

Abstract
Downstream success in Pharmaceutical Development requires thoughtful molecule design early in the lifetime of any potential therapeutic. Most therapeutic monoclonal antibodies are quite similar with respect to their developability properties. However, the properties of therapeutic peptides tend to be as diverse as the molecules themselves. Analysis of the primary sequence reveals sites of potential adverse posttranslational modifications including asparagine deamidation, aspartic acid isomerization, methionine, tryptophan, and cysteine oxidation and, potentially, chemical and proteolytic degradation liabilities that can impact the developability and manufacturability of a potential therapeutic peptide. Assessing these liabilities, both biophysically and functionally, early in a molecule’s lifetime can drive a more effective path forward in the drug discovery process. In addition to these potential liabilities, more complex peptides that contain multiple disulfide bonds can pose particular challenges with respect to production and manufacturability. Approaches to reducing the disulfide bond complexity of these peptides are often explored with mixed success. Proteolytic degradation is a major contributor to decreased half-life and efficacy. Addressing this aspect of peptide stability early in the discovery process increases downstream success. We will address aspects of peptide sequence analysis, molecule complexity, developability analysis, and manufacturing routes that drive the decision making processes during peptide therapeutic development.
KEY WORDS: developability, peptide, proteolysis, PTM risk
INTRODUCTION
Peptides have enormous potential for therapeutic development since small molecule therapeutics have inherent difficulties with respect to potency and off-target effects and larger biologics have high manufacturing costs, lower tissue penetration, and lower activity per unit mass. In fact, peptide-based therapeutics are one of the fastest growing classes of new therapeutic drugs and have unique pharmacokinetic properties compared to small molecule or large biologic drugs (1,2). Today, most, if not all, large pharmaceutical companies have peptide-based therapeutics programs and, with the advent of modern molecular biology and automated solid phase peptide chemistry, library design, and execution, these programs faciliate the generation of complex peptide libraries addressing uniqueness, potency, specificity, complexity, and developability.
While therapeutic peptides, like their larger protein counterparts, suffer from primary sequence liabilities during manufacturing, they are not without their own unique challenges as well. Although most peptides act quickly, they can be rapidly cleared. In contrast to antibody therapeutics, peptides are generally more susceptible to enzymatic and/or chemical degradation due, in large part, to their increased flexibility resulting in rapid clearance (3). In addition, peptides generally suffer from poor biophysical properties, such as low solubility, that make them inherently challenging to work with. As a result of these properties, peptides generally suffer short half-lives, limited bioavailability, and poor oral delivery (4–6).
Additionally, in contrast to protein therapeutics, candidate selection based on manufacturability processes may come into play early in molecule discovery. Solid phase peptide chemistry is relatively inexpensive if overall yields are high. On the other hand, recombinant expression is much more expensive as it entails the development of stable cell lines and complex manufacturing processes. Cell-free synthesis may be an attractive option, although it is not currently approved as a manufacturing process by the FDA, but that could change. In the following sections, we will have a closer look at the definition of a peptide and outline some of the challenges of peptides as therapeutics, followed by a description of engineering approaches to improve downstream success in pharmaceutical development. When reading through these sections, one must keep in mind that the application of engineering approaches, or changes made to an endogenous sequence, can affect bioassay results and the risk of immunogenicity.
HOW DO WE DEFINE A PEPTIDE?
Peptides are short polymers of amino acids that are generally less than 50 units long with a molecular weight of less than 10 kDa (7,8). Peptides have unique therapeutic advantages compared to larger biologics including a higher activity per unit mass, better tissue penetration, decreased potential for immunogenicity, and lower manufacturing costs when comparing solid phase peptide chemistry to the traditional recombinant protein expression methodologies required for larger protein therapeutics. Unlike larger biologics, peptides can be structurally flexible molecules with conformations that are highly environment dependent, leading to a structural and biophysical uniqueness that is as diverse as their targets.
For example, sunflower trypsin inhibitor (SFTI-1) is a 14-amino acid cyclic peptide isolated from sunflower seeds and is a potent inhibitor of trypsin. The three-dimensional structure of SFTI-1 was solved in complex with bovine β-trypsin and shown to have a novel, head-to-tail, covalently closed, cyclic structure (Fig. 1a) (9). The cyclic structure and intrachain disulfide bond restrict conformational flexibility, while greatly increasing the overall stability through a reduction in entropy. The structure of this inhibitor highlights clear similarities within the trypsin-reactive loop region of the Bowman-Birk family of serine protease inhibitors, including the amino acid sequence, structural conformation, and mechanism of inhibition. However, SFTI-1 has a clear difference from homologues in its size and head-to-tail cyclic structure, thereby highlighting the diversity of scaffolds of functionally similar peptides.
Fig. 1.
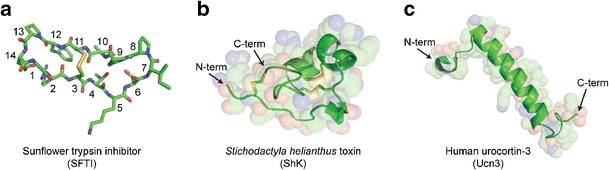
Examples of diversity among peptide tertiary structure. a The head-to-tail bicyclic structure of the 14-amino acid (Cα’s numbered from N-terminus to C-terminus) sunflower trypsin inhibitor (SFTI-1) is formed by a disulfide linkage between Cys3 and Cys11 (PDB 1SFI). b Three disulfide bonds stabilize the globular fold of the 35-amino acid peptide toxin, ShK, from the S. helianthus sea anemone (PDB 1ROO). c The 38-amino acid human urocortin-3 (Ucn-3) peptide is predominantly helical in DMSO solution (PDB 2RMH). Structures were rendered using PyMol, with carbon in green, nitrogen in blue, oxygen in red, and sulfur in yellow
An example of a peptide with a more canonical tertiary structure is the Stichodactyla helianthus (ShK) peptide toxin isolated from the venom of the sea anemone. This heavily disulfide bonded, 35-amino acid peptide is a potent inhibitor of voltage-gated potassium channels, with an IC50 in the low picomolar range for Kv1.3 (10). While this small peptide has limited secondary structure, the overall fold is maintained by 3-disulfide bonds in a manner similar to other venom peptides (Fig. 1b) (11,12). ShK interacts with all four subunits of the channel tetramer completely occluding the channel pore underlying its highly specific interaction with its target (11,13). As a result, ShK is widely regarded as a therapeutic target for preferential suppression of effector memory (CCR7−) T cells that mediate autoimmune diseases such as type 1 diabetes mellitus, rheumatoid arthritis, psoriasis, and multiple sclerosis (13).
Another example of peptide structural diversity is human urocortin-3 (Ucn-3), a 38-amino acid peptide predominantly comprised of alpha helical structure (Fig. 1c). The overall backbone is a helix-loop-helix motif with an amphipathic C-terminal helix that interacts with the first extracellular domain of the corticotropin releasing factor (CRF) receptor mainly through hydrophobic interactions and with an N-terminal helix that, once oriented, enables CRF activation through a unique binding interaction with the receptor. It is also suggested that the kink between the helix domains could play a key role in the overall ligand-receptor interaction. Interestingly, the structure was solved in DMSO as the molecule is unstructured in aqueous solution suggesting an induced fit mechanism (14).
WHY TURN TO PEPTIDES AS THERAPEUTICS?
As the biologics arm of a pharmaceutical company well versed in the discovery and development of antibody therapeutics, our initial approach against a soluble target such as a cytokine typically involves identification of a monoclonal antibody. Antigens for immunization or phage display are generally accessible and are also effective screening tools for affinity maturation. There are clear advantages to these large biologics such as high specificity and improved half-life through bypassing renal clearance or neonatal receptor-mediated recycling. For intracellular targets such as kinases, a small molecule approach is usually preferred, given the desirable cell and tissue-penetrating properties generally associated with small molecule therapeutics. Another potential benefit is the ability of certain peptides to cross the blood–brain barrier or intestinal epithelial cells for oral delivery which are particularly challenging for large biologics (15–17). Receptors or receptor complexes can be approached with either class of molecules when the target epitope is accessible.
However, there are more challenging classes of targets such as ion channels and G-protein coupled receptors (GPCRs). These transmembrane proteins lack single, defined epitopes and are generally recalcitrant to antibody generation by conventional means. Truncated domains or designs based on predicted linear, peptidic epitopes rarely retain native structural characteristics of the target in a cellular context. In addition, monoclonal antibodies are bulky and such targets generally do not provide sufficient protein-protein interaction surface area in the context of the whole cell. For small molecules, the lack of potency, poor selectivity, and off-target potential make these less viable approaches. In contrast, many cell surface receptors have peptidic hormones or cytokines as their endogenous ligands, underlying the potential for specific and potent targeting using a peptide-based platform. It is these types of targets that steer us towards developing peptide therapeutics as opposed to taking more typical monoclonal antibody or small molecule approaches.
CHALLENGES OF PEPTIDES AS THERAPEUTICS
Despite the potential for peptide therapeutics applied to otherwise difficult to target proteins, there are a number of considerations to make to ensure their success in a biological context. Even in the absence of structural or functional information, the primary sequence can provide early information on potential problems that can be encountered in pharmaceutical development. Posttranslational modification (PTM) risks can be gleaned from primary sequence motifs linked to the analysis of hydrolysis-sensitive peptide bonds, consensus sequences for asparagine deamidation, aspartate isomerization or N-linked glycosylation, or exposed methionine or tryptophan residues that could be susceptible to oxidation (Table I). Such chemical liabilities can adversely affect solution properties and/or functional behavior. Furthermore, free cysteine residues or disulfide bonds could result in misfolding or mixed disulfide pairings and can also adversely affect function and structural integrity. Finally, the high propensity for proteolytic degradation can be due to inherent flexibility or merely being a substrate for endogenous proteases
Table I.
Amino Acids at Risk for Posttranslational Modification
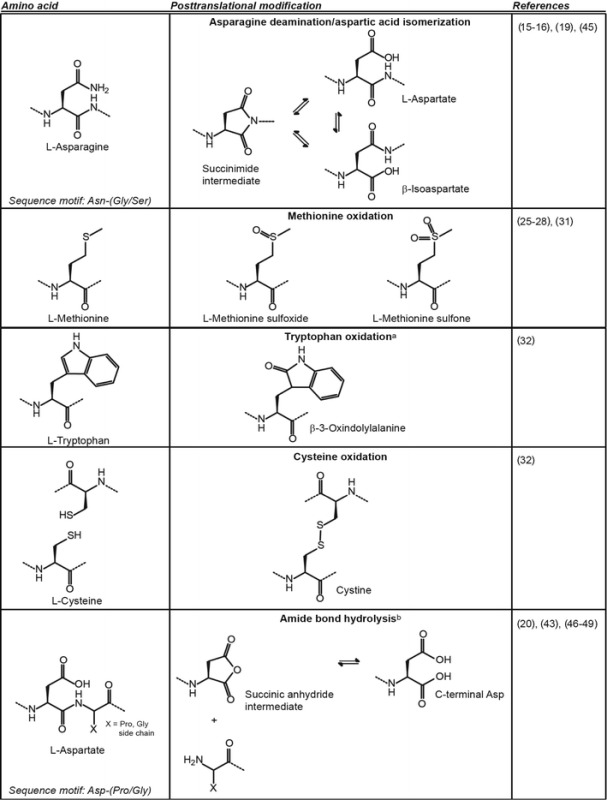
aOnly one of many possible oxidation products of tryptophan is shown
bAn analogous reaction can occur with asparagine leading to peptide bond hydrolysis through a succinimide intermediate
IMPROVING CHEMICAL AND PHYSICAL STABILITY
Asparagine Deamidation/Aspartic Acid Isomerization
At physiological temperature and pH, deamidation and isomerization are among the most prevalent chemical reactions known to affect proteins. Formation of β-isoaspartate (isoAsp) as a result of deamidation of Asn or isomerization of Asp is a major source of instability in peptides especially at neutral to alkaline pH (18,19). Although these reactions can occur for both asparaginyl/aspartyl and glutaminyl/glutamyl residues, the rate of reaction is much more rapid with asparaginyl/aspartyl residues and is generally the focus of PTM risk mitigation. In an examination of the temperature and sequence dependence of the deamidation of a series of Asn containing hexapeptides under physiological conditions at pH 7.4, Asn-Gly and Asn-Ser sequences were particularly labile (20–22). Not only can these modifications result in structural and functional changes, but it has also been suggested that the resulting l-isoaspartyl or d-aspartyl residues are substrates for mammalian carboxyl methyltransferases, further exacerbating peptide clearance (23). Cyclization of the amino terminal Gln residues to pyroglutamyl residues under mild acidic conditions is another frequent cause of peptide modification, something that is seen frequently in monoclonal antibodies (24,25).
As an example, amyloid formation underlines the adverse effects of deamidation that influence structure, stability, folding, and aggregation in peptides (26). The authors found that the deamidation of amylin, a causative agent in type 2 diabetes, accelerates amyloid formation and is able to seed amyloid formation by unmodified amylin. It has also been shown that all three aspartic acid residues in the Aβ polypeptide of Alzheimer’s disease patients isomerize to isoAsp where the non-canonical bonds of the peptide backbone cause structural disorder (26,27). In the context of therapeutics, if the primary sequence contains an Asn or Asp that has a potential PTM liability, the deamidation or isomerization propensity should be analyzed. However, it is critical to couple such results to a functional assay to determine how changes to the primary sequence, such as amino acid replacement or deletion, impact peptide function. If it is found that there are adverse effects on either the structure or function, these risks should instead be mitigated through library design using a systematic approach to address liabilities.
Methionine/Tryptophan Oxidation
Many diseases, as well as aging, have been linked to uncontrolled oxidative processes that lead to irreversible damage and ultimately death. Oxidative damage to proteins has been studied quite extensively, and although all amino acids can suffer some degree of oxidation under the right conditions, methionine and tryptophan are the most susceptible (28,29). The potential for methionine oxidation that leads to adverse changes in stability or function should be addressed as early as possible in a molecule’s lifetime. Oxidation risk assessment of methionine to generate methionine sulfoxide in peptides and proteins has been reviewed (30–34).
The best practice is to assess any potential liabilities experimentally. For example, the stability of ShK at different temperatures and pH values was investigated, and the analysis of by-products led to the design of additional stabilizing elements to address these liabilities (13). Several changes were made including addressing a methionine oxidation liability at position 21. In addition to creating a potential source of heterogeneity and instability, methionine sulfoxide creates a dipole and a new chiral center at the oxidized sulfur atom. To eliminate this PTM risk, the authors created a stable analogue through the replacement of Met with the hydrophobic isostere, norleucine.
Tryptophan side chains also pose a potential site of posttranslational modification risk (35). We have experienced first-hand many examples of binding and activity loss upon tryptophan oxidation in the context of monoclonal antibodies. As these residues are generally critical for binding or the formation of a stable hydrophobic core, positional changes can lead to loss of binding or function or both. Notwithstanding, positional changes are worth exploring when the residue is determined to be non-critical. However, if the residue cannot be changed without adverse effects on the molecule, the conversation changes to one of risk mitigation, and production and manufacturing schemes will have to be designed to minimize these potential liabilities.
Cysteine Oxidation
Another posttranslational modification liability often found in peptides is the proper formation of disulfide bonds. The potential for multiple disulfide bonded isomers increases considerably as the number of cystine bonds increase. For example, Pi4, a K+ channel blocker, contains eight cysteine residues that form four disulfide bonds (36). According to the equation for n = number of cysteine residues,
there are 105 possible combinations (P) of four disulfide bonds that can be formed (37). In cases where there is only one combination that is native and active, production, either in a recombinant sense or by solid phase peptide synthesis, often results in a complex mixture of misfolded isomers that need to be separated from the native isomer of interest. Approaches to decrease the complexity by decreasing the number of potential disulfide bonded isomers include directed disulfide bond formation or the removal or replacement of cysteine pairs. In a solid phase peptide approach, orthogonal blocking groups can be employed to selectively form disulfide pairs in a stepwise fashion, thus ensuring formation of the correct pairs (38). Disulfide bond replacement could be achieved by replacing the cysteine pairs with complimentary charged residues, creating a static pair, or with aliphatic pairs, creating hydrophobic interactions. One could also explore “stapling” techniques to replace the covalent disulfide bond with alternative covalent bonds thereby retaining the molecular stability of the molecule (39).
Although disulfide bridges play an important role in stabilizing proteins, it has been shown that in some peptides with multiple disulfide bridges not all of them are necessarily crucial for maintaining structural and functional integrity (40–42). From a manufacturing perspective, exploring the removal of disulfide bonds to reduce the complexity of the peptide can be initiated early in a molecule’s lifetime using standard biophysical techniques, such as nuclear magnetic resonance in combination with a functional assay, to evaluate the impact of these changes. The utility of disulfide bond deletion was demonstrated with μ-conotoxin KIIIA, a disulfide-rich venom peptide isolated from Conus kinoshitai that blocks mammalian neuronal voltage-gated sodium channels and is a potent analgesic (43). The authors systematically removed cysteine pairs, by making simple cysteine to alanine mutations, and screened for functional activity by testing the disulfide-deficient analogues against mammalian Nav1.2 and Nav1.4. It was shown that the removal of the Cys1-Cys9 disulfide bond (Fig. 2a) did not significantly alter the biological activity against either channel subtype, while removal of the Cys2-Cys15 disulfide bond resulted in a reduction in activity, and removal of the Cys4-Cys16 disulfide bond eliminated activity altogether. Amide proton chemical shift dispersion and solution structures were used to investigate the structural and functional impact of removing the Cys1-Cys9 disulfide bond in μ-conotoxin KIIIA (Fig. 2b) (44). In spite of the apparent flexibility of the N-terminus imparted by the removal of the disulfide bond, there was a minimal change in activity against Nav1.2 and only slight reduction in affinities for the Nav1.4 and Nav1.7 channel subtypes.
Fig. 2.
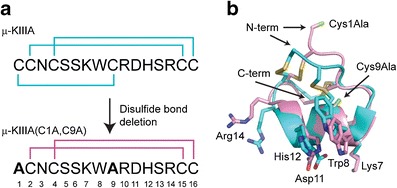
Cysteine editing to reduce peptide complexity. a The primary sequence of μ-conotoxin KIIIA (μ-KIIIA) from C. kinoshitai is shown with the native cystine bonding pattern. Cysteines at positions 1 and 9 were replaced with alanine, yielding the μ-KIIIA(C1A,C9A) mutant. b The overlay of μ-KIIIA (PDB 2LXG) and μ-KIIIA(C1A,C9A) (BMRB 20049) indicates only a slight perturbation of the structure with a pairwise RMSD of 0.95 Å. The backbone of μ-KIIIA is shown in teal and the backbone of μ-KIIIA(C1A,C9A) is shown in pink. The side chains of key amino acids responsible for interaction with sodium channels are labeled, with nitrogen colored blue, oxygen in red, and sulfur in yellow. The alanine mutations of μ-KIIIA(C1A,C9A) are highlighted in green
Mimicking disulfide bonds with other amino acid pairs, as opposed to the simple alanine replacement described above, is also an approach used to decrease the complexity of proteins while retaining structural integrity, function, and stability. As cystine is hydrophobic in nature, buried disulfide bonds within a peptide are prime candidates for disulfide bond replacement. This is generally accomplished with amino acids of similar size that are hydrophobic in nature to retain the hydrophobicity of the peptide core. Using a combinatorial approach, bovine pancreatic trypsin inhibitor, a 58-amino acid peptide with six disulfide bonds, was used as a model to develop a screen to identify stable disulfide bond replacement peptide mutants from a large number of sequences (45). The screen involved the analysis of amino acid pairs substituted for the disulfide bond formed between residues Cys14 and Cys38. They found that although no pair of mutations fully compensated for the destabilizing effect of removing the disulfide bond, some of these mutants had midpoint temperatures of thermal unfolding that were 12–17°C higher than those with the simple substitution of cysteine for alanine at both positions. The most favorable mutations involved combinations of C14G and C38V. Thermodynamic analysis showed that the enthalpy of unfolding of the C14G and C38V mutant groups differed considerably, suggesting different stabilizing mechanisms for these two groups. Thus while proper disulfide bond formation is often difficult in large-scale production of proteins, this approach can be employed as a means to improve these processes without considerable impact to structural and functional integrity.
PROTEOLYTIC DEGRADATION AND HYDROLYSIS
Historically, peptides have been considered to be poor therapeutic candidates due to their low oral bioavailability and their propensity to be readily metabolized. Unmodified peptides generally undergo extensive hydrolysis and proteolytic degradation, resulting in short plasma half-lives thereby impacting their utility as therapeutic agents. Therefore, when first exploring a peptide as a potential therapeutic, it is prudent to have some idea of how the peptide is eliminated in vivo whenever possible so that chemical modifications can be made if the peptide is rapidly cleared or function is lost. Unusually rapid clearance would suggest degradation by chemical or enzymatic means, and this hydrolysis is a key hurdle to overcome when discovering peptide therapeutics. Microbial contamination of a highly purified peptide preparation can often lead to loss of biological activity by proteolysis, particularly during long-term storage. In addition, contaminating proteases can cleave peptides but can be mitigated with the use of metal chelators such as EGTA/EDTA to inhibit the activity of metalloproteases: N-ethyl maleimide/iodoacetamide for sulfhydryl proteases and aprotonin/phenylmethanesulfonic acid/leupeptin for serine proteases. One must be careful that the chosen inhibitor does not interact adversely with the peptide therapeutic and is also amenable to human therapeutic formulations. In addition to enzymatic degradation, peptide bonds can also be cleaved spontaneously under physiological conditions when particularly labile sequence motifs are present. As with amino acid PTMs, these risks can potentially be removed through careful primary sequence analysis.
Amino Acid-Mediated Chemical Hydrolysis
Non-enzymatic degradation generally proceeds by the hydrolysis of peptide bonds between susceptible motifs within the peptide primary sequence. Work done on recombinant human macrophage colony-stimulating factor demonstrated that backbone degradation can occur under both acidic and alkaline conditions with distinctly different degradation product profiles (46). Under alkaline conditions, degradation proceeded via parallel cleavage and intramolecular cross-linking reactions, and a β-elimination mechanism was proposed to account for the degradation at high pH.
Analogous to the deamidation reactions described above, succinimide formation at Asn residues can result in spontaneous cleavage of peptides. In this case, the side chain amide nitrogen attacks the peptide bond to form a C-terminal succinimide residue and a newly formed amino terminus (47,48). Furthermore, preferential hydrolysis of peptide bonds at Asp occurs at the C-terminal side in peptides under acidic conditions (Table I). The carboxyl side chain of Asp initiates the cleavage by acting as a proton donor at pH values below the pKa of the carboxyl group (49). Peptide bonds comprised of Asp-Pro and Asp-Gly were shown to be the most susceptible to peptide backbone hydrolysis under acidic conditions (46,50–52). The mechanism of this cleavage is thought to proceed via intramolecular catalysis by carboxylate anion-mediated displacement of the protonated secondary nitrogen of the proline peptide bond, which has a greater basicity and is therefore preferentially protonated relative to other peptide backbone nitrogen residues. When analyzing the primary sequence of a potential peptide therapeutic, Asp-Pro and Asp-Gly motifs may cause downstream developability issues if the final formulation tends to be one of lower pH.
Enzymatic Hydrolysis
Proteolytic degradation is a natural means of clearing endogenous peptides. Aminopeptidases, endopeptidases, and carboxypeptidases all contribute to the degradation and clearance of peptides in vivo. Potential liabilities based on protease recognition motifs within a peptide primary sequence allows one to take a simple replacement approach, using naturally or non-naturally occurring amino acids, to engineer a more protease resistant peptide sequence.
Octreotide, a synthetic octapeptide analogue of somatostatin, is one example of this approach (50). First synthesized in 1979 by the chemist Wilfried Bauer, octreotide mimics the pharmacology of somatostatin but is a more potent inhibitor of growth hormone, insulin, and glucagon than native somatostatin. Experiments demonstrated that not only did this analogue have increased potency both in vitro and in vivo, but was also much longer lasting. Octreotide has a D-Phe residue at position 1 and a D-Trp residue at position 4 resulting in a molecule that has greater potency and a 50-fold increase in half-life as compared to the natural hormone (Fig. 3a) (51). The latter effect was attributed to a reduction in proteolytic degradation resulting in an increased functional lifetime in vivo.
Fig. 3.
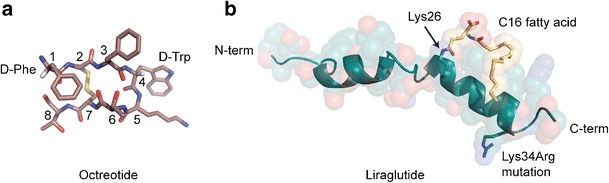
Methods for avoiding proteolytic degradation. a Octreotide contains two D-amino acids, which cannot be recognized by endogenous peptidases: D-Phe at position 1 and D-Trp at position 4 (PDB 1SOC). Carbon is colored pink, nitrogen in blue, oxygen in red, sulfur in yellow, and the Cα hydrogens of the D-amino acids are in white. b Liraglutide contains a C16 fatty acid chain added to Lys26, providing a physical barrier to enzymatic degradation. Additionally, a Lys34Arg mutation removes a protease recognition site (PDB 4APD). Amino acid carbons are colored teal and the C16 fatty acid carbons are colored yellow. Nitrogen is colored in blue and oxygen is red
Liraglutide is a peptide that shares 97% sequence identity with native GLP-1 (7-37). GLP-1 (7-37) has a half-life of approximately 2 min due to proteolytic degradation by the ubiquitous endogenous enzymes neutral endopeptidase (NEP) and dipeptidyl peptidase IV (DPP-IV). An R34K mutation and the addition of a C16 fatty acid chain at position 26 resulted in a molecule that is stable to degradation by both enzymes with a 14-h half-life, as compared to 1.2 h for GLP-1 (Fig. 3b) (53).
When peptide degradation occurs through the action of aminopeptidases or carboxypeptidases, plasma stability can be increased by blocking N- or C-terminal ends by N-acylation, N-pyroglutamate, C-amidation, or by the addition of carbohydrate. These methods can not only mask unwanted charge when the potential therapeutic peptide is an extraction from a larger protein but also protect against degradative exopeptidases.
Tesamorelin is a growth hormone-releasing hormone analogue (GHRH) that was approved for the reduction of excessive abdominal fat in HIV-infected patients with lipodystrophy (54). Initial studies supported the therapeutic potential for GHRH for treating visceral adiposity. However, the pharmacokinetic properties of GHRH did not warrant further development, partly due to its DPP-IV-mediated rapid clearance. Synthetic analogues of GHRH were explored to address this, leading to the development of TH9507 (tesamorelin) where the addition of a hexenoyl moiety attached to the amino terminus resulted in a molecule found to be resistant to DPP-IV cleavage (55).
As removing disulfide bonds from a peptide reduces the complexity of a peptide, from a production point of view, the incorporation of disulfide bonds, or disulfide bond “surrogates,” can increase the stability of the peptide, reduce conformational heterogeneity, and reduce susceptibility to proteolytic degradation of peptide therapeutics. Several examples exist where disulfide bonded peptides have increased stability to proteolysis and are stable enough for oral delivery including linaclotide, cyclosporine, and desmopressin (56–58).
Recent advances in organometallic catalyst design have opened the door for the use of hydrocarbon bridges to either cross-link side chains of specific residues or mimic intramolecular hydrogen bonds or disulfide bonds with carbon-carbon bonds. This approach uses the olefin metathesis reaction to create hydrocarbon stapled or hydrogen bond surrogate molecules that have increased resistance to proteolytic degradation since proteases generally bind their substrates in their extended, or relaxed, conformation (53,59).
Finally, molecule conjugation has proven to be a means that not only extends the half-life of a peptide but can also protect a peptide from proteolytic degradation. Polyethylene glycol (PEG) polymers have become widely used to improve the pharmacokinetics of therapeutic peptides by improving half-life and reducing the susceptibility to proteolytic degradation resulting in many commercial therapeutic peptide products (60–62). However, there are concerns about the toxicological effects (i.e., renal toxicity), the metabolic fate of the PEG moiety and immunogenicity (63,64). The latter having the opposite intended effect in that induced anti-PEG antibodies have been linked to enhanced blood clearance and reduced efficacy of the therapeutic. Fusions to the Fc domain of human gamma immunoglobulin (IgG) are another approach to increasing half-life that may also provide the advantage of protease protection. In fact, the proteolytic inactivation of GLP-1 by DPP-IV is reduced up to fivefold in a GLP-1/Fc fusion format (65). XTEN is another recombinant fusion partner that has been shown to increase the circulating half-life of GLP-2 while also providing protection from proteolytic degradation by DPP-IV (66). This is a new method of half-life extension with several important advantages over PEGylation including better biodegradability and lower immunogenicity (67).
MANUFACTURING PROCESSES
The size and nature of the therapeutic peptide often determines the most suitable manufacturing technology. Although there are many technologies to choose from, this review will touch on three main technologies: chemical synthesis, recombinant expression systems, and cell-free expression systems. With each technology comes certain advantages, as well as disadvantages.
Chemical Peptide Synthesis
Hundreds of synthetic therapeutic peptides are in clinical development today, and this method is especially suited for peptides up to 100 amino acids and less so for peptide fusions. There are even more examples of chemically synthesized peptides in advanced stages of preclinical development in numerous pharmaceutical companies. Chemical synthesis affords the use of unnatural and D-amino acids and pseudo-peptide bonds, providing a much broader chemical diversity than peptides derived from recombinant expression technologies, as well as the potential for unique intellectual space. Substitution of natural amino acids using this approach can also greatly reduce the potential for proteolytic cleavage resulting in greater plasma stability (3). Also, positional scanning synthetic combinatorial libraries (PS-SCL) represent a clear advance in the drug discovery process by allowing all possible combinations of building blocks to be incorporated at defined positions (individual SCLs) within the peptide backbone during synthesis, greatly reducing the time between library generation and lead candidate selection.
Peptide synthesis has come a long way from the early synthesis work of du Vigneaud (54). Today, techniques have been developed for more facile chemistry addressing coupling efficiencies, incorporation of non-native or D-amino acids, side chain protecting groups to add synthetic versatility and orthogonal blocking groups to aid in disulfide bond formation. This access to diverse chemical space coupled with the advent of automated peptide synthesizers allows peptide drug discovery to move at a much more accelerated rate than in the past. However, as peptide synthesis is a multistep process, the overall peptide yield is greatly impacted by coupling efficiencies. As the length of the peptide increases, poor coupling efficiencies have greater impact on the final yield of the pure peptide.
Recombinant Expression
When directed disulfide bond formation, non-native or D-amino acids, or specific termini blocking strategies are not required, a recombinant method can be the best manufacturing choice especially when the attachment of a fusion partner can be added directly to the N- or C-termini (HSA, Fc, XTEN, etc.). Production systems include transgenic plants and animals, bacteria, yeast, insect cells, and mammalian cells. The complexity, or lack thereof, of the peptide therapeutic drives the choice of production systems. There are already a number of peptide therapeutics manufactured by recombinant technologies commercially available including nesiritide, teriparatide, and salmon calcitonin (3).
Cell-Free Systems
Cell-free (or in vitro) translation technologies provide unique advantages over traditional cell-based methods for biotherapeutic discovery (68). These systems comprise all of the necessary components for protein translation and are typically derived from cell extracts of Escherichia coli S30, rabbit reticulocytes, or wheat germ, although Leischmanii, Thermus, and even human cell-free systems have recently been described. Without the limitations of cell walls or membranes, precise modulation of protein expression may be achieved by addition of exogenous factors, such as chaperones, isomerases, or posttranslational-modifying enzymes, to manipulate translation and folding. Furthermore, these systems eliminate cell viability constraints and allow more efficient use of reactor volume (69). Of particular note, the Protein synthesis Using Recombinant Elements (PURE) system, which is a reconstituted system of highly purified components, provides exquisite control for adding or subtracting components tailored specifically to production of the protein or peptide of interest. The PURE system has additionally enabled reprogramming of the genetic code by reassigning stop codons, four-base codons, or sense codons to direct the incorporation of non-canonical amino acids, which can impart drug-like properties and avoid the aforementioned PTM risks (70).
Until recently, cell-free systems have generally been used in small-scale peptide and protein preparations, with scale-up resulting in batch-to-batch variability and improperly folded protein. However, recent improvements have demonstrated this system to be linearly scalable under cGMP manufacturing processes, highlighting its potential to be an enabling technology at the industrial scale. For example, using batch processes in standard bioreactors, a biologically active and correctly disulfide-bonded human cytokine, granulocyte-macrophage colony-stimulating factor (GM-CSF), a 127 amino acid protein, was produced from a 100-L batch at titers of 700 mg/L in 10 h (71). Although there are currently only limited examples of biotherapeutic peptides produced using scaled-up cell-free methods, this versatile technology provides an interesting alternative when peptide generation is inaccessible by more traditional methods.
CONCLUSIONS
Peptides have unique niches for targeting proteins that have a paucity of small molecule and large molecule biologics therapeutics. Despite lacking a definitive tertiary structure and being conformationally diverse in solution, peptides have the potential of adapting to the relevant conformations required to generate a desired pharmacological response. However, a challenge for peptide therapeutics can be the assessment of manufacturing, stability, and safety. Developability assessment can be used to identify the risks that can be incorporated into the assignment of critical quality attributes early into the design of peptide therapeutics. Because of the absence of a definitive structure, such risks are often linked to the chemical composition of peptides whether they are derived recombinantly or via chemical synthesis. In both cases, most of the risks come from the non-enzymatic modification of the amino acids in the peptides. After lead selection, the candidates should be assessed for potential modifications in labile amino acids (Asp, Asn, Cys, Met, Trp). During candidate optimization, confirmation of the primary sequence is critical in establishing structure-function relationships for the scale-up process. Any engineering should be seen as an iterative process where in vitro and/or in vivo data not only drives an engineering path but also confirms that biological activity and stability are retained in conjunction with other engineered attributes. When liabilities cannot be engineered out, understanding the stability of these labile residues is critical for systematic development of the scale-up processes for peptide therapeutics. Nonetheless, there are still several hurdles to overcome: development of low-cost synthesis and purification; modifications that enhance pharmacokinetics and stability when introduced into the patient; control of metabolism and excretion to optimize the appropriate half-life to achieve therapeutic efficacy; and formulation to optimize their delivery and transport. Continual development on these points will foster better opportunities in expanding the potential of peptide therapeutics.
REFERENCES
- 1.Diao L, Meibohm B. Pharmacokinetics and pharmacokinetic-pharmacodynamic correlations of therapeutic peptides. Clin Pharmacokinet. 2013;52(10):855–68. doi: 10.1007/s40262-013-0079-0. [DOI] [PubMed] [Google Scholar]
- 2.Bruckdorfer T, Marder O, Albericio F. From production of peptides in milligram amounts for research to multi-tons quantities for drugs of the future. Curr Pharm Biotechnol. 2004;5(1):29–43. doi: 10.2174/1389201043489620. [DOI] [PubMed] [Google Scholar]
- 3.Vlieghe P, et al. Synthetic therapeutic peptides: science and market. Drug Discov Today. 2010;15(1–2):40–56. doi: 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 4.Adessi C, Soto C. Converting a peptide into a drug: strategies to improve stability and bioavailability. Curr Med Chem. 2002;9(9):963–78. doi: 10.2174/0929867024606731. [DOI] [PubMed] [Google Scholar]
- 5.Witt KA, et al. Peptide drug modifications to enhance bioavailability and blood–brain barrier permeability. Peptides. 2001;22(12):2329–43. doi: 10.1016/S0196-9781(01)00537-X. [DOI] [PubMed] [Google Scholar]
- 6.Renukuntla J, et al. Approaches for enhancing oral bioavailability of peptides and proteins. Int J Pharm. 2013;447(1–2):75–93. doi: 10.1016/j.ijpharm.2013.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sato AK, et al. Therapeutic peptides: technological advances driving peptides into development. Curr Opin Biotechnol. 2006;17(6):638–42. doi: 10.1016/j.copbio.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 8.Latham PW. Therapeutic peptides revisited. Nat Biotechnol. 1999;17(8):755–7. doi: 10.1038/11686. [DOI] [PubMed] [Google Scholar]
- 9.Luckett S, et al. High-resolution structure of a potent, cyclic proteinase inhibitor from sunflower seeds. J Mol Biol. 1999;290(2):525–33. doi: 10.1006/jmbi.1999.2891. [DOI] [PubMed] [Google Scholar]
- 10.Kalman K, et al. ShK-Dap22, a potent Kv1.3-specific immunosuppressive polypeptide. J Biol Chem. 1998;273(49):32697–707. doi: 10.1074/jbc.273.49.32697. [DOI] [PubMed] [Google Scholar]
- 11.Rauer H, et al. Structural conservation of the pores of calcium-activated and voltage-gated potassium channels determined by a sea anemone toxin. J Biol Chem. 1999;274(31):21885–92. doi: 10.1074/jbc.274.31.21885. [DOI] [PubMed] [Google Scholar]
- 12.Pennington MW, et al. Role of disulfide bonds in the structure and potassium channel blocking activity of ShK toxin. Biochemistry. 1999;38(44):14549–58. doi: 10.1021/bi991282m. [DOI] [PubMed] [Google Scholar]
- 13.Pennington MW, et al. Engineering a stable and selective peptide blocker of the Kv1.3 channel in T lymphocytes. Mol Pharmacol. 2009;75(4):762–73. doi: 10.1124/mol.108.052704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grace CR, et al. Common and divergent structural features of a series of corticotropin releasing factor-related peptides. J Am Chem Soc. 2007;129(51):16102–14. doi: 10.1021/ja0760933. [DOI] [PubMed] [Google Scholar]
- 15.Fasano A. Innovative strategies for the oral delivery of drugs and peptides. Trends Biotechnol. 1998;16(4):152–7. doi: 10.1016/S0167-7799(97)01170-0. [DOI] [PubMed] [Google Scholar]
- 16.Patel MM, et al. Getting into the brain: approaches to enhance brain drug delivery. CNS Drugs. 2009;23(1):35–58. doi: 10.2165/0023210-200923010-00003. [DOI] [PubMed] [Google Scholar]
- 17.Lalatsa A, Schatzlein AG, Uchegbu IF. Strategies to deliver peptide drugs to the brain. Mol Pharm. 2014;11(4):1081–93. doi: 10.1021/mp400680d. [DOI] [PubMed] [Google Scholar]
- 18.Geiger T, Clarke S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J Biol Chem. 1987;262(2):785–94. [PubMed] [Google Scholar]
- 19.Aswad DW. Deamidation and isoaspartate formation in peptides and proteins. In: Aswad DW, editor. CRC series in analytical Biotechnology. Boca Raton: CRC; 1994. [Google Scholar]
- 20.Stephenson RC, Clarke S. Succinimide formation from aspartyl and asparaginyl peptides as a model for the spontaneous degradation of proteins. J Biol Chem. 1989;264(11):6164–70. [PubMed] [Google Scholar]
- 21.Di Donato A, Galletti P, D’Alessio G. Selective deamidation and enzymatic methylation of seminal ribonuclease. Biochemistry. 1986;25(26):8361–8. doi: 10.1021/bi00374a005. [DOI] [PubMed] [Google Scholar]
- 22.Di Donato A, et al. Selective deamidation of ribonuclease A. Isolation and characterization of the resulting isoaspartyl and aspartyl derivatives. J Biol Chem. 1993;268(7):4745–51. [PubMed] [Google Scholar]
- 23.Johnson BA, Aswad DW. Fragmentation of isoaspartyl peptides and proteins by carboxypeptidase Y: release of isoaspartyl dipeptides as a result of internal and external cleavage. Biochemistry. 1990;29(18):4373–80. doi: 10.1021/bi00470a017. [DOI] [PubMed] [Google Scholar]
- 24.Blomback B. Derivatives of glutamine in peptides. Methods Enzymol. 1967;11:398–411. doi: 10.1016/S0076-6879(67)11046-X. [DOI] [Google Scholar]
- 25.Dick LW, Jr, et al. Determination of the origin of the N-terminal pyro-glutamate variation in monoclonal antibodies using model peptides. Biotechnol Bioeng. 2007;97(3):544–53. doi: 10.1002/bit.21260. [DOI] [PubMed] [Google Scholar]
- 26.Dunkelberger EB, et al. Deamidation accelerates amyloid formation and alters amylin fiber structure. J Am Chem Soc. 2012;134(30):12658–67. doi: 10.1021/ja3039486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ni W, et al. Analysis of isoaspartic acid by selective proteolysis with Asp-N and electron transfer dissociation mass spectrometry. Anal Chem. 2010;82(17):7485–91. doi: 10.1021/ac101806e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stadtman ER. Oxidation of free amino acids and amino acid residues in proteins by radiolysis and by metal-catalyzed reactions. Annu Rev Biochem. 1993;62:797–821. doi: 10.1146/annurev.bi.62.070193.004053. [DOI] [PubMed] [Google Scholar]
- 29.Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem. 1997;272(33):20313–6. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- 30.Brot N, Weissbach H. Biochemistry and physiological role of methionine sulfoxide residues in proteins. Arch Biochem Biophys. 1983;223(1):271–81. doi: 10.1016/0003-9861(83)90592-1. [DOI] [PubMed] [Google Scholar]
- 31.Swaim MW, Pizzo SV. Methionine sulfoxide and the oxidative regulation of plasma proteinase inhibitors. J Leukoc Biol. 1988;43(4):365–79. doi: 10.1002/jlb.43.4.365. [DOI] [PubMed] [Google Scholar]
- 32.Schechter Y. Selective oxidation and reduction of methionine residues in peptides and proteins by oxygen exchange between sulfoxide and sulfide. J Biol Chem. 1986;261:66–70. [PubMed] [Google Scholar]
- 33.Hiller KO, Asmus KD. Oxidation of methionine by X2.- in aqueous solution and characterization of some S therefore X three-electron bonded intermediates. A pulse radiolysis study. Int J Radiat Biol Relat Stud Phys Chem Med. 1981;40(6):583–95. doi: 10.1080/09553008114551571. [DOI] [PubMed] [Google Scholar]
- 34.Keck RG. The use of t-butyl hydroperoxide as a probe for methionine oxidation in proteins. Anal Biochem. 1996;236(1):56–62. doi: 10.1006/abio.1996.0131. [DOI] [PubMed] [Google Scholar]
- 35.Creed D. The photophysics and photochemistry of the near-uv absorbing amino acids—I. tryptophan and its simple derivatives. Photochem Photobiol. 1984;39(4):537–62. doi: 10.1111/j.1751-1097.1984.tb03890.x. [DOI] [Google Scholar]
- 36.M’Barek S, et al. Synthesis and characterization of Pi4, a scorpion toxin from Pandinus imperator that acts on K+ channels. Eur J Biochem. 2003;270(17):3583–92. doi: 10.1046/j.1432-1033.2003.03743.x. [DOI] [PubMed] [Google Scholar]
- 37.Benham CJ, Jafri MS. Disulfide bonding patterns and protein topologies. Protein Sci. 1993;2(1):41–54. doi: 10.1002/pro.5560020105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luo S, et al. Characterization of a novel alpha-conotoxin from conus textile that selectively targets alpha6/alpha3beta2beta3 nicotinic acetylcholine receptors. J Biol Chem. 2013;288(2):894–902. doi: 10.1074/jbc.M112.427898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kutchukian PS, et al. All-atom model for stabilization of alpha-helical structure in peptides by hydrocarbon staples. J Am Chem Soc. 2009;131(13):4622–7. doi: 10.1021/ja805037p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barnham KJ, et al. Role of the 6-20 disulfide bridge in the structure and activity of epidermal growth factor. Protein Sci. 1998;7(8):1738–49. doi: 10.1002/pro.5560070808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Flinn JP, et al. Role of disulfide bridges in the folding, structure and biological activity of omega-conotoxin GVIA. Biochim Biophys Acta. 1999;1434(1):177–90. doi: 10.1016/S0167-4838(99)00165-X. [DOI] [PubMed] [Google Scholar]
- 42.Carrega L, et al. The impact of the fourth disulfide bridge in scorpion toxins of the alpha-KTx6 subfamily. Proteins. 2005;61(4):1010–23. doi: 10.1002/prot.20681. [DOI] [PubMed] [Google Scholar]
- 43.Han TS, et al. Structurally minimized mu-conotoxin analogues as sodium channel blockers: implications for designing conopeptide-based therapeutics. ChemMedChem. 2009;4(3):406–14. doi: 10.1002/cmdc.200800292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khoo KK, et al. Structure of the analgesic mu-conotoxin KIIIA and effects on the structure and function of disulfide deletion. Biochemistry. 2009;48(6):1210–9. doi: 10.1021/bi801998a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hagihara Y, et al. Screening for stable mutants with amino acid pairs substituted for the disulfide bond between residues 14 and 38 of bovine pancreatic trypsin inhibitor (BPTI) J Biol Chem. 2002;277(52):51043–8. doi: 10.1074/jbc.M208893200. [DOI] [PubMed] [Google Scholar]
- 46.Schrier JA, et al. Degradation pathways for recombinant human macrophage colony-stimulating factor in aqueous solution. Pharm Res. 1993;10(7):933–44. doi: 10.1023/A:1018990001310. [DOI] [PubMed] [Google Scholar]
- 47.Clarke S, Stephenson RC, Lowenson JD. In: Stability of protein pharmaceuticals. Part A—chemical and physical pathways of protein degradation. Ahern TJ, Manning MC, editors. New York: Plenum; 1992. [Google Scholar]
- 48.Tyler-Cross R, Schirch V. Effects of amino acid sequence, buffers, and ionic strength on the rate and mechanism of deamidation of asparagine residues in small peptides. J Biol Chem. 1991;266(33):22549–56. [PubMed] [Google Scholar]
- 49.Inglis AS. Cleavage at aspartic acid. Methods Enzymol. 1983;91:324–32. doi: 10.1016/S0076-6879(83)91030-3. [DOI] [PubMed] [Google Scholar]
- 50.Bauer W, et al. SMS 201-995: a very potent and selective octapeptide analogue of somatostatin with prolonged action. Life Sci. 1982;31(11):1133–40. doi: 10.1016/0024-3205(82)90087-X. [DOI] [PubMed] [Google Scholar]
- 51.Maurer R, et al. Opiate antagonistic properties of an octapeptide somatostatin analog. Proc Natl Acad Sci U S A. 1982;79(15):4815–7. doi: 10.1073/pnas.79.15.4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oliyai C, Borchardt RT. Chemical pathways of peptide degradation. IV. Pathways, kinetics, and mechanism of degradation of an aspartyl residue in a model hexapeptide. Pharm Res. 1993;10(1):95–102. doi: 10.1023/A:1018981231468. [DOI] [PubMed] [Google Scholar]
- 53.Larsen PJ, et al. Systemic administration of the long-acting GLP-1 derivative NN2211 induces lasting and reversible weight loss in both normal and obese rats. Diabetes. 2001;50(11):2530–9. doi: 10.2337/diabetes.50.11.2530. [DOI] [PubMed] [Google Scholar]
- 54.Wang Y, Tomlinson B. Tesamorelin, a human growth hormone releasing factor analogue. Expert Opin Investig Drugs. 2009;18(3):303–10. doi: 10.1517/13543780802707658. [DOI] [PubMed] [Google Scholar]
- 55.Ferdinandi ES, et al. Non-clinical pharmacology and safety evaluation of TH9507, a human growth hormone-releasing factor analogue. Basic Clin Pharmacol Toxicol. 2007;100(1):49–58. doi: 10.1111/j.1742-7843.2007.00008.x. [DOI] [PubMed] [Google Scholar]
- 56.Busby RW, et al. Pharmacologic properties, metabolism, and disposition of linaclotide, a novel therapeutic peptide approved for the treatment of irritable bowel syndrome with constipation and chronic idiopathic constipation. J Pharmacol Exp Ther. 2013;344(1):196–206. doi: 10.1124/jpet.112.199430. [DOI] [PubMed] [Google Scholar]
- 57.Italia JL, et al. PLGA nanoparticles for oral delivery of cyclosporine: nephrotoxicity and pharmacokinetic studies in comparison to Sandimmune Neoral. J Control Release. 2007;119(2):197–206. doi: 10.1016/j.jconrel.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 58.Ilan E, et al. Improved oral delivery of desmopressin via a novel vehicle: mucoadhesive submicron emulsion. Pharm Res. 1996;13(7):1083–7. doi: 10.1023/A:1016023111248. [DOI] [PubMed] [Google Scholar]
- 59.Henchey LK, Jochim AL, Arora PS. Contemporary strategies for the stabilization of peptides in the alpha-helical conformation. Curr Opin Chem Biol. 2008;12(6):692–7. doi: 10.1016/j.cbpa.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Veronese FM, Pasut G. PEGylation, successful approach to drug delivery. Drug Discov Today. 2005;10(21):1451–8. doi: 10.1016/S1359-6446(05)03575-0. [DOI] [PubMed] [Google Scholar]
- 61.Yang BB, et al. Polyethylene glycol modification of filgrastim results in decreased renal clearance of the protein in rats. J Pharm Sci. 2004;93(5):1367–73. doi: 10.1002/jps.20024. [DOI] [PubMed] [Google Scholar]
- 62.Webster R, et al. PEGylated proteins: evaluation of their safety in the absence of definitive metabolism studies. Drug Metab Dispos. 2007;35(1):9–16. doi: 10.1124/dmd.106.012419. [DOI] [PubMed] [Google Scholar]
- 63.Rudmann DG, et al. High molecular weight polyethylene glycol cellular distribution and PEG-associated cytoplasmic vacuolation is molecular weight dependent and does not require conjugation to proteins. Toxicol Pathol. 2013;41(7):970–83. doi: 10.1177/0192623312474726. [DOI] [PubMed] [Google Scholar]
- 64.Schellekens H, Hennink WE, Brinks V. The immunogenicity of polyethylene glycol: facts and fiction. Pharm Res. 2013;30(7):1729–34. doi: 10.1007/s11095-013-1067-7. [DOI] [PubMed] [Google Scholar]
- 65.Kim DM, et al. Fc fusion to glucagon-like peptide-1 inhibits degradation by human DPP-IV, increasing its half-life in serum and inducing a potent activity for human GLP-1 receptor activation. BMB Rep. 2009;42(4):212–6. doi: 10.5483/BMBRep.2009.42.4.212. [DOI] [PubMed] [Google Scholar]
- 66.Alters SE, et al. GLP2-2G-XTEN: a pharmaceutical protein with improved serum half-life and efficacy in a rat Crohn’s disease model. PLoS One. 2012;7(11):e50630. doi: 10.1371/journal.pone.0050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haeckel A, et al. XTEN-annexin A5: XTEN allows complete expression of long-circulating protein-based imaging probes as recombinant alternative to PEGylation. J Nucl Med. 2014;55(3):508–14. doi: 10.2967/jnumed.113.128108. [DOI] [PubMed] [Google Scholar]
- 68.Murray CJ, Baliga R. Cell-free translation of peptides and proteins: from high throughput screening to clinical production. Curr Opin Chem Biol. 2013;17(3):420–6. doi: 10.1016/j.cbpa.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 69.Harris DC, Jewett MC. Cell-free biology: exploiting the interface between synthetic biology and synthetic chemistry. Curr Opin Biotechnol. 2012;23(5):672–8. doi: 10.1016/j.copbio.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hipolito CJ, Suga H. Ribosomal production and in vitro selection of natural product-like peptidomimetics: the FIT and RaPID systems. Curr Opin Chem Biol. 2012;16(1–2):196–203. doi: 10.1016/j.cbpa.2012.02.014. [DOI] [PubMed] [Google Scholar]
- 71.Zawada JF, et al. Microscale to manufacturing scale-up of cell-free cytokine production—a new approach for shortening protein production development timelines. Biotechnol Bioeng. 2011;108(7):1570–8. doi: 10.1002/bit.23103. [DOI] [PMC free article] [PubMed] [Google Scholar]