

Abstract
In situ immunization is based on the concept that it is possible to break immune tolerance by inducing tumor cell death in situ in a manner that provides antigen-presenting cells such as dendritic cells (DCs) with a wide selection of tumor antigens that can then be presented to the immune system and result in a therapeutic anticancer immune response. We designed a comprehensive approach to in situ immunization using poly(lactic-co-glycolic acid) (PLGA)-biodegradable microparticles (MPs) loaded with doxorubicin (Dox) and CpG oligodeoxynucleotides (CpG) that deliver Dox (chemotherapy) and CpG (immunotherapy) in a sustained-release fashion when injected intratumorally. Dox induces immunogenic tumor cell death while CpG enhances tumor antigen presentation by DCs. PLGA MPs allow their safe co-delivery while evading the vesicant action of Dox. In vitro, we show that Dox/CpG MPs can kill B and T lymphoma cells and are less toxic to DCs. In vivo, Dox/CpG MPs combined with antibody therapy to enhance and maintain the T cell response generated systemic immune responses that suppressed injected and distant tumors in a murine B lymphoma model, leading to tumor-free mice. The combination regimen was also effective at reducing T cell lymphoma and melanoma tumor burdens. In conclusion, Dox/CpG MPs represent an efficient and safe tool for in situ immunization that could provide a promising component of immunotherapy for patients with a variety of types of cancer.
KEY WORDS: cancer vaccine, CpG, doxorubicin, in situ immunization, microparticles, PLGA
INTRODUCTION
Therapeutic cancer vaccination aims at overcoming immune tolerance to tumor-associated antigens (TAAs) and generating potent antitumor immune responses, most commonly in the form of effector T cells (1,2). This can be achieved via different approaches that include using TAAs mixed with adjuvants and using dendritic cells (DCs) loaded with tumor lysates (DC vaccination). However, the heterogenous expression of TAAs by tumors, suboptimal preparation conditions of tumor lysates for DC loading, inefficient migration of DCs to tumor sites post-infusion, and immunosuppressive mechanisms employed by tumors are major barriers against the establishment of long-lived robust immune responses (3–5).
In situ immunization is designed to overcome these challenges. It involves utilizing the patient’s own tumor antigens by inducing tumor cell death in situ via intratumoral injection of cytotoxic agents. This potentially provides antigen-presenting cells such as DCs with a wide selection of tumor antigens (3). Moreover, the local delivery of cytotoxic agents assures a high concentration at the tumor environment and reduces systemic toxicity. An ideal in situ immunization design would include agents that not only kill tumor cells but also enhance DC maturation to ensure proper activation of antigen-specific T cells. To achieve this, we chose the combination of doxorubicin and unmethylated cytosine-phosphate-guanosine dinucleotides (CpG). Doxorubicin (Dox), a weak base with pKa 8.2, is a member of the anthracycline family of DNA-intercalating agents (6). It induces immunogenic tumor cell death by enhancing the expression of “eat-me” signals by tumor cells (most notably calreticulin), thus facilitating phagocytosis by DCs (7). Its dose-limiting cardiotoxicity, a major side effect of systemic administration (6), further strengthens the argument for its local delivery. In addition to immunogenic tumor cell death induced by Dox, administration of an adjuvant like CpG can further assist in the stimulation of a robust immune response. CpG mimics sequences found in bacterial DNA (8). It is a potent agonist of Toll-like receptor 9 (TLR9) that is expressed by B cells, monocytes, macrophages, and plasmacytoid DCs (pDCs). TLR9 stimulation induces DC maturation including upregulation of co-stimulatory molecules CD80 and CD86, allowing DCs to present tumor-derived antigens to T cells in an immunogenic instead of a tolerogenic context (9,10).
A major limitation for co-delivery of Dox and CpG is their opposite charges at physiological pH, which readily allow their aggregation in solution (11). This necessitates the development of a delivery system that can prevent the aggregation of Dox and CpG during co-administration. A recent attempt at developing a formulation for co-delivery of Dox and CpG included the complexation of Dox with a plasmid containing CpG motifs (12). Vaccination of mice carrying luciferase-expressing murine adenocarcinoma with Dox-CpG plasmid complexes showed reduced tumor proliferation as compared to mice treated with the Dox solution. However, this formulation failed to provide sustained release of Dox and CpG. Given the high potential for skin blistering (vesication) associated with local delivery of Dox (13), the development of a sustained-release delivery system is crucial to avoid this complication.
Biodegradable polymer particles are promising delivery systems for the development of injectable sustained-release formulations. Poly(lactic-co-glycolic acid) (PLGA) is one of the most successfully used biodegradable polymers. It is approved by the US FDA and European Medicine Agency (EMA) for various drug delivery systems in humans (14). Microparticles made of PLGA can be loaded with both Dox and CpG (Dox/CpG MPs) for efficient, intratumoral delivery of the two drugs without the risk of their precipitation. Moreover, the sustained release of Dox from PLGA particles should limit or eliminate vesication. Additionally, the immune adjuvant effect of PLGA itself has been shown in a number of reports (15). A size of 1 μm was selected for the MPs since it was reported to induce optimal NLRP3 inflammasome activation in DCs, which enhances cell-mediated immunity (16).
The in situ immunization approach utilized in this report is thus built to optimize intratumoral delivery of Dox/CpG MPs with the goal of breaking immune tolerance to tumor antigens and inducing an antitumor immune response. To maintain an activated T cell response, we also incorporated monoclonal antibodies that enhance T cell activation (anti-OX40) and overcome immunosuppression (anti-CTLA-4) in a comprehensive immunotherapy design that was tested both in vitro and in vivo.
MATERIALS AND METHODS
Fabrication of PLGA Particles Encapsulating Dox and CpG (Dox/CpG MPs)
One to three milligrams of Dox (Sigma, Allentown, PA) was dissolved in 75 μL of 1% poly(vinyl alcohol) (PVA; Mowiol®; Sigma, Allentown, PA) solution. The primary emulsion was prepared by emulsifying this solution in 750 μL of dichloromethane (DCM) containing 100 mg of PLGA (Resomer® RG 503; Boehringer Ingelheim KG, Germany) using Sonic Dismembrator (Model FB 120 equipped with an ultrasonic converter probe CL-18; Fisher Scientific, Pittsburgh, PA) at 40% amplitude for 30 s. Similarly, 1 to 4 mg of endotoxin-free CpG oligodeoxynucleotides (CpG ODN) (5′-TCCATGACGTTCCTGACGTT-3′, Integrated DNA Technologies, Coralville, IA) was dissolved in 75 μL of 1% PVA which was sonicated using the same conditions in 750 μL of DCM containing 100 mg of PLGA. The two primary emulsions were combined to get a compound primary emulsion, which was emulsified using the same settings in the Sonic Dismembrator into 8 mL of 1% PVA in 0.1 M ammonium acetate buffer (pH 8.4). This secondary emulsion was added to 22 mL of 1% PVA in 0.1 M ammonium acetate buffer (pH 8.4), which was stirred in the hood for 2 h for DCM to evaporate. Suspended particles were collected by centrifugation using Eppendorf Centrifuge 5804 R (Eppendorf, Westbury, NY) at 5,000 rpm (4,500×g) for 5 min, resuspended in 30 mL of nanopure water, and washed twice with nanopure water. Particles were then suspended in 5 mL of nanopure water which was frozen at −20°C for 4 h and lyophilized for 18 h with LABCONCO freeze dry system (FreeZone® 4.5 l, Model 7750020; Labconco Corporation, Kansas City, MO) at collector temperature of −53°C and 0.08 mBar pressure. The initial amount of Dox or CpG required for the preparation was varied to obtain Dox/CpG MPs with different ratios of Dox and CpG encapsulated in PLGA particles.
PLGA particles encapsulating Dox (Dox MPs) were prepared as mentioned above except that the primary emulsion was prepared by sonication of 150 μL of 1% PVA containing Dox into 1.5 mL of DCM containing 200 mg of PLGA. In addition, the procedure for the preparation of Dox MPs was used to prepare blank PLGA particles (blank MPs) without the drug.
Characterization of MPs
The morphology of the particles was examined using scanning electron microscope (SEM). Briefly, particle suspensions were placed on a silicon wafer mounted on SEM stubs. They were then coated with the gold-palladium by an argon beam K550 sputter coater (Emitech Ltd., Kent, England). Images were captured using the Hitachi S-4800 SEM at 5 kV accelerating voltage. The average size of particles was calculated from SEM images using ImageJ software (US National Institutes of Health, MD, USA) with n ≥ 100. Powder X-ray diffraction (XRD) patterns of PLGA particles were obtained using a Bruker D-5000 diffractometer (Bruker AXS, Karlsruhe, Germany). Differential scanning calorimetry (DSC) analysis was performed using PerkinElmer DSC 7 (Alameda, CA) to study the physical state of lyophilized particles.
Quantification of Dox in Dox MPs and Dox/CpG MPs
Quantification of Dox was performed using fluorescence spectroscopy. Briefly, different dilutions of Dox with known concentrations were prepared in DMSO. 100 μL of these standard solutions and samples were added to a 96-well plate. Fluorescence was measured at λex 470 nm and λem 585 nm using SpectraMax® M5 multi-mode microplate reader (Molecular Devices, Sunnyvale, CA). A standard curve of Dox was used to estimate the concentration of Dox in samples.
Quantification of CpG in Dox/CpG MPs
Quant-iT™ OliGreen® ssDNA Assay Kit (Invitrogen, Carlsbad, CA) was used according to the manufacturer’s protocol to quantify CpG. Briefly, in a 96-well plate 100 μL of working reagent was added to 100 μL of standard CpG solutions of different concentrations and samples with unknown CpG concentration. The plate was then incubated at room temperature for 5 min in the dark. Fluorescence was measured at λex 480 nm and λem 520 nm using a SpectraMax® M5 multi-mode microplate reader (Molecular Devices, Sunnyvale, CA). A standard curve of CpG was used to estimate the concentration of CpG in samples from the loading and release studies.
Loading of Dox and CpG in MPs
For estimation of Dox loading, 10 mg of particles from each batch was dissolved in 1 mL of DMSO. The concentration of Dox was estimated as described above. For estimation of CpG loading, 20 mg of particles from each batch was treated with 0.2 N NaOH for 12 h. Once a clear solution was obtained, it was neutralized using 0.2 N HCl. The concentration of CpG was estimated as described above. Loading was calculated using Eq. 1. Multiple batches of Dox/CpG MPs were combined, and weight average of loading (Eq. 2) was calculated to obtain 4:1, 1:1, and 1:4 ratios of Dox/CpG in Dox/CpG MPs.
| 1 |
where,
- Conc.
Concentration of drug in samples (μg/mL) as calculated from the standard curve
- Vol.
Volume of sample during the estimation of drug loading
| 2 |
where,
- WeightBatch1 and WeightBatch2
The weight of Dox/CpG MPs from batch 1 and batch 2, respectively
- LoadingBatch1 and LoadingBatch2
Loading (μg/mg) of the drug in MPs from batch 1 and batch 2, respectively
In Vitro Release of Dox and CpG from MPs
Release studies were performed in phosphate-buffered saline (PBS) at pH 7.4 in a 37°C incubator shaker at a speed of 200 rpm/min. Fifty milligrams of particles was added to 3 mL of PBS (optimal volume for performing repeated measurements of Dox and CpG release). Samples were collected at predetermined time points and the volumes removed were replaced by fresh PBS. Concentrations of Dox and CpG in samples were estimated as described above. At the end of the release study, the remaining drug in the PLGA particle matrix was extracted. The percentage released was calculated by normalizing the amount of drug released at each time point with the sum of the amount of drug release during the study and the amount of drug extracted from the particles at the end of the study. Percentage cumulative release of Dox and CpG was plotted with respect to time.
Evaluating the Cytotoxicity of Dox/CpG MPs in Tumor Cells and DCs In Vitro
Cell Lines
The A20 cell line (a BALB/c B cell lymphoma) and the EL4 cell line (a C57BL/6 T cell lymphoma) were purchased from ATCC (Manassas, VA). Tumor cells were cultured in RPMI-1640 medium (Gibco, Carlsbad, CA) supplemented with 10% heat-inactivated FCS (HyClone, Logan, UT), 100 U/mL penicillin, 100 μg/mL streptomycin, and 50 μM 2-ME (All from Gibco), as complete medium.
To generate bone marrow-derived DCs (BMDCs), bone marrow cells were flushed from the tibias and femurs of BALB/c mice with complete medium, and mononuclear cells were isolated using Ficoll gradient separation (Fico/Lite-LM, Atlanta Biologicals, Flowery Branch, GA). Cells were cultured in McCoy’s medium (Gibco) supplemented with 5 mL each Glutamax, MEM, and sodium pyruvate (all from Gibco) and 20 ng/mL each GM-CSF and IL-4 (PeproTech, Rocky Hill, NJ) for 7 days to enrich for DCs. After 7 days, the nonadherent cells were harvested and used. Cells were >70% DCs as determined by CD11c staining by flow cytometry.
Viability Assay
To determine the cytotoxic activity of MPs against A20 and EL4, the 3-(4,5 dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay for viability was conducted using CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI). Five thousand A20 and EL4 cells were separately incubated with soluble Dox or Dox/CpG MPs (4:1 loading) in 96-well plates for 24, 48, and 72 h (four wells per group) at a final Dox concentration of 4.5 μg/mL. Media or blank MPs at equivalent weights were used as negative controls. The MTS/PMS reagent was then added for 4 h at 37°C. Following centrifugation, 80 μL of supernatant was removed to another 96-well plate. Absorbance was read at 490 nm using Thermomax Microplate Reader (Molecular Devices, Sunnyvale, CA). To directly compare the viability of A20, EL4, and BMDCs, cells were separately incubated for 24 h as described with Dox/CpG MPs (1:1 loading) at final Dox concentrations ranging from 0.28125 to 4.5 μg/mL. Media or blank MPs (average equivalent weight for highest and lowest concentrations) were used as control. Viability was assessed as before.
Percent survival was expressed as the ratio of absorbance of treated cells relative to that of untreated cells in media group (after subtracting the absorbance of the blank from each) multiplied by 100. Wells with equivalent drug or MP concentrations in absence of cells were used as blanks.
Evaluating the Efficacy of Dox/CpG MPs in Murine Tumor Models
Mice
Mice (BALB/c and C57BL/six females, 6–8 weeks old) were purchased from Harlan Laboratories (Indianapolis, IN). All animal protocols used in these studies were approved by the Institutional Animal Care and Use Committee at the University of Iowa and complied with NIH Guidelines.
Tumor Models
To examine local and systemic tumor regression following in situ immunization, the two-tumor A20 lymphoma model was used in BALB/c mice (17). Tumor cells were injected subcutaneously on opposite sides of the animal, with one tumor used for in situ immunization (injection of Dox/CpG MPs) and the contralateral tumor observed to assess systemic immune responses.
Seven to nine million A20 tumor cells in 100 μL sterile PBS were injected subcutaneously in the right and left flanks. Treatment began when tumors reached 5–7 mm in largest diameter, which typically occurred at days 9–10 after tumor inoculation. Two treatment protocols were assessed. In one protocol, mice received PBS (100 μL) or Dox/CpG MPs (2 or 10 μg Dox; 4:1 loading) in the left tumor and six intraperitoneal doses of anti-CTLA-4 (50 μg) given every 3–4 days. In another protocol, mice received PBS (100 μL) or Dox/CpG MPs (2 μg Dox and 1.5 μg CpG; 1:1 loading) in the left tumor and four intraperitoneal doses of anti-CTLA-4 (50 μg) and anti-OX40 (200 μg) given every 3–4 days. Anti-CTLA4 (hamster IgG, clone UC10-4F10-11) and anti-OX40 (rat IgG1, clone OX86) were purchased from BioXCell (West Lebanon, NH). The doses used are 50% of the conventional published dose (17) to reduce systemic toxicity.
To examine the efficacy of Dox/CpG MPs at reducing tumor burdens, the EL4 T lymphoma and the B16F10 melanoma single-tumor models were used. Dox/CpG MPs were administered as part of a therapy regimen that also included anti-CTLA-4 and anti-OX40 antibodies.
For the EL4 model, C57BL/6 mice were subcutaneously inoculated with EL4 at a dose of one million cells in 100 μL sterile PBS in the right flank. On day 4 post-inoculation, Dox/CpG MPs (4:1 loading) in 100 μL PBS were injected into the tumor site at a Dox dose of 2, 10, 50, or 100 μg. PBS (100 μL) was given to control groups. Antibodies were administered as detailed for the A20 tumor model.
B16-fLUC cells (a luciferase-expressing B16 cell line) were a generous gift from Noah Craft (University of California, Los Angeles) and were used as previously described (18). C57BL/6 mice were subcutaneously inoculated with 5 × 104 B16-fLUC cells in 100 μL of a 1:1 PBS/Matrigel mixture in the right flank. Treatment was started when tumor growth was noted using bioluminescent imaging. Briefly, mice were shaved and depilated (Nair®, Church & Dwight Co., Ewing, NJ) one day prior to imaging. On the day of imaging, mice were injected intraperitoneally with 100 μL of 10 mg/mL of the luciferase substrate, d-luciferin (GoldBio.com, St. Louis, MO) in PBS, anesthetized via inhalation of oxygenated isoflurane, and imaged after 10 min using IVIS 200 (Caliper Life Sciences, Hopkinton, MA) with 4-min acquisition times using Living Image version 4.2 software (Caliper Life Sciences). Once tumors were established (∼day 6 post-inoculation), Dox/CpG MPs (70 μg Dox and 288 μg CpG; 1:4 loading) in 100 μL PBS were injected into the tumor site. Abs were administered as previously described except that the full dose was used (100 μg for anti-CTLA-4 and 400 μg for anti-OX40).
In all tumor models, tumor growth was monitored by calipers and expressed as tumor area (length by width in square millimeters; mm2). Mice were euthanized when tumors reached 20 mm in diameter in any direction.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism software, version 6.00 (San Diego, CA). Data were analyzed using paired or unpaired two-tailed Student’s t tests, where appropriate. Comparisons of means between more than two groups were done by one- or two-way analysis of variance (with a Bonferroni post hoc test). Significance at p < 0.05 is indicated by one asterisk; p < 0.01 is indicated by two asterisks; and p < 0.001 is indicated by three asterisks.
RESULTS AND DISCUSSION
Fabrication and Characterization of Dox/CpG MPs
A modified double emulsion solvent evaporation method was used for co-encapsulation of Dox and CpG in PLGA particles (Fig. 1a). All MPs demonstrated smooth morphology and a spherical shape (Fig. 1b). Examination of their release profile revealed that MPs showed burst release of encapsulated molecules followed by sustained release (Fig. 1c). PLGA particles with different loading ratios of Dox/CpG did not show differences in the percentage release of the encapsulated molecules for the range of loadings used for this work (data not shown). XRD patterns showed that all MPs are amorphous in nature, while DSC thermograms confirmed that Dox and CpG do not show any interaction with PLGA (data not shown). There was a significant difference in Dox release kinetics from Dox/CpG MPs and Dox MPs (p < 0.001). In contrast, there was no significant difference in CpG release kinetics from Dox/CpG MPs and CpG MPs. Since Dox and CpG dissolved in water have opposite charges at neutral pH, the delay in the release of Dox from Dox/CpG MPs could be due to the formation of Dox and CpG complexes within the PLGA particle matrix that retard the diffusion of Dox.
Fig. 1.
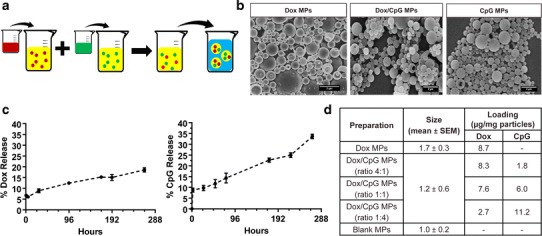
Fabrication and characterization of PLGA particles. a Modified double emulsion solvent evaporation procedure used for the preparation of Dox/CpG MPs. Dox (red) and CpG (green) solutions were emulsified separately in PLGA dissolved in dichloromethane forming w/o emulsions. The two emulsions were combined and w/o/w emulsion was prepared to obtain PLGA particles co-loaded with Dox and CpG. b SEM microphotographs of Dox, Dox/CpG, and CpG PLGA MPs. The scale bar on the lower right represents 2 μm length. c Percentage Dox and CpG release in PBS (pH 7.4) at 37°C from Dox/CpG MPs (mean ± SEM). Groups were compared using the paired t test (n = 3). d Table representing an average size (diameter) and an average loading (μg/mg particles) of Dox and CpG in different MPs
Three different loading formulations were prepared (4:1, 1:1, and 1:4 Dox/CpG) (Fig. 1d) and tested in three different tumor models. Loadings were adjusted to the tumor aggressiveness in order to optimize the generated antitumor responses by optimizing the delivered doses of chemotherapy (Dox) and immunotherapy (CpG).
Dox/CpG MPs Are Efficient at Killing Tumor Cells and Are Less Toxic to BMDCs
We studied the antitumor activity of Dox/CpG MPs in vitro by examining the viability of B cell (A20) and T cell (EL4) lymphoma cell lines post-exposure to the particles and comparing it to viability following exposure to the soluble drug (soluble Dox). The various statistical comparisons are summarized in Fig. 2. Within 24 h of incubation, A20 cells showed a dramatic decrease in viability in the presence of soluble Dox (Fig. 2a). A less dramatic but significant decrease was seen with Dox/CpG MPs. The sustained release of Dox from Dox/CpG MPs was evident in the slower decrease in viability of A20 incubated with Dox/CpG MPs over time (23% of A20 incubated with Dox/CpG MPs were viable on day 2 versus 61% on day 1; p < 0.05) which was not seen with soluble Dox (4% of A20 incubated with soluble Dox were viable on day 2 versus 26% on day 1; not significant). We also observed that blank MPs were nontoxic to tumor cells over the 3-day incubation period (Fig. 2a). Similar results were seen with EL4 cells (Fig. 2b). More importantly, Dox/CpG MPs were as efficient as soluble Dox in inducing tumor cell death of both A20 (Fig. 2a) and EL4 (Fig. 2b). Collectively, these data indicate that Dox/CpG MPs are fully capable of substituting for soluble Dox for inducing tumor cell death.
Fig. 2.
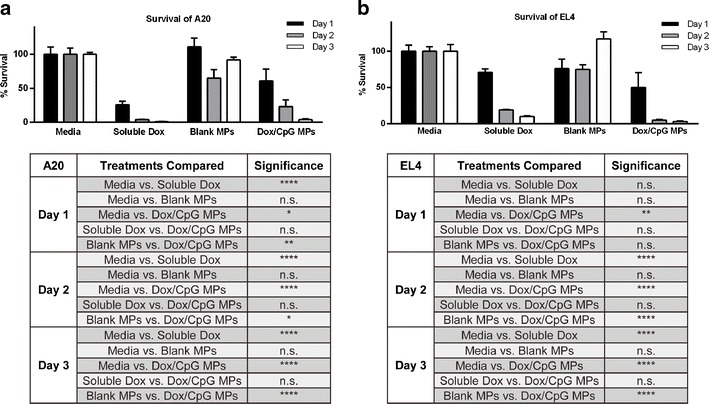
Dox/CpG MPs are efficient at killing tumor cells. A20 a and EL4 b cells were incubated with soluble Dox or Dox/CpG MPs (4:1 loading) in 96-well plates for 24, 48, and 72 h at a final Dox concentration of 4.5 μg/mL. Media or blank MPs at equivalent weights were used as negative controls. Viability was assessed by the MTS assay. Results are mean ± SEM (n = 4). Comparisons are summarized in the tables. **** p < 0.0001; ** p < 0.01; * p < 0.05; n.s. not significant
Given that Dox/CpG MPs are intended for delivery into the tumor which harbors tumor cells as well as infiltrating immune cells (most notably DCs), we evaluated the relative toxicity of Dox/CpG MPs to tumor cells and DCs (Fig. 3). Using BMDCs, we directly compared the viability of A20 and EL4 tumor cells to BMDCs over a 24-h incubation period with increasing concentrations of Dox/CpG MPs. Increasing concentrations of Dox/CpG MPs significantly reduced the viability of A20 and EL4 tumor cells (>70% survival at 4.5 μg/mL to <25% at 0.28125 μg/mL for both A20 and EL4; p < 0.0001). Dox/CpG MPs were less cytotoxic to BMDCs than to A20 or EL4, as reflected by higher percent survival at low to intermediate concentrations (93% survival for BMDCs versus 53% for EL4 and 39% for A20 at 1.125 μg/mL Dox; p < 0.001 for both). At high concentrations (4.5 μg/mL), Dox/CpG MPs were equally toxic to lymphoma cells and DCs (20% survival for BMDCs versus 19% for EL4 and 24% for A20; not significant for both). This suggests the injected dose and local concentration of Dox/CpG MPs have to be carefully considered, as higher concentrations may be detrimental to DCs that are responsible for initiating the antitumor immune response.
Fig. 3.
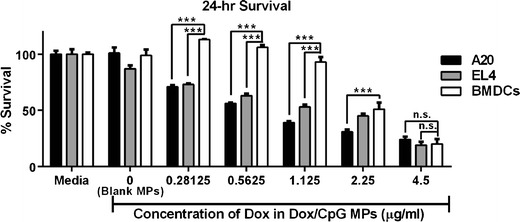
Dox/CpG MPs are less toxic to BMDCs. A20, EL4, and BMDCs were incubated for 24 h with Dox/CpG MPs (1:1 loading) at final Dox concentrations ranging 0.28125–4.5 μg/mL. Media or blank MPs (average equivalent weight for highest and lowest concentrations) were used as control. Viability was assessed by the MTS assay. Results are mean ± SEM (n = 4). *** p < 0.001; n.s. not significant
Low Doses of Dox/CpG MPs Are More Efficient at Reducing A20 Tumor Burdens
To validate the efficacy of Dox/CpG MPs in immunotherapy, we utilized Dox/CpG MPs in an in situ immunization regimen that impacts antitumor immune responses at multiple levels. This regimen consisted of intratumoral (i.t.) Dox/CpG MP injection combined with multiple injections of a systemic antibody that blocks CTLA-4 (anti-CTLA-4). Intratumorally, released Dox is expected to induce immunogenic tumor cell death, while released CpG enhances presentation of tumor-derived antigens by DCs. CTLA-4 is a co-inhibitory receptor that is constitutively expressed by regulatory T cells and is upregulated by T cells post-activation. A member of the immunoglobulin superfamily, it suppresses T cell activation and is employed by regulatory T cells to keep immune responses in check (19). As such, systemic anti-CTLA-4 administration can lead to long-lasting antitumor immunity by allowing activated tumor-specific T cells to remain activated longer.
We tested the designed therapy regimen in vivo in BALB/c mice using the previously described two-tumor A20 lymphoma model used for in situ immunization (17) (Fig. 4). Mice were inoculated subcutaneously with A20 tumor cells on both flanks. Once tumors formed, one tumor was used for a single i.t. injection of Dox/CpG MPs in addition to six intraperitoneal (i.p.) injections of anti-CTLA-4 at half the published dose (50 μg) (17). The other (distant) tumor received no therapy and was monitored for signs of regression, which can only be attributed to systemic immune responses generated locally at the injected tumor.
Fig. 4.
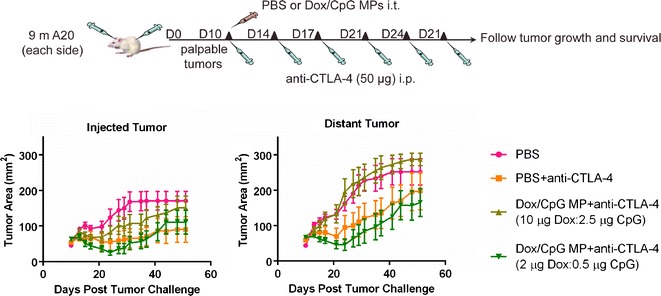
Low doses of Dox/CpG MPs are more efficient at reducing A20 tumor burdens. Nine million A20 tumor cells were injected subcutaneously in the right and left flanks of BALB/c mice. After 10 days, mice received PBS as control or Dox/CpG MPs (2 or 10 μg Dox; 4:1 loading) in the left tumor and six intraperitoneal doses of anti-CTLA-4 (50 μg) given every 3–4 days. Mice were monitored for tumor growth of both injected and distant tumors and survival. Tumor areas are mean ± SEM
We tested two doses of Dox/CpG MPs that delivered either 2 μg Dox (low-dose therapy) or 10 μg Dox (high-dose therapy) at a (4:1) loading (Fig. 4). Low-dose therapy with Dox/CpG MPs significantly reduced the injected tumor burden (p < 0.01; low-dose therapy versus PBS between days 29 and 35 post-tumor challenge). Low-dose therapy also reduced distant tumor burdens (p < 0.01; low-dose therapy versus PBS after day 29), suggesting that they are capable of inducing a systemic immune response. On the other hand, high-dose therapy was inefficient at reducing both local and distant tumor burdens. These data are in agreement with our in vitro data (Fig. 3) showing that high doses of Dox/CpG MPs can be lethal to DCs, thus abrogating the generation of an antitumor immune response.
Dox/CpG MPs Combined with Ab Generate Systemic Immune Responses that Eradicate Distant Tumors
While low-dose therapy was more efficient than high-dose therapy, its beneficial effect was transient as tumors regrew (Fig. 4; low-dose tumors not significantly different from PBS tumors after day 44). This represented a limitation, since the goal was to generate long-lasting antitumor immune responses. Moreover, low-dose therapy was comparable to Ab therapy (PBS+Ab) at reducing both injected and distant tumors (p > 0.05; not significant). As such, we sought to optimize our therapy design by increasing the CpG dose delivered to DCs via use of Dox/CpG MPs at the same optimized Dox dose (2 μg) but at (1:1) loading. This increased the delivered CpG dose threefold to 1.5 μg instead of 0.5 μg. We also enhanced the activated T cell immune responses by adding anti-OX40 to anti-CTLA-4. OX40 (CD134) is expressed by activated T cells, whose proliferation and survival can be enhanced using agonistic antibodies to OX40 (anti-OX40) (17). Furthermore, the combination of anti-CTLA-4 and anti-OX40 has been shown to enhance antitumor immune responses in murine lymphoma models (17). Since the activity of anti-CTLA-4 was being augmented by anti-OX40, and since our goal was to illustrate the benefits of local therapy without increasing systemic toxicity, we reduced the number of anti-CTLA-4 doses administered with anti-OX40 to four instead of six.
Using the A20 two-tumor model (Fig. 5), our optimized therapy of i.t. Dox/CpG MPs combined with i.p. anti-CTLA-4 and anti-OX40 antibodies (referred to as Ab) resulted in regression of both injected and distant tumors (100% of mice in Dox/CpG MP+Ab group became tumor-free). Ab therapy alone (PBS+Ab) was also capable of inducing regression of the injected tumor, as were Dox/CpG MPs administered with Ab therapy (p < 0.0001 for all groups versus PBS starting day 16 post-tumor challenge). These data point to the antibodies (the common denominator in all groups) as the major contributor to the tumor regression seen. Indeed, Dox/CpG MPs administered without Ab therapy were incapable of inducing antitumor immune responses (data not shown).
Fig. 5.
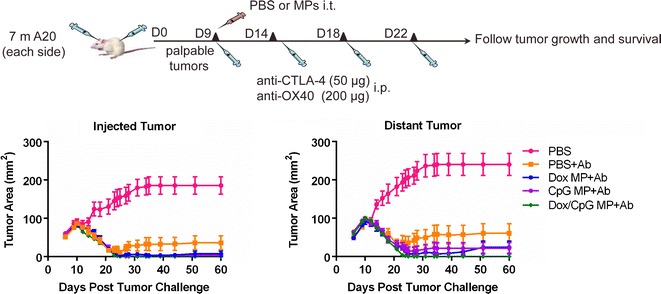
Dox/CpG MPs combined with Ab generate systemic immune responses that eradicate distant tumors. Seven million A20 tumor cells were injected subcutaneously in the right and left flanks of BALB/c mice. Treatment began when tumors reached 5–7 mm in largest diameter, which typically occurred at days 9–10 after tumor inoculation. Mice received PBS as control, Dox MPs (2 μg Dox), CpG MPs (1.5 μg CpG), or Dox/CpG MPs (2 μg Dox and 1.5 μg CpG; 1:1 loading) in the left tumor and three intraperitoneal doses of anti-CTLA-4 (50 μg) and anti-OX40 (200 μg) given every 3–4 days. Mice were monitored for tumor growth of both injected and distant tumors and survival. Tumor areas are mean ± SEM. Results shown are pooled from two experiments (15–20 mice/group)
However, Dox/CpG MP+Ab was significantly better than Ab therapy at eradicating distant tumors (p < 0.01 by day 60 post-tumor challenge). This indicates that our optimized combination regimen with Dox/CpG MPs is more efficient than Ab therapy alone at generating long-lasting antitumor responses. Indeed, mice (n = 5) that received Dox/CpG MP+Ab and became tumor-free were rechallenged with 10 million A20 tumor cells implanted subcutaneously at a different site from the MP-injected tumor at day 51 post-tumor challenge. None of the mice developed any tumors 22 days later (data not shown).
The combination of anti-CTLA-4 and anti-OX40 was shown by Houot el al. (17) to be effective as part of an in situ immunization regimen that uses multiple injections of soluble CpG i.t. totaling 500 μg. While the Houot therapy regimen similarly led to complete regression of both injected and contralateral tumors, our regimen used only 1.5 μg of CpG, an over 300-fold reduction in the CpG dose that was delivered in just one i.t. injection.
Dox/CpG MPs Combined with Ab Are Efficient at Reducing EL4 Tumor Burdens
To validate the efficiency of Dox/CpG MPs in other tumor models, we tested Dox/CpG MPs in combination with anti-CTLA-4 and anti-OX40 in an EL4 T cell lymphoma tumor model (Fig. 6). Given that EL4 tumors are more aggressive than A20 tumors, higher MP doses (up to 100 μg Dox) were tested. Moreover, a single rather than a two-tumor model was used due to the fast growth rate of the distant tumor that necessitated sacrifice of the mice before the effect of a systemic immune response could be observed (data not shown).
Fig. 6.
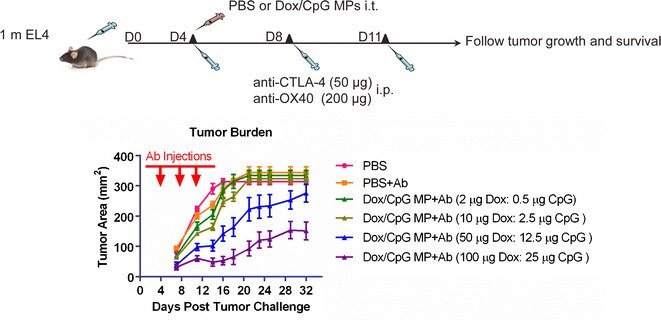
Dox/CpG MPs combined with Ab are efficient at reducing EL4 tumor burdens. C57BL/6 mice (5–10 mice/group) were subcutaneously inoculated with EL4 at a dose of one million cells in the right flank. On day 4 post-inoculation, Dox/CpG MPs (4:1 loading) were injected into the tumor site at a Dox dose of 2, 10, 50, or 100 μg (corresponding CpG dose of 0.5, 2.5, 12.5, or 25 μg). PBS was given to control groups. Anti-CTLA4 (50 μg) and anti-OX40 (200 μg) (collectively referred to as Ab) were administered by intraperitoneal injections of three doses given every 3–4 days, starting from day 1 of treatment. Mice were monitored for tumor growth and survival. Tumor areas are mean ± SEM
The previously tested doses of Dox/CpG MPs (2 and 10 μg Dox) were inefficient at reducing EL4 tumor burdens, most likely due to the fast tumor progression rate. Using 50 μg Dox/CpG MPs combined with Ab therapy, the tumor burden was significantly reduced (p < 0.0001; 50 μg Dox/CpG MP+Ab versus PBS between days 14 and 16 post-tumor challenge). 50 μg Dox/CpG MPs combined with Ab therapy were even more efficient than Ab therapy alone (p < 0.0001; 50 μg Dox/CpG MP+Ab versus Ab therapy between days 16 and 18 post-tumor challenge). However, this impact was transient as tumors regrew after day 18. In contrast, using 100 μg Dox/CpG MPs combined with Ab therapy had a longer-lasting antitumor effect (significant differences between 100 μg Dox/CpG MP+Ab and PBS were seen until day 32 post-tumor challenge; p < 0.001). 100 μg Dox/CpG MPs combined with Ab therapy was also significantly better than Ab therapy alone at reducing tumor burdens (p < 0.0001; 100 μg Dox/CpG MP+Ab versus Ab therapy on day 32 post-tumor challenge) and was also more efficient than 50 μg Dox/CpG MPs (p < 0.01; 100 μg Dox/CpG MP+Ab versus 50 μg Dox/CpG MP+Ab on day 32 post-tumor challenge). Collectively, these data indicate that in a more aggressive tumor model, higher doses of Dox/CpG MPs may be needed to control the rapidly dividing tumor cells.
Dox/CpG MPs Combined with Ab Are Efficient at Reducing Melanoma Tumor Burdens
We also tested Dox/CpG MPs in a murine B16 melanoma tumor model (Fig. 7). While doxorubicin is not part of the standard care for melanoma patients as it is for lymphoma patients, our goal was to show that local chemotherapy can be used as part of an immunotherapy regimen to potentiate antitumor immune responses.
Fig. 7.
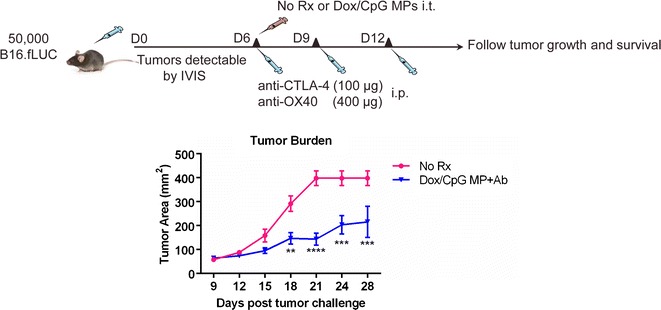
Dox/CpG MPs combined with Ab are efficient at reducing B16 tumor burdens. C57BL/6 mice (five per group) were subcutaneously inoculated with 5 × 104 B16-fLUC (stable luciferase-expressing B16 cells) in a 1:1 PBS/Matrigel mixture in the right flank. Treatment was commenced when tumors were established (∼day 6 post-inoculation), which was verified using bioluminescent imaging (IVIS). Mice received either no treatment (no Rx) or intratumoral Dox/CpG MPs (70 μg Dox and 288 μg CpG; 1:4 loading). Anti-CTLA-4 (100 μg) and anti-OX40 (400 μg) (collectively referred to as Ab) were administered i.p. in three doses as previously described except that the full dose was used. Mice were monitored for tumor growth and survival. Tumor areas are mean ± SEM. * p < 0.05; ** p < 0.01; *** p < 0.001
Because melanoma is an aggressive tumor, we used a single-tumor model similar to EL4. We also modified our therapy regimen by using 1:4 Dox/CpG MPs delivering a Dox dose of 70 μg and CpG dose of 288 μg and increased the doses of anti-CTLA-4 and anti-OX40 to 100% their published dose rather 50%. These adjustments were made to enhance the generated immune responses by further potentiating DC (via CpG) and T cell (via Ab) responses against the poorly immunogenic melanoma tumors.
Due to limitations of caliper measurements to detect established subcutaneous tumors (a palpable tumor may be measurable only when well over 106 cells are present (18)), we evaluated the potential of our therapy using a B16 tumor line expressing firefly luciferase (B16-fLUC). This allows for verification of tumor establishment by bioluminescent imaging even before the tumor is palpable, which was necessary to confirm before treatment could be commenced.
Similar to the EL4 tumor model, Ab therapy (anti-CTLA-4+anti-OX40) alone tested in pilot experiments was inefficient at reducing tumor burdens at both 50 and 100% conventional dose (data not shown). On the other hand, Dox/CpG MPs combined with Ab therapy significantly reduced B16 tumor burdens as compared to untreated control mice.
CONCLUSION
As we learn more about cancer immunotherapy, and various immune manipulations are found to be effective not only in the laboratory but also in the clinic, studies exploring optimal ways to combine such treatments will be increasingly important. In the current study, we evaluated the potential value of MPs that contain both Dox and CpG as a component of cancer immunotherapy. Pilot studies were conducted to explore a large number of variables including drug doses, drug ratios, loading, polymer size, particle size, timing of exposure, etc. These studies were done in parallel in various systems including in vitro and in vivo evaluation. Given the large number of variables, it was not possible to optimize every parameter individually in every system. The overall goal of these studies was to highlight the feasibility of the overall approach. Once we identified reasonable parameters in each system, additional studies were done in that system using those parameters. Therefore, some of the details, such as loading, varied from model to model. Before this concept is translated to the clinic, additional optimization will need to be done to select the best single approach for each variable.
In vitro, we showed that Dox/CpG MPs are efficient at killing tumor cells and are less toxic to BMDCs. Using a B lymphoma two-tumor model, we found that Dox/CpG MPs combined with Ab therapy (anti-CTLA-4 and anti-OX40) are efficient at eradicating both local and distant tumors. We further validated the antitumor efficacy of this design in T cell lymphoma and melanoma tumor models, demonstrating that Dox/CpG MPs can reduce tumor burdens more efficiently than Ab therapy alone.
In situ immunization is appealing based on reduced systemic toxicity, potential for developing an immune response against a variety of endogenous tumor antigens, and ability to generate antigen-specific responses right at the tumor site (and so evade problems associated with trafficking of immune cells to the tumor). On the other hand, a major limitation is that it requires accessibility to the tumor site (20). It will also be important to assess the efficacy of in situ immunization, and duration of response, in de novo models where the tumors have greater heterogeneity and eventually in clinical trials.
In conclusion, our studies indicate MPs containing both Dox and CpG have promise as a component of in situ immunization and deserve further evaluation as a component of cancer immunotherapy.
ACKNOWLEDGMENTS
We gratefully acknowledge support from the National Cancer Institute at the National Institutes of Health (P50 CA97274/UI Mayo Clinic Lymphoma SPORE grant and P30 CA086862 Cancer Center support grant).
Conflict of Interest
The authors have declared that no conflict of interest exists.
REFERENCES
- 1.Topalian SL, Weiner GJ, Pardoll DM. Cancer immunotherapy comes of age. J. Clin.Oncol Off J Am Soc Clin Oncol. 2011;29(36):4828–36. doi: 10.1200/JCO.2011.38.0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galluzzi L, Vacchelli E, Eggermont A, Fridman WH, Galon J, Sautes-Fridman C, et al. Trial watch: adoptive cell transfer immunotherapy. Oncoimmunology. 2012;1(3):306–15. doi: 10.4161/onci.19549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–9. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vacchelli E, Martins I, Eggermont A, Fridman WH, Galon J, Sautes-Fridman C, et al. Trial watch: peptide vaccines in cancer therapy. Oncoimmunology. 2012;1(9):1557–76. doi: 10.4161/onci.22428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galluzzi L, Senovilla L, Vacchelli E, Eggermont A, Fridman WH, Galon J, et al. Trial watch: dendritic cell-based interventions for cancer therapy. Oncoimmunology. 2012;1(7):1111–34. doi: 10.4161/onci.21494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hortobagyi GN. Anthracyclines in the treatment of cancer. An overview. Drugs. 1997;54(Suppl 4):1–7. doi: 10.2165/00003495-199700544-00003. [DOI] [PubMed] [Google Scholar]
- 7.Vacchelli E, Galluzzi L, Fridman WH, Galon J, Sautes-Fridman C, Tartour E, et al. Trial watch: chemotherapy with immunogenic cell death inducers. Oncoimmunology. 2012;1(2):179–88. doi: 10.4161/onci.1.2.19026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krieg AM. From A to Z on CpG. Trends Immunol. 2002;23(2):64–5. doi: 10.1016/S1471-4906(01)02150-0. [DOI] [PubMed] [Google Scholar]
- 9.Galluzzi L, Vacchelli E, Eggermont A, Fridman WH, Galon J, Sautes-Fridman C, et al. Trial watch: experimental toll-like receptor agonists for cancer therapy. Oncoimmunology. 2012;1(5):699–716. doi: 10.4161/onci.20696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang H, Liu L, Yu D, Kandimalla ER, Sun HB, Agrawal S, et al. An in situ autologous tumor vaccination with combined radiation therapy and TLR9 agonist therapy. PLoS One. 2012;7(5):e38111. doi: 10.1371/journal.pone.0038111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geary SM, Krishnamachari Y, Lemke C, Salem AK, Weiner GJ. Biodegradable particulate formulations. Google Patents; 2012.
- 12.Mizuno Y, Naoi T, Nishikawa M, Rattanakiat S, Hamaguchi N, Hashida M, et al. Simultaneous delivery of doxorubicin and immunostimulatory CpG motif to tumors using a plasmid DNA/doxorubicin complex in mice. J Control Release Off J Control Release Soc. 2010;141(2):252–9. doi: 10.1016/j.jconrel.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 13.Conde-Estevez D, Mateu-de AJ. Treatment of anthracycline extravasations using dexrazoxane. Clin Transl Oncol Off Publ Fed Span Oncol Soc Natl Cancer Inst Mexico. 2014;16(1):11–7. doi: 10.1007/s12094-013-1100-7. [DOI] [PubMed] [Google Scholar]
- 14.Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Preat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release Off J Control Release Soc. 2012;161(2):505–22. doi: 10.1016/j.jconrel.2012.01.043. [DOI] [PubMed] [Google Scholar]
- 15.Yoshida M, Babensee JE. Poly(lactic-co-glycolic acid) enhances maturation of human monocyte-derived dendritic cells. J Biomed Mater Res A. 2004;71(1):45–54. doi: 10.1002/jbm.a.30131. [DOI] [PubMed] [Google Scholar]
- 16.Sharp FA, Ruane D, Claass B, Creagh E, Harris J, Malyala P, et al. Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc Natl Acad Sci U S A. 2009;106(3):870–5. doi: 10.1073/pnas.0804897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Houot R, Levy R. T-cell modulation combined with intratumoral CpG cures lymphoma in a mouse model without the need for chemotherapy. Blood. 2009;113(15):3546–52. doi: 10.1182/blood-2008-07-170274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Craft N, Bruhn KW, Nguyen BD, Prins R, Liau LM, Collisson EA, et al. Bioluminescent imaging of melanoma in live mice. J Investig Dermatol. 2005;125(1):159–65. doi: 10.1111/j.0022-202X.2005.23759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolchok JD, Yang AS, Weber JS. Immune regulatory antibodies: are they the next advance? Cancer J. 2010;16(4):311–7. doi: 10.1097/PPO.0b013e3181eb3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crittenden MR, Thanarajasingam U, Vile RG, Gough MJ. Intratumoral immunotherapy: using the tumour against itself. Immunology. 2005;114(1):11–22. doi: 10.1111/j.1365-2567.2004.02001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]