

Abstract
Although glucagon (GLU) plays a pivotal role in glucose homeostasis, its role in the regulation of fetal growth and maturation is poorly understood. These issues were examined in a line of mice with a global deletion of the GLU receptor (Gcgr−/−), which are characterized by lower blood glucose levels and by α- and δ-cell hyperplasia in adults. Ablation of Gcgr was deleterious to fetal survival; it delayed β-cell differentiation and perturbed the proportion of β- to α-cells in embryonic islets. In adults, the mutation inhibited the progression of α-cells to maturity, affected the expression of several β-cell-specific genes, and resulted in an augmentation of the α-, β-, and δ-cell mass. This increase was due to an augmentation in both islet number and in the rate of proliferation of cells expressing GLU or insulin. These findings suggest that GLU participates in a feedback loop that regulates the proportion of the different endocrine cell types in islets, the number of islets per pancreas, and development of the mature α-cell phenotype.
The islets of Langerhans play a central role in glucose homeostasis. Insulin (IN) released from β-cells after a meal promotes the storage of glucose into target organs. In adults, the action of IN is counterbalanced by glucagon (GLU), a hormone produced by the α-cells that acts on the liver to stimulate glycogenolysis and gluconeogenesis. Although the effects of a glucoregulatory failure due to IN deficiency on embryonic development had received considerable attention (1-3), there is scant information on disturbances caused by the lack of GLU signaling on fetal growth. In this study, the role of GLU was examined in a line of mice with a global deletion of the GLU receptor (Gcgr−/−) (4). In the first part of this study, we asked whether the lack of GLU signaling affected fetal survival.
The second goal of this study was to use the Gcgr−/− strain to uncover the effect of GLU signaling on islet cell development and maturation. The process that guides the differentiation of the endocrine cells of the pancreas is believed to involve the sequential expression of a number of transcription factors (5, 6). However, little is known on the role of pancreatic hormones in islet cell differentiation and growth during pre- and postnatal development. Mice lacking prohormone convertase (PC) 2−/−, which fail to convert proinsulin, proglucagon, and prosomatostatin to the mature peptide hormones (7), showed a delay in β-cell differentiation during development and in α-cell maturation in adults (8). PC2−/− mice were also characterized by an increase in the number of islets per pancreas and non-β-cell hyperplasia (7, 8). The increase in non-β-cell number appeared to be due to the absence of GLU because the infusion of mature GLU to adult PC2−/− mice normalized islet cell composition (9). Because the lack of PC2 affects the processing not only of GLU but also of other pancreatic hormones and extrapancreatic neuropeptides (10-13), those studies left unresolved the question of whether the altered phenotype was due to effect(s) of one or more hormones affected by the mutation.
In the present study, we used previously described Gcgr−/− mice (4) to test whether the islet phenotype characteristic of PC2−/− mice could be reproduced by the ablation of the GLU signaling cascade. Significantly, islets of mature Gcgr−/− mice, like those of PC2−/− mice, display non-β-cell hyperplasia (4), suggesting similarities in the properties of the two mutant mouse strains. Studies described here demonstrate that the lack of GLU signal transduction has a profound effect on fetal survival and on growth and maturation of pancreatic islets.
Materials and Methods
Generation of Gcgr−/− mice
As previously reported (4), exons 3–6 of the murine Gcgr gene were deleted using homologous recombination and embryonic stem cell technology. Heterozygous (Gcgr+/−) and homozygous (Gcgr−/−) matings yielded null (Gcgr−/−) mice in a Mendelian ratio that were genotyped by Southern blot and/or PCR analysis. Mice backcrossed (F6 and F7) onto the C57-BL6J background and litter mate controls were studied. Animals were fed ad libitum with free access to water and maintained in a murine hepatitis virus-free barrier facility on a 12-h light, 12-h dark cycle. All protocols were approved by the institutional Animal Care and Use Committees.
Timed pregnancies
The following combinations of mice were mated: Gcgr+/+ females with Gcgr+/+ males, Gcgr−/− females with Gcgr−/− males, and Gcgr+/− females with Gcgr−/− males. Cages were monitored at 0800 h for the presence of vaginal plugs.
Embryo and pup collection
Mice were killed on embryonic day (e) 10.5, e15.5, and e18.5 of gestation, and postnatal day (P) 1, P7, P15, and P21. Embryos and pups were weighed and placed in 4% buffered paraformaldehyde for further analysis.
Tissue processing
Three-month-old adult mice were perfused through the heart with a solution of 4% paraformadehyde in 0.1 m phosphate buffer and post-fixed for several hours in the same fixative. Embryos were fixed overnight by immersion in the same fixative solution. Fixed tissues were infiltrated in 30% sucrose, mounted in embedding matrix (Lipshaw Co., Pittsburgh, PA), and 20-μm cryostat sections were mounted onto glass slides coated with a solution of 1% gelatin containing 0.05% chromium potassium sulfate.
Antibodies
Guinea pig antibodies to bovine IN was purchased from Linco Research, Inc. (Eureka, MO). Rabbit antibodies to human GLU and to Pax6 were purchased from Calbiochem, Inc. (San Diego, CA) and the Developmental Studies Hybridoma Bank (University of Iowa, Ames, IA), respectively. Rabbit antihuman pancreatic polypeptide (PP) and somatostatin (SOM) sera were obtained from Peninsula Labs, Inc. (Belmont, CA). Antirabbit GLUT2 sera were purchased from Chemicon Inc. (Temecula, CA). Monoclonal antibodies to IN and GLU were purchased from Sigma-Aldrich Inc. (St. Louis, MO). Rabbit antiserum to Pdx-1, Nkx2.2, Isl-1, Ngn3, PC3/1, PC2, and Maf-1 were a generous gift from C.V.E. Wright (Vanderbilt University, Nashville, TN), T. Jessel (Columbia University, New York, NY), M. German (University of California, San Francisco, CA), D. F. Steiner (University of Chicago, Chicago, IL), and R. Stein (Vanderbilt University), respectively. Antibodies were used at the following dilutions: guinea pig antibody to IN, 1:400; monoclonal antibody to GLU, 1:6000; rabbit antisera to human GLU, 1:4000 and to GLUT2, 1:1000; rabbit antihuman SOM and PP, 1:8000 and 1:10,000, respectively; rabbit antimouse Pdx-1, 1:5000; rabbit antiserum to Pax6, 1:3000; and rabbit antisera to Nkx2.2, Isl-1, PC2, PC3/1 Maf-1, and Ngn3, 1:1000. For secondary antibodies, biotinylated goat antirabbit IgG and avidin-labeled peroxidase were purchased from Vector Laboratories, Inc. (Burlingame, CA). Alexa fluor 488 antimouse, antirat, and antirabbit IgG and Alexa fluor 594 antiguinea pig, antirabbit, and antimouse IgG were purchased from Molecular Probes, Inc. (Eugene, OR). Sections immunostained for visualization of PC2 or PC3/1 were counterstained with the DNA-binding dye 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich Inc.) diluted 1:1000 in Tris-saline buffer (pH 7.4).
Immunostaining
These techniques have been previously described (7). In brief, the sections were incubated sequentially in an empirically derived optimal dilution of control serum or primary antibody raised in species X overnight and with a 1:200 dilution of the secondary antibodies. After completion of the staining procedure, sections were covered with two to three drops of Vectashield Solution (Vector Laboratories, Inc.).
Confocal microscopy
Confocal images were obtained using a Radiance 2000 confocal microscope (Bio-Rad, Hercules, CA) attached to a Zeiss Axioskop microscope (Carl Zeiss Inc., Thornwood, NY). Images of 540 × 540 pixels were obtained and processed using Adobe Photoshop 6.0 (Adobe Systems, Mountain View, CA).
Determination of cell proliferation in vivo
5-Bromo-2′ deoxyuridine (BrdU) was injected ip (200 mg/kg), the animals were perfused 2 h later with fixative solution, and the pancreas was removed and cryosectioned. Sections were incubated with 2 n HCl for 20 min and with 0.1 n HCl containing 0.5% pepsin (Sigma-Aldrich Inc.) for 20 min at 37 C. Then, sections were sequentially incubated with 10% goat serum in 0.1 m phosphate buffer, a 1:40 dilution of a monoclonal antibody to BrdU (Dako, Carpinteria, CA), donkey-antimouse antibody linked to Alexa green, and processed for visualization of IN or GLU.
Determination of apoptosis
Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end labeling (TUNEL) staining was carried as indicated in the manufacturer’s instructions (Chemicon). After completion of this step, the slides were processed for visualization of IN or GLU.
Morphometry
The relative number of α-, β-, and δ-cells per islet was determined in sections immunostained for GLU (GLU+), IN (IN+), and SOM (SOM+) by the point sampling method (14) using a 300-point ocular grid at a total magnification of 400×. The average number of stained cells per islet volume was calculated according to the formula: F = h/n, in which h is the number of hits over stained cells, and n is the number of points scored over islets. At least 5000 points were scored in 30 islets/strain. The relative GLU+, IN+, and SOM+ cell volume per section was calculated by dividing the number of points over immunostained cells over the number of points scored for that section. At least 10,000 points from three pancreata/age/matched strain were quantified for each experiment. The total α-, β-, and δ-cell mass was calculated by multiplying the relative cell volume per section by pancreas weight for individual animals.
To determine the number of islets per pancreas, consecutive 20-μm sections were obtained from each pancreas (three pancreases/genotype/age), and one section of every 10 was examined. The number of islets was determined in 10 sections per pancreas. We chose to examine one section of adult pancreas every 200 μm because recent studies indicate that large, hyperplastic islets achieve a volume of 600 μm3, giving a value of 200 μm for each axis (15, 16). This protocol avoids counting the same islet more than once. To measure islet cell size and islet area, sections were projected on the screen of a video monitor, and the area was measured using the National Institutes of Health Image program for Macintosh (http://rsb.info.nih.gov./nih-image). The area of GLU+ or IN+ cells was measured in at least 100 cells from 25 islets/mouse/three mice per line/age. The perimeter of at least 25 islets were visualized and measured with a 20× objective in slides immunostained with a cocktail of antibodies to IN, SOM, and GLU using the same program.
Determination of blood glucose and hormone levels
Blood was obtained either by decapitation at e18 and P1, P7, P15, and P21 or from retroorbital blood vessels in adult mice and spun at 5000 × g for 5 min. Fed glucose levels were determined with the Precision QID Glucose monitoring kit (gift from Abbott Laboratories, Chicago, IL) using 15 μl peripheral blood. Blood glucose levels are represented as an average ± sem. Fed IN levels were determined with murine ELISAs (Crystal Chem, Chicago, IL). GLU levels were determined by RIA from Linco Research, Inc. (St. Charles, MO).
RNA isolation and semiquantitative RT-PCR
Total RNA was isolated from islets using TRIzol reagent (Invitrogen, Carlsbad, CA) and further purified with RNAeasy Kit (QIAGEN, Valencia, CA). The concentration and purity of the RNA was determined by spectrophotometry. The RNA integrity was verified by electrophoresis. RNA preparations were treated with recombinant DNAse-1 (DNA-free kit, Ambion). One microgram of total RNA was transcribed using SuperScript III reverse transcriptase (Invitrogen). For each PCR, an equal amount of cDNA (1/10 of the reaction) was used. PCR was performed in 20- to 50-μl reactions containing 0.2 μ m each primer and 2.5 U Platinum Pfx DNA polymerase (Invitrogen). The number of cycles was optimized depending on the particular mRNA abundance and chosen to select PCR amplification on the linear portion of the curve to avoid saturation effect. Aliquots (10–20 μl) were analyzed by electrophoresis, and the bands were quantified by densitometric scanning of band intensities and normalized to the levels of the housekeeping gene 18S using Image J 1.32 gel analysis software (National Institutes of Health, Bethesda, MD). Information about genes is as follows: Pdx-1, amplicon size 169 bp, forward primer CGGACATCTCCCCATACG, reverse primer AAAGGGAGATGAACGGG (17), GenBank accession no. NM_022852.1; Ins-1, amplicon size 288 bp, forward primer TAGTGACCAGCTATAATCAGAG, reverse primer ACGCCAAGGTCTGAAGGTCC (18), GenBank accession no. NM_008386.2; MafA, amplicon size 405 bp, forward primer CACCACGTGCGCTTGG, reverse primer CAGAAAGAAGTCGGGTG (19), GenBank accession no. NM_026859.2; 18s rRNA, amplicon size 68 bp, forward primer AGTCCCTGCCCTTTGTACACA, reverse primer GATCCGAGGGCCTCACTAAAC, GenBank accession no. BK000964; PC2, amplicon size 414 bp, forward primer TGACTTCAGCAGCAATGACC, reverse primer CTTGAAGCATAGCCGTCACA, GenBank accession no. NM_008792; and PC3/1, amplicon size 264 bp, forward primer CCTGTAGGCACCTGGACATT, reverse primer ATTGCTGCTGCTGGAGTTTT, GenBank accession no. NM_013628.
Real-time RT-PCR
Islets were isolated and transferred to a 1.5-ml sterile tube. Total RNA was extracted using the RNeasy mini kit (QIAGEN), and agarose gel electrophoresis was used to analyze the quality of total RNA. Real-time RT-PCR was performed with One-step RT-PCR Master Mix reagents kit (TaqMan, Branchburg, NJ). Primers and 5-carboxyfluorescein/5-carboxytetramethylrhodamine-(Fam/Tamra)labeled probes were designed for target genes INS-1, Maf-A, Pdx-1, PC2, and PC3/1 and for the housekeeping gene TBP using Primer Express software and purchased from Sigma Genosys (St. Louis, MO; sequence of primers and probes are available upon request).
The cycling conditions for RT-PCR were as follows: 48 C for 30 min and 95 C for 10 min followed by 40 cycles of 95 C for 10 sec and 60 C for 1 min. PCR was run on ABI prism 7700 Sequence detection System (Applied Biosysytems, Foster City, CA). Quantitative values were obtained as threshold PCR cycle number (Ct) when the increase in fluorescent signal of PCR product showed exponential amplification. Target gene mRNA level was normalized to that of TBP in the same sample. In brief, the relative expression level of the target gene compared with that of TBP was calculated as 2 −ΔCt, where ΔCt = Ct target gene − Ct (TBP). The ratio of relative expression of the target gene in Gcgr−/− islets to that in Gcgr+/+ islets was then calculated as 2{Δ}{Δ}Ct, where {Δ}{Δ}Ct ={Δ}Ct Gcgr−/− islet − {Δ}Ct control Gcgr +/+ islet. Each sample was measured in triplicate.
Statistical analysis
All values indicate the mean ± sem. For comparison between groups, the unpaired Student’s t test (two tail) or ANOVA analysis was used. P < 0.05 was considered significant unless stated otherwise.
Results
Maternal genotype affects fetal growth and survival
Embryos derived from Gcgr+/+, Gcgr+/−, and Gcgr−/− matings were examined at e15.5 and e18.5, and results are indicated in Table 1. No differences in litter size were found between embryos derived from Gcgr+/+ and Gcgr+/− mothers. In contrast, only 50% of embryos derived from Gcgr−/− mothers survived until birth with fetal death starting after e16, with the remaining pups dying at P1. Although the weight of pups from Gcgr+/+, Gcgr+/−, and Gcgr−/− mothers was similar at e15.5 of gestation, the weight of e18 fetuses from Gcgr−/− mothers was lower than those from Gcgr+/− and Gcgr+/+ mothers (Table 1). These differences persisted at P1 of life (Table 1). Although smaller in size, Gcgr−/− pups from Gcgr−/− mothers did not have other macroscopic phenotypic abnormalities.
TABLE 1.
Effects of Gcgr mutation on fetal development
| Gcgr+/+ embryos | Gcgr−/− embryos from Gcgr−/− mothers | Gcgr−/− embryos from Gcgr+/− mothers | |
|---|---|---|---|
| Litter size | n = 15 | n = 19 | n = 14 |
| e15 | 8.5 ± 1.0 | 7.3 ± 1.0 | 8.0 ± 0.5 |
| e18 | 8.5 ± 0.3 | 3.7 ± 0.5a | 7.8 ± 0.8 |
| Body weight (g) | |||
| e15 | 0.4 ± 0.1 | 0.4 ± 0.1 | 0.4 ± 0.2 |
| e18 | 1.2 ± 0.1 | 0.8 ± 0.1a | 1.3 ± 0.1 |
| P1 | 1.7 ± 0.3 | 1.1 ± 0.1a | 1.6 ± 0.4 |
| Glycemia (mg/dl) | |||
| e18 | 90 ± 20 | 20 ± 5a | 75 ± 12 |
| P14 | 126 ± 48 | NA | 89 ± 26b |
| IN (ng/dl) | |||
| e18 | 0.99 ± 0.05 | 1.22 ± 0.2 | 1.1 ± 0.1 |
| P14 | 0.94 ± 0.2 | NA | 0.48 ± 0.02b |
e, Embryonic day; n, number of embryos/pregnancy. NA, not applicable.
P < 0.01 vs. Gcgr+/+ embryos and Gcgr−/−embryos from Gcgr+/− mothers.
P < 0.005 vs. Gcgr+/+.
Embryos from Gcgr+/+, Gcgr+/−, and Gcgr−/− mothers were born at e19.5. However, Gcgr−/− pups from Gcgr−/− mothers died 24 h after birth. To determine whether the observed lethality was related to differences in maternal glycemia, blood glucose levels were measured daily from d 12–19 of pregnancy. Pregnant Gcgr−/− had significantly lower blood glucose levels (99.7 ± 3.6 mg/dl) than pregnant Gcgr+/− and Gcgr+/+ litter mates (141.5 ± 2.99 mg/dl, P < 0.001). The genotype of the mother affected the glycemic levels of the embryos. e18 and P1 Gcgr+/− and Gcgr−/− embryos and pups from Gcgr+/− mothers displayed normal blood glucose levels, whereas blood glucose of the Gcgr−/− embryos from Gcgr−/− mothers was significantly decreased (Table 1). Interestingly, random IN levels at e18 were not significantly different in Gcgr−/− and Gcgr+/+ pups from Gcgr−/− or Gcgr+/− mothers, suggesting that hypoglycemia in utero does not affect IN secretion (Table 1).
Immediately after birth, Gcgr−/− pups from Gcgr−/− mothers exhibited severe hypoglycemia (26.72 ± 14 mg/dl, n = 36), and blood glucose levels dropped sharply just before death (<10 mg/dl, n = 26). In contrast, age-matched Gcgr+/− and Gcgr−/− pups born to Gcgr+/− mothers were normoglycemic at birth (Gcgr+/− pups, 85.67 ± 12 mg/dl, n = 26; Gcgr−/− pups, 80.72 ± 11 mg/dl, n = 32). These results indicate that litter size and fate of embryos and pups were determined by the genotype of the mother.
Gcgr−/− pups born to Gcgr+/− mothers maintained normoglycemia during the 1st week of life (Gcgr+/+ pups, 60.70 ± 20 mg/dl, n = 16; Gcgr−/− pups, 74.75 ± 15 mg/dl, n = 8). However, at P14, Gcgr−/− pups born to Gcgr+/− mothers displayed lower glucose and IN levels (Table 1) and a decreased body weight compared with Gcgr+/+ mice (Gcgr+/+ pups, 7.2 ± 0.6 g, n = 14; Gcgr−/− pups, 6.0 ± 0.8 g, n = 9; P < 0.005), indicating that the phenotype characteristic of this line is established 2 wk after birth.
Deletion of Gcgr delays β-cell differentiation
To determine whether deletion of Gcgr affects islet cell development, pancreata of Gcgr+/+ and Gcgr−/− embryos were examined for the presence of cells containing endocrine hormones. Pancreata of e10.5 Gcgr+/+ embryos contained IN+ cells (Fig. 1A). In contrast, pancreata of e10.5 Gcgr−/− embryos lacked cells immunostained for IN (Fig. 1B). The lack of IN+ cells was not due to the absence of the enzymes involved in IN maturation. IN is synthesized as proinsulin, and its processing to IN is mediated by the enzymes PC2 and PC3/1 (7, 20). PC2 also participates in the conversion of proglucagon to GLU in pancreatic α-cells (7). Pancreata of e10 Gcgr−/− embryos contained clusters of PC2 (Fig. 1C) and PC3/1 (Fig. 1E) cells. Similar results were obtained for Gcgr+/+ embryos (data not shown). Moreover, PC 3/1 was also found at e10 in the epithelial lining of the pancreatic primordia in both mouse lines (Fig. 1D), indicating that this enzyme is a marker of endocrine precursor cells. IN-expressing cells were first observed in pancreata of Gcgr−/− mice from Gcgr+/− and Gcgr−/− mothers at e13.5 (Fig. 1F), and, at e15.5, pancreata of both Gcgr+/+ and Gcgr−/− embryos from Gcgr+/− and Gcgr−/− mothers contained a significant number of IN+ cells (Fig. 1, G and H) respectively. These results provide evidence that the absence of GLU signaling in the embryo delayed the appearance of IN+ cells during development. In agreement with results reported elsewhere for other mouse strains (21-23), SOM+ - and PP+-containing cells first appeared at e15 and P1, respectively, in both Gcgr+/+ and Gcgr−/− embryos (data not shown).
Fig. 1.
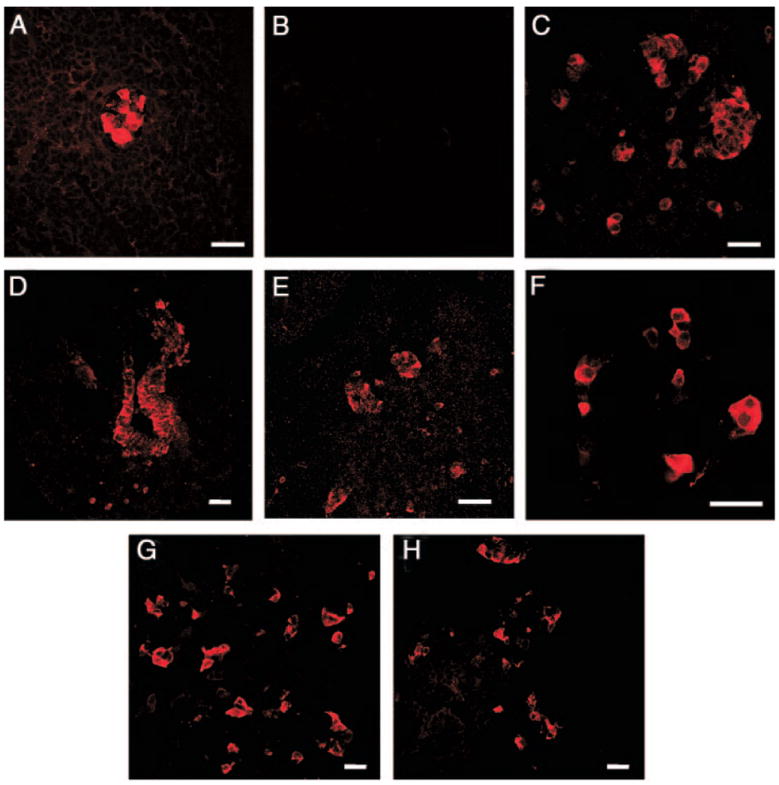
Delayed appearance of IN+ cells in pancreas of Gcgr−/− mice during development. Photomicrographs illustrate confocal microscopic images. A, Pancreas of e10 Gcgr+/+ embryo immunostained for IN shows expression of the hormone. B, Section of pancreas of an e10 Gcgr−/− embryo incubated with IN antibodies demonstrates the absence of immunopositive cells. C, Section of pancreas of an e10 Gcgr−/− embryo incubated with antibodies to PC2 demonstrates the presence of immunopositive cells. Photomicrographs D and E illustrate the presence of PC3/1 in the epithelium (D) and in a cell cluster (E) of e10 pancreatic primordia. Photomicrograph of sections of pancreata of e13.5 (F) and at e15 (G) Gcgr−/− mice and of e15 Gcgr+/+ litter mates (H) illustrates the presence of IN+ cells at these stages of development. Bars, 40 μm.
Deletion of Gcgr results in α- and δ-cell hyperplasia and increased islet number
To ascertain whether deletion of the Gcgr affected islet cell composition during development, we examined pancreata of e15.5 and e18.5 Gcgr−/− embryos. Endocrine cells of e15 Gcgr−/− embryos from Gcgr−/− mothers were dispersed as single cells within the exocrine parenchyma (Fig. 2E). Three days later, the endocrine cells form aggregates (Fig. 2F), but these aggregates retained an elongated configuration reminiscent of islets of younger stage embryos. Islets were generally larger in Gcgr−/− (5564.9 ± 1138 μm2) than in Gcgr+/+ litter mates (5166 ± 1033 μm2) (P < 0.01). Pancreata of e18 Gcgr−/− embryos from Gcgr−/− and Gcgr+/− mothers (Fig. 2, D–F) showed increased percentage of α-cells per islet compared with Gcgr+/+ embryos (30 ± 3% Gcgr−/− from Gcgr−/− and Gcgr+/− mothers vs. 15 ± 7% Gcgr+/+; P < 0.05) (Fig. 2B). Similarly, the percentage of SOM+ cells per islet was higher in pancreas of Gcgr−/− embryos than in Gcgr+/+ litter mates (8.5 ± 2.2%vs. 1.6 ± 0.5; P < 0.001), indicating that the lack of GLU signaling resulted in an increase in GLU+ and SOM+ cell numbers during prenatal development.
Fig. 2.
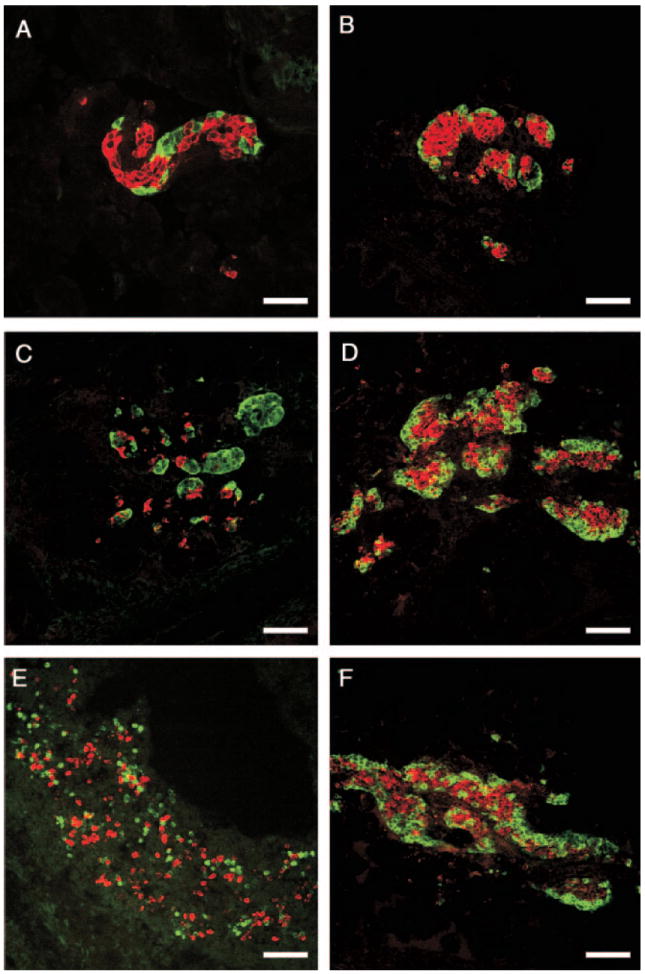
Lack of GLU signaling affects the morphology of embryonic islets. Photomicrographs of confocal microscope images illustrate islets of embryos at e15 (A, C, and E) and e18 (B, D, and F) of development. Gcgr+/+ embryos (A and B), Gcgr−/− embryos from Gcgr+/− mothers (C and D) and Gcgr−/− embryos from Gcgr−/− mother (E and F) immunostained for IN (red) and GLU (green). Pancreas of Gcgr−/− mice exhibit an increased number of α-cells at e15 (C and E) and e18 (D and F) compared with Gcgr+/+ litter mates (A and B). Also note the lack of aggregation of islet cells in pancreata of e15 Gcgr−/− embryos from Gcgr−/− mothers. Bars, 80 μm.
The increased number of α- and δ-cells in islets during development became more prominent during postnatal life. In agreement with results previously described (4), islets of Gcgr−/− mice were comprised of a thick mantle of α- and δ-cells that surround a core of IN+ cells (Fig. 3, B, D, and F). Morphometric analysis confirmed that mean number of GLU+ and SOM+ cells per section and per pancreas was more than 2-fold higher in Gcgr−/− than in Gcgr+/+ litter mates (Table 2), whereas the relative β-cell volume per section was similar in both lines. In addition to an increase in number, δ-cells in islets of Gcgr−/− mice exhibited an abnormal distribution because they were no longer localized to the mantle zone; rather, they were scattered in the core of the islet (Fig. 3F). The average PP-cell number per section was similar in islets of Gcgr−/− and Gcgr+/+ mice (5.4 ± 0.10 Gcgr−/− vs. 5.3 ± 0.12 Gcgr+/−; over 5000 points scored/pancreas/strain, n = 3).
Fig. 3.
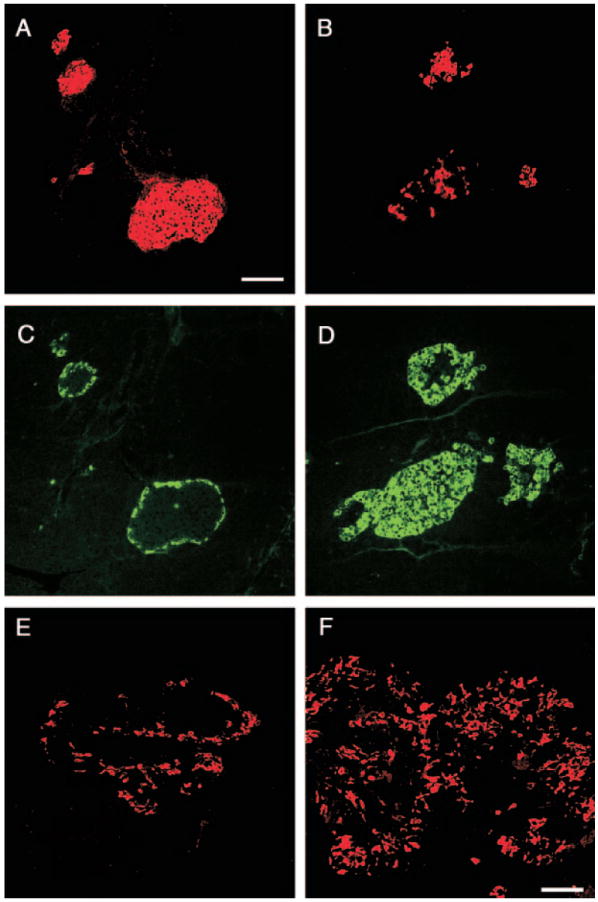
Islets of adult Gcgr−/− mice display non-β-cell hyperplasia. Photomicrographs of confocal microscope images illustrate islets from 3-month-old mice immunostained for visualization of IN (A and B), GLU (C and D), and SOM (E and F). A, C, and E, Gcgr+/+. B, D, and F, Gcgr−/− mice, respectively. Note that the number of GLU+ and SOM+ cells in islets of Gcgr−/− mice is higher than in Gcgr+/+ litter mates. Bar, 80 μm.
TABLE 2.
Effects of Gcgr mutation in adult mice
| Gcgr+/+ (n = 5) | Gcgr−/− (n = 8) | |
|---|---|---|
| Glycemia (mg/dl) | 115 ± 6 | 100 ± 4a |
| Pancreas weight (g) | 0.34 ± 0.1 | 0.48 ± 0.1b |
| IN (ng/dl) | 0.51 ± 0.2 | 0.42 ± 0.1 |
| GLU (pg/ml) | 128 ± 86 | 26,000 ± 14,000b |
| Number of islets/section | ||
| Small | 6 ± 3 | 19 ± 5b |
| Large | 12 ± 5 | 29 ± 5b |
| Relative cell volume/section (%) | ||
| β-Cell | 5.5 ± 0.5 | 6 ± 1.6 |
| α-Cell | 4.5 ± 0.7 | 12.6 ± 1.5b |
| δ-Cell | 2.1 ± 0.4 | 6.1 ± 1.1b |
| Relative cell volume/islet (%) | ||
| β-Cell | 80 ± 5 | 20 ± 15b |
| α-Cell | 15 ± 8 | 70 ± 20b |
| δ-Cell | 4 ± 1 | 10 ± 5b |
| Total cell mass/pancreas | ||
| β-Cell | 1.9 ± 0.4 | 2.8 ± 0.3a |
| α-Cell | 1.6 ± 0.4 | 6.1 ± 0.2b |
| δ-Cell | 0.8 ± 0.2 | 2.9 ± 0.3b |
Adult Gcgr−/− mice represent the offspring of Gcgr−/− mothers. The number of islets was determined in 10 sections per pancreas, three pancreata/strain. Relative α-, β-, or δ-cell volume/section was determined as the ratio of the number of points over stained cells divided by the total number of points in sections. At least 10,000 points were scored/strain. Relative cell volume per islet was determined as the total number of points scored over stained cells divided by the total number of points over islets. At least 5000 points were scored in 30 islets/strain. Total cell mass/pancreas is the relative cell volume/section multiplied by pancreas weight (grams).
P < 0.05 vs. Gcgr+/+.
P < 0.01 vs. Gcgr+/+.
To determine whether GLU signaling affects islet number, the number of islets in pancreas of Gcgr+/+ and Gcgr−/− mice was compared. This analysis revealed that pancreata of adult Gcgr−/− contained 3 times more small (two to 10 endocrine cells) and medium/large islets (more than 10 endocrine cells/islet) than pancreata of Gcgr+/+ litter mates (Table 2). Both types of islets were evenly distributed throughout the pancreas. The presence of more islets per pancreas in Gcgr−/− than in Gcgr+/+ mice contributed to the augmentation in the total β-cell mass that was determined in the Gcgr−/− line (Table 2). Deletion of the Gcgr gene also resulted in development of α-cell hypertrophy in adults. The mean area of GLU+ cells in mutant Gcgr−/− mice was almost twice that of Gcgr+/+ mice (Gcgr−/−, 109.71 ± 6.34 μm2; Gcgr+/+, 69.46 ± 5.42 μm2; P < 0.001)
Increased rate of α- and β-cell proliferation in Gcgr−/− mice during the perinatal period
To ascertain the role of cell division in the development of α-cell hyperplasia and in the increase in the β-cell mass characteristic of pancreas of Gcgr−/− mice, the rate of proliferation of GLU+ and IN+ cells was determined in e18.5, P1, P7, and 3-month-old Gcgr−/− and litter mate Gcgr+/+ mice. The rate of GLU+ cell proliferation was higher in Gcgr−/− than in Gcgr+/+ mice in embryos at e18.5 and in pups during the 1st week after birth (Fig. 4). In contrast, the rate of IN+ cells increased in Gcgr−/− mice above Gcgr+/+ litter mate levels only after birth (Fig. 4). The replication rate of both IN+ and GLU+ cells decreased to very low levels at 3 months in both genotypes (<0.01%). In contrast to proliferation, the rate of apoptosis was very low in islets of P1, P7, P21, and adult Gcgr+/+ and Gcgr−/− mice (data not shown).
Fig. 4.
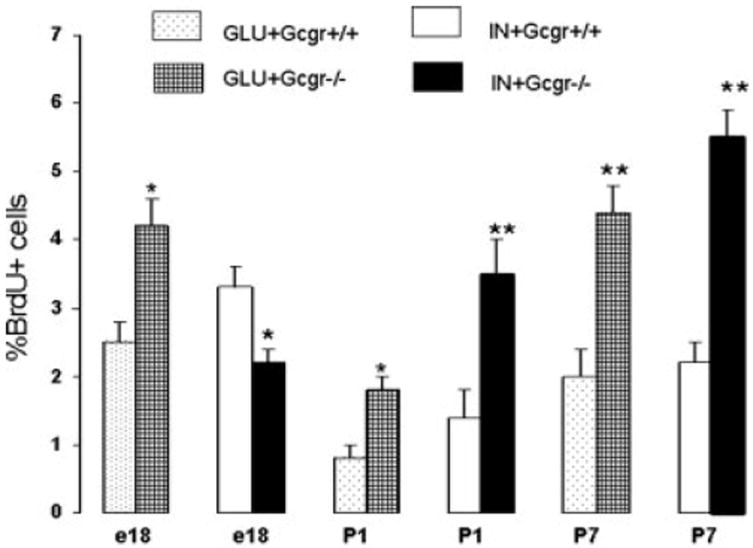
Increased rate of α- and β-cell proliferation in islets of Gcgr−/− mice during the perinatal period. Pregnant mice, pups, or adult mice were injected with BrdU, and the number of cells expressing the thymidine analog and a hormone was determined as indicated in Materials and Methods. The labeling index was determined as the number of GLU+ and IN+ cells containing the BrdU per total number of GLU+ or IN+ cells scored. Over 500 cells were scored per antibody/age/genotype (n = 3). Note the increased rate of α-cell proliferation in fetuses and young pups. In contrast, the rate of IN+ cell proliferation increased only after birth. In adults, the rate of IN+ and GLU+ cell replication in both strains was very low (data not shown). *, Less than 0.05; **, less than 0.01.
Islets cells of Gcgr−/− mice express embryonal traits
Deletion of the Gcgr in α-cells resulted in the presence of α-cells expressing embryonic markers. In control embryos, GLUT2 is expressed by both α- and β-cells during development (24), where it is localized in the cytoplasm of GLU+ cells and in the cell membrane of β-cells (8, 24). α-Cells of Gcgr−/− mice also expressed GLUT2 during development (Fig. 5A). In Gcgr+/+ adult mice, GLUT2 was found only in the cell membrane of IN+ cells and was absent from GLU+ cells (Fig. 5B). In contrast to Gcgr+/+ mice, GLUT2 was present in the cytoplasm of α-cells of islets of Gcgr−/− mice throughout life (Fig. 5C). Deletion of Gcgr also affected the expression of the transcription factor Pdx-1. In Gcgr+/+ mice, the transcription factor Pdx-1 is expressed by β-cells and by a subset of δ-cells throughout life and by α-cells of e10 to e14 embryos but not of older embryos or adults (Fig. 5D) (22). Examination of pancreata of Gcgr−/− mice revealed the presence of a subset of GLU+ cells expressing Pdx-1 in islets of adults (Fig. 5E). No difference among e15.5, e18.5, and 3-month-old Gcgr+/+ and Gcgr−/− mice was found in the distribution and pattern of expression of the islet cell factors Pax-6, Isl-1, Nkx2.2, and Ngn3 (data not shown), which play key roles in the specification of the islet precursor cells to the endocrine fate (5, 6). Islets of adult Gcgr−/− mice also contain scattered cells co-expressing IN+ and GLU+ (Fig. 6, B and C). As previously reported by us and others (21, 25), IN+ GLU+ cells were transiently found in pancreas of Gcgr+/+ from e10 to e13.5, but not at later stages of development or adults (Fig. 6A).
Fig. 5.
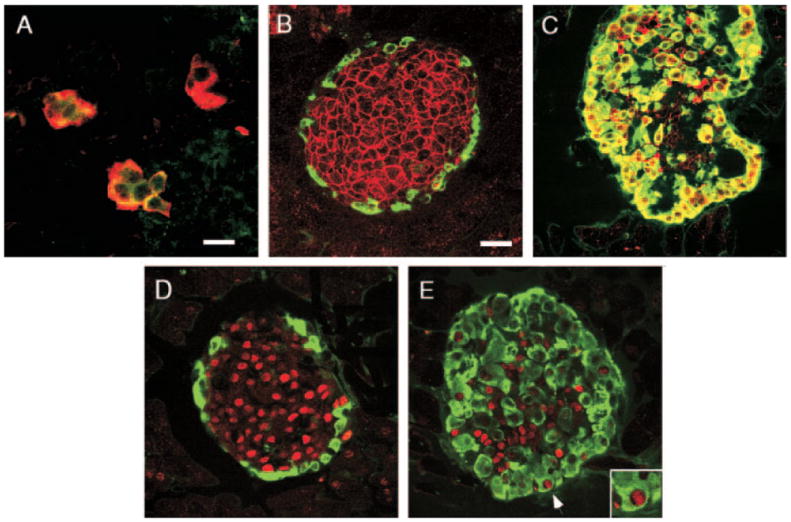
GLU+ cells of Gcgr−/− mice express immature traits. Photomicrographs illustrate confocal microscope images. A, Pancreas of e15 Gcgr−/− embryos stained for visualization of GLUT2 (green) and GLU (red). Note the presence of GLUT2+/GLU+ cells (yellow). B and C, Islets of 3-month-old mice immunostained for visualization of GLUT2 (red) and GLU (green). B, Gcgr+/+ mice; C, Gcgr−/− mice. Note that α-cells of Gcgr+/+ mice lack GLUT2. In contrast, most α-cells of Gcgr−/− mice express the transporter. D and E, Photomicrographs illustrate islets of 3-month-old mice immunostained for visualization of Pdx1 (red) and GLU (green). D, In Gcgr+/+, GLU+ cells did not express Pdx-1. E, In Gcgr−/− mice, a small subset of GLU+ cells coexpressed Pdx-1. Insert in E, Expression of Pdx-1 by an α-cell (indicated by an arrow) of adult. Bars, 20 μm.
Fig. 6.
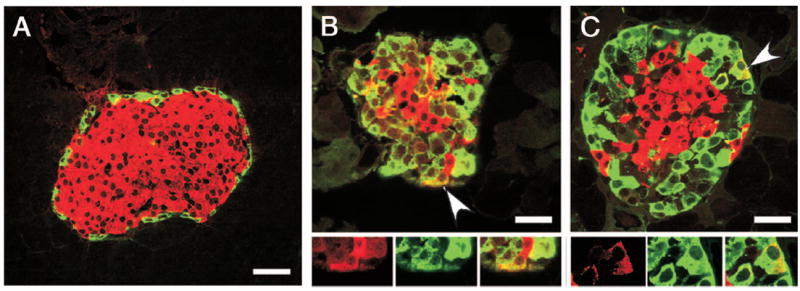
Coexpression of GLU and IN in pancreatic islets of Gcgr−/− mice. A, In Gcgr+/+ mice, cells expressing IN (red) did not immunostain for GLU (green). Bar, 60 μm. In contrast, IN+GLU+ cells, indicated by arrowheads, were found in islets of Gcgr−/− mice (B and C). Photomicrographs illustrate confocal microscope images. Bar, 40 μm.
Lack of GLU signaling affects the level of expression of β-cell-specific genes
To ascertain whether ablation of the Gcgr affected the β-cell phenotype, pancreata of adult Gcgr+/+ and Gcgr−/− mice were double immunostained for IN and Pdx-1, GLUT2, or MafA, a transcription factor proposed to be involved in the glucose-mediated increase in IN gene transcription (26-28). In agreement with other reports (26), MafA expression was restricted to IN+ cells (Fig. 7A) and was not coexpressed in GLU+, SOM+, PP+, or exocrine cells of any of the lines of mice examined (data not shown). In contrast to Gcgr+/+, only a subset of IN+ cells from pancreata of Gcgr−/− mice coexpressed Maf-1 (Fig. 7B). Immunohistochemical analysis revealed that the level of expression of Pdx-1 (Fig. 7D) and of membrane-localized GLUT2 (Fig. 7, E–G) was lower in β-cells of Gcgr−/− than in those of Gcgr+/+ mice (Figs. 7C and 5B). Deletion of Gcgr affected the level of expression of PC3/1, the converting enzyme that plays a critical role in the conversion of proinsulin to IN (29). Examination of tissues using PC3/1-specific antibodies indicated a significant reduction in the level of immunolabeling in β-cells of islets of Gcgr−/− (Fig. 8D) than β-cells of Gcgr+/+ litter mate controls (Fig. 8C). The mutation also affected the level of INS-1, Maf-A, Pdx-1, and PC3/1 mRNA, which were lower in islets of Gcgr−/− mice than in islets of Gcgr+/+ litter mates (Fig. 9). In contrast, ablation of Gcgr did not affect the level of PC2 immunolabeling or mRNA levels, which were similar in islets of Gcgr+/+ and Gcgr−/− mice (Figs. 8, A and B, and 9).
Fig. 7.
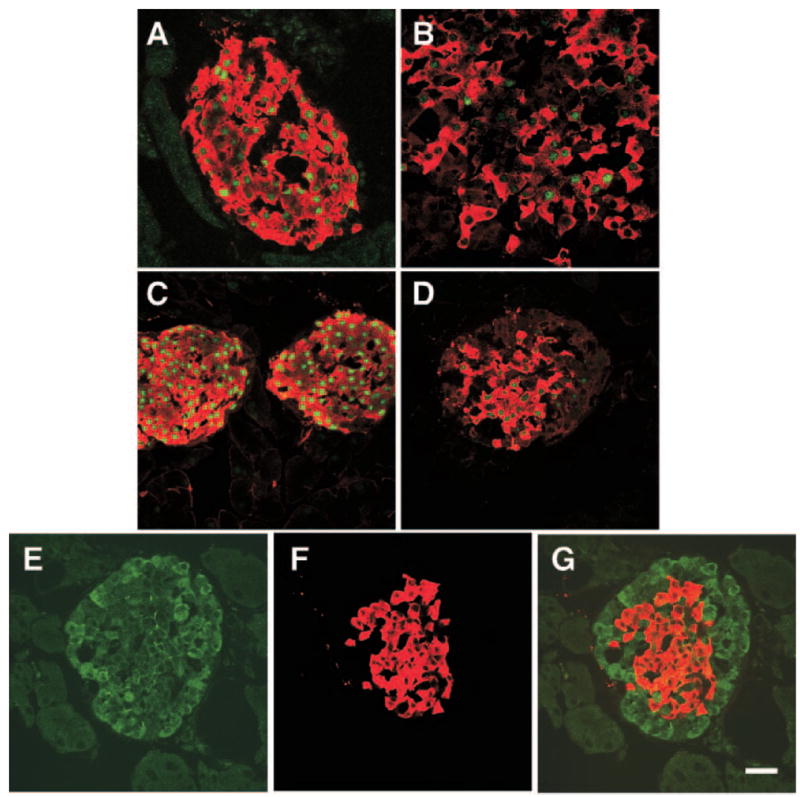
β-Cells of Gcgr−/− mice express low levels of Pdx-1, Maf-A, and GLUT2. Confocal microscope analysis of islets immunostained for IN (red) and either Maf-A (A and B, green) or PDX-1 (C and D, green). A, Photomicrograph illustrates an islet of adult Gcgr+/+ mice. Note that most, if not all, β-cells coexpress IN and Maf-A. B, In islets of Gcgr−/− mice, only a subset of islet IN+ cells express Maf-A. C and D, Islets immunostained for Pdx-1 and IN. Note that Pdx-1 expression in β-cells is higher in islets of Gcgr+/+ mice (C) than in islets of Gcgr−/− mice (D). E and G, Photomicrograph of islets of Gcgr−/− immunostained for GLUT2 (E) and IN (F) and GLUT2/IN. G, Decreased GLUT2 immunoreactivity in β-cells when compared with litter mate controls (depicted in Fig. 5B). Bar, 20 μm.
Fig. 8.
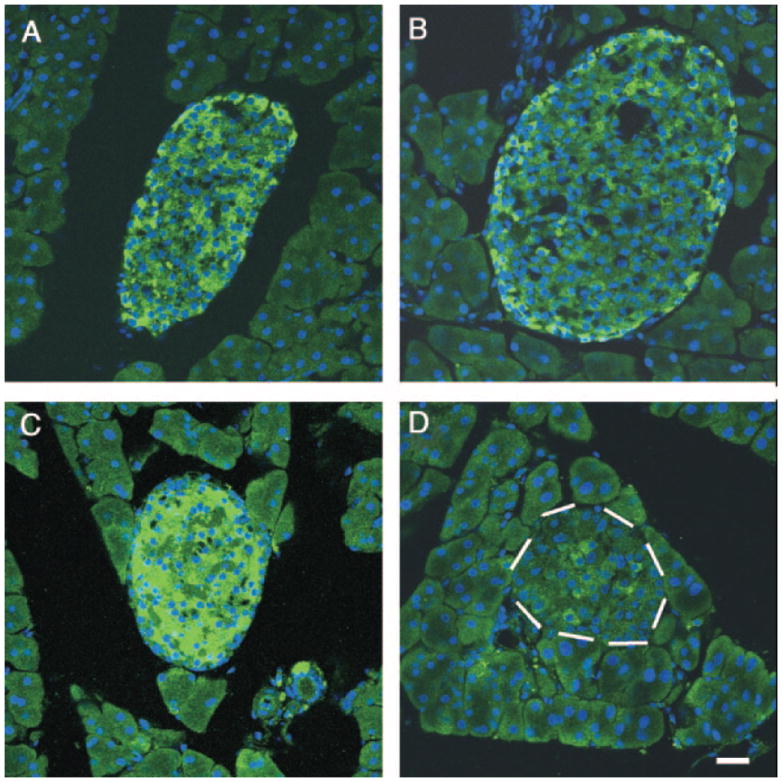
Ablation of Gcgr decreases expression of protein convertase PC3/1 in β-cells. Confocal microscope analysis of islets of 3-month-old Gcgr+/+ mice (A) and of Gcgr−/− mice (B) immunostained for visualization of PC2 documents similar levels of labeling in both tissues. In contrast, the level of PC3/1 is significantly lower in islets of Gcgr−/− (D) than in islets of Gcgr+/+ mice (C). White dashes, Location of the islet in D. Nuclei of cells were visualized using 4′,6-diamidino-2-phenylindole. Bars, 50 μm.
Fig. 9.
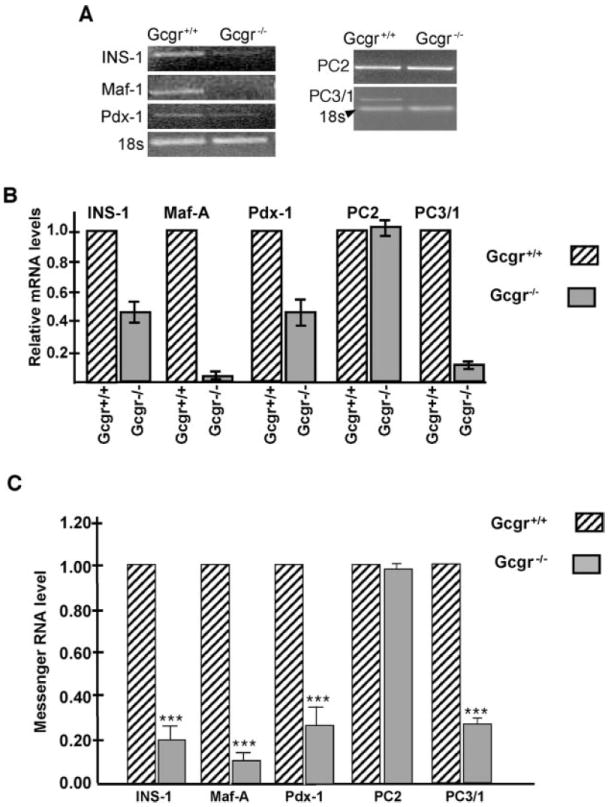
β-Cells of Gcgr−/− mice have low levels of expression of β-cell-specific genes. A, Semiquantitative RT-PCR analysis was performed on RNA isolated from islets of Gcgr+/+ and Gcgr−/− mice with Maf-A, Pdx-1, PC2, PC3/1, and IN-specific primers. Note that the levels of IN, Pdx-1, PC3/1, and Maf-A mRNA are lower in islets of Gcgr−/− mice than in islets of litter mate controls. In contrast, PC2 levels are similar in both strains. B, Histogram illustrates relative mRNA levels normalized to 18sRNA. C, Real-time RT-PCR analysis of IN, Maf-A, Pdx-1, PC2, and PC3/1 expression in islets isolated from Gcgr+/+ and Gcgr−/− mice, respectively. Note the significant reduction in the levels of IN, Maf-A, Pdx-1, and PC3/1 mRNA levels in islets of Gcgr−/− mice. ***, P < 0.0001.
Discussion
In assessing the role of pancreatic hormones on development, it is important to distinguish their putative roles in proliferation and/or differentiation in islets from their function in glucose metabolism. Mice lacking the Gcgr gene (4) provide a unique system in which to test the effects of this mutation on fetal growth and survival. We found that the number of Gcgr−/− embryos derived from Gcgr−/− was lower than those derived from Gcgr+/− mothers, indicating that total ablation of the Gcgr gene was deleterious to fetal survival.
The fact that Gcgr+/− mothers generated Gcgr−/− embryos with a normal Mendelian ratio indicates that the decrease in litter size from Gcgr−/− mothers was not due to an innate defect in Gcgr−/− embryos. Rather, the survival of the mutant embryos was associated with the genotype of the mother. Gcgr−/− embryos from Gcgr−/− mothers were also smaller than embryos from Gcgr+/− mothers, in agreement with the known effect of maternal milieu on embryonic size (30). Interestingly, embryonic death occurs during late gestation, supporting previous evidence (31, 32) indicating that this period is highly susceptible to maternal metabolic disorders. It remains to be determined whether fetal lethality in Gcgr−/− embryos from Gcgr−/− mothers is a direct effect of an abnormal uterine environment or whether the lack of GLU signaling disrupts the transport of other required nutrients from the mother to the fetus. This question is of particular importance when considering the severe low serum glucose levels found in the e18 Gcgr−/− embryos when compared with the maternal glycemia of Gcgr−/− mothers. Although glucose is known to be transported through the placenta (33), fetal glucose concentration in blood appears to be a reflection of both the maternal concentration and the degree of placental transfer. Previous studies of human fetuses with growth retardation have shown the presence of maternal-fetal glucose gradient, suggesting an altered placental fetal transfer (34). In agreement with those reports, preliminary findings indicate that deletion of Gcgr−/− results in altered placentation (35), supporting the hypothesis that placental defects causes the hypoglycemia determined in e18 Gcgr−/− embryos from Gcgr−/− mothers. The difference in lethality between Gcgr−/− pups from null and heterozygote mothers and Gcgr+/+ pups also suggests that the rise in GLU-induced glucose release from the liver that occurs soon after birth (36) is blunted in the mutants, leading to the demise of the hypoglycemic Gcgr−/− pups from Gcgr−/− mothers. Interestingly, during the perinatal period, Gcgr−/− pups from Gcgr+/− mothers are able to maintain normal glucose levels during the 1st week of life but become hypoglycemic during the 2nd week of life. This observation suggests that the lack of GLU signaling leads to inadequate energy supply, which is translated into a defect in weight gain in Gcgr−/− compared with Gcgr+/+ pups.
Because GLU is the first hormone to appear during development (23, 36-38), we hypothesized that the hormone may have autocrine or paracrine interactions with neighboring cells in the embryonic pancreas. Conceivably, the lack of Gcgr would modify the microenvironment of the islet precursor cells, affecting the differentiation of other endocrine cell types. In agreement with this possibility, ablation of the Gcgr gene inhibited the differentiation of IN cells during the protodifferentiated stage, which is considered to extend from early development until postnatal d 14 in mice (38). Cells expressing IN were detected in pancreases of Gcgr−/− embryos after e14, which is equivalent to the second wave of IN+ cell differentiation in Gcgr+/+ mice, supporting the view that GLU is required for the differentiation of the first wave of IN cells (8, 39, 40). The absence of the early group of IN+ cells in Gcgr−/− mice did not affect the time table for differentiation of δ or PP cells presumably because these two cell types first appear after midgestation.
Ablation of the Gcgr gene affected the development of a mature islet phenotype. During development in Gcgr+/+ mice, the different islet cell types appear in clusters of one cell type or as individual cells intermixed with other islet cells (23, 38). Endocrine cells achieve a typical islet configuration during late gestation, with a core of β-cells surrounded by non-β-cells (23, 38). In contrast, islet cells in pancreata of e15 Gcgr−/− embryos from Gcgr−/− mothers were dispersed as single cells. At e18, these cells formed elongated aggregates but failed to adopt a mature cytoarchitecture until after birth. It is believed that the acquisition of a mature islet configuration is determined by the presence of different cell adhesion molecules in β- and non-β-cells (41-44), a process that may be delayed in Gcgr−/− embryos derived from Gcgr−/− mothers. The fact that this delay in maturation was seen in Gcgr−/− embryos from Gcgr−/− but not from Gcgr+/− mothers indicates that it was not directly related to the deletion of GLU signaling. Rather, the perturbed morphology of the islet cell clusters in Gcgr−/− embryos may be due to an altered intrauterine environment in Gcgr−/− mothers.
The absence of GLU signaling reversed the normal proportion of β- and non-β-cells in islets. Although in Gcgr+/+ mice the β- and non-β-cells comprise approximately 80 and 20%, respectively, of the islet population, islets of Gcgr−/− mice contained nearly 80% non-β-cells. The increase in the non-β-cell population was due primarily to larger numbers of GLU+ and SOM+ cells, whereas the percentage of PP+ cells remained similar to Gcgr+/+ values. The deviation from normal type was even more striking in small islets, which in many instances were comprised mostly of α-cells with occasional β-cells. The increase in the mass of the non-β-cell population was already evident during late prenatal development in Gcgr−/− fetus generated by Gcgr+/− and Gcgr−/− mothers. Growth in the number of non-β-cells was partly due to increased rates of GLU+ cell proliferation in islets of embryos and pups. This enlargement persisted in adults, where the rate of non-β-cell proliferation was very low, suggesting that non-β-cells generated early in life persisted for a long period of time.
One possible explanation for the increase in the non-β-cell population in pancreases from Gcgr−/− mice is that it is caused by the mild hypoglycemia characteristic of this mutant strain. According to this hypothesis, a decrease in circulating blood sugar levels would signal an augmentation in the number of GLU cells and GLU secretion, which would normally lead to a rise in gluconeogenesis and euglycemia. Alternatively, the development of α-cell hyperplasia may be due to the disruption of an autocrine loop regulating α-cell number in Gcgr−/− mice. This possibility is supported by the observation that suppression of Gcgr expression by antisense oligonucleotide results in α-cell hyperplasia (45). In addition, the administration of GLU to PC2−/− mice induced an increase in α-cell apoptosis and led to the restoration of normal islet morphology (9).
An additional role of GLU is its involvement in α-cell maturation. The lack of GLU signaling did not affect the pattern of expression of the proendocrine gene Ngn3, which was transiently expressed during early gestation or of Pax6, Nkx2.2, and Isl-1, which were expressed by all islet cells of embryos and adults in the two strains examined (Gcgr+/+ and Gcgr−/−). However, deletion of the Gcgr gene prevented the development of a mature α-cell phenotype. In control embryos, Pdx-1 and GLUT2 are expressed by both α- and β-cells and later become restricted to β-cells of adults (8, 22, 24). In contrast, pancreatic islets of embryonic and adult Gcgr−/− mice contained GLU+ cells coexpressing Pdx-1 or GLUT2.
The molecular profile of β-cells was also affected by the global deletion of the Gcgr gene. IN+ cells of Gcgr−/− mice had low levels of Pdx-1, GLUT2, and MafA, which are molecules involved in the regulation of IN expression (26-28, 46, 47). Accordingly, the level of IN mRNA was low in Gcgr−/− mice, and this decrease was coordinated with a down-regulation in the level of PC3/1. The decrease in the level of IN expression could be due to the lower blood glucose levels characteristic of the mutant strain (48). Alternatively, it is possible that the regulation of IN mRNA levels is mediated by IN itself because IN has been shown to regulate its own level of expression (49, 50). Presumably, the overabundance of β-cells in pancreases of Gcgr−/− mice is responsible for a decrease in the amount of IN produced per cell and of the factors regulating its expression. This proposed compensatory mechanism is supported by the presence of similar circulating IN levels in Gcgr+/+ and Gcgr−/− mice and by findings that other lines of mice with augmented β-cell mass also have normal circulating IN levels (51, 52 and references therein).
The precise mechanisms underlying the regulation of the number of the different endocrine cells per islet and of islets per pancreas by GLU remain to be elucidated. Although a subset of α-, β-, and δ-cells have been reported to express Gcgr (53), it remains to be determined whether the receptor is widely distributed in all the different islet cells but remains undetected. Alternatively, regulation of β- and non-β-cell number could be mediated by the neurotransmitters GABA and glutamate, which have been shown to be involved in the induction of precursor populations in the central nervous system and other organs and in signaling between islet cells (54-57). The level of expression of these neurotransmitters may be altered by the deletion of Gcgr. Finally, it is possible that the pancreatic phenotype of Gcgr−/− mice is due to the lack of GLU signaling in α-cells acting in concert with the ensuing lower glucose levels.
In conclusion, we provide evidence supporting a novel role of GLU in fetal survival, pancreatic islet cell development, and maturation. Our findings demonstrate that GLU plays an important role in the regulation of α- and β-cell proliferation, in the establishment of islet phenotype, and in the progression of α-cells to maturity. Elucidation of the mechanisms underlying the action of GLU is likely to provide insights into therapeutic tools for the management of diabetes.
Acknowledgments
The authors acknowledge the expert technical assistance of Carlos Vargas.
This work was supported by the National Institutes of Health (Grants DK47425 to M.J.C., DK71949 to G.T., HL58119 to M.J.C., and KO8 HD042172 to P.V.), by the American Diabetes Association (Grant 1-02-RA-101 to GT), by the Albert Einstein College of Medicine Comprehensive Cancer Center (to M.J.C.), and by the Albert Einstein College of Medicine Diabetes Center (to M.J.C.). P.M.V., M.H.K., L.C., Y.G., M.N., M.J.C., and G.T. have nothing to declare. R.W.G. has equity in Novo-Nordisk and is employed by Metabolex.
Abbreviations
- BrdU
5-Bromo-2′deoxyuridine
- Ct
threshold PCR cycle number
- e
embryonic day
- Gcgr
GLU receptor
- GLU
glucagon
- IN
insulin
- P
postnatal day
- PC
prohormone convertase
- PP
pancreatic polypeptide
- SOM
somatostatin
References
- 1.Joshi RL, Lamothe B, Cordonnier N, Mesbah K, Monthioux E, Jami J, Bucchini D. Targeted disruption of the insulin receptor gene in the mouse results in neonatal lethality. EMBO J. 1996;15:1542–1547. [PMC free article] [PubMed] [Google Scholar]
- 2.Duvillie B, Currie C, Chrones T, Bucchini D, Jami J, Joshi RL, Hill DJ. Increased islet cell proliferation, decreased apoptosis, and greater vascularization leading to β-cell hyperplasia in mutant mice lacking insulin. Endocrinology. 2002;143:1530–1537. doi: 10.1210/endo.143.4.8753. [DOI] [PubMed] [Google Scholar]
- 3.Accili D, Drago J, Lee EJ, Johnson MD, Cool MH, Salvatore P, Asico LD, Jose PA, Taylor SI, Westphal H. Early neonatal death in mice homozygous for a null allele of the insulin receptor gene. Nat Genet. 1996;12:106–109. doi: 10.1038/ng0196-106. [DOI] [PubMed] [Google Scholar]
- 4.Gelling RW, Du XQ, Dichmann DS, Rømer J, Huang H, Cui L, Obici S, Tang B, Holst JJ, Fledelius C, Johansen PB, Rossetti L, Jelicks LA, Serup P, Nishimura E, Charron MJ. Lower blood glucose, hyperglucagonemia, and pancreatic α cell hyperplasia in glucagon receptor knockout mice. Proc Natl Acad Sci USA. 2003;100:1438–1443. doi: 10.1073/pnas.0237106100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jensen J. Gene regulatory factors in pancreatic development. Dev Dynamics. 2004;229:176–200. doi: 10.1002/dvdy.10460. [DOI] [PubMed] [Google Scholar]
- 6.Wilson ME, Scheel D, German MS. Gene expression cascades in pancreatic development. Mech Dev. 2003;120:65–80. doi: 10.1016/s0925-4773(02)00333-7. [DOI] [PubMed] [Google Scholar]
- 7.Furuta M, Yano H, Zhou A, Rouille Y, Holst JJ, Carroll R, Ravazzola M, Orci L, Furuta H, Steiner DF. Defective prohormone processing and altered pancreatic islet morphology in mice lacking active SPC2. Proc Natl Acad Sci USA. 1997;94:6646–6651. doi: 10.1073/pnas.94.13.6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vincent M, Guz Y, Rozenberg M, Webb G, Furuta M, Steiner D, Teitelman G. Abrogation of protein convertase 2 activity results in delayed islet cell differentiation and maturation, increased α-cell proliferation, and islet neogenesis. Endocrinology. 2003;144:4061–4069. doi: 10.1210/en.2003-0088. [DOI] [PubMed] [Google Scholar]
- 9.Webb GC, Akbar MS, Zhao C, Swift HH, Steiner DF. Glucagon replacement via micro-osmotic pump corrects hypoglycemia and α-cell hyperplasia in prohormone convertase 2 knockout mice. Diabetes. 2002;51:398–405. doi: 10.2337/diabetes.51.2.398. [DOI] [PubMed] [Google Scholar]
- 10.Furuta M, Carroll R, Martin SD, Swift HH, Ravazzola M, Orci L, Steiner DF. Incomplete processing of proinsulin to insulin accompanied by elevation of Des-31,32 proinsulin intermediates in islets of mice lacking active PC2. J Biol Chem. 1998;273:3431–3437. doi: 10.1074/jbc.273.6.3431. [DOI] [PubMed] [Google Scholar]
- 11.Steiner DF. The proprotein convertases. Curr Opin Chem Biol. 1998;2:31–39. doi: 10.1016/s1367-5931(98)80033-1. [DOI] [PubMed] [Google Scholar]
- 12.Villeneuve P, Feliciangeli S, Croissandeau G, Seidah NG, Mbikay M, Kitabgi P, Beaudet A. Altered processing of the neurotensin/neuromedin N precursor in PC2 knock down mice: a biochemical and immuno-histochemical study. J Neurochem. 2002;82:783–793. doi: 10.1046/j.1471-4159.2002.00988.x. [DOI] [PubMed] [Google Scholar]
- 13.Winsky-Sommerer R, Grouselle D, Rougeot C, Laurent V, David JP, Delacourte A, Dournaud P, Seidah NG, Lindberg I, Trottier S, Epelbaum J. The proprotein convertase PC2 is involved in the maturation of prosomatostatin to somatostatin-14 but not in the somatostatin deficit in Alzheimer’s disease. Neuroscience. 2003;122:437–447. doi: 10.1016/s0306-4522(03)00560-8. [DOI] [PubMed] [Google Scholar]
- 14.Hellestrom C, Swenne I. Growth pattern of pancreatic islet cells in animals. In: Volk B, Arquilla E, editors. The diabetic pancreas. 2. New York and London: Plenum Medical Book Co.; 1985. pp. 53–80. [Google Scholar]
- 15.Bock T, Pakkenberg B, Buschard K. Increased islet volume but unchanged islet number in ob/ob mice. Diabetes. 2003;52:1717–1722. doi: 10.2337/diabetes.52.7.1716. [DOI] [PubMed] [Google Scholar]
- 16.Bock T, Pakkenberg B, Buschard K. Genetic background determines the size and structure of the endocrine pancreas. Diabetes. 2005;54:133–137. doi: 10.2337/diabetes.54.1.133. [DOI] [PubMed] [Google Scholar]
- 17.Sharma A, Zangen DH, Reitz P, Taneja M, Lissauer ME, Miller CP, Weir GC, Habener JF, Bonner-Weir S. The homeodomain protein IDX-1 increases after an early burst of proliferation during pancreatic regeneration. Diabetes. 1999;48:507–513. doi: 10.2337/diabetes.48.3.507. [DOI] [PubMed] [Google Scholar]
- 18.Lumelsky N, Blondel O, Laeng P, Velasco I, Ravin R, McKay R. Differentiation of embryonic stem cells to insulin secreting structures similar to pancreatic islets. Science. 2001;292:1389–1394. doi: 10.1126/science.1058866. [DOI] [PubMed] [Google Scholar]
- 19.Kajihara M, Sone H, Amemiya M, Katoh Y, Isogai M, Shimano H, Yamada N, Takahashi S. Mouse MafA, homologue of zebrafish somite Maf 1, contributes to the specific transcriptional activity through the insulin promoter. Biochem Biophys Res Commun. 2003;312:831–842. doi: 10.1016/j.bbrc.2003.10.196. [DOI] [PubMed] [Google Scholar]
- 20.Zhu X, Zhou A, Dey A, Norrbom C, Carroll R, Zhang C, Laurent V, Lindberg I, Ugleholdt R, Holst JJ, Steiner DF. Disruption of PC1/3 expression in mice causes dwarfism and multiple neuroendocrine peptide processing defects. Proc Natl Acad Sci USA. 2002;99:10293–10298. doi: 10.1073/pnas.162352599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alpert S, Hanahan D, Teitelman G. Hybrid insulin genes reveal a developmental lineage for pancreatic endocrine cells and imply a relationship with neurons. Cell. 1988;53:295–308. doi: 10.1016/0092-8674(88)90391-1. [DOI] [PubMed] [Google Scholar]
- 22.Guz Y, Montminy MR, Stein R, Leonard J, Gamer LW, Wright CVE, Teitelman G. Expression of murine STF-1, a putative insulin gene transcription factor, in β cells of pancreas, duodenal epithelium and pancreatic exocrine and endocrine progenitors during ontogeny. Development. 1995;121:11–18. doi: 10.1242/dev.121.1.11. [DOI] [PubMed] [Google Scholar]
- 23.Herrera PL, Huarte J, Sanvito F, Meda P, Orci L, Vassalli JD. Embryogenesis of the murine endocrine pancreas; early expression of the pancreatic polypeptide gene. Development. 1991;113:1257–1265. doi: 10.1242/dev.113.4.1257. [DOI] [PubMed] [Google Scholar]
- 24.Pang K, Mukonoweshuro C, Wong GC. β Cells arise from glucose transporter type 2 (Glut-2) expressing epithelial cells of the developing rat pancreas. Proc Natl Acad Sci USA. 1994;91:9559–9563. doi: 10.1073/pnas.91.20.9559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Krijger RR, Aanstoot HJ, Kranenburg G, Reinhard M, Visser WJ, Bruining G. The midgestational human fetal pancreas contains cells co-expressing islet hormones. Dev Biol. 1992;153:368–375. doi: 10.1016/0012-1606(92)90121-v. [DOI] [PubMed] [Google Scholar]
- 26.Matsuoka T, Artner I, Henderson E, Means A, Sander M, Stein R. The MafA transcription factor appears to be responsible for tissue-specific expression of insulin. Proc Natl Acad Sci USA. 2004;101:2930–2933. doi: 10.1073/pnas.0306233101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsuoka T, Zhao L, Artner I, Jarrett HW, Friedman D, Means A, Stein R. Members of the large Maf transcription family regulate insulin gene transcription in islet β cells. Mol Cell Biol. 2003;23:6049–6062. doi: 10.1128/MCB.23.17.6049-6062.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kataoka K, Han SI, Shioda S, Hirai M, Nishizawa M, Handa H. MafA is a glucose-regulated and pancreatic β-cell-specific transcriptional activator for the insulin gene. J Biol Chem. 2002;2002:49903–49910. doi: 10.1074/jbc.M206796200. [DOI] [PubMed] [Google Scholar]
- 29.Zhu X, Orci L, Carroll R, Norrbom C, Ravazzola M, Steiner DF. Severe block in processing of proinsulin to insulin accompanied by elevation of des-64,65 proinsulin intermediates in islets of mice lacking prohormone convertase 1/3. Proc Natl Acad Sci USA. 2002;99:10299–10304. doi: 10.1073/pnas.162352799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vuguin P. Animal models for assessing the consequences of intrauterine growth restriction on subsequent glucose metabolism of the offspring: a review. J Matern Fetal Neonatal Med. 2002;11:254–257. doi: 10.1080/jmf.11.4.254.257. [DOI] [PubMed] [Google Scholar]
- 31.Petrik J, Pell JM, Arany E, McDonald TJ, Dean WL, Reik W, Hill DJ. Overexpression of insulin-like growth factor-II in transgenic mice is associated with pancreatic islet cell hyperplasia. Endocrinology. 1999;140:2353–2363. doi: 10.1210/endo.140.5.6732. [DOI] [PubMed] [Google Scholar]
- 32.Petrik J, Reusens B, Arany E, Remacle C, Coelho C, Hoet JJ, Hill DJ. A low protein diet alters the balance of islet cell replication and apoptosis in the fetal and neonatal rat and is associated with a reduced pancreatic expression of insulin-like growth factor-II. Endocrinology. 1999;140:4861–4873. doi: 10.1210/endo.140.10.7042. [DOI] [PubMed] [Google Scholar]
- 33.Boileau P, Mrejen C, Girard J, Hauguel-de Mouzon S. Overexpression of GLUT3 placental glucose transporter in diabetic rats. J Clin Invest. 1995;96:309–317. doi: 10.1172/JCI118036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hubinont C, Nicolini U, Fisk NM, Tannirandorn Y, Rodeck CH. Endocrine pancreatic function in growth-retarded fetuses. Obstet Gynecol. 1991;77:541–544. [PubMed] [Google Scholar]
- 35.Ouilal S, Cui L, Gelling R, Cohen P, Russell R, Santoro N, Charron MJ. Impaired reproduction in glucagon receptor knockout mice localized to abnormalities in placentation and pregnancy maintenance is corrected by reversal of hypoglycemia. J Soc Gynecol Investig. 2003;10:91A. Abstract. [Google Scholar]
- 36.Sperling MA, Ganguli S, Leslie N, Landt K. Fetal-perinatal catecholamine secretion: role in perinatal glucose homeostasis. Am J Physiol. 1984;247:E69–E74. doi: 10.1152/ajpendo.1984.247.1.E69. [DOI] [PubMed] [Google Scholar]
- 37.Gittes GK, Rutter WJ. Onset of cell-specific gene expression in the developing mouse pancreas. Proc Natl Acad Sci USA. 1992;89:1128–1132. doi: 10.1073/pnas.89.3.1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pictet R, Rutter WJ. Development of the embryonic pancreas. In: Steiner DF, Frenkel N, editors. Handbook of physiology, section 7. Washington, DC: American Physiological Society; 1972. pp. 25–66. [Google Scholar]
- 39.Prasadan K, Daume E, Preuett B, Spilde T, Bhatia A, Kobayashi H, Hembree M, Manna P, Gittes GK. Glucagon is required for early insulin-positive differentiation in the developing mouse pancreas. Diabetes. 2002;51:3229–3236. doi: 10.2337/diabetes.51.11.3229. [DOI] [PubMed] [Google Scholar]
- 40.St-Onge L, Sosa-Pineda B, Chowdhury K, Mansouri A, Gruss P. Pax 6 is required for differentiation of glucagon-producing α cells in the mouse pancreas. Nature. 1997;387:406–409. doi: 10.1038/387406a0. [DOI] [PubMed] [Google Scholar]
- 41.Cirulli V, Baetens D, Rutishauser U, Halban PA, Orci L, Rouiller DG. Expression of neural cell adhesion molecule (N-CAM) in rat islets and its role in islet cell type segregation. J Cell Sci. 1994;107:1429–1436. doi: 10.1242/jcs.107.6.1429. [DOI] [PubMed] [Google Scholar]
- 42.Dahl U, Sjodin A, Semb H. Cadherins regulate aggregation of pancreatic β-cells in vivo. Development. 1996;122:2895–2902. doi: 10.1242/dev.122.9.2895. [DOI] [PubMed] [Google Scholar]
- 43.Esni F, Taljedal IB, Perl AK, Cremer H, Christofori G, Semb H. Neural cell adhesion molecule (N-CAM) is required for cell type segregation and normal ultrastructure in pancreatic islets. J Cell Biol. 1999:144. doi: 10.1083/jcb.144.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamaoka T, Itakura M. Development of pancreatic islets (review) Int J Mol Med. 1999;3:247–261. doi: 10.3892/ijmm.3.3.247. [DOI] [PubMed] [Google Scholar]
- 45.Sloop KW, Cao JX, Siesky AM, Zhang HY, Bodenmiller DM, Cox AL, Jacobs JS, Owens RA, Showalter AD, Brenner MB, Raap A, Gromada J, Berridge BR, Monteith DK, Porksen N, McKay RA, Monia BP, Bhanot S, Watts LM, Michael MD. Hepatic and glucagon-like peptide-1-mediated reversal of diabetes by glucagon receptor antisense oligonucleotide inhibitors. J Clin Invest. 2004;113:1571–1581. doi: 10.1172/JCI20911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guillam MT, Hummler E, Schaerer E, Yeh JI, Birnbaum MJ, Beermann F, Schmidt A, Deriaz N, Thorens B, Wu JY. Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat Genet. 1997;17:327–330. doi: 10.1038/ng1197-327. [DOI] [PubMed] [Google Scholar]
- 47.Stein R. Insulin gene transcription: the factors involved in cell-type-specific and glucose-regulated expression in islet β cells are also essential during pancreatic development. In: Cherrington A, Jefferson J, editors. Handbook of physiology, section 7. The endocrine system II. Washington, DC: American Physiology Society; 2001. pp. 25–78. [Google Scholar]
- 48.Giddings SJ, Carnaghi LR, Shalwitz RA. Hypoglycemia but not hyperglycemia induces rapid changes in pancreatic β-cell gene transcription. Am J Physiol. 1993;265:E259–E266. doi: 10.1152/ajpendo.1993.265.2.E259. [DOI] [PubMed] [Google Scholar]
- 49.Aspinwall CA, Lakey JR, Kennedy RT. Insulin-stimulated insulin secretion in single pancreatic β cells. J Biol Chem. 1999;274:6360–6365. doi: 10.1074/jbc.274.10.6360. [DOI] [PubMed] [Google Scholar]
- 50.Khan FA, Goforth PB, Zhang M, Satin LS. Insulin activates ATP-sensitive K(+) channels in pancreatic β-cells through a phosphatidylinositol 3-kinase-dependent pathway. Diabetes. 2001;50:2192–2198. doi: 10.2337/diabetes.50.10.2192. [DOI] [PubMed] [Google Scholar]
- 51.Zaiko M, Estreicher A, Ritz-Laser B, Herrera P, Favor J, Meda P, Philippe J. Pax2 mutant mice display increased number and size of islets of Langerhans but no change in insulin and glucagon content. Eur J Endocrinol. 2004;150:389–395. doi: 10.1530/eje.0.1500389. [DOI] [PubMed] [Google Scholar]
- 52.Zhang X, Gaspard JP, Mizukami Y, Li J, Graeme-Cook F, Chung DC. Overexpression of cyclin D1 in pancreatic β-cells in vivo results in islet hyperplasia without hypoglycemia. Diabetes. 2005;54:712–719. doi: 10.2337/diabetes.54.3.712. [DOI] [PubMed] [Google Scholar]
- 53.Kieffer TJ, Heller RS, Unson CG, Weir GC, Habener JF. Distribution of glucagon receptors on hormone-specific endocrine cells of rat pancreatic islets. Endocrinology. 1996;37:5119–5125. doi: 10.1210/endo.137.11.8895386. [DOI] [PubMed] [Google Scholar]
- 54.Satin LS, Kinard TA. Neurotransmitters and their receptors in the islets of Langerhans of the pancreas: what messages do acetylcholine, glutamate, and GABA transmit? Endocrine. 1998;8:213–223. doi: 10.1385/ENDO:8:3:213. [DOI] [PubMed] [Google Scholar]
- 55.Lauder JM, Liu J, Devaud L, Morrow AL. GABA as a trophic factor for developing monoamine neurons. Perspect Dev Neurobiol. 1998;5:247–259. [PubMed] [Google Scholar]
- 56.Owens DF, Kriegstein AR. Is there more to GABA than synaptic inhibition? Nat Rev Neurosci. 2002;3:715–727. doi: 10.1038/nrn919. [DOI] [PubMed] [Google Scholar]
- 57.Geigerseder C, Doepner RF, Thalhammer A, Krieger A, Mayerhofer A. stimulation of TM3 Leydig cell proliferation via GABA(A) receptors: a new role for testicular GABA. Reprod Biol Endocrinol. 2004;2:13. doi: 10.1186/1477-7827-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]