

Abstract
As one targeting strategy of prodrug delivery, gene-directed enzyme prodrug therapy (GDEPT) promises to realize the targeting through its three key features in cancer therapy—cell-specific gene delivery and expression, controlled conversion of prodrugs to drugs in target cells, and expanded toxicity to the target cells’ neighbors through bystander effects. After over 20 years of development, multiple GDEPT systems have advanced into clinical trials. However, no GDEPT product is currently marketed as a drug, suggesting that there are still barriers to overcome before GDEPT becomes a standard therapy. In this review, we first provide a general introduction of this prodrug targeting strategy. Then, we utilize the four most thoroughly studied systems to illustrate components, mechanisms, preclinical and clinical results, and further development directions of GDEPT. These four systems are herpes simplex virus thymidine kinase/ganciclovir, cytosine deaminase/5-fluorocytosine, cytochrome P450/oxazaphosphorines, and nitroreductase/CB1954 system. Later, we focus our discussion on bystander effects including local and distant bystander effects. Lastly, we discuss carriers that are used to deliver genes for GDEPT including virus carriers and non-virus carriers. Among these carriers, the stem cell-based gene delivery system represents one of the newest carriers under development, and may brought about a breakthrough to the gene delivery issue of GDEPT.
KEY WORDS: bystander effects, gene delivery, gene-directed enzyme, prodrug, stem cell-based targeting
INTRODUCTION
Increased attention has been focused on developing novel strategies which can more effectively target prodrugs to tumor cells for enhanced efficacy and reduced toxicity. Gene-directed enzyme prodrug therapy (GDEPT) is one of the most important and successful prodrug delivery approaches and has shown great promise in cancer therapy. GDEPT utilizes transgenes which encode enzymes that can convert prodrugs into active therapeutic metabolites. There are several other names in the literature for this approach, including virus-directed enzyme prodrug therapy (VDEPT), suicide gene therapy, and gene prodrug activation therapy, among others. The concept of GDEPT has existed for over 20 years. However, the clinical importance of the GDEPT approach has only begun to emerge in the last 5 years with more and more GDEPT systems entering clinical trials.
GDEPT usually comprises a three-component system: an inactive drug (prodrug), a gene coding for an enzyme that converts inactive prodrug to an active drug, and a carrier. Figure 1 illustrates the basic mechanisms of the GDEPT system for the treatment of cancer. In the first step, the coding gene is cloned into a vector and delivered to a tumor cell with or without carriers. In the second step, the gene is transcribed into an mRNA which later is translated into the enzyme inside the tumor cell. In the third step, a prodrug is administered systemically and absorbed by the same cell; the prodrug can then be converted to a cytotoxic drug by the enzyme inside the cell. Because gene expression may be controlled by tumor cell-specific promoters, the enzyme and its associated enzymatic reaction can be precisely predisposed to tumor cells, leaving other cells unaffected even if they engulf the gene and the prodrug (1–4). As the result of this preferential conversion of prodrugs to drugs, toxic drugs are only produced in tumor cells while having minimal exposure to healthy cells. In this way, the therapeutic index of prodrugs can be much higher than regular cancer chemotherapeutics (5). What makes the GDEPT an attractive therapy also includes a bystander effect. The effect is achieved via different mechanisms (e.g., passive diffusion) to achieve meaningful tumor regression and durable clinical response.
Fig. 1.
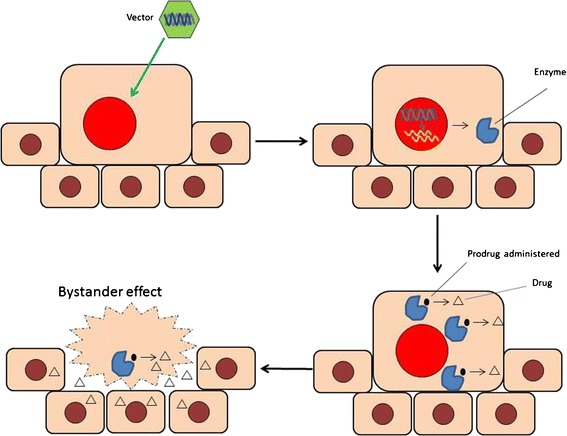
The mechanism of GDEPT systems
To be considered as an enzyme for GDEPT, the enzyme should be expressed either exclusively or with a relatively high ratio in tumor cells compared to healthy cells, and it should have high catalytic activity, so that tumor cells can convert prodrugs even at low substrate concentration. An ideal prodrug for GDEPT should be non-toxic or minimally toxic prior to activation by enzyme but highly toxic after enzymatic activation. Furthermore, prodrugs should be effectively taken up by tumor cells with high affinity to the transduced enzyme and low affinity to irrelevant endogenous enzymes. For effective tumor cell killing, the cytotoxic metabolite of prodrugs should possess a long half-life. Since not all tumor cells are able to uptake one copy of the gene and produce the foreign enzyme, a bystander cytotoxic effect is favored, whereby the toxic form of the drug diffuses out from one tumor cell and is taken up by surrounding cells via active transport.
Many enzyme/prodrug systems have been investigated in the last two decades (Table I). The most extensively studied pairs are herpes simplex virus thymidine kinase (HSV-TK) with ganciclovir (GCV), cytosine deaminase (CD) of Escherichia coli with 5-fluorocytosine (5-FC), cytochrome P450 with cyclophosphamide/ifosfamide (CPA/IFA), and nitroreductase with CB1954. This review article will provide in-depth discussion about these enzyme/prodrug systems.
Table I.
Major GDEPT Systems
| Enzyme | Prodrug | Drug | Mechanism |
|---|---|---|---|
| Thymidine kinase | Ganciclovir (GCV) | Ganciclovir triphosphate | Inhibit DNA polymerase, GT incorporate into replicating cell DNA leading to replication failure and cell death. |
| Cytosine deaminase | 5-fluorocytosine (5-FC) | 5-fluorouracil (5-FU) | Inhibition of thymidylate synthase, 5-FU forms complex with DNA and RNA leading to inhibition of protein synthesis and DNA breakdown. |
| Cytochrome P450 | Cyclophosphamide Ifosfamide |
4-OH cyclophosphamide 4-OH ifosfamide |
The 4-OH derivatives decomposes to phosphoramide mustard which generates a highly electrophilic arizidinium species that forms DNA cross-links. |
| Nitroreductase | CB1954 | N-acetoxy derivatives | Induce rapid cell death by forming interstrand DNA cross-links. |
HERPES SIMPLEX VIRUS THYMIDINE KINASE/GANCICLOVIR SYSTEM
One of the most commonly used experimental and clinical models for gene therapy involves the use of the HSV-TK gene transfection into tumor cells, followed by treatment with the prodrug, GCV. The expression of the HSV-TK gene leads to the synthesis of viral thymidine kinases. Viral thymidine kinases convert GCV into GCV monophosphate which is then converted into a toxic triphosphate form by cellular kinases. Although human cells express both cytosolic and mitochondrial thymidine kinase enzymes, these endogenous enzymes have a much lower ability to convert ganciclovir compared to HSV-TK (6). The tumor cell lysis occurs when GCV triphosphate which is analogous to 2’-deoxyguanosine triphosphate (dGTP) inhibits DNA polymerase or is incorporated into the replicating DNA, causing premature stand termination, replication failure, and apoptosis (7). The mechanism of cell death caused by HSV-TK plus GCV has been widely attributed to apoptosis rather than a direct chemical effect (8–11).
In vivo antitumor activity of the HSV-TK/GCV system has been demonstrated in several animal tumor models, including glioma (12,13), leukemia (14), bladder cancer (15), liver cancer (16), colon adenocarcinoma (17,18), and oral cancer (19). The encouraging results obtained in the preclinical studies led to its application in a number of clinical trials against different types of cancers (20–25). Positive results have been obtained in these clinical trials; however, it is far from a perfect system because gene targeting and drug delivery to the tumor cells remains challenging. Although the thymidine kinase gene is overly expressed in rapidly dividing cells such as cancer cells, only a fraction of cells are dividing at any given time, which prevents efficient viral vector transfection and leads to low levels of prodrug activation. Furthermore, in the case of solid tumors, only about 10% of cells can be transduced. On the prodrug delivery side, the primary disadvantage of GCV is its high diffusibility, which limits its concentration in tumor cells. Moreover, GCV has limited clinical usefulness because it can only be administered at lower concentrations because of its adverse effects on non-target tissues such as bone marrow cells (26). Furthermore, GCV’s active metabolite, GCV triphosphate is membrane insoluble and has low diffusibility, which hinders GCV triphosphate’s passive diffusion to surrounding cells. Therefore, the only mode for transporting GCV triphosphate is through gap junction, limiting its bystander effect. Additionally, some tumor cells, like side population cells in glioblastoma, pump out small molecule drugs like GCV, rendering it resistant to the therapy (27). Combination therapy with HSV-TK/GCV and other agents have also been explored to improve the diffusion of the gene delivery systems using trypsin or collagenase/dispase to degrade extracellular matrix and enhance the tumor penetration of the delivery system (28). Radiation therapy has been used to up-regulate promoters to increase gene expression (29) and to enhance the cytotoxic effects of prodrug (30). A number of modifications to the active (nucleoside binding) site of HSV-TK has been attempted to increase its affinity toward GCV, which may allow lower administration of GCV to patients (31).
A few other substrates have been studied in experimental and clinical studies. Acyclovir is the most frequently used alternative prodrug with HSV-TK gene. In a study using ovarian cancer cell lines to compare acyclovir to GCV, acyclovir showed equal or higher cell killing efficacy and bystander effect (32). Valacyclovir, a prodrug form of acyclovir, has also been studied. The main advantage of valacyclovir over acyclovir is that it can be given orally because of high lipid solubility and bioavailability (33). A recent phase IB study by Chiocca EA et al. suggested that HSV-TK plus valacyclovir can be used safely in combination with surgery and accelerated radiation in newly diagnosed malignant gliomas patients. In addition, therapy resulted in encouraging 2- and 3-year patient survival rates, which were 33and 25%, respectively (34). HSV-TK plus valacyclovir was also studied in localized and metastatic hormone refractory prostate cancer with limited success (35). Results of a phase I clinical trial of HSV-TK and valacyclovir showed stabilization of hepatocellular tumor (36).
There is considerable excitement about the HSV-TK system. Clinical trials are underway or recruiting patients for hematological malignancies (NCT00423124), recurrent prostate cancer (NCT01913106), high risk acute leukemia (NCT00914628), and malignant gliomas (NCT00751270) (www.clinicaltrials.gov).
CYTOSINE DEAMINASE/5-FLUOROCYTOSINE SYSTEM
CD is another well-studied enzyme for GDEPT. This enzyme, found only in bacteria and fungi, catalyzes the deamination of cytosine into uracil and is an important member of the pyrimidine nucleotide salvage pathway. GDEPT therapy using CD has focused almost entirely on one prodrug 5-FC. 5-FU has been widely used for cancer chemotherapy, but high doses are required for anticancer activity and those high doses are not tolerated by patients. Compared to 5-FU, 5-FC is less toxic. Thus, the CD/5-FC system mitigates systemic toxicity that could be caused by the usage of 5-FU alone. Because CD is able to convert 5-FC to 5-FU, the toxicity of 5-FU can be only directed to tumor cells expressing CD. The resulted 5-FU can be further converted by cellular enzymes into potent pyrimidine antimetabolites (5-fluorodeoxyuridine 5-monophosphate [5-FdUMP], 5-fluorodeoxyuridine 5-triphosphate [5-FdUTP], and 5-fluorouridine 5-triphosphate [5-FUTP]). Cell death is mediated by one of the three distinct pathways, including the inhibition of thymidylate synthase, and the formation of 5-FU RNA and DNA complexes (37,38). The primary mechanism of cytotoxicity induced by the CD/5-FC system is similar to HSV-TK/GCV and involves apoptosis (11).
CD/5-FC therapy has been studied in a wide variety of in vitro and in vivo animal models of cancers. It has been shown that MDA-MB-231 breast carcinoma cells transfected with E. coli CD were 1000-fold more sensitive to 5-FC than control. Only 10% of cell transfection is needed to induce complete cytotoxicity in co-cultures with non-infected cells. Intratumoral administration of adenovirus encoded CD along with systemic 5-FC led to control of MDA-MB 231 breast carcinoma xenografts in nude mice and intracranial human glioma xenografts in severe combined immunodeficiency (SCID) mice (39). Similar studies have shown the benefits of systemic CD/5-FC in hepatic metastases of colon carcinoma (40) and prostate cancer (41). Furthermore, it has been suggested that CD/5-FC therapy in solid tumor models can generate complete cures even if only 4% of the tumor cell mass expresses the enzyme (42).
A number of studies have compared the efficacy of CD/5-FC with HSV-Tk/GCV system. Both appeared to have similar efficacy in hepatocellular carcinoma (43), but CD/5-FC was clearly better than HSV-TK/GCV for renal carcinoma (44) and colorectal carcinoma (45). The superior effect of CD/5-FC may be attributed to its greater bystander effect. The bystander effect of GCV is mainly dependent on gap-junction whereas effects of 5-FU are mediated by passive diffusion.
The CD/5-FC system has been further improved by the incorporation of E. coli uracil phosphoribosyltransferase (UPRT) along with CD. UPRT is absent in mammalian cells and directly converts 5-FU to 5-FdUMP. The combination has shown synergistic activity in 9L glioma cells (46) and in human colon cancer cells (47). Further, this approach has been shown to be effective against 5-FU-resistant human primary cancer cells because these cells are susceptible to 5-FdUMP (48). Co-expression of both CD and HSV-TK enzymes was shown to be synergistic in vitro (49) and in vivo in rat 9L glioma tumors in nude mice (50). Combination of radiotherapy along with CD/5-FC also shows improvement in the outcome of experimental models of human cancer xenograft of squamous cell carcinoma (51), choliangiocarcinoma (52), and colon carcinoma (53). In vivo antitumor activity of the CD/5-FC system has been further demonstrated in several animal models including fibrosarcomas (54), carcinomas (55–59), gliomas (60), and metastasis of different origins (40,61).
Several clinical trials using the CD/5-FC system have reported limited success. A phase I clinical trial conducted for specific targeting to erB-2 expressed in breast cancer cells showed safety to patients and significant levels of suicide gene expression restricted to erB-2 positive cells by the use of the tumor-specific erbB-2 promoter (62). Nemunaitis et al. demonstrated the use of attenuated Salmonella bacterium to deliver E. coli CD gene successfully to malignant tissue retaining gene functionality (63). Another phase I clinical trial consisting of 75 newly diagnosed intermediate to high risk prostate cancer patients used a different delivery system, an oncolytic adenovirus containing the HSV-TK fusion gene (64). A combination of radiation and gene therapy resulted in significant decline in prostate specific antigen (PSA) in all patients, and it was shown to be a safe approach (64). Currently, there are three clinical trials are underway. One active pilot feasibility study is for treatment of recurrent high-grade glioma (NCT01172964), whereas two others are recruiting patients for solid tumors (NCT01562626) and for malignant brain tumor (NCT01470794) (www.clinicaltrials.gov).
CYTOCHROME P450/OXAZAPHOSPHORINES SYSTEM
Cytochrome P450 (CYP) enzymes play an important role in hepatic drug metabolism and have been utilized to activate several established anticancer prodrugs. The advantages of P450-based GDEPT include the feasibility of using human P450 genes to limit host immune response and its compatibility with existing anti-cancer drugs (65). The most studied prodrugs are the oxazaphosphorines including cyclophosphamide and ifosfamide. CPA and IFA are isomeric alkylating agents which are inactive before being metabolically activated by specific CYP enzymes into the bioactive and cytotoxic metabolites. Primarily, both CPA and IFA are activated through hydroxylation to yield 4-hydoxyl derivatives, which further yield to cytotoxic phosphoramide mustard (key therapeutically active metabolite) and acrolein. CPA 4-hydroxylation is primarily catalyzed by CYP2B6 while IFA is majorly catalyzed by CYP3A4. Secondly, CPA and IFA can also be catalyzed by CYP3A4 to form another cytotoxic metabolite chloroacetaldehyde via N-dechloroethylation which contributes to not only the anti-tumor therapeutic activity but also severe neurotoxicity and urotoxicity. This second pathway may consume up to 50% of the IFA dose but only about 10% of the CPA dose (66). Therefore, although CPA and IFA are chemical isomers, they exhibit a spectrum of anti-tumor activities and side effects due to different fractions of cytotoxic metabolites. CPA is mostly used for the treatment of lymphoma, leukemias, multiple myeloma, neuroblastoma, retinoblastoma, ovarian cancer, breast cancer, and endometrial cancer. IFA is commonly used for the treatment of soft tissues sarcomas, testicular, ovarian, and breast cancer. The key active oxazaphosphorine metabolite, phosphoramide mustard, is however unable to cross cell membrane for producing an effective local tumor concentration and a strong bystander effect if the drug is activated in the liver (66). The P450 GDEPT approach overcomes this obstaclet by targeting P450-expressing genes to tumor cells which generates cell permeable 4-hydroxy metabolites in the tumor cells. 4-OH-CPA and 4-OH-IFA exhibit different cytotoxic activities. 4-OH-CPA shows several-fold higher intrinsic cytotoxicity than 4-OH-IFA due to distinctly different DNA cross-links induced by each drug. In addition, 4-OH-CPA induces apoptosis while 4-OH-IFA induces necrosis (67) or apoptosis (68). A few strategies have been successfully used to enhance the effectiveness of P450 GDEPT in cancer treatment. For example, a P450 reductase gene has been coexpressed to facilitate P450-dependent prodrug activation and cytotoxicity (69). Inhibition of hepatic P450 reductase activity using anti-thyroid drugs such as propylthiouracil and methimazole have also been used along with the GDEPT approach to improve the prodrug activation in the tumor cells (70). Furthermore, anti-apoptotic factors have been utilized to delay tumor cell death which can in turn increase and prolong the bystander effect of the GDEPT systems (71). A phase 1 clinical trial was conducted utilizing the MetXia-P450 recombinant retrovirus vector that encodes CYP2B6 and oral cyclophosphamide to treat advanced breast cancer or melanoma. The initial trial results not only confirmed the safety of this approach but also demonstrated consistent levels of gene expression in the cancer cells. The promising results have led to ongoing further clinical trials (72).
NITROREDUCTASE/CB1954 SYSTEM
Nitroreductases metabolize aromatic nitro groups to hydroxylamines by generating massive electronic change, which is a very large “switch” that can result in potent cytotoxicity (73). As nitroreductase is not found in human cells, the enzyme used for GDEPT is typically the nfsB gene product from E. coli, an oxygen-insensitive flavin mononucleotide nitroreductase. Four classes of prodrugs for the nitroreductase GDEPT system have been described: dinitroaziridinylbenzamides, dinitrobenzamide mustards, 4-nitrobenzylcarbamates, and nitroindolines. The most successful prodrug used for GDEPT in conjunction with nitroreductase is 5-aziridinyl-2,4-dinitrobenzamide (CB 1954, or Prolarix), which is the prototype of the dinitrobenzamide family of prodrugs (73). The metabolites of CB1954 are potent alkylating agents which kill both dividing and non-diving tumor cells. This is an advantage compared to some other GDEPT systems which only target one type of cells. The metabolites are highly cell-permeable which showed a strong bystander effect for killing the adjacent tumor cells (74). The efficacy of this GDEPT system has been demonstrated in a few clinical trials for treatment of liver and prostate cancers (75,76). However, the immunogenicity of the bacterial nitroreductase remains a major drawback for this GDEPT system (73).
BYSTANDER EFFECT
The delivery of genes to a sufficient number of tumor cells which is required to cause tumor regression remains a challenge for using the GDEPT approach to treat cancer (20). Gene transfer efficiencies are usually less than 10% in the target tissue. Therefore, the bystander effect is critically important for GDEPT to achieve successful tumor regression. The bystander effect is described as the death of non-transfected cells due to indirect effects caused by their neighboring transfected cells, causing more widespread cell death than if transfected cells alone were killed (55,77,78). Thus, bystander effects can amplify the toxic effects of drug several-fold. In order to achieve bystander effects, the active metabolite should have diffusible properties or be actively transported to neighboring cells through gap junctions. Furthermore, cytotoxic effects should be cell cycle independent in order to kill non-dividing neighboring cells. Other hypothesized and studied mechanisms for bystander effects are endocytosis of apoptotic vesicles, release of soluble factors, and stimulation of the immune system in vivo (79).
There are two types of bystander effects, local and distant, which help in tumor regression. The local effect is known to induce cell death and tumor regression although only fractions of tumor cells are transfected. The distant bystander effect is observed in vivo and consists of regression of tumors distant from the tumors expressing transfected genes (79). The CD/5-FC system shows strong local bystander effects through a non-facilitated diffusion, and it does not require cell-to-cell contact. In vitro experiments conducted using mixed cell populations of both transfected and non-transfected cells exposed to 5-FC showed that 1–30% of transfected cells could generate sufficient 5-FU to inhibit the growth of neighboring non-transfected cells (55,80). On the other hand, the local bystander effect of HSV-TK/GCV GDEPT system is mainly observed through gap junction as the active metabolite GCV-triphosphate is highly charged and unable to passively permeate through the cell membranes. Transfer of soluble factors such as GCV-monophosphate derivative to neighboring cells where it is converted to active GCV-triphosphate also plays a role in the bystander effect (81). Recently, expression of E-cadherin, which is involved in the formation and function of gap junctions, was shown to play an important role in the HSV-TK/GCV bystander effect (82).
When HSV-TK transfected cells and non-transfected cells were seeded at low density and then treated with GCV, the result was lower non-transfected cell killing. However, if the mixture was plated at high density, greater non-transfected cell killing was achieved (83). Significant local bystander effects are also reported in preclinical in vivo studies. Treatment of nude mice bearing tumor xenografts generated by CD-positive and negative human WiDr colorectal carcinoma cells caused tumor regression even when only 4% of the tumor cells expressed the cytosine deaminase encoding genes (55). Local bystander effects have also been observed by using cellular vehicles expressing a suicide gene using mouse embryonic endothelial progenitor cells (84), human adipose tissue-derived mesenchymal stem cells (85), and apoptotic bodies generated from dying thymidine kinase-expressing cells and phagocytosed by unmodified neighboring cells (78).
GDEPT transfected cells can cause inflammation and stimulate a systemic anti-tumor immune-mediated response. This distant bystander effect is especially important for treatment of metastatic cancers as the immune response is effective in killing tumor cells outside of the primary tumor. The involvement of the immune system (86–89) and soluble factors (90) have been proposed to explain the thymidine kinase related distant anti-tumor effect, while lymphocytes and natural killer cells are thought to be involved in mediating distant bystander effects in the CD/5-FC GDEPT system (79).
Bystander effects can be a double-edged sword. While they are helpful in amplifying the effect of toxic drug several fold, they may also exert toxic effects on normal healthy cells, limiting the clinical usefulness of GDEPT (91) For example, ganciclovir can only be administered in low doses because high doses lead to adverse effects on non-target tissues such as bone marrow cells (26). Fortunately, in most cases, bystander effects are limited and thus cause marginal off-target toxicity. It is noteworthy that the magnitude of the off-target toxicity can be affected by genes of GDEPT. Cell cycle-independent suicide systems carry higher off-target toxicity compared to cell cycle dependent GDEPT systems. In addition, host immunity can be modulated to augment bystander effects and hence their potential toxicity. Tumor antigens released from GDEPT-caused dying cells can stimulate the immune system, which, in turn, help to eliminate other cells through distant bystander effects or vaccination effects (92). Co-expression of immunomodulators such as IL-2, IL-12, IFN-γ, and TNF-α has been used to augment the bystander effects (93). The relationship between the bystander effects of GDEPT and host immunity is also influenced by the immunogenicity of the GDEPT enzymes. If the enzymes are immunogenic, the chance of off-target toxicity is lower (93). In summary, off-target toxicity of the bystander effect is not a concern in most cases. However, caution should be used when immunomodulators are used to enhance immune-responses against tumor antigens.
GDEPT DELIVERY SYSTEMS
One of the challenges of GDEPT is the development of efficient gene delivery systems to optimize enzyme gene expression and improve the efficacy of GDEPT. A variety of delivery systems have been explored for targeting GDEPT systems into tumors, including viral vectors, liposomes, nanoparticles, mesenchymal stem cells (MSCs), naked DNA, etc.
Viruses are excellent vectors for delivery of the therapeutic genes required for GDEPT. Commonly used viral vectors for GDEPT include retroviruses, adenoviruses, herpes simplex virus (HSV), and lentiviruses. Most retroviruses used in gene therapy are rendered replication defective and the integration is limited to dividing cells. Because retrovirus vectors produce stable integration, low immunogenicity, and long-term transgene expression, they have been most widely used and are involved in more than one-third of GDEPT clinical trials. The inability to infect non-dividing cells and achieve high titers are the major drawbacks of retrovirus vectors for gene delivery (94). Lentiviruses are currently under preclinical development for GDEPT as they possess an attractive ability to infect both dividing and non-dividing cells, resulting in much greater transduction efficiencies compared to retroviruses (94). Adenovirus vectors allow transient transgene expression and offer a higher transduction efficiency than retroviruses. However, the use of adenoviruses in GDEPT has been restricted by its immunogenicity. HSV amplicon vectors, which are conditionally replication competent oncolytic vectors, have also shown great efficacy in animal studies for a variety of GDEPT systems. The safety and effectiveness of HSV vectors are still under investigation in clinical trials (94).
Safety concerns associated with viral vectors prompted extensive research in developing non-viral vectors for gene delivery. Non-viral vectors are generally less efficient than viral vectors due to short-term therapeutic gene expression. Naked DNAs have been used in clinical trials, however low cellular uptake and rapid clearance remain the major obstacle for them to be an effective delivery vector. The most studied non-viral delivery systems are cationic liposomes and polymers are also widely studied as they have the ability to bind DNA. In addition, nanoparticles have also been shown to successfully deliver therapeutic DNA into cancer cells.
Mesenchymal stem cells (MSC) are a group of stem cells that were first discovered in the bone marrow and later in other anatomical locations. They can be expanded in vitro and possess a tumor tropism property (95), which render them an attractive carrier to specifically deliver therapeutic genes and proteins to tumors (96). Consequently, MSCs were also used to deliver genes for GDEPT (96). In one of these attempts, MSCs or neural stem cells were used to deliver the CD gene to glioma; the gene expression product of this GDEPT, CD, was shown to convert 5-FC to 5-FU in vitro (97). Further, this system has demonstrated its effect to inhibit tumor growth and extend the survival of tumored mice. Overall, this MSC-based GDEPT has achieved reasonable efficacy so that it has been approved for the first-in-human clinical trial of MSC-based GDEPT (97). Similarly, MSCs were also employed to deliver a GDEPT gene coding carboxylesterase that can convert a prodrug, irinotecan to its toxic metabolite, an inhibitor to topoisomerase I (98).
It is noteworthy that the MSCs as gene carriers have current limitations. First, the tumor tropism efficiency is limited, which hindered the efficiency of gene delivery to tumors. In the above two examples, the therapeutic efficacy was only reported when the MSCs were intracranially injected although systematic livery of the MSCs did lead to some accumulation of the cells in glioma (97,98). To overcome this limitation, methods to enhance the MSC’s tumor tropism have been proposed and tested including modification of the tumor microenvironment and modification of MSC surface molecules for better adhesion and tumor infiltration (99,100). The second limitation originates from the relationship between MSCs and tumor growth. Whether or not MSCs promote tumor growth remains controversial (101–103). Thus, for safety reasons, suicidal genes are the most desirable payload for the MSCs as they can destroy the cell carriers after the delivery. In summary, MSCs are a very promising carrier for GDEPT if their limitations can be overcome. Its application perspective will soon be revealed by ongoing clinical trials.
CONCLUSION
In the last two decades, significant advancements have been achieved with the GDEPT approach. Many enzyme/prodrug systems have shown effectiveness in preclinical and clinical experiments. Numerous new delivery systems have also been investigated to improve the efficacy of GDEPT systems. The GDEPT principle represents an emerging opportunity in the area of cancer treatment that has yet to be fully investigated. Substantial work still needs to be dedicated to this field in order to exploit this promising cancer treatment option.
Acknowledgments
Disclaimer
This paper is a result of the Dr. Jin Zhang’s independent research and does not reflect the views of U.S. Food and Drug Administration.
Contributor Information
Jin Zhang, Email: jinzhang526@gmail.com.
Mingnan Chen, Email: mingnan.chen@utah.edu.
References
- 1.Saukkonen K, Hemminki A. Tissue-specific promoters for cancer gene therapy. Expert Opin Biol Ther. 2004;4(5):683–96. doi: 10.1517/14712598.4.5.683. [DOI] [PubMed] [Google Scholar]
- 2.Maitland NJ, Stanbridge LJ, Dussupt V. Targeting gene therapy for prostate cancer. Curr Pharm Des. 2004;10(5):531–55. doi: 10.2174/1381612043453252. [DOI] [PubMed] [Google Scholar]
- 3.Lo HW, Day CP, Hung MC. Cancer-specific gene therapy. Adv Genet. 2005;54:235–55. doi: 10.1016/S0065-2660(05)54010-0. [DOI] [PubMed] [Google Scholar]
- 4.Both GW. Recent progress in gene-directed enzyme prodrug therapy: an emerging cancer treatment. Curr Opin Mol Ther. 2009;11(4):421–32. [PubMed] [Google Scholar]
- 5.Hamstra DA, et al. The use of 19F spectroscopy and diffusion-weighted MRI to evaluate differences in gene-dependent enzyme prodrug therapies. Mol Ther. 2004;10(5):916–28. doi: 10.1016/j.ymthe.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 6.Aghi M, Hochberg F, Breakefield XO. Prodrug activation enzymes in cancer gene therapy. J Gene Med. 2000;2(3):148–64. doi: 10.1002/(SICI)1521-2254(200005/06)2:3<148::AID-JGM105>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 7.Balzarini J, Bohman C, De Clercq E. Differential mechanism of cytostatic effect of (E)-5-(2-bromovinyl)-2′-deoxyuridine, 9-(1,3-dihydroxy-2-propoxymethyl)guanine, and other antiherpetic drugs on tumor cells transfected by the thymidine kinase gene of herpes simplex virus type 1 or type 2. J Biol Chem. 1993;268(9):6332–7. [PubMed] [Google Scholar]
- 8.Beltinger C, et al. Herpes simplex virus thymidine kinase/ganciclovir-induced apoptosis involves ligand-independent death receptor aggregation and activation of caspases. Proc Natl Acad Sci U S A. 1999;96(15):8699–704. doi: 10.1073/pnas.96.15.8699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei SJ, et al. S- and G2-phase cell cycle arrests and apoptosis induced by ganciclovir in murine melanoma cells transduced with herpes simplex virus thymidine kinase. Exp Cell Res. 1998;241(1):66–75. doi: 10.1006/excr.1998.4005. [DOI] [PubMed] [Google Scholar]
- 10.Tomicic MT, Thust R, Kaina B. Ganciclovir-induced apoptosis in HSV-1 thymidine kinase expressing cells: critical role of DNA breaks, Bcl-2 decline and caspase-9 activation. Oncogene. 2002;21(14):2141–53. doi: 10.1038/sj.onc.1205280. [DOI] [PubMed] [Google Scholar]
- 11.Fischer U, et al. Mechanisms of thymidine kinase/ganciclovir and cytosine deaminase/ 5-fluorocytosine suicide gene therapy-induced cell death in glioma cells. Oncogene. 2005;24(7):1231–43. doi: 10.1038/sj.onc.1208290. [DOI] [PubMed] [Google Scholar]
- 12.Ribot EJ, et al. In vivo MR tracking of therapeutic microglia to a human glioma model. NMR Biomed. 2011;24(10):1361–8. doi: 10.1002/nbm.1699. [DOI] [PubMed] [Google Scholar]
- 13.Staquicini FI, et al. Systemic combinatorial peptide selection yields a non-canonical iron-mimicry mechanism for targeting tumors in a mouse model of human glioblastoma. J Clin Invest. 2011;121(1):161–73. doi: 10.1172/JCI44798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bondanza A, et al. IL-7 receptor expression identifies suicide gene-modified allospecific CD8+ T cells capable of self-renewal and differentiation into antileukemia effectors. Blood. 2011;117(24):6469–78. doi: 10.1182/blood-2010-11-320366. [DOI] [PubMed] [Google Scholar]
- 15.Tang W, et al. A novel Bifidobacterium infantis-mediated TK/GCV suicide gene therapy system exhibits antitumor activity in a rat model of bladder cancer. J Exp Clin Cancer Res. 2009;28:155. doi: 10.1186/1756-9966-28-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kakinoki K, et al. Prevention of intrahepatic metastasis of liver cancer by suicide gene therapy and chemokine ligand 2/monocyte chemoattractant protein-1 delivery in mice. J Gene Med. 2010;12(12):1002–13. doi: 10.1002/jgm.1528. [DOI] [PubMed] [Google Scholar]
- 17.Chen LS, et al. Efficient gene transfer using the human JC virus-like particle that inhibits human colon adenocarcinoma growth in a nude mouse model. Gene Ther. 2010;17(8):1033–41. doi: 10.1038/gt.2010.50. [DOI] [PubMed] [Google Scholar]
- 18.Ambade AV, Joshi GV, Mulherkar R. Effect of suicide gene therapy in combination with immunotherapy on antitumour immune response & tumour regression in a xenograft mouse model for head & neck squamous cell carcinoma. Indian J Med Res. 2010;132:415–22. [PubMed] [Google Scholar]
- 19.Greish K, et al. Silk-elastinlike protein polymers improve the efficacy of adenovirus thymidine kinase enzyme prodrug therapy of head and neck tumors. J Gene Med. 2010;12(7):572–9. doi: 10.1002/jgm.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rainov NG. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum Gene Ther. 2000;11(17):2389–401. doi: 10.1089/104303400750038499. [DOI] [PubMed] [Google Scholar]
- 21.Voges J, et al. Imaging-guided convection-enhanced delivery and gene therapy of glioblastoma. Ann Neurol. 2003;54(4):479–87. doi: 10.1002/ana.10688. [DOI] [PubMed] [Google Scholar]
- 22.Nasu Y, et al. Suicide gene therapy with adenoviral delivery of HSV-tK gene for patients with local recurrence of prostate cancer after hormonal therapy. Mol Ther. 2007;15(4):834–40. doi: 10.1038/sj.mt.6300096. [DOI] [PubMed] [Google Scholar]
- 23.Xu F, et al. Phase I and biodistribution study of recombinant adenovirus vector-mediated herpes simplex virus thymidine kinase gene and ganciclovir administration in patients with head and neck cancer and other malignant tumors. Cancer Gene Ther. 2009;16(9):723–30. doi: 10.1038/cgt.2009.19. [DOI] [PubMed] [Google Scholar]
- 24.Li N, et al. Adjuvant adenovirus-mediated delivery of herpes simplex virus thymidine kinase administration improves outcome of liver transplantation in patients with advanced hepatocellular carcinoma. Clin Cancer Res. 2007;13(19):5847–54. doi: 10.1158/1078-0432.CCR-07-0499. [DOI] [PubMed] [Google Scholar]
- 25.Freytag SO, et al. Phase I trial of replication-competent adenovirus-mediated suicide gene therapy combined with IMRT for prostate cancer. Mol Ther. 2007;15(5):1016–23. doi: 10.1038/mt.sj.6300120. [DOI] [PubMed] [Google Scholar]
- 26.Hasegawa Y, et al. Avoidance of bone marrow suppression using A-5021 as a nucleoside analog for retrovirus-mediated herpes simplex virus type I thymidine kinase gene therapy. Cancer Gene Ther. 2000;7(4):557–62. doi: 10.1038/sj.cgt.7700134. [DOI] [PubMed] [Google Scholar]
- 27.Hu W, Liu W. Side populations of glioblastoma cells are less sensitive to HSV-TK/GCV suicide gene therapy system than the non-side population. In Vitro Cell Dev Biol Anim. 2010;46(6):497–501. doi: 10.1007/s11626-010-9274-6. [DOI] [PubMed] [Google Scholar]
- 28.Kuriyama N, et al. Protease pretreatment increases the efficacy of adenovirus-mediated gene therapy for the treatment of an experimental glioblastoma model. Cancer Res. 2001;61(5):1805–9. [PubMed] [Google Scholar]
- 29.Marples B, et al. Molecular approaches to chemo-radiotherapy. Eur J Cancer. 2002;38(2):231–9. doi: 10.1016/s0959-8049(01)00367-7. [DOI] [PubMed] [Google Scholar]
- 30.Nishihara E, et al. Retrovirus-mediated herpes simplex virus thymidine kinase gene transduction renders human thyroid carcinoma cell lines sensitive to ganciclovir and radiation in vitro and in vivo. Endocrinology. 1997;138(11):4577–83. doi: 10.1210/endo.138.11.5509. [DOI] [PubMed] [Google Scholar]
- 31.Black ME, Kokoris MS, Sabo P. Herpes simplex virus-1 thymidine kinase mutants created by semi-random sequence mutagenesis improve prodrug-mediated tumor cell killing. Cancer Res. 2001;61(7):3022–6. [PubMed] [Google Scholar]
- 32.Tong XW, et al. Improvement of gene therapy for ovarian cancer by using acyclovir instead of ganciclovir in adenovirus mediated thymidine kinase gene therapy. Anticancer Res. 1998;18(2A):713–8. [PubMed] [Google Scholar]
- 33.Hasenburg A, et al. Thymidine kinase (TK) gene therapy of solid tumors: valacyclovir facilitates outpatient treatment. Anticancer Res. 1999;19(3B):2163–5. [PubMed] [Google Scholar]
- 34.Chiocca EA, et al. Phase IB study of gene-mediated cytotoxic immunotherapy adjuvant to up-front surgery and intensive timing radiation for malignant glioma. J Clin Oncol. 2011;29(27):3611–9. doi: 10.1200/JCO.2011.35.5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kubo H, et al. Phase I dose escalation clinical trial of adenovirus vector carrying osteocalcin promoter-driven herpes simplex virus thymidine kinase in localized and metastatic hormone-refractory prostate cancer. Hum Gene Ther. 2003;14(3):227–41. doi: 10.1089/10430340360535788. [DOI] [PubMed] [Google Scholar]
- 36.Sangro B, et al. A phase I clinical trial of thymidine kinase-based gene therapy in advanced hepatocellular carcinoma. Cancer Gene Ther. 2010;17(12):837–43. doi: 10.1038/cgt.2010.40. [DOI] [PubMed] [Google Scholar]
- 37.Springer CJ, Niculescu-Duvaz I. Gene-directed enzyme prodrug therapy (GDEPT): choice of prodrugs. Adv Drug Deliv Rev. 1996;22(3):351–64. doi: 10.1016/s0169-409x(97)00032-x. [DOI] [PubMed] [Google Scholar]
- 38.Kurozumi K, et al. Apoptosis induction with 5-fluorocytosine/cytosine deaminase gene therapy for human malignant glioma cells mediated by adenovirus. J Neurooncol. 2004;66(1–2):117–27. doi: 10.1023/b:neon.0000013494.98345.80. [DOI] [PubMed] [Google Scholar]
- 39.Li Z, et al. Enzyme/prodrug gene therapy approach for breast cancer using a recombinant adenovirus expressing Escherichia coli cytosine deaminase. Cancer Gene Ther. 1997;4(2):113–7. [PubMed] [Google Scholar]
- 40.Topf N, et al. Regional ‘pro-drug’ gene therapy: intravenous administration of an adenoviral vector expressing the E. coli cytosine deaminase gene and systemic administration of 5-fluorocytosine suppresses growth of hepatic metastasis of colon carcinoma. Gene Ther. 1998;5(4):507–13. doi: 10.1038/sj.gt.3300611. [DOI] [PubMed] [Google Scholar]
- 41.O’Keefe DS, et al. Prostate-specific suicide gene therapy using the prostate-specific membrane antigen promoter and enhancer. Prostate. 2000;45(2):149–57. doi: 10.1002/1097-0045(20001001)45:2<149::aid-pros9>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 42.Huber BE, Richards CA, Austin EA. VDEPT: an enzyme/prodrug gene therapy approach for the treatment of metastatic colorectal cancer. Adv Drug Deliv Rev. 1995;17:279–92. [Google Scholar]
- 43.Kuriyama S, et al. Comparison of gene therapy with the herpes simplex virus thymidine kinase gene and the bacterial cytosine deaminase gene for the treatment of hepatocellular carcinoma. Scand J Gastroenterol. 1999;34(10):1033–41. doi: 10.1080/003655299750025156. [DOI] [PubMed] [Google Scholar]
- 44.Shirakawa T, et al. Cytotoxicity of adenoviral-mediated cytosine deaminase plus 5-fluorocytosine gene therapy is superior to thymidine kinase plus acyclovir in a human renal cell carcinoma model. J Urol. 1999;162(3 Pt 1):949–54. doi: 10.1097/00005392-199909010-00096. [DOI] [PubMed] [Google Scholar]
- 45.Trinh QT, et al. Enzyme/prodrug gene therapy: comparison of cytosine deaminase/5-fluorocytosine versus thymidine kinase/ganciclovir enzyme/prodrug systems in a human colorectal carcinoma cell line. Cancer Res. 1995;55(21):4808–12. [PubMed] [Google Scholar]
- 46.Adachi Y, et al. Experimental gene therapy for brain tumors using adenovirus-mediated transfer of cytosine deaminase gene and uracil phosphoribosyltransferase gene with 5-fluorocytosine. Hum Gene Ther. 2000;11(1):77–89. doi: 10.1089/10430340050016175. [DOI] [PubMed] [Google Scholar]
- 47.Koyama F, et al. Combined suicide gene therapy for human colon cancer cells using adenovirus-mediated transfer of Escherichia coli cytosine deaminase gene and Escherichia coli uracil phosphoribosyltransferase gene with 5-fluorocytosine. Cancer Gene Ther. 2000;7(7):1015–22. doi: 10.1038/sj.cgt.7700189. [DOI] [PubMed] [Google Scholar]
- 48.Richard C, et al. Sensitivity of 5-fluorouracil-resistant cancer cells to adenovirus suicide gene therapy. Cancer Gene Ther. 2007;14(1):57–65. doi: 10.1038/sj.cgt.7700980. [DOI] [PubMed] [Google Scholar]
- 49.Aghi M, et al. Synergistic anticancer effects of ganciclovir/thymidine kinase and 5-fluorocytosine/cytosine deaminase gene therapies. J Natl Cancer Inst. 1998;90(5):370–80. doi: 10.1093/jnci/90.5.370. [DOI] [PubMed] [Google Scholar]
- 50.Rogulski KR, et al. Glioma cells transduced with an Escherichia coli CD/HSV-1 TK fusion gene exhibit enhanced metabolic suicide and radiosensitivity. Hum Gene Ther. 1997;8(1):73–85. doi: 10.1089/hum.1997.8.1-73. [DOI] [PubMed] [Google Scholar]
- 51.Hanna NN, et al. Virally directed cytosine deaminase/5-fluorocytosine gene therapy enhances radiation response in human cancer xenografts. Cancer Res. 1997;57(19):4205–9. [PubMed] [Google Scholar]
- 52.Pederson LC, et al. Molecular chemotherapy combined with radiation therapy enhances killing of cholangiocarcinoma cells in vitro and in vivo. Cancer Res. 1997;57(19):4325–32. [PubMed] [Google Scholar]
- 53.Gabel M, et al. Selective in vivo radiosensitization by 5-fluorocytosine of human colorectal carcinoma cells transduced with the E. coli cytosine deaminase (CD) gene. Int J Radiat Oncol Biol Phys. 1998;41(4):883–7. doi: 10.1016/s0360-3016(98)00125-4. [DOI] [PubMed] [Google Scholar]
- 54.Mullen CA, et al. Tumors expressing the cytosine deaminase suicide gene can be eliminated in vivo with 5-fluorocytosine and induce protective immunity to wild type tumor. Cancer Res. 1994;54(6):1503–6. [PubMed] [Google Scholar]
- 55.Huber BE, et al. Metabolism of 5-fluorocytosine to 5-fluorouracil in human colorectal tumor cells transduced with the cytosine deaminase gene: significant antitumor effects when only a small percentage of tumor cells express cytosine deaminase. Proc Natl Acad Sci U S A. 1994;91(17):8302–6. doi: 10.1073/pnas.91.17.8302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huber BE, et al. In vivo antitumor activity of 5-fluorocytosine on human colorectal carcinoma cells genetically modified to express cytosine deaminase. Cancer Res. 1993;53(19):4619–26. [PubMed] [Google Scholar]
- 57.Ohwada A, Hirschowitz EA, Crystal RG. Regional delivery of an adenovirus vector containing the Escherichia coli cytosine deaminase gene to provide local activation of 5-fluorocytosine to suppress the growth of colon carcinoma metastatic to liver. Hum Gene Ther. 1996;7(13):1567–76. doi: 10.1089/hum.1996.7.13-1567. [DOI] [PubMed] [Google Scholar]
- 58.Kanai F, et al. In vivo gene therapy for alpha-fetoprotein-producing hepatocellular carcinoma by adenovirus-mediated transfer of cytosine deaminase gene. Cancer Res. 1997;57(3):461–5. [PubMed] [Google Scholar]
- 59.Bentires-Alj M, et al. Cytosine deaminase suicide gene therapy for peritoneal carcinomatosis. Cancer Gene Ther. 2000;7(1):20–6. doi: 10.1038/sj.cgt.7700093. [DOI] [PubMed] [Google Scholar]
- 60.Ichikawa T, et al. In vivo efficacy and toxicity of 5-fluorocytosine/cytosine deaminase gene therapy for malignant gliomas mediated by adenovirus. Cancer Gene Ther. 2000;7(1):74–82. doi: 10.1038/sj.cgt.7700086. [DOI] [PubMed] [Google Scholar]
- 61.Consalvo M, et al. 5-Fluorocytosine-induced eradication of murine adenocarcinomas engineered to express the cytosine deaminase suicide gene requires host immune competence and leaves an efficient memory. J Immunol. 1995;154(10):5302–12. [PubMed] [Google Scholar]
- 62.Pandha HS, et al. Genetic prodrug activation therapy for breast cancer: a phase I clinical trial of erbB-2-directed suicide gene expression. J Clin Oncol. 1999;17(7):2180–9. doi: 10.1200/JCO.1999.17.7.2180. [DOI] [PubMed] [Google Scholar]
- 63.Nemunaitis J, et al. Pilot trial of genetically modified, attenuated Salmonella expressing the E. coli cytosine deaminase gene in refractory cancer patients. Cancer Gene Ther. 2003;10(10):737–44. doi: 10.1038/sj.cgt.7700634. [DOI] [PubMed] [Google Scholar]
- 64.Freytag SO, et al. Phase I study of replication-competent adenovirus-mediated double-suicide gene therapy in combination with conventional-dose three-dimensional conformal radiation therapy for the treatment of newly diagnosed, intermediate- to high-risk prostate cancer. Cancer Res. 2003;63(21):7497–506. [PubMed] [Google Scholar]
- 65.Kan O, et al. Direct retroviral delivery of human cytochrome P450 2B6 for gene-directed enzyme prodrug therapy of cancer. Cancer Gene Ther. 2001;8(7):473–82. doi: 10.1038/sj.cgt.7700329. [DOI] [PubMed] [Google Scholar]
- 66.Roy P, Waxman DJ. Activation of oxazaphosphorines by cytochrome P450: application to gene-directed enzyme prodrug therapy for cancer. Toxicol In Vitro. 2006;20(2):176–86. doi: 10.1016/j.tiv.2005.06.046. [DOI] [PubMed] [Google Scholar]
- 67.Karle P, et al. Necrotic, rather than apoptotic, cell death caused by cytochrome P450-activated ifosfamide. Cancer Gene Ther. 2001;8(3):220–30. doi: 10.1038/sj.cgt.7700290. [DOI] [PubMed] [Google Scholar]
- 68.Schwartz PS, Waxman DJ. Cyclophosphamide induces caspase 9-dependent apoptosis in 9L tumor cells. Mol Pharmacol. 2001;60(6):1268–79. doi: 10.1124/mol.60.6.1268. [DOI] [PubMed] [Google Scholar]
- 69.Tychopoulos M, et al. A virus-directed enzyme prodrug therapy (VDEPT) strategy for lung cancer using a CYP2B6/NADPH-cytochrome P450 reductase fusion protein. Cancer Gene Ther. 2005;12(5):497–508. doi: 10.1038/sj.cgt.7700817. [DOI] [PubMed] [Google Scholar]
- 70.Ross AD, et al. Effect of propylthiouracil treatment on NADPH-cytochrome P450 reductase levels, oxygen consumption and hydroxyl radical formation in liver microsomes from rats fed ethanol or acetone chronically. Biochem Pharmacol. 1995;49(7):979–89. doi: 10.1016/0006-2952(95)00007-m. [DOI] [PubMed] [Google Scholar]
- 71.Schwartz PS, Chen CS, Waxman DJ. Enhanced bystander cytotoxicity of P450 gene-directed enzyme prodrug therapy by expression of the antiapoptotic factor p35. Cancer Res. 2002;62(23):6928–37. [PubMed] [Google Scholar]
- 72.Braybrooke JP, et al. Phase I study of MetXia-P450 gene therapy and oral cyclophosphamide for patients with advanced breast cancer or melanoma. Clin Cancer Res. 2005;11(4):1512–20. doi: 10.1158/1078-0432.CCR-04-0155. [DOI] [PubMed] [Google Scholar]
- 73.Denny WA. Nitroreductase-based GDEPT. Curr Pharm Des. 2002;8(15):1349–61. doi: 10.2174/1381612023394584. [DOI] [PubMed] [Google Scholar]
- 74.Bridgewater JA, et al. The bystander effect of the nitroreductase/CB1954 enzyme/prodrug system is due to a cell-permeable metabolite. Hum Gene Ther. 1997;8(6):709–17. doi: 10.1089/hum.1997.8.6-709. [DOI] [PubMed] [Google Scholar]
- 75.Patel P, et al. A phase I/II clinical trial in localized prostate cancer of an adenovirus expressing nitroreductase with CB1954 [correction of CB1984] Mol Ther. 2009;17(7):1292–9. doi: 10.1038/mt.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Palmer DH, et al. Virus-directed enzyme prodrug therapy: intratumoral administration of a replication-deficient adenovirus encoding nitroreductase to patients with resectable liver cancer. J Clin Oncol. 2004;22(9):1546–52. doi: 10.1200/JCO.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 77.Dachs GU, et al. Bystander or no bystander for gene directed enzyme prodrug therapy. Molecules. 2009;14(11):4517–45. doi: 10.3390/molecules14114517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Freeman SM, et al. The “bystander effect”: tumor regression when a fraction of the tumor mass is genetically modified. Cancer Res. 1993;53(21):5274–83. [PubMed] [Google Scholar]
- 79.Duarte S, et al. Suicide gene therapy in cancer: where do we stand now? Cancer Lett. 2012;324(2):160–70. doi: 10.1016/j.canlet.2012.05.023. [DOI] [PubMed] [Google Scholar]
- 80.Domin BA, Mahony WB, Zimmerman TP. Transport of 5-fluorouracil and uracil into human erythrocytes. Biochem Pharmacol. 1993;46(3):503–10. doi: 10.1016/0006-2952(93)90527-4. [DOI] [PubMed] [Google Scholar]
- 81.Mesnil M, Yamasaki H. Bystander effect in herpes simplex virus-thymidine kinase/ganciclovir cancer gene therapy: role of gap-junctional intercellular communication. Cancer Res. 2000;60(15):3989–99. [PubMed] [Google Scholar]
- 82.Garcia-Rodriguez L, et al. E-cadherin contributes to the bystander effect of TK/GCV suicide therapy and enhances its antitumoral activity in pancreatic cancer models. Gene Ther. 2011;18(1):73–81. doi: 10.1038/gt.2010.114. [DOI] [PubMed] [Google Scholar]
- 83.Moolten FL. Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a prospective cancer control strategy. Cancer Res. 1986;46(10):5276–81. [PubMed] [Google Scholar]
- 84.Wei J, et al. Embryonic endothelial progenitor cells armed with a suicide gene target hypoxic lung metastases after intravenous delivery. Cancer Cell. 2004;5(5):477–88. doi: 10.1016/s1535-6108(04)00116-3. [DOI] [PubMed] [Google Scholar]
- 85.Kucerova L, et al. Adipose tissue-derived human mesenchymal stem cells mediated prodrug cancer gene therapy. Cancer Res. 2007;67(13):6304–13. doi: 10.1158/0008-5472.CAN-06-4024. [DOI] [PubMed] [Google Scholar]
- 86.Bi W, et al. An HSVtk-mediated local and distant antitumor bystander effect in tumors of head and neck origin in athymic mice. Cancer Gene Ther. 1997;4(4):246–52. [PubMed] [Google Scholar]
- 87.Kianmanesh AR, et al. A “distant” bystander effect of suicide gene therapy: regression of nontransduced tumors together with a distant transduced tumor. Hum Gene Ther. 1997;8(15):1807–14. doi: 10.1089/hum.1997.8.15-1807. [DOI] [PubMed] [Google Scholar]
- 88.Wilson KM, et al. HSV-tk gene therapy in head and neck squamous cell carcinoma. Enhancement by the local and distant bystander effect. Arch Otolaryngol Head Neck Surg. 1996;122(7):746–9. doi: 10.1001/archotol.1996.01890190042011. [DOI] [PubMed] [Google Scholar]
- 89.Wei MX, et al. Suicide gene therapy of chemically induced mammary tumor in rat: efficacy and distant bystander effect. Cancer Res. 1998;58(16):3529–32. [PubMed] [Google Scholar]
- 90.Dilber MS, et al. Suicide gene therapy for plasma cell tumors. Blood. 1996;88(6):2192–200. [PubMed] [Google Scholar]
- 91.Herraiz M, et al. Liver failure caused by herpes simplex virus thymidine kinase plus ganciclovir therapy is associated with mitochondrial dysfunction and mitochondrial DNA depletion. Hum Gene Ther. 2003;14(5):463–72. doi: 10.1089/104303403321467225. [DOI] [PubMed] [Google Scholar]
- 92.Pierrefite-Carle V, et al. Cytosine deaminase/5-fluorocytosine-based vaccination against liver tumors: evidence of distant bystander effect. J Natl Cancer Inst. 1999;91(23):2014–9. doi: 10.1093/jnci/91.23.2014. [DOI] [PubMed] [Google Scholar]
- 93.Portsmouth D, Hlavaty J, Renner M. Suicide genes for cancer therapy. Mol Aspects Med. 2007;28(1):4–41. doi: 10.1016/j.mam.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 94.Kay MA, Glorioso JC, Naldini L. Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nat Med. 2001;7(1):33–40. doi: 10.1038/83324. [DOI] [PubMed] [Google Scholar]
- 95.Kidd S, et al. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells. 2009;27(10):2614–23. doi: 10.1002/stem.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dwyer RM, et al. Advances in mesenchymal stem cell-mediated gene therapy for cancer. Stem Cell Res Ther. 2010;1(3):25. doi: 10.1186/scrt25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Aboody KS, et al. Neural stem cell-mediated enzyme/prodrug therapy for glioma: preclinical studies. Sci Transl Med. 2013;5(184):184ra59. doi: 10.1126/scitranslmed.3005365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Metz MZ, et al. Neural stem cell-mediated delivery of irinotecan-activating carboxylesterases to glioma: implications for clinical use. Stem Cells Transl Med. 2013;2(12):983–92. doi: 10.5966/sctm.2012-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Klopp AH, et al. Tumor irradiation increases the recruitment of circulating mesenchymal stem cells into the tumor microenvironment. Cancer Res. 2007;67(24):11687–95. doi: 10.1158/0008-5472.CAN-07-1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sato H, et al. Epidermal growth factor receptor-transfected bone marrow stromal cells exhibit enhanced migratory response and therapeutic potential against murine brain tumors. Cancer Gene Ther. 2005;12(9):757–68. doi: 10.1038/sj.cgt.7700827. [DOI] [PubMed] [Google Scholar]
- 101.Klopp AH, et al. Concise review: dissecting a discrepancy in the literature: do mesenchymal stem cells support or suppress tumor growth? Stem Cells. 2011;29(1):11–9. doi: 10.1002/stem.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Waterman RS, Henkle SL, Betancourt AM. Mesenchymal stem cell 1 (<italic>MSC1</italic>)-based therapy attenuates tumor growth whereas <italic>MSC2-</italic>treatment promotes tumor growth and metastasis. PLoS One. 2012;7(9):e45590. doi: 10.1371/journal.pone.0045590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang T, et al. Bone marrow-derived mesenchymal stem cells promote growth and angiogenesis of breast and prostate tumors. Stem Cell Res Ther. 2013;4(3):70. doi: 10.1186/scrt221. [DOI] [PMC free article] [PubMed] [Google Scholar]