

Abstract
The combination of lipid drug delivery systems with prodrugs offers several advantages including improved pharmacokinetics, increased absorption, and facilitated targeting. Lipidization and use of lipid carriers can increase the pharmacological half-life of the drug, thus improving pharmacokinetics and allowing less frequent dosing. Lipids also offer advantages such as increased absorption through the intestines for oral drug absorption and to the CNS for brain delivery. Furthermore, the use of lipid delivery systems can enhance drug targeting. Endogenous proteins bind lipids in the blood and carry them to the liver to enable targeting of this organ. Drugs with significant side effects in the stomach can be specifically delivered to enterocytes by exploiting lipases for prodrug activation. Finally, lipids can be used to target the lymphatic system, thus bypassing the liver and avoiding first-pass metabolism. Lymphatic targeting is also important for antiviral drugs in the protection of B and T lymphocytes. In this review, both lipid-drug conjugates and lipid-based carriers will be discussed. An overview, including the chemistry and assembly of the systems, as well as examples from the clinic and in development, will be provided.
KEY WORDS: drug delivery, fatty acids, glyceride, phospholipid, prodrug
INTRODUCTION
Currently, approximately 40% of marketed prodrugs are activated by enzymatic hydrolysis, with alkyl esters being the most common type of prodrug. Lipid modification of a drug has several advantages. First, lipid modification can result in an improved pharmacokinetic (PK) profile, an important determining factor for the success of a drug. Non-protein-bound, small-molecule drugs are subjected to rapid renal clearance from the blood following filtration at the glomerulus. Depending on several factors including the solubility and pKa of the drug and the pH of the urine, drugs may be reabsorbed across the tubular epithelium or excreted in the urine. While hydrophilic compounds are quickly excreted, lipophilic drugs tend to be easily reabsorbed across the tubular epithelium back into circulation, thus prolonging their circulation time (1). Renal clearance of lipophilic drugs is further reduced by the increased binding to plasma proteins, mainly albumin (2,3). Lipid modification also has the advantage of increased absorption across biological barriers, most importantly the gastrointestinal epithelium and blood-brain barrier. Improved oral absorption is commonly achieved by acylation, particularly for antibiotic prodrugs including pivampicillin, talampicillin, and bacampicillin (4). The absorption of these prodrugs is increased from <50% for the parent drug, ampicillin, to 98–99% (5). For enhanced delivery to the brain, glyceride analogs, such as GABA glycerides and l-dopa diglycerides, have been utilized to increase the passive diffusion through the blood-brain barrier. Additionally, lipid modification can also be used to achieve a controlled release property. For example, highly lipophilic prodrugs of steroids and neuroleptics are slowly released into circulation from the injection site, resulting in a prolonged duration of action (6). In this review, the final advantage of lipid modification, enabling drug targeting, will be discussed.
LIPID MOIETIES IN TARGETED PRODRUG DESIGN
In this type of prodrug approach, the lipid moiety is covalently attached to the active drug. These prodrugs are designed to target specific sites either via selective absorption, retention, or release of active drug at the target site. In this section, several different types of lipids, conjugation strategies, and target sites will be discussed.
Types of Lipids and Conjugation Strategies
Various natural lipids carriers are commonly used in the design of lipid prodrugs, including fatty acids, glycerides, and phospholipids. In the design of fatty acid-linked conjugates, the drugs are linked either to the free carboxylate group or to the ω-position at the end of the carbon chain. In the conjugation to the carboxylate group, a drug containing an alcohol or amino group is linked to the fatty acid, resulting in an ester or amide-linked conjugate, respectively (Fig. 1a). These conjugation strategies generally involve the use of an activating agent, such as N,N′-carbonyldiimidazole or carbodiimide, to convert the poor –OH leaving group to a better one, followed by the addition of an amine- or alcohol-containing drug (7). This approach is the most common method of linking fatty acids and has been utilized for many parent drugs including NSAIDs, ACE inhibitors, angiotensin, nucleosides, and testosterone (8,9). While the conjugation chemistries required for this reaction are relatively straightforward and simple, this approach does not take advantage of the natural fatty acid chemistries. The carboxylate group of a fatty acid is essential for binding to the fatty acid binding site of albumin and for recognition by the fatty acid binding protein for internalization at the cell surface (3). Therefore, conjugation to the ω-position is preferable in cases where increased albumin binding and cell membrane transporter properties are preferred. For this method, an ω-modified fatty acid, such as amino (10) or thiol (11) analog, is utilized to link to the parent drug. In addition to deciding on the drug linkage site, the chain length of the fatty acid is also an important parameter in the design of prodrugs. Fatty acids have differences in chain lengths (i.e., short chain (<C10), medium chain (C10–C12), and long chain (>C12)), which impact various properties of the conjugate. For example, longer-chain fatty acids (>C14) generally show increased lymphatic absorption and stability in the circulation compared to shorter chains (12).
Fig. 1.
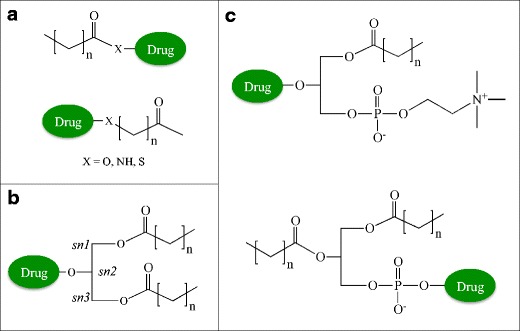
Types of lipids and conjugation sites used in prodrug delivery. a Fatty acid conjugates are linked to prodrugs via the carboxylate or the ω-carbon of the lipid chain (preferred). b Glyceride conjugates are typically modified in the 2-position. c Phospholipid conjugates are linked via the phosphate group or in the 2-position of the glycerol backbone
Similar to fatty acids, glyceride prodrugs also take advantage of natural biosynthetic pathways. Glyceride conjugates are linked to orally administered drugs in order to (i) reduce gastric damage of certain drugs (e.g., NSAIDs including aspirin, indomethacin, ibuprofen (13–15)) by preventing their release in the stomach, (ii) reduce enzymatic degradation in the intestines (16), (iii) target the lymphatic route (9), or (iv) enhance their delivery through the blood-brain barrier (17). The sn-2 monoglyceride, specifically, remains intact in the intestines prior to absorption and is therefore the major site of drug attachment (Fig. 1b). In this approach, drugs containing a carboxylate group are linked to the glyceride via an ester bond. The conjugation chemistry is also similar to fatty acid conjugation, involving the use of an activating agent or acid halide (e.g., chloride) derivative (18,19).
In phospholipid-linked prodrugs, the drugs are either attached to the phosphate group or to the glycerol backbone (Fig. 1c). Drugs linked to the phosphate group are generally antiviral nucleoside analogs. These prodrugs take advantage of the release of the drug inside the cell in a monophosphate form, bypassing possible deficiencies in nucleoside kinase activity. Examples of this type of approach will be discussed in “Tumor Targeting of Anticancer Drugs.” Drugs attached to the glycerol backbone utilize the natural absorption pathway for phospholipids to cross the intestinal lumen or blood-brain barrier (20). Following absorption, the prodrug can be incorporated into the lipoprotein assembly pathway, which will also be described in a later section.
Tumor Targeting of Anticancer Drugs
Examples for the use of lipid modification to generate prodrugs for anticancer therapies are slightly different from other targeting methods in that they are not using active targeting. Since a majority of anticancer agents are hydrophilic molecules, they rely on active ligand transport mechanisms to be efficiently internalized into cells (e.g., folate receptor, nucleoside transporters). However, rapid development of resistance to therapy often occurs via downregulation of these receptors (21). Lipid modification can enhance passive transport of these hydrophilic drugs mediated by the lipid moiety (22). The resultant drugs no longer require active ligand transport mechanisms and are therefore able to overcome transport resistance barriers. Another drug resistance mechanism is enhanced the efflux of certain drugs by transporters including the p-glycoprotein (P-gp) or multidrug resistance (MDR) transporters (21). Lipid conjugation can also lead to a reduction in the amount of drug effluxed out of the cell by these transporters, resulting in increased accumulation compared to free drug (23–25). Therefore, increased antitumor efficacy can be achieved using lipid conjugation strategies by increasing cell permeability and retention of anticancer agents (Fig. 2a).
Fig. 2.
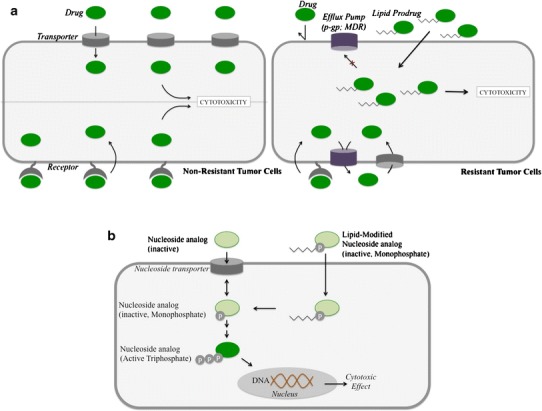
Improved tumor targeting. a Enhanced penetration and reduced efflux. Lipid prodrugs can improve drug targeting by overcoming resistance mechanisms, including receptor downregulation, and drug efflux. Small-molecule antitumor drugs generally depend on surface transporters or receptors for internalization. However, tumor resistance involves downregulation of these surface molecules, and therefore, the drug can no longer accumulate inside tumor cells to exert cytotoxicity. Since lipid prodrugs can be internalized via passive absorption, this issue can be overcome. Another tumor resistance mechanism is the expression of drug efflux transporters, including p-gp and MDR. Lipid prodrugs may have reduced substrate recognition by these transporters, leading to increased accumulation compared to the free drug. b Enhanced activity of nucleoside monophosphate conjugates. Nucleoside analogs are typically internalized via the nucleoside transporter and subsequently activated through a series of steps to the mono-, di-, and triphosphate forms. The active triphosphate form interacts with DNA in the nucleus to exert its cytotoxic effect. Lipid monophosphate nucleoside conjugates can improve efficacy by (1) enhancing passive internalization, independent of the nucleoside transporter, and (2) bypassing the first and rate-limiting monophosphorylation step in the activation process
Nucleoside analogs are one of the most common classes of anticancer drugs combined with lipids to enhance their therapeutic effect. AraC, for example, is internalized into cells via the nucleoside transporter where it is converted via three different kinases into mono-, di-, and triphosphate forms. Cytosine arabinoside triphosphate, the active form, damages the DNA of rapidly dividing cells and inhibits DNA and RNA polymerases and nucleotide reductase enzymes important for DNA synthesis. The drug has a limited activity against solid tumors due to the inactivation by cytidine deaminase, exonuclease degradation of the monophosphate form as well as the low retention and rapid elimination of the active triphosphate form (26–28). AraC lipid conjugates include N4-acyl derivatives, 5′-esters, and phospholipid conjugates (Fig. 3). The N4-acyl derivatives of AraC, with C15–C22 chain lengths, have shown improved antitumor activity attributed to the increased cell internalization that is independent of the nucleoside transporter and to the decreased deactivation by the deaminase enzyme. The conjugation of lipids (C16–C20) to the 5′-position of the sugar moiety was also developed in order to prolong the retention of the prodrug inside the cell. In order to be activated via phosphorylation, these derivatives need to be converted back to AraC inside the cells. In structure-activity studies comparing carbon chain lengths from C16 to C22, it was found that the prodrug conjugates with the shorter, unsaturated chains tested were more efficacious due to lower intracellular hydrolysis rates, leading to longer intracellular retention (29,30). One C18-AraC 5′-modified analog, CP-4055, showed success in vivo and was tested in clinical trials (31). Finally, phospholipid-linked AraC conjugates were also developed. Following enzymolysis, these prodrugs release the monophosphate form of AraC (32–34). Although the mechanism of increased efficacy is still unknown, it has been shown that they lead to a more prolonged intracellular retention of the triphosphate (active) form than the parent AraC drug.
Fig. 3.
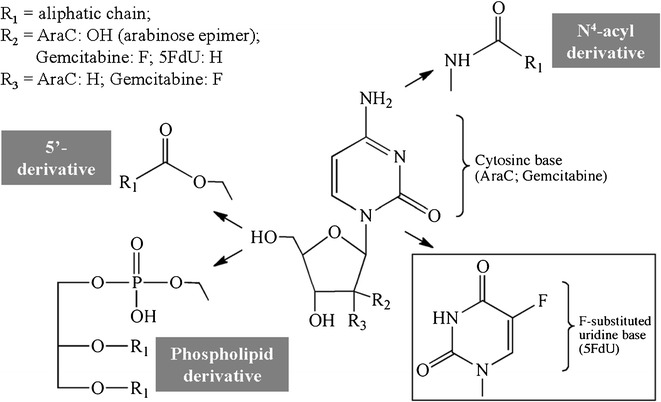
Nucleoside analog conjugates. Nucleoside analogs are modified with lipids via their N4-acyl site or via the 5′-hydroxyl group
Other nucleoside analog conjugates have also been made, including 5-fluoro-2′-deoxyuridine (5FdU, Fig. 3) (35,36), gemcitabine (CP-4126, Fig. 3) (37–40), and troxacitabine-lipid conjugates (41). Similar to studies on AraC, results of these lipid prodrugs showed increased efficacy in various types of in vitro and in vivo tumor models. In these cases, the increased efficacy was attributed to the increased passive diffusion through cell membranes.
Other than nucleoside analogs, lipid conjugation strategies have also been applied to several other types of anticancer agents. Antibiotic derivatives, including mitomycin C (MMC) and doxorubicin (docosahexaenoic acid-DOX conjugates), have been linked to lipids via thiol (dithiobenzyl linker) (42) or pH-sensitive (hydrazine) linkers, respectively (43). A variety of taxane-lipid conjugates, involving modification at 2′ or 7-OH positions of a series of second-generation taxoids (paclitaxel, docetaxel, SB-T-1103, SB-T-1104, SB-T-1213, SB-T-1214, SB-T-1216, and SB-T-1217), have also been tested (44).
Liver Targeting
One approach in liver targeting is to exploit the physiological fate of lipids, which naturally accumulate in this organ. This approach is used in designing lipid prodrugs for treatment of chronic liver diseases such as viral hepatitis, cancer, and steatohepatitis. A majority of liver-targeted lipid prodrugs focus on nucleoside analogs to treat viral hepatitis, the most common chronic liver disease. Nucleoside analogs are known to exhibit not only high antiviral activity but also show many extrahepatic side effects. Thus, there is a strong rationale to improve their targeting to the liver. One of the first examples of lipid prodrugs for liver targeting was reported for the antiviral drug, acyclovir. The bioactive form of acyclovir, and of most nucleoside drugs, is the triphosphate form. However, the conversion of acyclovir to the nucleoside triphosphate (NTP) involves a series of phosphorylation steps and is relatively inefficient. In order to overcome this issue, a series of phospholipid prodrugs were tested. The acylated prodrug, modified at the 5′-phospho AZT position, is deacetylated by phospholipases and phosphodiesterases to form the monophosphate form of AZT, thus bypassing the first step in activation of AZT. The monophosphate form is then converted into the active NTP (Fig. 2b) (45). Another example of a nucleoside lipid prodrug is YNK-01, an AraC analog (46). Although there are examples of their success, results from studies utilizing lipid modification in liver targeting tend to show extrahepatic activity. These types of prodrugs are not only effective in treating hepatic viral diseases but also non-hepatic viral targets, indicating that the drugs are cleaved at other sites. This outcome is not surprising since most of the enzymes (primarily esterases) involved in prodrug conversion are also present in the blood, kidney, and other tissues. Therefore, the success of these types of prodrugs relies more on the high liver accumulation than the specific activation in this organ.
Lymphatic Targeting
Access to systemic circulation through the gastrointestinal (GI) epithelium can occur by absorption into portal blood or through the intestinal lymphatic system. The predominant pathway following oral drug administration is into the portal blood, mainly due to the high amount of blood flow in comparison to intestinal lymph flow. On the other hand, the lymphatic system is the main route of transport for dietary lipids, including triglycerides and lipid-soluble vitamins. These analytes are hydrolyzed in the GI lumen to their corresponding monoglycerides and fatty acids, and then resynthesized into triglycerides and assembled into lipoproteins in enterocytes. The resultant lipoproteins are then preferentially taken up into the lymphatic system (Fig. 4). Utilizing this natural pathway, hydrophobic lipid-modified drugs are also preferentially absorbed by the lymphatic system. This process can be advantageous from a PK perspective, since absorption into the lymph system rather than portal blood bypasses the liver and avoids first-pass metabolism (22). This benefit is exploited for several drugs, such as orally administered testosterone (testosterone undecanoate prodrug) (47–49), that are ineffective due to their extensive first-pass metabolism. Alternatively, from a drug targeting perspective, preferential absorption into the lymphatic system can be used to target drugs to treat lymphatic cancers (50,51) and is also important for antiviral medication for protection of B and T lymphocytes (52–54).
Fig. 4.
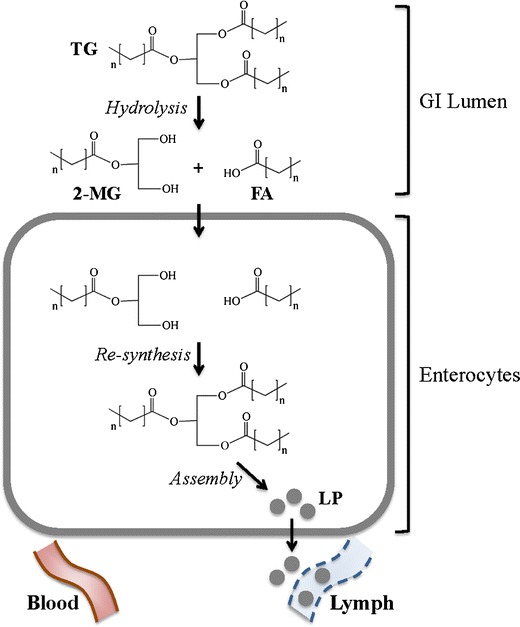
Targeting the lymph system by fatty acid modification. Triglycerides (TG) are hydrolyzed in the lumen of the GI tract to form the respective 2-monoglyceride (2-MG) and fatty acid (FA). Following internalization of 2-MG and FA into enterocytes, they are resynthesized into TG, which are subsequently packaged into lipoproteins (LP). LPs have preferential access to the lymph, where they avoid first-pass metabolism and have access to lymphocytes
There are many examples of using lipid modification, in the form of lipophilic esters or ethers, glycerides, and phospholipids, to target the lymphatic system following oral delivery (Table I). The simplest form of the three, lipophilic esters/ethers, also has the most limited success. The results often show that the extensive pre-absorptive hydrolysis circumvents lymphatic transport; however, some success has been noted for delivery of highly potent hormone drugs such as testosterone (48,49,55). In strategies utilizing glycerides to enhance lymphatic transport, the site of drug attachment to the glyceride is an important consideration. In the natural lipid biosynthetic pathway, triglycerides are hydrolyzed to produce fatty acids along with sn-2 monoglycerides (Fig. 1). Both of these components are internalized into enterocytes in the GI tract, re-acylated, and incorporated into lymph lipoproteins (Fig. 4). Since the sn-2 monoglycerides remain intact, the majority of studies utilize this site of attachment for prodrug design. Similarly, for phospholipid-modified prodrugs, the most common drug attachment site is the phosphate group of the phospholipid backbone. This design allows for the absorption of the drug-lysophospholipid analog through the natural phospholipid processing pathway (Fig. 4).
Table I.
Lipid Prodrugs for Targeting the Lymphatic System
| Parent drug | Lipid | Lipid modification site | Linkage | Reference(s) |
|---|---|---|---|---|
| AZT | Fatty acid (butyrate, C4; laurate, C12; oleate, C18) | Carboxylate group | Ester | (78) |
| Retinol | Fatty acid (palmitate, C16) | Carboxylate group | Ester | (79) |
| Testosterone | Fatty acid (undecylate, C10) | Carboxylate group | Ester | (48,49,55,80) |
| l-dopa | 1,3-Dihexadecanoylpropane-1,2,3-triol glyceride | sn-2 | Ether | (19) |
| Melphalan | 1,3-Dipalmitoyl glyceride | sn-2 | Ether | (81,82) |
| Chlorambucil | 1,3-Dipalmitoyl glyceride | sn-2 | Ether | (18) |
| Aspirin | 1,3-Bis(alkanoyl)-2-(O-acetylsalicyloyl)glyceride | sn-1 | Ether | (13,83) |
| Fluorouridine | Dipalmitoylphosphate | sn-3 | Phosphodiester | (84) |
LIPID-BASED CARRIERS TO IMPROVE PRODRUG TARGETING
Lipid carriers are an important technology in drug delivery. These types of carriers can be utilized to overcome inadequacies in drug absorption or targeting, to enhance stability, or to improve poor aqueous solubility. Prodrugs, in particular, can benefit from lipid carriers in cases where the bond between the drug and promoiety are too unstable under storage conditions or in vivo. Further, the lipid prodrugs described in “LIPID MOIETIES IN TARGETED PRODRUG DESIGN” suffer poor aqueous solubility and cannot be formulated at high enough concentrations for clinical application. Finally, lipid-based drug carriers can be combined with prodrugs to develop a “double prodrug” approach, where the carrier can aid in the targeting or delivery of a prodrug to its active site via either increased stability and/or retention or the combination of active targeting approaches. In this section, these advantages will be explored using two common lipid-based carriers: liposomes and lipoproteins.
Liposomes
The application of liposomes in drug delivery is a well-established field, with several liposomal drug formulation approved in the clinic over the last 20 years. Hydrophobic drugs or lipid-modified drugs can be incorporated directly into the lipid membrane of a liposome, while hydrophilic prodrugs can be encapsulated within its aqueous core. Due to advancements in liposome technology, including stabilization of lipids and maintenance of a relatively small size (~100 nm), modern liposomes generally show long blood circulation times (t1/2 > 40 h), with only 10–15% of the dose delivered to the liver (56,57).
In order to overcome formulation issues for hydrophobic prodrugs, particularly the lipid prodrugs described in “LIPID MOIETIES IN TARGETED PRODRUG DESIGN”, liposomes are commonly utilized. The advantage of combining lipid prodrugs with liposomes is twofold, where the entrapment efficiency of the lipid prodrug is increased compared to the free drug and the prodrug is protected from hydrolysis and enzymatic cleavage while encapsulated in the carrier (58). Liposomes have been used to enhance the delivery of phospholipid prodrugs for antiviral agents (45,59), 6-mercaptopurine glycerol monostearate analogs (60), AraC analogs (61,62), and gemcitabine-acyl derivatives (38). Liposomes have also been applied for delivery of ocular prodrugs. Lipid prodrugs for ocular agents are challenging because the prodrug needs to be stable, particularly against hydrolysis, at the ocular surface, but must be rapidly converted once absorbed. For example, liposome carriers have been utilized for pilocarpine prodrugs to prevent against hydrolysis at the ocular surface. Due to its poor permeability across the corneal membrane, various mono- and di-ester prodrugs of pilocarpane have been generated. These prodrugs undergo rapid hydrolysis and therefore have instability issues in storage solution and at the ocular surface (63). As shown by Burke et al., liposome formulations can protect against hydrolysis and enhance stability at the ocular surface (64).
As mentioned, the combination of prodrugs with liposomes can also enable a double prodrug approach for targeting applications (65). Lipid prodrugs generally provide no active antitumor targeting and enhance bioactivity via increased passive absorption. Therefore, several different strategies utilized to enable the targeting capabilities of liposomes, which were recently reviewed in detail (66), can also be applied to lipid prodrugs. The two main approaches are the addition of targeting ligands or antibodies on the liposome surface (i.e., active targeting methods) (67,68) or incorporation of a bio-responsive modality to specifically release the liposome contents at the target site (56,66,69). The ligands or antibodies utilized in active targeting methods target overexpressed or uniquely expressed antigens or receptors located in the target site. More traditionally, monoclonal antibodies were utilized as targeting molecules (i.e., “immunoliposomes”). However, several drawbacks in their application have been noted, such as triggering of immunogenic responses and reduced affinity of the antibody after incorporation into the liposome. Recent alternatives include the incorporation of smaller molecules, such as hormones, peptide aptamers, or small-molecule ligands.
Compared to actively targeted liposomes, bio-responsive liposomes represent a more recent targeting technology. These techniques can incorporate physical targeting methods, which use external stimuli to enhance targeting. For example, thermosensitive liposomes use externally applied heat to release the liposome content near the target site. The thermoresponsiveness is imparted via either the use of lipid mixtures with a desirable lipid chain melting temperature (Tm) which will destabilize the lipid membrane or the incorporation of thermosensitive polymers which undergo hydrophilic to hydrophobic transitions in response to temperature (70). Similarly, liposomes can be destabilized using light by photooxidation-induced phase transitions in the liposomal bilayer, by photodeprotection where photocleavable stabilizers are released, and by photoisomerization where light-induced isomerization interferes with bilayer packing (69). Alternatively, the contents of liposomes can be released following exposure to a specific microenvironment. For example, pH-responsive liposomes are designed to release their contents at a mildly acidic pH, which is encountered at the surface of tumors or in the endosome. The destabilization of pH-sensitive liposomes involves acid catalyzed hydrolysis of bilayer-stabilizing lipids (71).
Lipoproteins
Lipoproteins are particles formed by the aggregation of lipids (i.e., triglycerides, phospholipids) and cholesterol of cholesteryl esters. The main advantages of lipoproteins as prodrug carriers are their lack of generating an immune response due to their endogenous nature, long circulation half-life, and relatively small particle size which aids in increasing diffusion from vascular to extravascular sites (72). These particles are also quite versatile, where the lipid core incorporates hydrophobic drugs, and, depending on the subtype, can be internalized by receptors overexpressed on some types of cancer cells. The different types of lipoproteins are categorized based on their composition, size, and their assembly origin (Fig. 5). Each type has distinct advantages, and there are many examples of their applications in drug delivery (Table II).
Fig. 5.
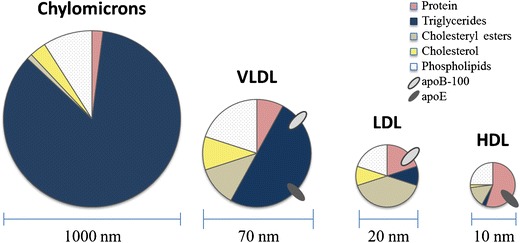
Size and composition of lipoproteins. Lipoproteins exhibit differences in their size; in their composition of proteins, triglycerides, cholesterol esters, cholesterol, and phospholipids; and in the types of surface protein molecules
Table II.
Drug Delivery Using Lipoproteins
| Lipoprotein | Advantage(s) | Example(s) |
|---|---|---|
| Chylomicrons | Routed via intestinal lymphatics (i.e., serve as natural carriers for transport through the lymph) | Gene delivery (85,86); iododeoxyuridine (87) |
| VLDL | Have ApoE (overexpressed on some types of cancer) as a protein ligand; high drug loading capacity due to low protein/high triglyceride composition | Boron neutron capture therapy (88); 5-FU, IUdR, doxorubicin, vindesine (89) |
| LDL | Internalized by LDL-R-mediated endocytosis; long serum half-life (2–4 days) | Doxorubicin (90); 5-FU, IUdR, vindesine (89); dexamethasone (91); fluorophore (diagnostics) (92); gene delivery (93,94) |
| HDL | Small size (5–25 nm); rapid internalization by cancer cells | Taxol (95); iododeoxyuridine (96) |
VLDL very-low-density lipoprotein, LDL low-density lipoprotein, HDL high-density lipoprotein, 5-FU 5-fluorouracil, IUdR idoxuridine
Similar to liposome carriers, lipoproteins are also utilized to overcome formulation issues with lipid-based prodrugs. In general, low-density lipoprotein (LDL) nanoparticles are most commonly combined with anticancer prodrugs due to the added advantage of the upregulation of LDL receptors on the surface of tumors (73,74). Examples include paclitaxel oleate (75), N4-octadecyl-AraC (76), and doxorubicin-lipid conjugates (77). These studies show that the toxicity of the lipid prodrugs is improved after incorporation into LDLs and that the toxicity occurs in an LDL receptor-dependent mechanism.
CONCLUSION
Lipid modification is a great asset in drug delivery applications. Commonly, lipid modification is utilized to enhance the pharmacokinetic properties of a drug, where the subsequent lipid conjugate exhibits a longer circulation half-life and increased serum protein binding. Other advantages of lipid modification that are exploited when applied to prodrugs include enabling targeting tumor sites, the liver, or the lymphatic system. Many different types of lipid molecules are available, from simple fatty acid conjugation to complex lipoprotein particles, for use in drug delivery. Therefore, the numerous advantages and diverse forms of lipid modification and lipid carriers offer a significant benefit in improving prodrug delivery.
References
- 1.Smith DA, Brown K, Neale MG. Chromone-2-carboxylic acids: roles of acidity and lipophilicity in drug disposition. Drug Metab Rev. 1985;16(4):365–388. doi: 10.3109/03602538508991440. [DOI] [PubMed] [Google Scholar]
- 2.van der Vusse GJ. Albumin as fatty acid transporter. Drug Metab. Pharmacokinet. 2009;24(4):300–307. doi: 10.2133/dmpk.24.300. [DOI] [PubMed] [Google Scholar]
- 3.Hackett MJ, Zaro JL, Shen WC, Guley PC, Cho MJ. Fatty acids as therapeutic auxiliaries for oral and parenteral formulations. Adv Drug Deliv Rev. 2013;65(10):1331–1339. doi: 10.1016/j.addr.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wermuth CG GJ-C, Marchandeau C. Designing prodrugs and bioprecursors I: carrier prodrugs. In: CG W, editor. The practice of medicinal chemistry. London: Academic; 1996. pp. 671–696. [Google Scholar]
- 5.Clayton JP, Cole M, Elson SW, Ferres H. BRL.8988 (talampicillin), a well-absorbed oral form of ampicillin. Antimicrob Agents Chemother. 1974;5(6):670–671. doi: 10.1128/AAC.5.6.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minto CF, Howe C, Wishart S, Conway AJ, Handelsman DJ. Pharmacokinetics and pharmacodynamics of nandrolone esters in oil vehicle: effects of ester, injection site and injection volume. J. Pharmacol. Exp. Ther. 1997;281(1):93–102. [PubMed] [Google Scholar]
- 7.Christie WW, editor. Advances in lipid methodology. Dundee: Oily Press; 1993. [Google Scholar]
- 8.Taylor MD. Improved passive oral delivery via prodrugs. Adv Drug Deliv Rev. 1996;19(2):131–148. doi: 10.1016/0169-409X(95)00104-F. [DOI] [Google Scholar]
- 9.Yanez JA, Wang SW, Knemeyer IW, Wirth MA, Alton KB. Intestinal lymphatic transport for drug delivery. Adv Drug Deliv Rev. 2011;63(10–11):923–942. doi: 10.1016/j.addr.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Charbon V, Latour I, Lambert DM, Buc-Calderon P, Neuvens L, De Keyser JL, et al. Targeting of drug to the hepatocytes by fatty acids. Influence of the carrier (albumin or galactosylated albumin) on the fate of the fatty acids and their analogs. Pharm Res. 1996;13(1):27–31. doi: 10.1023/A:1016012913664. [DOI] [PubMed] [Google Scholar]
- 11.Sasson K, Marcus Y, Lev-Goldman V, Rubinraut S, Fridkin M, Shechter Y. Engineering prolonged-acting prodrugs employing an albumin-binding probe that undergoes slow hydrolysis at physiological conditions. J. Control. Release : Off J. Control. Release Society. 2010;142(2):214–220. doi: 10.1016/j.jconrel.2009.10.028. [DOI] [PubMed] [Google Scholar]
- 12.Caliph SM, Charman WN, Porter CJ. Effect of short-, medium-, and long-chain fatty acid-based vehicles on the absolute oral bioavailability and intestinal lymphatic transport of halofantrine and assessment of mass balance in lymph-cannulated and non-cannulated rats. J Pharm Sci. 2000;89(8):1073–1084. doi: 10.1002/1520-6017(200008)89:8<1073::AID-JPS12>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 13.Paris GY, Garmaise DL, Cimon DG, Swett L, Carter GW, Young P. Glycerides as prodrugs. 2. 1,3-Dialkanoyl-2-(2-methyl-4-oxo-1,3-benzodioxan-2-yl)glycerides (cyclic aspirin triglycerides) as antiinflammatory agents. J Med Chem. 1980;23(1):79–82. doi: 10.1021/jm00175a015. [DOI] [PubMed] [Google Scholar]
- 14.Paris GY, Garmaise DL, Cimon DG, Swett L, Carter GW, Young P. Glycerides as prodrugs. 3. Synthesis and antiinflammatory activity of [1-(p-chlorobenzoyl)-5-methoxy-2-methylindole-3-acetyl]glycerides (indomethacin glycerides) J Med Chem. 1980;23(1):9–13. doi: 10.1021/jm00175a003. [DOI] [PubMed] [Google Scholar]
- 15.Cullen E. Novel anti-inflammatory agents. J Pharm Sci. 1984;73(5):579–589. doi: 10.1002/jps.2600730503. [DOI] [PubMed] [Google Scholar]
- 16.Delie F, Couvreur P, Nisato D, Michel JB, Puisieux F, Letourneux Y. Synthesis and in vitro study of a diglyceride prodrug of a peptide. Pharm Res. 1994;11(8):1082–1087. doi: 10.1023/A:1018916311111. [DOI] [PubMed] [Google Scholar]
- 17.Scriba GK, Lambert DM. Bioavailability of phenytoin and anticonvulsant activity after oral administration of phenytoin-bis-hydroxyisobutyrate to rats. Pharm Res. 1997;14(2):251–253. doi: 10.1023/A:1012073332415. [DOI] [PubMed] [Google Scholar]
- 18.Garzonaburbeh A, Poupaert JH, Claesen M, Dumont P, Atassi G. 1,3-Dipalmitoylglycerol ester of chlorambucil as a lymphotropic, orally administrable anti-neoplastic agent. J Med Chem. 1983;26(8):1200–1203. doi: 10.1021/jm00362a021. [DOI] [PubMed] [Google Scholar]
- 19.Garzonaburbeh A, Poupaert JH, Claesen M, Dumont P. A lymphotropic prodrug of L-dopa—synthesis, pharmacological properties, and pharmacokinetic behavior of 1,3-dihexadecanoyl-2-[(S)-2-amino-3-(3,4-dihydroxyphenyl)propanoyl]propane-1,2,3-triol. J Med Chem. 1986;29(5):687–691. doi: 10.1021/jm00155a018. [DOI] [PubMed] [Google Scholar]
- 20.Lambert DM. Rationale and applications of lipids as prodrug carriers. Eur. J. Pharm. Sci : Off. J. Eur. Fed Pharm. Sci. 2000;11(Suppl 2):S15–S27. doi: 10.1016/S0928-0987(00)00161-5. [DOI] [PubMed] [Google Scholar]
- 21.Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 22.Prodrugs: challenges and rewards. New York: Springer; 2007.
- 23.Ojima I, Slater JC, Michaud E, Kuduk SD, Bounaud PY, Vrignaud P, et al. Syntheses and structure-activity relationships of the second-generation antitumor taxoids: exceptional activity against drug-resistant cancer cells. J Med Chem. 1996;39(20):3889–3896. doi: 10.1021/jm9604080. [DOI] [PubMed] [Google Scholar]
- 24.Raub TJ. P-glycoprotein recognition of substrates and circumvention through rational drug design. Mol Pharm. 2006;3(1):3–25. doi: 10.1021/mp0500871. [DOI] [PubMed] [Google Scholar]
- 25.Ruchelman AL, Houghton PJ, Zhou N, Liu A, Liu LF, LaVoie EJ. 5-(2-aminoethyl)dibenzo[c, h][1,6]naphthyridin-6-ones: variation of n-alkyl substituents modulates sensitivity to efflux transporters associated with multidrug resistance. J Med Chem. 2005;48(3):792–804. doi: 10.1021/jm049447z. [DOI] [PubMed] [Google Scholar]
- 26.Wechter WJ, Johnson MA, Hall CM, Warner DT, Berger AE, Wenzel AH, et al. ara-Cytidine acrylates. Use of drug design predictors in structure-activity relationship correlation. J Med Chem. 1975;18(4):339–344. doi: 10.1021/jm00238a003. [DOI] [PubMed] [Google Scholar]
- 27.Neil GL, Wiley PF, Manak RC, Moxley TE. Antitumor effect of 1-beta-D-arabinofuranosylcytosine 5′-adamantoate (NSC 117614) in L1210 leukemic mice. Cancer Res. 1970;30(4):1047–1054. [PubMed] [Google Scholar]
- 28.Wechter WJ, Gish DT, Greig ME, Gray GD, Moxley TE, Kuentzel SL, et al. Nucleic acids. 16. Orally active derivatives of ara-cytidine. J Med Chem. 1976;19(8):1013–1017. doi: 10.1021/jm00230a007. [DOI] [PubMed] [Google Scholar]
- 29.Bergman AM, Kuiper CM, Noordhuis P, Smid K, Voorn DA, Comijn EM, et al. Antiproliferative activity and mechanism of action of fatty acid derivatives of gemcitabine in leukemia and solid tumor cell lines and in human xenografts. Nucleosides Nucleotides Nucleic Acids. 2004;23(8–9):1329–1333. doi: 10.1081/NCN-200027579. [DOI] [PubMed] [Google Scholar]
- 30.Peters GJ, Voorn DA, Kuiper CM, van der Wilt CL, Noordhuis P, Smid K, et al. Cell specific cytotoxicity and structure-activity relationship of lipophilic 1-B-D-arabinofuranosylcytosine (ara-C) derivatives. Nucleosides Nucleotides. 1999;18(4–5):877–878. doi: 10.1080/15257779908041589. [DOI] [PubMed] [Google Scholar]
- 31.Breistol K, Balzarini J, Sandvold ML, Myhren F, Martinsen M, De Clercq E, et al. Antitumor activity of P-4055 (elaidic acid-cytarabine) compared to cytarabine in metastatic and s.c. human tumor xenograft models. Cancer Res. 1999;59(12):2944–2949. [PubMed] [Google Scholar]
- 32.Jordheim LP, Cros E, Gouy MH, Galmarini CM, Peyrottes S, Mackey J, et al. Characterization of a gemcitabine-resistant murine leukemic cell line: reversion of in vitro resistance by a mononucleotide prodrug. Clin. Cancer Res : Off J. Am. Assoc. Cancer Res. 2004;10(16):5614–5621. doi: 10.1158/1078-0432.CCR-04-0506. [DOI] [PubMed] [Google Scholar]
- 33.Tobias SC, Borch RF. Synthesis and biological evaluation of a cytarabine phosphoramidate prodrug. Mol Pharm. 2004;1(2):112–116. doi: 10.1021/mp034019v. [DOI] [PubMed] [Google Scholar]
- 34.Raetz CR, Chu MY, Srivastava S, Turcotte JG. A phospholipid derivative of cytosine arabinoside and its conversion to phosphatidylinositol by animal tissue. Science. 1977;196(4287):303–305. doi: 10.1126/science.191910. [DOI] [PubMed] [Google Scholar]
- 35.Ludwig PS, Schwendener RA, Schott H. Synthesis and anticancer activities of amphiphilic 5-fluoro-2′-deoxyuridylic acid prodrugs. Eur J Med Chem. 2005;40(5):494–504. doi: 10.1016/j.ejmech.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 36.Peghini PA, Zahner R, Kuster H, Schott H, Schwendener RA. In vitro anti-human immunodeficiency virus and anti-hepatitis B virus activities and pharmacokinetic properties of heterodinucleoside phosphates containing AZT or ddC. Antivir Chem Chemother. 1998;9(2):117–126. doi: 10.1177/095632029800900203. [DOI] [PubMed] [Google Scholar]
- 37.Galmarini CM, Myhren F, Sandvold ML. CP-4055 and CP-4126 are active in ara-C and gemcitabine-resistant lymphoma cell lines. Br J Haematol. 2009;144(2):273–275. doi: 10.1111/j.1365-2141.2008.07467.x. [DOI] [PubMed] [Google Scholar]
- 38.Immordino ML, Brusa P, Rocco F, Arpicco S, Ceruti M, Cattel L. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. J Control Release : off. J Controll Release Soc. 2004;100(3):331–346. doi: 10.1016/j.jconrel.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 39.Ali SM, Khan AR, Ahmad MU, Chen P, Sheikh S, Ahmad I. Synthesis and biological evaluation of gemcitabine-lipid conjugate (NEO6002) Bioorg Med Chem Lett. 2005;15(10):2571–2574. doi: 10.1016/j.bmcl.2005.03.046. [DOI] [PubMed] [Google Scholar]
- 40.Chen P, Chien PY, Khan AR, Sheikh S, Ali SM, Ahmad MU, et al. In-vitro and in-vivo anti-cancer activity of a novel gemcitabine-cardiolipin conjugate. Anti-Cancer Drugs. 2006;17(1):53–61. doi: 10.1097/01.cad.0000185182.80227.48. [DOI] [PubMed] [Google Scholar]
- 41.Adema AD, Radi M, Daft J, Narayanasamy J, Hoebe EK, Alexander LE, et al. Troxacitabine prodrugs for pancreatic cancer. Nucleosides Nucleotides Nucleic Acids. 2007;26(8–9):1073–1077. doi: 10.1080/15257770701515591. [DOI] [PubMed] [Google Scholar]
- 42.Senter PD, Pearce WE, Greenfield RS. Development of a drug-release strategy based on the reductive fragmentation of benzyl carbamate disulfides. J Org Chem. 1990;55(9):2975–2978. doi: 10.1021/jo00296a082. [DOI] [Google Scholar]
- 43.Wang Y, Li L, Jiang W, Yang Z, Zhang Z. Synthesis and preliminary antitumor activity evaluation of a DHA and doxorubicin conjugate. Bioorg Med Chem Lett. 2006;16(11):2974–2977. doi: 10.1016/j.bmcl.2006.02.066. [DOI] [PubMed] [Google Scholar]
- 44.Kuznetsova L, Chen J, Sun L, Wu XY, Pepe A, Veith JA, et al. Syntheses and evaluation of novel fatty acid-second-generation taxoid conjugates as promising anticancer agents. Bioorg Med Chem Lett. 2006;16(4):974–977. doi: 10.1016/j.bmcl.2005.10.089. [DOI] [PubMed] [Google Scholar]
- 45.Hostetler KY, Beadle JR, Hornbuckle WE, Bellezza CA, Tochkov IA, Cote PJ, et al. Antiviral activities of oral 1-O-hexadecylpropanediol-3-phosphoacyclovir and acyclovir in woodchucks with chronic woodchuck hepatitis virus infection. Antimicrob Agents Chemother. 2000;44(7):1964–1969. doi: 10.1128/AAC.44.7.1964-1969.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suto T, Miyazawa J, Watanabe Y, Suto K, Yoshida Y, Sakata Y. The effect of YNK-01 (an oral prodrug of cytarabine) on hepatocellular carcinoma. Semin Oncol. 1997;24(6):S6-122–S6-9. [PubMed] [Google Scholar]
- 47.Tauber U, Schroder K, Dusterberg B, Matthes H. Absolute bioavailability of testosterone after oral administration of testosterone-undecanoate and testosterone. Eur J Drug Metab Pharmacokinet. 1986;11(2):145–149. doi: 10.1007/BF03189840. [DOI] [PubMed] [Google Scholar]
- 48.Horst HJ, Holtje WJ, Dennis M, Coert A, Geelen J, Voigt KD. Lymphatic absorption and metabolism of orally administered testosterone undecanoate in man. Klin Wochenschr. 1976;54(18):875–879. doi: 10.1007/BF01483589. [DOI] [PubMed] [Google Scholar]
- 49.Coert A, Geelen J, de Visser J, van der Vies J. The pharmacology and metabolism of testosterone undecanoate (TU), a new orally active androgen. Acta Endocrinol (Copenh) 1975;79(4):789–800. doi: 10.1530/acta.0.0790789. [DOI] [PubMed] [Google Scholar]
- 50.Cense HA, van Eijck CH, Tilanus HW. New insights in the lymphatic spread of oesophageal cancer and its implications for the extent of surgical resection. Best Pract Res Clin Gastroenterol. 2006;20(5):893–906. doi: 10.1016/j.bpg.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 51.Garzon-Aburbeh A, Poupaert JH, Claesen M, Dumont P, Atassi G. 1,3-dipalmitoylglycerol ester of chlorambucil as a lymphotropic, orally administrable antineoplastic agent. J Med Chem. 1983;26(8):1200–1203. doi: 10.1021/jm00362a021. [DOI] [PubMed] [Google Scholar]
- 52.Pantaleo G, Graziosi C, Fauci AS. The role of lymphoid organs in the immunopathogenesis of HIV infection. AIDS. 1993;7(Suppl 1):S19–S23. doi: 10.1097/00002030-199301001-00003. [DOI] [PubMed] [Google Scholar]
- 53.Pantaleo G, Graziosi C, Demarest JF, Cohen OJ, Vaccarezza M, Gantt K, et al. Role of lymphoid organs in the pathogenesis of human immunodeficiency virus (HIV) infection. Immunol Rev. 1994;140:105–130. doi: 10.1111/j.1600-065X.1994.tb00867.x. [DOI] [PubMed] [Google Scholar]
- 54.Umeda M, Marusawa H, Seno H, Katsurada A, Nabeshima M, Egawa H, et al. Hepatitis B virus infection in lymphatic tissues in inactive hepatitis B carriers. J Hepatol. 2005;42(6):806–812. doi: 10.1016/j.jhep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 55.Shackleford DM, Faassen WA, Houwing N, Lass H, Edwards GA, Porter CJH, et al. Contribution of lymphatically transported testosterone undecanoate to the systemic exposure of testosterone after oral administration of two andriol formulations in conscious lymph duct-cannulated dogs. J Pharmacol Exp Ther. 2003;306(3):925–933. doi: 10.1124/jpet.103.052522. [DOI] [PubMed] [Google Scholar]
- 56.Allen TM, Hansen C. Pharmacokinetics of stealth versus conventional liposomes: effect of dose. Biochim Biophys Acta. 1991;1068(2):133–141. doi: 10.1016/0005-2736(91)90201-I. [DOI] [PubMed] [Google Scholar]
- 57.Newman MS, Colbern GT, Working PK, Engbers C, Amantea MA. Comparative pharmacokinetics, tissue distribution, and therapeutic effectiveness of cisplatin encapsulated in long-circulating, pegylated liposomes (SPI-077) in tumor-bearing mice. Cancer Chemother Pharmacol. 1999;43(1):1–7. doi: 10.1007/s002800050855. [DOI] [PubMed] [Google Scholar]
- 58.Liposome technology. Boca Raton: CRC Press; 1992
- 59.Korba BA, Xie H, Wright KN, Hornbuckle WE, Gerin JL, Tennant BC, et al. Liver-targeted antiviral nucleosides: enhanced antiviral activity of phosphatidyl-dideoxyguanosine versus dideoxyguanosine in woodchuck hepatitis virus infection in vivo. Hepatology. 1996;23(5):958–963. doi: 10.1002/hep.510230503. [DOI] [PubMed] [Google Scholar]
- 60.Taneja D, Namdeo A, Mishra PR, Khopade AJ, Jain NK. High-entrapment liposomes for 6-mercaptopurine—a prodrug approach. Drug Dev Ind Pharm. 2000;26(12):1315–1319. doi: 10.1081/DDC-100102315. [DOI] [PubMed] [Google Scholar]
- 61.Tokunaga Y, Iwasa T, Fujisaki J, Sawai S, Kagayama A. Liposomal sustained-release delivery systems for intravenous injection V. Biological disposition of liposome-entrapped lipophilic prodrug of 1-beta-D-arabinofuranosylcytosine. Chem Pharm Bull. 1988;36(10):4060–4067. doi: 10.1248/cpb.36.4060. [DOI] [PubMed] [Google Scholar]
- 62.Tokunaga Y, Iwasa T, Fujisaki J, Sawai S, Kagayama A. Liposomal sustained-release delivery systems for intravenous injection. IV. Antitumor activity of newly synthesized lipophilic 1-beta-D-arabinofuranosylcytosine prodrug-bearing liposomes. Chem Pharm Bull. 1988;36(9):3574–3583. doi: 10.1248/cpb.36.3574. [DOI] [PubMed] [Google Scholar]
- 63.Bundgaard H, Falch E, Larsen C, Mikkelson TJ. Pilocarpine prodrugs. 1. Synthesis, physicochemical properties and kinetics of lactonization of pilocarpic acid-esters. J Pharm Sci. 1986;75(1):36–43. doi: 10.1002/jps.2600750109. [DOI] [PubMed] [Google Scholar]
- 64.Burke TG, Mishra AK, Wani MC, Wall ME. Lipid bilayer partitioning and stability of camptothecin drugs. Biochemistry. 1993;32(20):5352–5364. doi: 10.1021/bi00071a010. [DOI] [PubMed] [Google Scholar]
- 65.Bundgaard H. The double prodrug concept and its applications. Adv Drug Deliv Rev. 1989;3(1):39–65. doi: 10.1016/0169-409X(89)90004-5. [DOI] [Google Scholar]
- 66.Arouri A, Hansen AH, Rasmussen TE, Mouritsen OG. Lipases, liposomes and lipid-prodrugs. Curr. Opin. Colloid In. 2013;18(5):419–431. doi: 10.1016/j.cocis.2013.06.001. [DOI] [Google Scholar]
- 67.Park YS. Tumor-directed targeting of liposomes. Biosci Rep. 2002;22(2):267–281. doi: 10.1023/A:1020190606757. [DOI] [PubMed] [Google Scholar]
- 68.Noble GT, Stefanick JF, Ashley JD, Kiziltepe T, Bilgicer B. Ligand-targeted liposome design: challenges and fundamental considerations. Trends Biotechnol. 2014;32(1):32–45. doi: 10.1016/j.tibtech.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 69.Shum P, Kim JM, Thompson DH. Phototriggering of liposomal drug delivery systems. Adv Drug Deliv Rev. 2001;53(3):273–284. doi: 10.1016/S0169-409X(01)00232-0. [DOI] [PubMed] [Google Scholar]
- 70.Lee H, Messersmith P. Bio-inspired nanomaterials for a new generation of medicine. In: Vo-Dihn T, editor. Nanotechnology in biology and medicine: methods, devices, and applications. Boca Raton, Florida: CRC Press; 2007. pp. 3–1–3–20. [Google Scholar]
- 71.Ganta S, Devalapally H, Shahiwala A, Amiji M. A review of stimuli-responsive nanocarriers for drug and gene delivery. J Control Release. 2008;126(3):187–204. doi: 10.1016/j.jconrel.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 72.Sabnis N, Lacko AG. Drug delivery via lipoprotein-based carriers: answering the challenges in systemic therapeutics. Ther Deliv. 2012;3(5):599–608. doi: 10.4155/tde.12.41. [DOI] [PubMed] [Google Scholar]
- 73.Firestone RA. Low-density lipoprotein as a vehicle for targeting antitumor compounds to cancer cells. Bioconjug Chem. 1994;5(2):105–113. doi: 10.1021/bc00026a002. [DOI] [PubMed] [Google Scholar]
- 74.Yen CF, Kalunta CI, Chen FS, Kaptein JS, Lin CK, Lad PM. Flow cytometric evaluation of LDL receptors using DiI-LDL uptake and its application to B and T lymphocytic cell lines. J Immunol Methods. 1994;177(1–2):55–67. doi: 10.1016/0022-1759(94)90143-0. [DOI] [PubMed] [Google Scholar]
- 75.Nikanjam M, Gibbs AR, Hunt CA, Budinger TF, Forte TM. Synthetic nano-LDL with paclitaxel oleate as a targeted drug delivery vehicle for glioblastoma multiforme. J. Control. Release : offi J Controll Release Soc. 2007;124(3):163–171. doi: 10.1016/j.jconrel.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 76.Koller-Lucae SK, Schott H, Schwendener RA. Low density lipoprotein and liposome mediated uptake and cytotoxic effect of N4-octadecyl-1-beta-D-arabinofuranosylcytosine in Daudi lymphoma cells. Br J Cancer. 1999;80(10):1542–1549. doi: 10.1038/sj.bjc.6690558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Versluis AJ, Rensen PC, Rump ET, Van Berkel TJ, Bijsterbosch MK. Low-density lipoprotein receptor-mediated delivery of a lipophilic daunorubicin derivative to B16 tumours in mice using apolipoprotein E-enriched liposomes. Br J Cancer. 1998;78(12):1607–1614. doi: 10.1038/bjc.1998.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bibby DC, Charman WN, Charman SA, Iskander MN, Porter CJH. Synthesis and evaluation of 5′ alkyl ester prodrugs of zidovudine for directed lymphatic delivery. Int J Pharm. 1996;144(1):61–70. doi: 10.1016/S0378-5173(96)04720-5. [DOI] [Google Scholar]
- 79.Fernandez E, Borgstrom B. Intestinal-absorption of retinol and retinyl palmitate in the Rat—effects of tetrahydrolipstatin. Lipids. 1990;25(9):549–552. doi: 10.1007/BF02537163. [DOI] [PubMed] [Google Scholar]
- 80.Noguchi T, Charman WNA, Stella VJ. The effect of drug lipophilicity and lipid vehicles on the lymphatic absorption of various testosterone esters. Int J Pharm. 1985;24(2–3):173–184. doi: 10.1016/0378-5173(85)90018-3. [DOI] [Google Scholar]
- 81.Deverre JR, Loiseau P, Puisieux F, Gayral P, Letourneux Y, Couvreur P, et al. Synthesis of the orally macrofilaricidal and stable glycerolipidic prodrug of melphalan, 1,3-dipalmitoyl-2-(4′(bis(2″-chloroethyl)amino)phenylalaninoyl)glycerol. Arzneimittelforschung. 1992;42–2(9):1153–1156. [PubMed] [Google Scholar]
- 82.Loiseau PM, Deverre JR, Elkihel L, Gayral P, Letourneux Y. Study of lymphotropic targeting and macrofilaricidal activity of a melphalan prodrug on the Molinema-dessetae model. J Chemother. 1994;6(4):230–237. doi: 10.1080/1120009x.1994.11741157. [DOI] [PubMed] [Google Scholar]
- 83.Carter GW, Young PR, Swett LR, Paris GY. Pharmacological studies in the rat with [2-(1,3-didecanoyloxy)-propyl]2-acetyloxybenzoate (a-45474)—an aspirin pro-drug with negligible gastric irritation. Agents Actions. 1980;10(3):240–245. doi: 10.1007/BF02025942. [DOI] [PubMed] [Google Scholar]
- 84.Sakai A, Mori N, Shuto S, Suzuki T. Deacylation reacylation cycle—a possible absorption mechanism for the novel lymphotropic antitumor agent dipalmitoylphosphatidylfluorouridine in rats. J Pharm Sci. 1993;82(6):575–578. doi: 10.1002/jps.2600820606. [DOI] [PubMed] [Google Scholar]
- 85.Hara T, Liu F, Liu DX, Huang L. Emulsion formulations as a vector for gene delivery in vitro and in vivo. Adv Drug Deliv Rev. 1997;24(2–3):265–271. doi: 10.1016/S0169-409X(96)00467-X. [DOI] [Google Scholar]
- 86.Hara T, Tan Y, Huang L. In vivo gene delivery to the liver using reconstituted chylomicron remnants as a novel nonviral vector. Proc Natl Acad Sci U S A. 1997;94(26):14547–14552. doi: 10.1073/pnas.94.26.14547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rensen PC, de Vrueh RL, van Berkel TJ. Targeting hepatitis B therapy to the liver. Clinical pharmacokinetic considerations. Clin Pharmacokinet. 1996;31(2):131–155. doi: 10.2165/00003088-199631020-00005. [DOI] [PubMed] [Google Scholar]
- 88.Shawer M, Greenspan P, OI S, Lu DR. VLDL-resembling phospholipid-submicron emulsion for cholesterol-based drug targeting. J Pharm Sci. 2002;91(6):1405–1413. doi: 10.1002/jps.10117. [DOI] [PubMed] [Google Scholar]
- 89.Kader A, Pater A. Loading anticancer drugs into HDL as well as LDL has little affect on properties of complexes and enhances cytotoxicity to human carcinoma cells. J. Control. Release : off J Controll Release Soc. 2002;80(1–3):29–44. doi: 10.1016/S0168-3659(01)00536-3. [DOI] [PubMed] [Google Scholar]
- 90.Chu AC, Tsang SY, Lo EH, Fung KP. Low density lipoprotein as a targeted carrier for doxorubicin in nude mice bearing human hepatoma HepG2 cells. Life Sci. 2001;70(5):591–601. doi: 10.1016/S0024-3205(01)01441-2. [DOI] [PubMed] [Google Scholar]
- 91.Tauchi Y, Takase M, Zushida I, Chono S, Sato J, Ito K, et al. Preparation of a complex of dexamethasone palmitate-low density lipoprotein and its effect on foam cell formation of murine peritoneal macrophages. J Pharm Sci. 1999;88(7):709–714. doi: 10.1021/js980422v. [DOI] [PubMed] [Google Scholar]
- 92.Li H, Zhang Z, Blessington D, Nelson DS, Zhou R, Lund-Katz S, et al. Carbocyanine labeled LDL for optical imaging of tumors. Acad Radiol. 2004;11(6):669–677. doi: 10.1016/j.acra.2004.01.016. [DOI] [PubMed] [Google Scholar]
- 93.Kim JS, Kim BI, Maruyama A, Akaike T, Kim SW. A new non-viral DNA delivery vector: the terplex system. J Control Release. 1998;53(1–3):175–182. doi: 10.1016/S0168-3659(97)00251-4. [DOI] [PubMed] [Google Scholar]
- 94.Kim JS, Maruyama A, Akaike T, Kim SW. In vitro gene expression on smooth muscle cells using a terplex delivery system. J Control Release. 1997;47(1):51–59. doi: 10.1016/S0168-3659(96)01615-X. [DOI] [Google Scholar]
- 95.Lacko AG, Nair M, Paranjape S, Johnso S, McConathy WJ. High density lipoprotein complexes as delivery vehicles for anticancer drugs. Anticancer Res. 2002;22(4):2045–2049. [PubMed] [Google Scholar]
- 96.Bijsterbosch MK, Schouten D, van Berkel TJ. Synthesis of the dioleoyl derivative of iododeoxyuridine and its incorporation into reconstituted high density lipoprotein particles. Biochemistry. 1994;33(47):14073–14080. doi: 10.1021/bi00251a016. [DOI] [PubMed] [Google Scholar]