogenesis and cell proliferation [Kreuger et al., 2006; Nangia-Makker et al., 2000; Stringer, 2006]. Interaction with HS provides a means of retaining and concentrating these proteins in the extracellular matrix where they may later be released by the actions of proteinases and/or heparanases [Mott and Werb, 2004; Vlodavsky and Friedmann, 2001]. In the latter case, this activity releases HS-bound growth factors without hydrolysis of the protein cores, thereby better retaining the structural integrity of the extracellular matrix. Heparanase (HPSE) activity is weak at physiological pH which may provide for slow growth factor release [Freeman and Parish, 1998]. Nonetheless, it appears that HPSE retains HS binding activity at physiological pH and even may support HS-dependent aspects of cell-cell or cell-extracellular matrix adhesion [Goldshmidt et al., 2003]. Various growth factor receptors, including those for the FGFs, VEGF and HGF, require interactions with both the growth factor and HS in a ternary complex to provide optimal receptor activation and/or to coordinate binding with the growth factor [Ashikari-Hada et al., 2005; Kan et al., 1999; Presta et al., 2005; Rubin et al., 2001; Selleck, 2006]. Moreover, we recently demonstrated that FGF2 binding, signaling and angiogenesis are modulated by HPSE in metastatic melanoma cells [Reiland et al., 2006].
HIP/RPL29 is a small, highly basic polyanion binding protein with multiple functions. The viability of mice and yeast cells null for HIP/RPL29 indicates an accessory function [DeLabre et al., 2002; Kirn-Safran et al., 2002]. Human HIP/RPL29 was identified at the cell surface where it can bind HS [Jacobs et al., 1997; Rohde et al., 1998; Rohde et al., 1996]. Domain mapping studies indicate that most of the HIP/RPL29 polypeptide participates in HS binding, an interaction that substantially alters the structure of this protein [Hoke et al., 2000]. HIP/RPL29 supports cell attachment [Liu et al., 1997a] and modulates blood coagulation activities [Liu et al., 1997b]. A synthetic peptide sequence of HIP/RPL29 inhibits HPSE activity in vitro [Marchetti et al., 1997], an activity that can alter HS-binding growth factor responsiveness. In a previous study [Ta et al., 2002], we determined that exogenously added HIP/RPL29 inhibited FGF2, but not IGF1, stimulated proliferative activity of human fibroblasts. The effects of HIP/RPL29 are associated with HS interactions, although the mechanism is unclear. Possibilities include inhibition of growth factor binding to or displacement from HS proteoglycans, inhibition of HPSE-dependent HBGF release or inhibition of proper formation of ternary complexes among HS-binding growth factors, their receptors and HS required for receptor activation. To discriminate among these alternatives, we conducted a systematic study of HIP/RPL29 interactions with each of these components including the ability to terminate signaling associated with ternary complex formation.
Materials and Methods
Materials
Human FGF2 and VEGF-165 were obtained from R&D Systems (Minneapolis, MN). Matrigel® was purchased from BD Biosciences (San Jose, CA). BME-1 endothelial cells were obtained from Dr. Carlton Cooper (University of Delaware). Cell culture reagents were obtained from Invitrogen (Carlsbad, CA). Recombinant HIP/RPL29 and perlecan domain I were expressed and purified as described previously [Ta et al., 2002; Yang et al., 2005]. HPSE was partially purified from cultured human 70W melanoma cells. Briefly, purification consisted of sequential chromatography on heparin-agarose and conconavalin-A agarose yielding an approximately 120-fold enrichment in activity [Marchetti et al., 1997]. These preparations displayed a HPSE specific activity similar to that of recombinant human HPSE [McKenzie et al., 2003; Murry et al., 2006]. All chemicals used were reagent grade or better.
Tube Formation Assay
Endothelial cell tube formation assays were performed and analyzed as described previously [Muir et al., 2006]. Briefly, undiluted growth factor reduced Matrigel® (250 μl/well) was used in each well of a 24-well tissue culture plate, and incubated at 37°C for 30 min to allow time to gel. BME-1 cells (100,000 cells/well) were plated in treatment medium (500 µl containing exogenous growth factors [30 ng/ml each] and HIP/RPL29 or lysozyme [1 or 2.5 μg/ml]) for 15 min prior to addition to Matrigel® coated wells. After 3 hrs of incubation at 37°C, cells were stained with Crystal Violet, then photographed. Images were converted to black and white, then thresholded. Images were subjected to a Gaussian blur, and skeletonized with a cutoff of 10 pixels using Adobe Photoshop. Measurements of total length of skeletonized pictures also were performed using Adobe Photoshop. Experiments were performed in duplicate, and four representative images were selected from different areas of each sample for analysis. Results represent at least three independent experiments.
Aortic Outgrowth Assay
The procedure of Zhu et al. [Zhu et al., 2003] was followed with minor modifications. Briefly, aortae were dissected from 6–8 week old C57BL6 mice and placed in serum-free endothelial basal medium (EBM, Clonetics, San Diego, CA). The aortae then were cross-cut into 1 mm-long strips with Noyes scissors and rinsed with serum-free EBM. These strips were placed on the bottom of 16 mm wells in 4-well NUNC dishes with the luminal axis of each strip lying parallel to the bottom of the culture dish. Each strip was covered with 25 μl of Matrigel® with a pipette tip to form a uniform thin disc of approximately 6 mm in diameter around each explant. Once the gel had set (5–10min at 37°C), 0.4 ml of serum-free EBM was added to each well with or without FGF2 (40 ng/ml), or VEGF-165 (40 ng/ml) and/or HIP/RPL29 (5–40 μg/ml). Medium was changed every 2 days maintaining the growth factor or HIP/RPL29 supplements. Angiogenic sprouting was photographed after 8 days of culture.
To quantify sprouting, images were captured with a Nikon digital camera (DXM1200F) mounted on a Nikon microscope (SMZ 1500). Software [Zeiss LSM Imagine Browser, version 3.2.0.115] was used to measure the area covered by microvessels (mm2) as well as the length of sprouts from the aortic strip (μm) according to published methods [Berger et al., 2004; Brill et al., 2004; Zhu et al., 2003] with some modifications. Nine points on triplicate aortic cultures were selected to measure the length of sprouted vessels. The mean length was calculated from these points. Images of aortic cultures were analyzed by manually encircling the outgrowth area. Mean area was computed from triplicate samples of outgrowths.
MAPK activation
BME-1 cells were serum starved in high glucose DMEM medium in a T75 flask at 80% confluency for 24 hrs. The cells then were trypsinized and treated as described in each experiemnt for 30 min in suspension (300,000 cells in a 1 ml volume of the same medium) before extracting protein for Western blotting. VEGF and FGF2 were used at 30 ng/ml each, HIP/RPL29 and lysozyme were used at 2.5, l, 25 or 50 μg/ml.
Cells were solubilized with 200 μl RIPA buffer (50mM Tris pH8.0, 150mM NaCl, 0.5% [w/v] deoxycholate, 1% (v/v) NP-40 and 0.1% [w/v] SDS) on ice for 10 min, centrifuged at 13,000 rpm for 15 seconds and the resultant supernatant used for Western blotting. Cell associated protein (20 μg) were separated by SDS-PAGE, transferred to nitrocellulose and the transfer blocked with 3% (w/v) BSA −0.1% (v/v) Tween-20 in PBS overnight at 4°C with constant rotary agitation. The membranes then were probed with p44/42 MAP kinase (MAPK) antibody (Cell Signaling, Danvers, MA, cat. no. 9120) or Phospho-p44/p42 MAPK antibody (Cell Signaling, cat. no. 9101) each diluted 1:1000 overnight in 3% (w/v) BSA-0.1% (v/v) Tween-20 in PBS at 4°C with constant rotary agitation. The blots then were rinsed several times with PBS and subsequently incubated with goat anti rabbit 1gG (Jackson ImmunoResearch, West Grove, PA, cat. no. 111-065-144), diluted 1:200,000 in 3% (w/v) BSA in PBS −0.1% (v/v) Tween-20 at room temperature for one hour with constant rotary agitation. Excess secondary antibody was removed by several rinses of the membrane with PBS −0.1 (v/v) Tween-20 and the signal developed using ECL reagent (Pierce, Rockford, IL) according to the manufacturer’s instructions. Signals subsequently were quantified by densitometry and expressed as the ratio of phospho-MAPK to total MAPK for duplicate samples in each treatment group.
VEGFR-2 receptor activation
Serum-starved (24 hr) BME-1 cells were stimulated with VEGF-165 (30 ng/ml) for 15 min at 37°C either in the presence or absence of HIP/RPL29. After 15 min, the supernatant was removed and cells were rinsed with PBS. Cell lysates were collected in a buffer containing 50 mM Tris pH 7.2, 5 mM EDTA, 1% (v/v) Triton X-100 and 150 mM sodium chloride. Protease and phosphatase inhibitors were added to the cell lysates at 1:100 dilution. Protein concentrations on trichloroacetic acid precipitates were determined as described [Lowry et al., 1951]. Total protein extracts (30 μg) were mixed with Laemmli sample buffer (BioRad Laboratories, Hercules, CA) in a 1:1 (v/v) ratio and boiled for 5 min. Protein samples were electrophoresed through acrylamide on a 10% (w/v) Porzio and Pearson gel [Porzio and Pearson, 1977] for 2 h at 100 V. Gels were transferred to Protein Pure Nitrocellulose and Immobilization Membrane (Schleicher and Schuell Bioscience Inc, Keene, NH) for 5 h at 40 V in a cold room (4°C). After the transfer, the blot was blocked in 5% (w/v) nonfat dry milk prepared in 0.1% (v/v) Tween 20/Tris buffered saline (TBS-T) at 4°C to prevent non-specific binding. The membrane was incubated overnight at 4°C with polyclonal VEGFR-2 antibody (Cell Signaling) at a 1:1000 dilution prepared in 3% (w/v) BSA in TBS-T. Unbound antibody was removed by rinsing three times in TBS-T for 5 min at room temperature. The blot then was incubated for 2 h at 4°C with donkey anti-rabbit IgG horseradish peroxidase conjugate (Jackson ImmunoResearch Lab. Inc.) at a final dilution of 1:200,000 in 3% (w/v) BSA in TBS-T. Unbound antibody was removed by rinsing three times in TBS-T for 5 min at room temperature. The signal was developed using Enhanced Chemiluminesence reagent (ECL) (Pierce). The blot also was immunoblotted with phospho-VEGFR-2 (Y951) antibody (Cell Signaling) at a 1:1000 dilution prepared in 3% (w/v) BSA in TBS-T. Unbound antibody was removed by rinsing three times in TBS-T for 5 min at room temperature. The blot then was incubated for 2 h at 4°C with donkey anti-rabbit IgG horseradish peroxidase conjugate (Jackson ImmunoResearch Lab. Inc.) at a final dilution of 1:200,000 in 3% (w/v) BSA in TBS-T. Unbound antibody was removed by rinsing three times in TBS-T for 5 min at room temperature. The signal was developed using Enhanced Chemiluminesence reagent (ECL) (Pierce, Rockford, IL).
Preparation of Radiolabeled Extracellular Matrix (ECM)-HSPGs
35S-Labeled HSPGs were prepared from WiDr ce1ls, an adenocarcinoma cell line which produces large amounts of perlecan [Iozzo, 1984]. Briefly, WiDr cells were cultured in Eagle’s Minimum Essential Medium supplemented with 10% (v/v) FBS, 2 mM L-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin sulfate. After the second passage, the cells were plated in a 24-well plate. When the cells were 80% confluent, the media was removed and the cells were rinsed with low-sulfate media containing RPMI-1640 (Invitrogen), 3.3 mM magnesium chloride, 1.5 mM HEPES, 1.2 g/L sodium bicarbonate, and 0.05% (v/v) penicillin/streptomycin solution. Cells were cultured in one ml of low-sulfate medium containing 100 μCi of Na235SO4. After 48 h, cells were washed four times with magnesium/calcium-free PBS to remove unincorporated Na235SO4. The wells then were treated with 0.5% (v/v) Triton X-100 and 20 mM ammonium hydroxide in PBS for 10 min to solubilize the cell layer followed by four washes with magnesium/calcium-free PBS. The ECM-H[35S]PGs are defined as those that remain firmly attached to the tissue culture wells under these conditions. The plates were used immediately to test for HPSE activity.
HPSE Activity Assay
The protocol for HPSE activity assay was modified from Marchetti and Nicolson [Marchetti and Nicolson, 1997]. Briefly, 50 μg of day 8 endometrial extracts, a robust source of heparanase activity [D’Souza et al., 2007] was incubated on sulfate-labeled ECM-H[35S]PG-coated, 1.5-mm dishes in 0.5 ml of heparanase reaction buffer (50 mM sodium acetate; pH 5.0) for 24 h at 37°C either in the absence of the presence of HIP/RPL29 (5 g/ml or 40 μg/ml). The incubation medium containing sulfate-labeled degradation fragments released from the ECM-H[35S]PG was analyzed by molecular exclusion column chromatography on a Superose 12 PC 10/300 GL column with dimensions of 1.0 × 30 cm (Amersham Biosciences, Piscataway, NJ). Fractions (1.0 ml) were eluted with PBS containing 0.02% (w/v) sodium azide. HS degradation fragments eluted near the Vt (total volume) of the column. HS identity was validated by nitrous acid degradation and β-elimination. Briefly, β-elimination was performed by incubation with 0.05 M NaOH and 1 M sodium borohydride at 45°C for 48 h followed by neutralization with acetic acid. The reaction mixture was lyophilized, dissolved in PBS, and analyzed by gel filtration on a Sephadex G-75 column (1.0 × 40 cm) (Amersham Biosciences). One ml fractions were collected and the radioactivity was counted using a scintillation counter. Nitrous acid degradation was performed by incubation with a 0.5:0.5:1.0 (v/v/v) ratio of 20% (v/v) N-butyl-nitrite in 100% ethanol, 1 N HCl, and double distilled H2O, respectively, at 25°C for 4 h. The reaction mixture then was lyophilized, dissolved in PBS, and analyzed by a Sephadex G-75 column chromatography as described above. Radioactivity in the fractions was counted using a scintillation counter. Dextran blue and potassium dichromate were used to determine Vo (void volume) and Vt, respectively.
HS protection assay
Bovine intestinal mucosa HS (30 μg) (Sigma) was pre-incubated with or without HIP/RPL29 (5 or 40 μg/ml) in 20mM TrisCl pH7.2 for 15 min at 37°C. The heparitinase enzyme mixture containing heparinase I, heparinase II and heparinase III (Sigma) (0.25 U/ml) was added to the reaction to initiate HS digestion in a total reaction volume of 20 μl. The reaction was incubated for 2 h at 37°C after which the tube was placed at 93°C for 2 min to stop the reaction. Preliminary experiments revealed that HS was quantitatively digested to fragments smaller than hexasaccharides under these conditions. The reaction mixture then was incubated with heparin-agarose (100 μl) for 5 min to separate HIP/RPL29 from HS. HIP/RPL29 remained bound to the heparin-agarose and HS was eluted with 20mM TrisCl pH 7.2 buffer (100 μl). Preliminary experiments determined that HIP/RPL29 quantitatively bound and virtually no HS bound to the resin under these conditions. The eluate then was lyophilized and the pellet was resuspended in 20mM TrisCl pH7.2 buffer (10 μl). Samples were prepared by adding loading buffer (2 μl) containing 80% (w/v) sucrose, 0.1% (w/v) bromophenol blue and 0.1% (w/v) xylene cyanol. Samples were electrophoresed on a 15% (w/v) acrylamide gel. The gel was stained with 0.5% (w/v) Alcian blue prepared in 3% (v/v) glacial acetic acid followed by destaining in water until clear bands were visible.
HIP/RPL29 solid phase 3H-heparin binding assay
HIP/RPL29 (1 μg) was added to each well and incubated to dryness at 37°C overnight. The next day the wells were rinsed with PBS three times. The wells were blocked with 0.1% (w/v) of heat-denatured BSA (100 μl) for 1 h at 37°C. The wells were rinsed three times with PBS. [3H]-Heparin (typically 1 × 105 dpm) then was added to each well in PBS (final volume 50 μl) and incubated overnight at 37°C. The next day the wells were rinsed three times with PBS. Bound [3H]-heparin was extracted with extraction buffer (100 l) consisting of 4 M guanidine HCl, 25 mM Tris-HCl, pH 8.0, 2.5 mM EDTA and 0.02% (w/v) sodium azide, by incubating overnight at 37°C. The extract (50 μl) was counted on a Beckman scintillation counter. For the competition assays, either a ten- or hundred-fold molar excess of the competitor (heparin or modified heparins) was added at the same time as [3H]-heparin.
HBGF displacement assay
To determine if HIP competed for HBGF binding to perlecan domain I, a solid-phase binding assay was performed essentially as described previously [25,35]. Briefly, after blocking 96-well microplates pre-coated with perlecan domain I with 3% (w/v) BSA prepared in PBS, FGF2 or VEGF-165 were added into the wells and preincubated for 1 hour at room temperature. The 96-well microplate surfaces coated with BSA served as controls. Following washing three times with 0.05% (v/v) Tween 20 in PBS, 100 μl of HIP/RPL29 in blocking buffer was added at increasing concentrations (0, 0.05, 0.5, 5, 10, 20, 40 and 80g/ml) to each well of a 96-well microplate and incubated for 2 hours at room temperature. After washing three times with 0.05% (v/v) Tween 20 in PBS, the bound FGF2 and VEGF-165 were identified with 2 μg/ml biotinylated anti-recombinant human FGF2 antibody (R&D Systems, BAM-233) and 3 μg/ml biotinylated anti-recombinant human VEGF antibody (R&D Systems, BAF-293), respectively. After incubation with 0.1 μg/ml of horse radish peroxidase-conjugated NeutrAvidin (Pierce, Rockford, IL) in SuperBlock Blocking Buffer (Pierce, Rockford, IL) for 30 min, the wells were again rinsed several times as above and then reacted with 200 μl of TMB solution followed by washing with PBS. The reaction was stopped with 10 μl 2M sulfuric acid. The optical density was measured at 450 nm.
To investigate if pH affected FGF2 displacement by HIP/RPL29, two kinds of media were used: a PBS buffer at pH 7.2 and a sodium acetate buffer at pH 5.0. HIP/RPL29 was added in these buffers to substrates containing prebound FGF2. FGF2 binding to perlecan domain I was measured by the solid binding assay described above. BSA coating was employed as negative control.
FGF2 release from perlecan domain I after HPSE digestion was evaluated further by the ELISA method as described above. FGF2 perlecan domain complex on the surface of a 96-well microplate was treated with 100 μl of HPSE (2 μg/ml or 5 μg/ml) in 1% (w/v) BSA in PBS on a shaker (120 rpm) for 24 hours at 37°C. After washing three times with PBS, remained FGF2 binding to perlecan domain I was tested by ELISA as described above. FGF2 perlecan domain complexes treated with buffer only, and BSA coating (without perlecan domain I) were used as positive and negative controls, respectively.
Results
HIP/RPL29 inhibits VEGF and FGF2-driven angiogenic processes
We determined the ability of recombinant human HIP/RPL29 to modulate endothelial cell responses to the HBGFs, VEGF-165 and FGF2, in two assays: capillary-like tube formation and aortic ring outgrowth. As shown in Figure 1, both growth factors greatly stimulated capillary-like tube formation. Inclusion of HIP/RPL29 at concentrations of 1–2.5 μg/ml (ca. 60 – 150 nM) reduced tube formation to a level similar to that observed in the absence of growth factors. In contrast, lysozyme, a protein of similar size and isoelectric point as HIP/RPL29, had no effect in this assay at concentrations up to 50 μg/ml (data not shown). Similar results were obtained using an aortic outgrowth assay. As shown in Figure 2, both FGF2 and VEGF-165 stimulated robust outgrowth which was inhibited markedly (>90 %) by the inclusion of HIP/RPL29 at 40 µg/ml (ca. 2.5 μM). Little effect of HIP/RPL29 was observed at 5 or 10 μg/ml (data not shown). Thus, HIP/RPL29 was effective in antagonizing HBGF activity in two independent assays of endothelial cell function, although it was much more active in the shorter tube formation assay.
Figure 1. HIP/RPL29 inhibits endothelial tube formation.
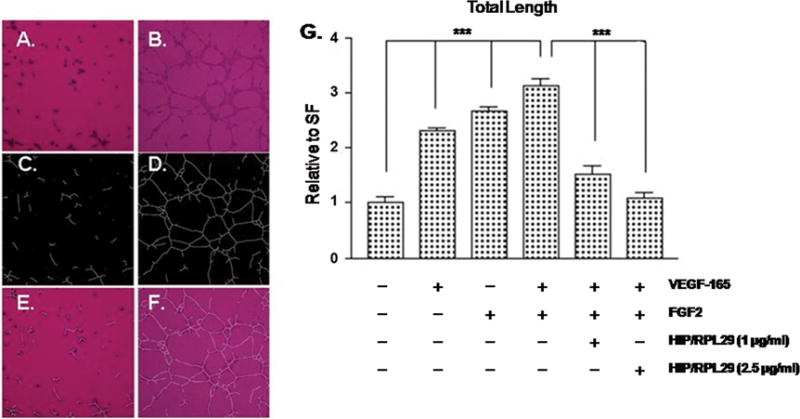
Endothelial cells (BME-1) were cultured on Matrigel®-coated surfaces and incubated either without (A, C and E) or with the addition of FGF2 and VEGF-165 (B, D and F) for 3 hrs. Panels A and B show phase contrast images, panels C and D show computer-generated projections of panels A and B used for quantification and panels E and F show the superimposed images. Panel G shows quantification of tube length (mean ± SEM of triplicate determinations in each case) as indicated. *** p < .001 relative to cells receiving no growth factors (left four bars) or to cells receiving growth factors without HIP/RPL29 (right three bars).
Figure 2. HIP/RPL29 inhibits aortic explant outgrowth.
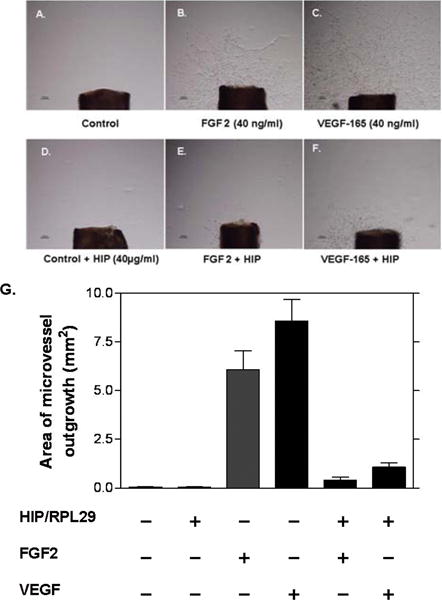
Mouse aortic outgrowth assays were performed and quantified by computer-based morphometric analyses as described in Materials and Methods. Panels A–F show 8 day outgrowths from cultures grown in the presence of EBM with the following additions: A, none (control); B. 40 ng/ml FGF2; C, 40 ng/ml VEGF-165; D, 40 μg/ml HIP/RPL29; E, 40 ng/ml FGF2 plus 40 μg/ml HIP/RPL29; F, 40 ng/ml VEGF-165 plus 40 μg/ml HIP/RPL29. Panel G shows the quantitation of results of these types of assays and demonstrates near complete inhibition of outgrowth in the presence of HIP/RPL29 in all cases. * p < 0.001 vs. corresponding growth factor treatment in the absence of HIP/RPL29.
HIP/RPL29 actions on growth factor receptor activation
To determine if HIP/RPL29 inhibited acute HBGF receptor activation, MAPK activation was examined. In preliminary experiments, we determined that maximal MAPK phosphorylation was observed 8 min after VEGF + FGF2 treatment (data not shown). In several experiments, we found that neither VEGF-165 nor FGF2-stimulated MAPK phosphorylation was inhibited by HIP/RPL29 at concentrations up to 40 μg/ml (ca. 2.5 μM) (data not shown). Likewise, lysozyme had no effect on MAPK phosphorylation at any dose tested. In a second series of experiments, we examined HIP/RPL29 effects on VEGF-stimulated phosphorylation of VEGF receptor-2, the VEGF receptor primarily associated with endothelial cell migration activities [Cebe-Suarez et al., 2006; Rahimi, 2006; Zeng et al., 2001]. Even at 40 μg/ml, HIP/RPL29 had no effect on phosphorylation of tyrosine-996 of VEGF receptor 2; however, a partial (40%) reduction in phosphorylation at tyrosine-951 was observed (Figure 3). The latter site is associated with stimulation of cell migration [Zeng et al., 2001]. It thus appears that at high, i.e., 40 μg/ml, but not low, concentrations HIP/RPL29 partially antagonize VEGF receptor activation, but not MAPK activation.
Figure 3. HIP/RPL29 partially inhibits VEGF receptor-2 activation.
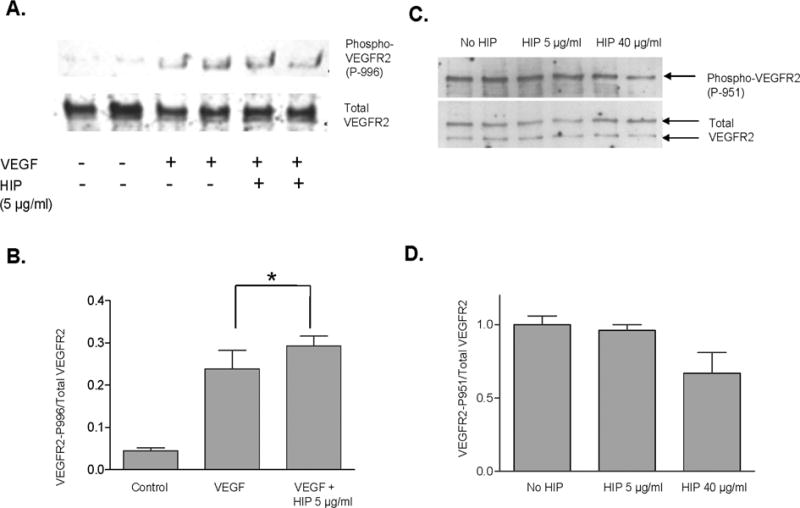
Western blotting for total VEGF receptor-2 (VEGFR2) and phospho-VEGFR2 was performed as described in Materials and Methods. Panels A and C show representative blots for duplicate treatment groups for phosphorylation at Y-996 and Y-951, respectively. Panels B and D show the corresponding densitometirc analyses of the ratio of VEGFR2 phosphorylation at Y-996 and Y-951, respectively, relative to total VEGFR2. *, p<0.05 relative to control (no VEGF or HIP/RPL29).
HIP/RPL29 releases HBGFs from HS-bearing substrates
We considered that HIP/RPL29 antagonized HBGF activity by inhibiting binding or causing displacement of HBGFs from HS-bearing substrates. We used recombinant perlecan domain I preparations that are well decorated with HS [Yang et al., 2005] as a solid phase matrix to support HBGF binding. In previous studies, it was demonstrated that these substrates retain HBGFs for several days [Yang et al., 2006; Yang et al., 2005]. As shown in Figure 4, lysozyme at concentrations up to 800 μg/ml failed to displace either FGF2 (Figure 4A) or VEGF-165 (Figure 4B, open symbols). In contrast, HIP/RPL29 displaced either growth factor, with 50% displacement occurring around 20 μg/ml (Figures 4A, 4B, solid symbols). Similar results were obtained regardless of whether HIP/RPL29 was preincubated with the substrate or if it was added after the growth factor was already bound (data not shown). Given that displacement involves disruption of charge-dependent interactions, we considered that FGF2 dissociation from HS might be pH dependent. However, we found that HIP/RPL29, when added alone at concentrations up to 40 μg/ml, displaced FGF2 similarly at pH 5.0 and pH 7.2 (Figure 5).
Figure 4. HIP/RPL29 displaces growth factors from HS-bearing substrates.
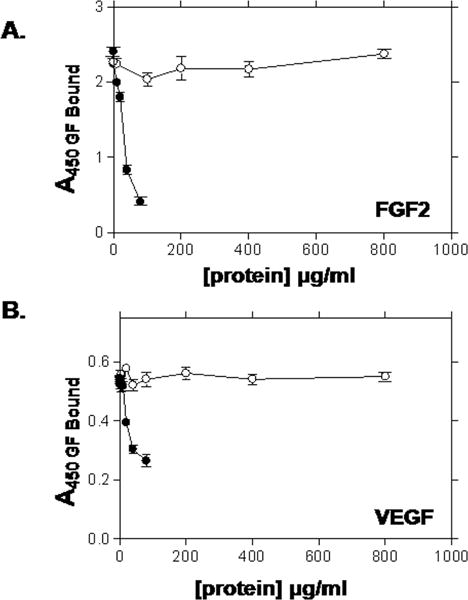
FGF2 (A) or VEGF-165 (B) were preincubated with perlecan domain I in a solid phase assay, unbound growth factor rinsed off and the substrates subsequently incubated with the indicated concentrations of HIP/RPL29 (HIP; filled circles) or lysozyme (LYS; open circles) for 2 hr. The surface was rinsed again and bound growth factor determined by ELISA as described in Materials and Methods. The points indicate the means ± SEM of triplicate determinations from a representative experiment.
Figure 5. HIP/RPL29 displacement of FGF2 from perlecan domain I is similar at pH 5.0 and pH 7.2.
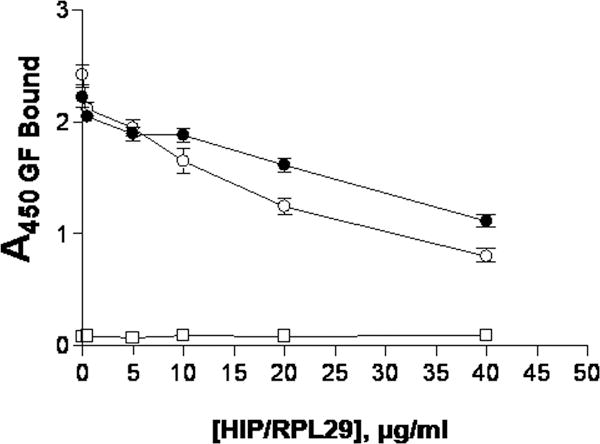
HIP/RPL29 displacement was performed as described in the legend to Figure 5. In one series (open circles) a sodium acetate, pH 5.0 buffer used for heparanase incubations was used and in a second series (closed circles) PBS at pH 7.2 was used along with the indicated concentrations of HIP/RPL29. The data points indicate the means +/− SEM of triplicate determinations in each case. Single determinations (negative control; open squares) at each concentration are the values obtained when perlecan domain I substrate only was used.
Given previous data indicating that a HIP/RPL29 synthetic peptide inhibits HPSE activity in vitro [Marchetti et al., 1997], we next sought to determine if intact HIP/RPL29 also inhibited HPSE-dependent growth factor release from an HS-bearing substrate. Again, recombinant HS-bearing perlecan domain I was used for this purpose. As expected, HPSE released FGF2 from these substrates in a dose-dependent fashion with almost 70% of the specifically bound FGF2 released by the addition of 5 μg/ml of the enzyme (Figure 6A). We next asked if FGF2 release required the enzymatic activity of HPSE. Experiments using the HS mimetic HPSE inhibitors, PI88 and heparin [Ferro et al., 2007; Vlodavsky et al., 2007], revealed that these inhibitors by themselves displaced FGF2 by >90%, making it difficult to determine the actions in combination with HPSE (data not shown). Nonetheless, increasing the pH to 7.2 reduced HPSE catalytic activity by 80–90% [Freeman and Parish, 1998] and inhibited FGF2 release to a similar extent (Figure 6B). Thus, FGF2 release was greatly enhanced by the presence of enzymatically active heparanase. Next, we examined the ability of HIP/RPL29 to inhibit HPSE activity at pH 5. HIP/RPL29 inhibited 35SO4-HS release from a prelabeled solid phase substrate by 64% and 77% at doses of 5 and 40 μg/ml, respectively, indicating that HIP/RPL29 was an effective antagonist of HPSE catalytic activity even at low concentrations (Figure 7).
Figure 6. HPSE releases FGF2 from perlecan domain I in a dose- and pH-dependent manner.
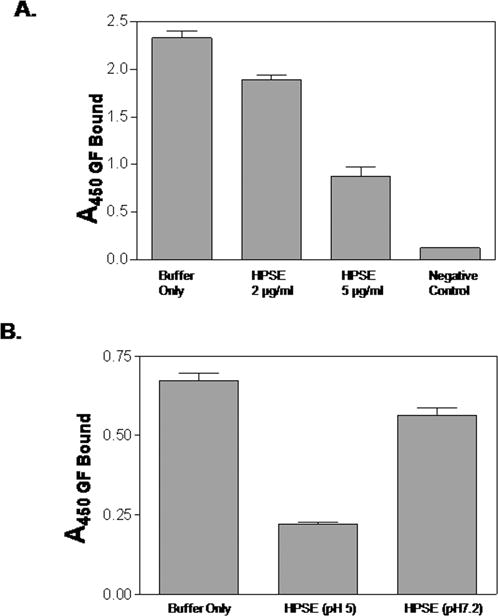
FGF2 was bound to a solid phase perlecan domain I substrate as described in Materials and Methods and Figure 5. In panel A, complexes subsequently were incubated with HPSE at the indicated concentrations for 24 hr, rinsed to remove unbound/released FGF2 and bound FGF2 measured by ELISA as described in Materials and Methods. The negative control was the perlecan domain I substrate not exposed to FGF2. In panel B, 5 μg/ml HPSE was incubated in the presence of the FGF2 bound to perlecan domain I for 24 hr at pH 5 or pH 7.2 as indicated and FGF2 release assayed by ELISA as described in Materials and Methods. FGF2 that remained bound at either pH 5.0 or 7.2 in the absence of HPSE served as a buffer only control. The bars represent the means ± SEM of triplicate determinations in each case.
Figure 7. HIP/RPL29 inhibits HPSE activity.
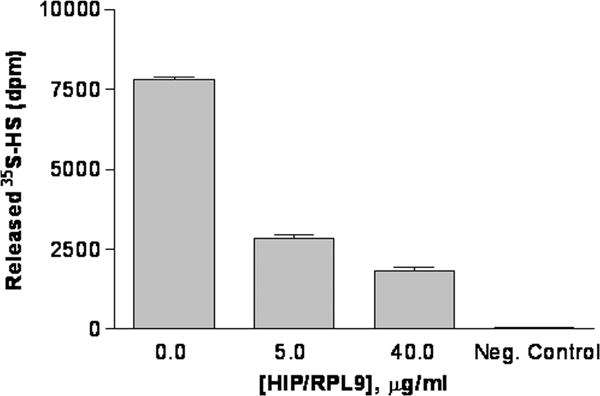
HPSE activity was measured as release of 35S-labeled HS from a solid phase assay as described in Materials and Methods. Day 8 pregnant mouse endometrial extracts (50 μg/well) protein were added to all assays in the presence of the indicated amount of HIP/RPL29 (HIP). Release observed in the absence of HPSE or HIP/RPL29 is indicated (Buffer only). The bars indicate the means +/− SEM of triplicate determinations in each case.
HIP/RPL29 and HPSE then were combined in an FGF2 release assay to determine if HIP/RPL29 could inhibit HPSE-dependent release. In spite of the fact that HIP/RPL29- inhibited 64–77% of HPSE activity at concentrations between 5 and 40 μg/ml, no inhibition of free FGF2 release was observed when these proteins were used in combination (Figure 8). These assays were performed at pH 5.0, the optimum for HPSE activity. Thus, when both proteins are present, FGF2 dissociation at pH 5.0 under conditions when HPSE is largely inhibited still can occur. At low concentrations of HIP/RPL29 (0.5–5 μg/ml), residual HPSE activity (36% of total) that persists in the presence of HIP/RPL29 may account for FGF2 release. At high HIP/RPL29 concentrations (40 μg/ml) both residual HPSE activity (23% of total) and displacement of free HBGFs by HIP/RPL29 may contribute to FGF2 release from an HS-bearing substrate. The enzymatically released FGF2 is expected to be bound in complex with HS, whereas displaced FGF2 is expected to be free.
Figure 8. HIP/RPL29 does not inhibit HPSE-dependent FGF2 release from perlecan domain I.
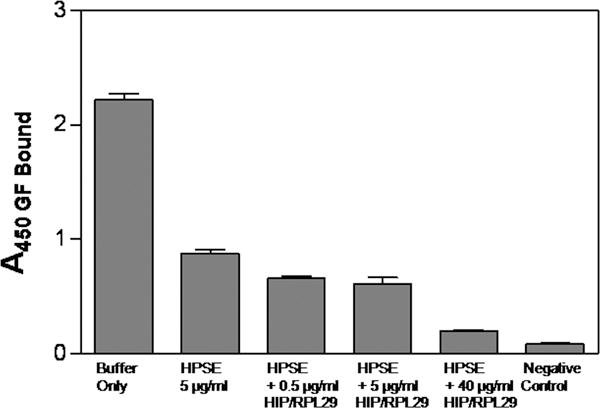
FGF2 was bound to a solid phase perlecan domain I substrate as described in the legend to Figure 5. This complex subsequently was incubated with HPSE (5 μg/ml) alone or in the presence of 0.5, 5 or 40 μg/ml HIP/RPL29 as indicated on the figure. After 24 hr, the surfaces were rinsed to remove unbound/released FGF2 and bound FGF2 measured by ELISA as described in Materials and Methods. The negative control was the perlecan domain I substrate not exposed to FGF2. The bars represent the means ± SEM of triplicate determinations from a representative experiment in each case.
HS binding to HIP/RPL29 requires multiple structural features of HS
In light of the ability of HIP/RPL29 to both displace HBGFs from and inhibit HPSE action on HS-bearing substrates, we sought to determine the structural features of HS involved in HIP/RPL29 interactions to determine if any were shared with those known for HBGF or HPSE recognition. Initially, we used HIP/RPL29 in solution with HS and a mixture of bacterial heparitinases to determine if we could identify and characterize HS fragments that were bound by HIP/RPL29 and “protected” from digestion by heparitinases; however, we discovered that essentially the full-length HS polysaccharides were protected in these assays indicating that multiple complex interactions took place (Figure 9A). These observations indicated that disaccharide analyses of the protected fragments would generate the same profile as the starting material and, therefore, be uninformative. In addition, it suggested that HIP/RPL29 binds to many regions in HS and, therefore, was likely to recognize multiple structural features in these polysaccharides. As a direct test of this hypothesis, we used a series of HS derivatives as competitors in [3H]-heparin binding assays to identify the critical structural features involved in HIP/RPL29 binding. As shown in Figure 9B, intact heparin was an excellent competitor, as expected from the displacement studies. Fully N-sulfated or N-acetylated heparin, N-desulfated or 2-O-desulfated heparins were the next best inhibitors, although substantially less effective than intact heparin. These studies indicated that a combination of both N-acetylated and N-sulfated glucosamines participated in the interaction as well as 2-O-sulfates. Carboxyl reduction, complete O-desulfation or selective 6-O-desulfation almost completely destroyed competitive activity, most evident at 10 μg/ml of competitor. These latter observations demonstrated key roles for carboxy groups as well as 6-O-sulfates in HIP/RPL29 binding. Nonetheless, other structural features also contributed to HIP/RPL29 binding.
Figure 9. HIP/RPL29 binding involves multiple structural features of HS.
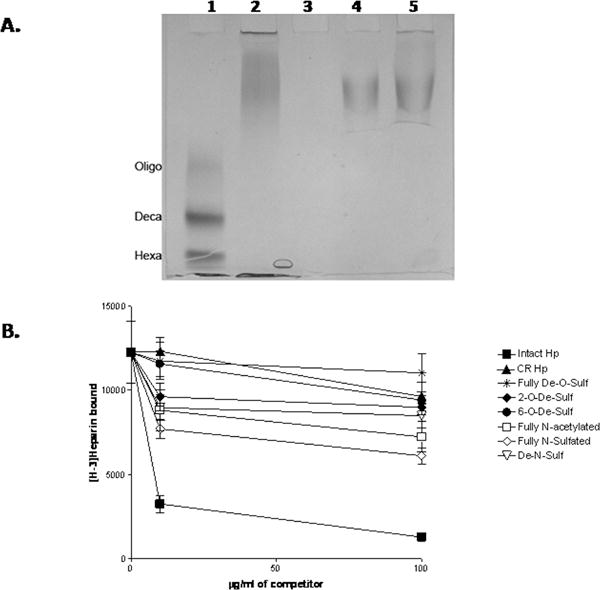
Panel A: HS heparitinase protection assays and gel electrophoresis were performed as described in Materials and Methods. Lane 1 shows the migration position of HS MW standards including an HS oligosaccharide mixture (Oligo) with a median MW of 4.2 KDa, an HS decasaccharide (Deca) and an HS hexasaccharide (Hexa). Lane 2 is HS incubated with buffer only. Lane 3 shows the products obtained after HS digestion with heparitinases. Lanes 4 and 5 are the products obtained after heparitinases digestion in the presence of 5 μg/ml and 40 μg/ml HIP/RPL29, respectively. Note that the products are essentially the same size as the undigested HS. Panel B: [3H]-Heparin was bound to HIP/RPL29 in a solid phase binding assay in the presence of the indicated concentrations of heparin derivatives and bound [3H]-heparin measured as described in Materials and Methods. The graph points each indicate the average +/− SD of duplicate determinations in each case. Symbols: ▼, fully D-O-sulfated; ▲, carboxyl reduced; ●, 6-0-de-sulfated; ◆, 2-0-desulfated; ▽, De-N-sulfated; □, fully N-acetylated, △, fully N-sulfated; ■, intact.
Discussion
In various human cells, HIP/RPL29 is detected at the cell surface, including uterine epithelial, trophoblastic and various tumor cell lines [Jacobs et al., 1997; Rohde et al., 1998; Rohde et al., 1996]. HIP/RPL29 may be released during cell lysis or via unconventional pathways such as those suggested for FGF2 and certain cytokines that, like HIP/RPL29, lack signal sequences [Rubartelli et al., 1990; Taverna et al., 2003]. HIP/RPL29 displays a restricted pattern of expression in adult tissues and increases in certain instances, e.g., during uterine stromal cell differentiation (decidualization; [Julian et al., 2001]). All known extracellular activities described for HIP/RPL29 rely upon interactions with glycosaminoglycans, particularly HS to which HIP/RPL29 binds with higher affinity and selectivity [Liu et al., 1997a]. Thus, local release of HIP/RPL29 can impact HS-dependent processes at these sites.
Previously, we found that HIP/RPL29 antagonized FGF2-stimulated proliferation of human gingival fibroblasts, but not those of a non-HS binding growth factor, IGF-I [Ta et al., 2002]. Given the key roles played by HBGFs in endothelial cell activities and the general importance of vascularization in many normal and pathological processes, we sought to determine if HIP/RPL29 also antagonized HBGF-driven processes in endothelial cells. HIP/RPL29 inhibited both FGF2- and VEGF-stimulated endothelial tube formation and aortic outgrowth. These represent primarily cell migratory events because the former requires only a few hours to occur while the latter is not dependent upon cell proliferation [Zeng et al., 2001]. In both cases, these processes involve activation of intracellular signal transduction events that trigger cell motility as well as cell surface interactions with extracellular matrix components that provide a support for cell migration, including HS proteoglycans [Iivanainen et al., 2003; Lake et al., 2006; Presta et al., 2003]. The inhibitory effects on tube formation occurred at lower doses than those required for inhibition of aortic outgrowth, but this could be due to the longer period of time required for the latter process (8 days) relative to the former (a few hours) to occur. At low concentrations (≤ 5 μg/ml), HIP/RPL29 had little effect on FGF2-triggered MAPK activation or VEGF-stimulated VEGF receptor 2 phosphorylation. At relatively high concentrations (≥ 40 μg/ml), HIP/RPL29 partially antagonized both FGF2- and VEGF-stimulated intracellular signaling responses, particularly those affecting endothelial cell migration. Thus, HIP/RPL29 can antagonize HBGF-driven signal transduction responses over a significant period of time, presumably by interfering either with HBGF binding to cell surface HS or formation of requisite active HS:HBGF: HBGF receptor ternary complexes [Ashikari-Hada et al., 2005; Kan et al., 1999; Presta et al., 2005; Rubin et al., 2001; Selleck, 2006].
In solid phase binding assays, HIP/RPL29 displayed a similar dose-dependence for growth factor displacement or inhibition of initial binding with a 50% inhibition at approximately 20 μg/ml; however, it should be noted that the stoichiometry of HIP/RPL29 to HBGF is high, i.e., approximately 200 – 1000:1, likely due to the necessity to saturate excess binding sites on HS in the assay. The amount of HS associated with perlecan domain I per assay is approximately 1 μg. HIP/RPL29 is approximately the same molecular weight as an average HS chain. Therefore, at a HIP/RPL29 concentration of 20 μg/ml in a 50 μl incubation volume, the concentration at which 50% displacement of growth factors is observed, HIP/RPL29 and HS are approximately equimolar. Given that virtually all of the HIP/RPL29 polypeptide participates in HS binding [Hoke et al., 2000], the results of the displacement assays are in good agreement with a model in which HIP/RPL29 binds to very large areas of HS chains. Consistent with this, we found that almost the entire length of HS was protected by HIP/RPL29 in a heparitinase protection assay and that multiple HS structural features contributed to HIP/RPL29 binding. Thus, it appears that HBGFs preferentially bind to more selective, higher affinity sites within HS until the HIP/RPL29 concentration becomes sufficiently high to saturate these sites along with other HS regions not involved in growth factor binding.
HPSE is an enzyme proposed to mediate aspects of cell migration and HBGF release in both normal contexts and in cancer progression [Vlodavsky and Friedmann, 2001]. Previous studies indicated that a synthetic peptide designed from the HIP/RPL29 primary sequence inhibit HPSE activity [Marchetti et al., 1997]. We confirmed that intact HIP/RPL29 substantially (64–77%) inhibited HPSE activity in an enzymatic assay and that HPSE stimulated FGF2 release from an HS-bearing substrate in a fashion that was enhanced by HPSE enzymatic activity. Nonetheless, inclusion of low concentrations of HIP/RPL29 (0.5–5 μg/ml) in this assay did not inhibit residual HPSE-stimulated FGF2 release. At high HIP/RPL29 concentrations (40 μg/ml), HIP/RPL29 alone caused substantial growth factor release by displacement. Of interest, the release of free FGF2 not expected to be complexed with HS produced occur under these conditions clearly is not able to stimulate outgrowth from aortic explants as was clearly evident in the experiments shown in Figure 2. These findings are consistent with the notion that stable activation of FGF receptors requires the formation of a ternary complex containing HS in addition to the free growth factor [Kan et al., 1999].
Finally, we considered that HIP/RPL29 might inhibit endothelial cell migratory activities by direct interference with cell surface adhesive interactions with HS required for cell migration. Previous studies have shown the HS dependence of cell binding to HIP/RPL29 [Liu et al., 1997a]. As discussed above and further demonstrated in this report, the structural requirements for HIP/RPL29:HS binding are complex and HIP/RPL29 can interact with virtually all regions of HS chains. Therefore, consistent with earlier studies in which HIP/RPL29 was shown to support HS-dependent cell adhesion, we conclude that interference with endothelial cell HS-dependent adhesion involved in cell migration also contributes to HIP/RPL29’s ability to inhibit angiogenesis associated events in these assays. Figure 10 depicts a model for the formation of active HS-containing complexes, the multiple routes by which they may be formed, and the potential mechanism by which HIP/RPL29 can disrupt formation of ternary complexes to inhibit angiogenesis.
Figure 10. Model of HIP/RPL29 interference with HS-binding growth factor actions.
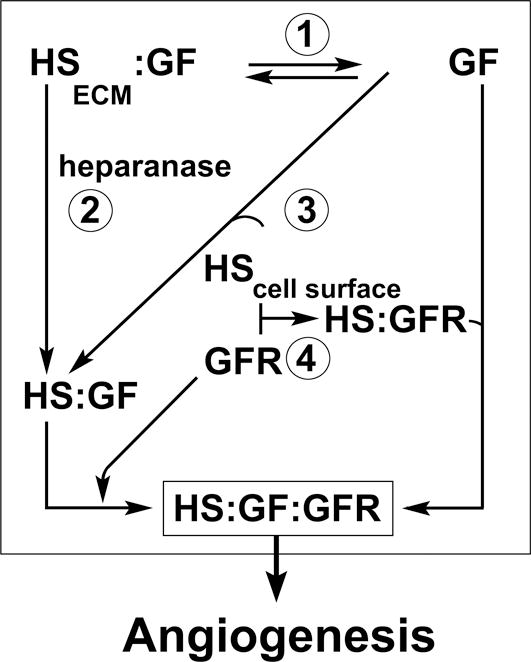
The model presents several steps involved in HS-binding growth factor actions in the context of angiogenesis. The model is predicated on the requirement for ternary complexes of growth factors (GF) with HS and GF receptors (GFR) to properly trigger signaling, i.e., formation of HS:GF:GFR. GFs are retained by HS in the extracellular matrix (HSECM) and can be released from this site via displacement by HIP/RPL29 (1). GFs also may be released from HSECM through the activity of HPSE as a HS:GF complex. HPSE activity also is inhibited by HIP/RPL29 (2). GFs that are released in free form may become complexed with HS fragments or HS attached to cell surface proteoglycans which also can be blocked by HIP/RPL29 (3). Similarly, GFRs may perform complexes with cell surface HS that, in turn, can bind free GF. HIP/RPL29 also is expected to block GFR:HS interactions at the cell surface (4). Direct evidence for HIP/RPL29 interference with each of these steps (except for HS:GFR) is presented in this paper.
In conclusion, we have shown that HIP/RPL29 effectively antagonizes HBGF driven responses of endothelial cells via interference with HS interactions required both for triggering intracellular signal transduction events as well as cell surface interactions with HS directly involved in cell migration processes. These observations indicate that HIP/RPL29 has potential use as an antagonist to restrict HBGF (FGF2, VEGF):HS-dependent processes including angiogenesis. Ongoing studies will expand these studies to pre-clinical animal models including effects on placentation or tumor growth.
Acknowledgments
The authors are grateful to Dr. Catherine Kirn-Safran, Ms. Lynn Opdenaker, Dr. Leland Chung (Winship Cancer Center, Emory University) and the members of the PO1 team in the Chung and Petros laboratories (Emory) for many useful discussions and Ms. Sharron Kingston for her preparation of the manuscript and many of the figures. This work was supported by NIH grants R37 HD25235 (to DDC), NCI P01 CA098912, Project 2 (to MCFC and DDC) R01 CA86832 and R21 CA103955 (to DM).
Abbreviations
- HIP/RPL29
heparin/heparan sulfate interacting protein/ribosomal protein L29
- HS
heparan sulfate
- HBGF
heparin-binding growth factor
- VEGF
vascular endothelial growth factor
- FGF2
fibroblast growth factor 2
- HPSE
heparanase
References
- Ashikari-Hada S, Habuchi H, Kariya Y, Kimata K. Heparin regulates vascular endothelial growth factor165-dependent mitogenic activity, tube formation, and its receptor phosphorylation of human endothelial cells. Comparison of the effects of heparin and modified heparins. J Biol Chem. 2005;280:31508–15. doi: 10.1074/jbc.M414581200. [DOI] [PubMed] [Google Scholar]
- Berger AC, Wang XQ, Zalatoris A, Cenna J, Watson JC. A murine model of ex vivo angiogenesis using aortic disks grown in fibrin clot. Microvasc Res. 2004;68:179–87. doi: 10.1016/j.mvr.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Brill A, Elinav H, Varon D. Differential role of platelet granular mediators in angiogenesis. Cardiovasc Res. 2004;63:226–35. doi: 10.1016/j.cardiores.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Cebe-Suarez S, Zehnder-Fjallman A, Ballmer-Hofer K. The role of VEGF receptors in angiogenesis; complex partnerships. Cell Mol Life Sci. 2006;63:601–15. doi: 10.1007/s00018-005-5426-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza SS, Daikoku T, Farach-Carson MC, Carson DD. Heparanase expression and function during early pregnancy in mice. Biol Reprod. 2007;77:433–41. doi: 10.1095/biolreprod.107.061317. [DOI] [PubMed] [Google Scholar]
- DeLabre ML, Kessl J, Karamanou S, Trumpower BL. RPL29 codes for a non-essential protein of the 60S ribosomal subunit in Saccharomyces cerevisiae and exhibits synthetic lethality with mutations in genes for proteins required for subunit coupling. Biochim Biophys Acta. 2002;1574:255–61. doi: 10.1016/s0167-4781(01)00372-4. [DOI] [PubMed] [Google Scholar]
- Ferro V, Dredge K, Liu L, Hammond E, Bytheway I, Li C, Johnstone K, Karoli T, Davis K, Copeman E, Gautam A. PI-88 and novel heparan sulfate mimetics inhibit angiogenesis. Semin Thromb Hemost. 2007;33:557–68. doi: 10.1055/s-2007-982088. [DOI] [PubMed] [Google Scholar]
- Freeman C, Parish CR. Human platelet heparanase: purification, characterization and catalytic activity. Biochem J. 1998;330(Pt 3):1341–50. doi: 10.1042/bj3301341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldshmidt O, Zcharia E, Cohen M, Aingorn H, Cohen I, Nadav L, Katz BZ, Geiger B, Vlodavsky I. Heparanase mediates cell adhesion independent of its enzymatic activity. Faseb J. 2003;17:1015–25. doi: 10.1096/fj.02-0773com. [DOI] [PubMed] [Google Scholar]
- Hoke DE, LaBrenz SR, Hook M, Carson DD. Multiple domains contribute to heparin/heparan sulfate binding by human HIP/L29. Biochemistry. 2000;39:15686–94. doi: 10.1021/bi0009688. [DOI] [PubMed] [Google Scholar]
- Iivanainen E, Kahari VM, Heino J, Elenius K. Endothelial cell-matrix interactions. Microsc Res Tech. 2003;60:13–22. doi: 10.1002/jemt.10238. [DOI] [PubMed] [Google Scholar]
- Iozzo RV. Biosynthesis of heparan sulfate proteoglycan by human colon carcinoma cells and its localization at the cell surface. J Cell Biol. 1984;99:403–17. doi: 10.1083/jcb.99.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs AL, Julian J, Sahin AA, Carson DD. Heparin/heparan sulfate interacting protein expression and functions in human breast cancer cells and normal breast epithelia. Cancer Res. 1997;57:5148–54. [PubMed] [Google Scholar]
- Julian J, Das SK, Dey SK, Baraniak D, Ta VT, Carson DD. Expression of heparin/heparan sulfate interacting protein/ribosomal protein l29 during the estrous cycle and early pregnancy in the mouse. Biol Reprod. 2001;64:1165–75. doi: 10.1095/biolreprod64.4.1165. [DOI] [PubMed] [Google Scholar]
- Kan M, Wu X, Wang F, McKeehan WL. Specificity for fibroblast growth factors determined by heparan sulfate in a binary complex with the receptor kinase. J Biol Chem. 1999;274:15947–52. doi: 10.1074/jbc.274.22.15947. [DOI] [PubMed] [Google Scholar]
- Kirn-Safran CB, Julian J, Fongemie JE, Hoke DE, Czymmek KJ, Carson DD. Changes in the cytologic distribution of heparin/heparan sulfate interacting protein/ribosomal protein L29 (HIP/RPL29) during in vivo and in vitro mouse mammary epithelial cell expression and differentiation. Dev Dyn. 2002;223:70–84. doi: 10.1002/dvdy.1226. [DOI] [PubMed] [Google Scholar]
- Kreuger J, Spillmann D, Li JP, Lindahl U. Interactions between heparan sulfate and proteins: the concept of specificity. J Cell Biol. 2006;174:323–7. doi: 10.1083/jcb.200604035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake AC, Vassy R, Di Benedetto M, Lavigne D, Le Visage C, Perret GY, Letourneur D. Low molecular weight fucoidan increases VEGF165-induced endothelial cell migration by enhancing VEGF165 binding to VEGFR-2 and NRP1. J Biol Chem. 2006;281:37844–52. doi: 10.1074/jbc.M600686200. [DOI] [PubMed] [Google Scholar]
- Liu S, Hoke D, Julian J, Carson DD. Heparin/heparan sulfate (HP/HS) interacting protein (HIP) supports cell attachment and selective, high affinity binding of HP/HS. J Biol Chem. 1997a;272:25856–62. doi: 10.1074/jbc.272.41.25856. [DOI] [PubMed] [Google Scholar]
- Liu S, Zhou F, Hook M, Carson DD. A heparin-binding synthetic peptide of heparin/heparan sulfate-interacting protein modulates blood coagulation activities. Proc Natl Acad Sci U S A. 1997b;94:1739–44. doi: 10.1073/pnas.94.5.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–75. [PubMed] [Google Scholar]
- Marchetti D, Liu S, Spohn WC, Carson DD. Heparanase and a synthetic peptide of heparan sulfate-interacting protein recognize common sites on cell surface and extracellular matrix heparan sulfate. J Biol Chem. 1997;272:15891–7. doi: 10.1074/jbc.272.25.15891. [DOI] [PubMed] [Google Scholar]
- Marchetti D, Nicolson GL. Human melanoma cell invasion: selected neurotrophin enhancement of invasion and heparanase activity. J Investig Dermatol Symp Proc. 1997;2:99–105. doi: 10.1038/jidsymp.1997.19. [DOI] [PubMed] [Google Scholar]
- McKenzie E, Young K, Hircock M, Bennett J, Bhaman M, Felix R, Turner P, Stamps A, McMillan D, Saville G, Ng S, Mason S, Snell D, Schofield D, Gong H, Townsend R, Gallagher J, Page M, Parekh R, Stubberfield C. Biochemical characterization of the active heterodimer form of human heparanase (Hpa1) protein expressed in insect cells. Biochem J. 2003;373:423–35. doi: 10.1042/BJ20030318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott JD, Werb Z. Regulation of matrix biology by matrix metalloproteinases. Curr Opin Cell Biol. 2004;16:558–64. doi: 10.1016/j.ceb.2004.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir C, Chung LW, Carson DD, Farach-Carson MC. Hypoxia increases VEGF-A production by prostate cancer and bone marrow stromal cells and initiates paracrine activation of bone marrow endothelial cells. Clin Exp Metastasis. 2006;23:75–86. doi: 10.1007/s10585-006-9021-2. [DOI] [PubMed] [Google Scholar]
- Murry BP, Blust BE, Singh A, Foster TP, Marchetti D. Heparanase mechanisms of melanoma metastasis to the brain: Development and use of a brain slice model. J Cell Biochem. 2006;97:217–25. doi: 10.1002/jcb.20714. [DOI] [PubMed] [Google Scholar]
- Nangia-Makker P, Baccarini S, Raz A. Carbohydrate-recognition and angiogenesis. Cancer Metastasis Rev. 2000;19:51–7. doi: 10.1023/a:1026540129688. [DOI] [PubMed] [Google Scholar]
- Porzio MA, Pearson AM. Improved resolution of myofibrillar proteins with sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Biochim Biophys Acta. 1977;490:27–34. doi: 10.1016/0005-2795(77)90102-7. [DOI] [PubMed] [Google Scholar]
- Presta M, Dell’Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005;16:159–78. doi: 10.1016/j.cytogfr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Presta M, Leali D, Stabile H, Ronca R, Camozzi M, Coco L, Moroni E, Liekens S, Rusnati M. Heparin derivatives as angiogenesis inhibitors. Curr Pharm Des. 2003;9:553–66. doi: 10.2174/1381612033391379. [DOI] [PubMed] [Google Scholar]
- Rahimi N. Vascular endothelial growth factor receptors: molecular mechanisms of activation and therapeutic potentials. Exp Eye Res. 2006;83:1005–16. doi: 10.1016/j.exer.2006.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiland J, Kempf D, Roy M, Denkins Y, Marchetti D. FGF2 binding, signaling, and angiogenesis are modulated by heparanase in metastatic melanoma cells. Neoplasia. 2006;8:596–606. doi: 10.1593/neo.06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde LH, Janatpore MJ, McMaster MT, Fisher S, Zhou Y, Lim KH, French M, Hoke D, Julian J, Carson DD. Complementary expression of HIP, a cell-surface heparan sulfate binding protein, and perlecan at the human fetal-maternal interface. Biol Reprod. 1998;58:1075–83. doi: 10.1095/biolreprod58.4.1075. [DOI] [PubMed] [Google Scholar]
- Rohde LH, Julian J, Babaknia A, Carson DD. Cell surface expression of HIP, a novel heparin/heparan sulfate binding protein, of human uterine epithelial cells and cell lines. J Biol Chem. 1996;271:11824–30. doi: 10.1074/jbc.271.20.11824. [DOI] [PubMed] [Google Scholar]
- Rubartelli A, Cozzolino F, Talio M, Sitia R. A novel secretory pathway for interleukin-1 beta, a protein lacking a signal sequence. Embo J. 1990;9:1503–10. doi: 10.1002/j.1460-2075.1990.tb08268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin JS, Day RM, Breckenridge D, Atabey N, Taylor WG, Stahl SJ, Wingfield PT, Kaufman JD, Schwall R, Bottaro DP. Dissociation of heparan sulfate and receptor binding domains of hepatocyte growth factor reveals that heparan sulfate-c-met interaction facilitates signaling. J Biol Chem. 2001;276:32977–83. doi: 10.1074/jbc.M105486200. [DOI] [PubMed] [Google Scholar]
- Selleck SB. Signaling from across the way: transactivation of VEGF receptors by HSPGs. Mol Cell. 2006;22:431–2. doi: 10.1016/j.molcel.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Stringer SE. The role of heparan sulphate proteoglycans in angiogenesis. Biochem Soc Trans. 2006;34:451–3. doi: 10.1042/BST0340451. [DOI] [PubMed] [Google Scholar]
- Ta TV, Baraniak D, Julian J, Korostoff J, Carson DD, Farach-Carson MC. Heparan sulfate interacting protein (HIP/L29) negatively regulates growth responses to basic fibroblast growth factor in gingival fibroblasts. J Dent Res. 2002;81:247–52. doi: 10.1177/154405910208100405. [DOI] [PubMed] [Google Scholar]
- Taverna S, Ghersi G, Ginestra A, Rigogliuso S, Pecorella S, Alaimo G, Saladino F, Dolo V, Dell’Era P, Pavan A, Pizzolanti G, Mignatti P, Presta M, Vittorelli ML. Shedding of membrane vesicles mediates fibroblast growth factor-2 release from cells. J Biol Chem. 2003;278:51911–9. doi: 10.1074/jbc.M304192200. [DOI] [PubMed] [Google Scholar]
- Vlodavsky I, Friedmann Y. Molecular properties and involvement of heparanase in cancer metastasis and angiogenesis. J Clin Invest. 2001;108:341–7. doi: 10.1172/JCI13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlodavsky I, Ilan N, Naggi A, Casu B. Heparanase: structure, biological functions, and inhibition by heparin-derived mimetics of heparan sulfate. Curr Pharm Des. 2007;13:2057–73. doi: 10.2174/138161207781039742. [DOI] [PubMed] [Google Scholar]
- Yang W, Gomes RR, Brown AJ, Burdett AR, Alicknavitch M, Farach-Carson MC, Carson DD. Chondrogenic differentiation on perlecan domain I, collagen II, and bone morphogenetic protein-2-based matrices. Tissue Eng. 2006;12:2009–24. doi: 10.1089/ten.2006.12.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WD, Gomes RR, Jr, Alicknavitch M, Farach-Carson MC, Carson DD. Perlecan domain I promotes fibroblast growth factor 2 delivery in collagen I fibril scaffolds. Tissue Eng. 2005;11:76–89. doi: 10.1089/ten.2005.11.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Sanyal S, Mukhopadhyay D. Tyrosine residues 951 and 1059 of vascular endothelial growth factor receptor-2 (KDR) are essential for vascular permeability factor/vascular endothelial growth factor-induced endothelium migration and proliferation, respectively. J Biol Chem. 2001;276:32714–9. doi: 10.1074/jbc.M103130200. [DOI] [PubMed] [Google Scholar]
- Zhu WH, Iurlaro M, MacIntyre A, Fogel E, Nicosia RF. The mouse aorta model: influence of genetic background and aging on bFGF- and VEGF-induced angiogenic sprouting. Angiogenesis. 2003;6:193–9. doi: 10.1023/B:AGEN.0000021397.18713.9c. [DOI] [PubMed] [Google Scholar]