

Abstract
We reported that suramin is an effective chemosensitizer at noncytotoxic concentrations (<50 μM); this effect was observed in multiple types of human xenograft tumors in vitro and in vivo. Clinical evaluation of noncytotoxic suramin is ongoing. Because (a) suramin inhibits reverse transcriptase, (b) telomerase is a reverse transcriptase, and (c) inhibition of telomerase enhances tumor chemosensitivity, we studied the pharmacodynamics of noncytotoxic suramin on telomerase activity and telomere length in cultured cells and tumors grown in animals. In three human cancer cells that depend on telomerase for telomere maintenance (pharynx FaDu, prostate PC3, breast MCF7), suramin inhibited telomerase activity in cell extracts and intact cells at concentrations that exhibited no cytotoxicity (IC50 of telomerase was between 1 and 3 μM vs. >60 μM for cytotoxicity), and continuous treatment at 10–25 μM for 6 weeks resulted in gradual telomere shortening (maximum of 30%) and cell senescence (measured by β-galactosidase activity and elevation of mRNA levels of two senescence markers p16 and p21). In contrast, noncytotoxic suramin did not shorten the telomere in telomerase-independent human osteosarcoma Saos-2 cells. In mice bearing FaDu tumors, treatment with noncytotoxic suramin for 6 weeks resulted in telomere erosion in >95% of the tumor cells with an average telomere shortening of >40%. These results indicate noncytotoxic suramin inhibits telomerase, shortens telomere and induces cell senescence, and suggest telomerase inhibition as a potential mechanism of its chemosensitization.
Key words: noncytotoxic suramin, pharmacodynamics, senescence, telomerase, telomere
INTRODUCTION
Telomeres are specific DNA structures at the ends of chromosomes that protect chromosomes from end-to-end fusion, maintain chromosome integrity, reversibly repress transcription of neighboring genes, and play a role in chromosome positioning in the nucleus [1]. Due to the inability to replicate the 3′ end of chromosomes by DNA polymerases, telomeres shorten by 50 to 200 bp per cell division in normal somatic cells. Shortened telomeres triggers replicative and accelerated senescence and induces apoptosis [2–9], whereas telomere elongation in human fibroblast IDH4 and prostate tumor DU145 cells resulted in resistance to apoptosis caused by serum depletion [10]. Senescent cells show changes in gene expression of known cell-cycle inhibitors or activators, including the cyclin-dependent kinase inhibitors p21 and p16, which are components of the p53 and pRB tumor suppressor signaling cascades [11].
Telomerase, a ribonucleoprotein, functions as a reverse transcriptase to synthesize telomeric repeats de novo [12]. Telomerase is present in nearly all immortal cell lines, germ-line cells, stem cells and about 90% of human tumors, but is seldom present in normal somatic cells [13, 14]. The selective expression of telomerase in tumor cells makes telomerase an attractive therapeutic target, and several agents, including an oligonucleotide targeting the active site of telomerase and several immunotherapeutics against telomerase peptide fragments, have been in clinical trials [14].
We and others have shown telomerase inhibition and telomere shortening enhance the chemosensitivity of tumors that depend on telomerase for telomere maintenance [8, 15, 16]. For example, telomerase inhibitors (e.g., antisense to telomerase’s RNA component, reverse transcriptase inhibitor 3′-azido-3′-deoxythymidine which inhibits telomerase’s protein component hTERT) inhibit telomerase activity, shorten telomere length, reduce cell growth rate, and significantly enhance tumor sensitivity to conventional cytotoxics (e.g., paclitaxel in human pharynx FaDu tumor cells and xenograft tumors, cisplatin in cisplatin-resistant human glioblastoma U251-MG cells, and agents that induce double-stranded DNA breaks and cause telomere fragmentation). In contrast, telomerase inhibitors had no activity in telomerase-independent osteosarcoma Saos-2 cells.
Suramin, a polysulfonated naphthylurea, was first discovered in 1916. The emergence of the AIDS epidemic in early 1980s led to the discovery that suramin is a reverse transcriptase inhibitor. Suramin was tested in AIDS patients and later abandoned due to life-threatening toxicities. Nonetheless, these trials revealed its antitumor properties. Subsequent studies showed that suramin inhibits multiple growth factors. This, in turn, sparked considerable interests and efforts in developing it as a conventional cytotoxic agent [17]. In mouse tumor models [18–20], antitumor activity of suramin was observed at doses between 100 and 260 mg/kg per week with corresponding plasma concentrations exceeding 100 μM. Clinically, suramin was evaluated in a wide variety of solid tumors, either as single agent or in combination with other chemotherapeutics, and showed modest activity at plasma concentrations of 100–250 μM; these cytotoxic concentrations resulted in significant toxicities (summarized in 21). Our laboratory has since discovered that suramin, at noncytotoxic doses that yield five to ten times lower plasma concentrations between 10 and 50 μM and no antitumor activity as a single agent, significantly enhances the efficacy of chemotherapeutics in primary or metastatic human prostate, breast, lung, and colon xenograft tumors in immunodeficient mice [21–25]. These findings have led to several phase I/II clinical trials, including an ongoing randomized trial, of combinations of noncytotoxic suramin with conventional chemotherapeutics [26–35].
The mechanisms of suramin’s in vivo chemosensitization effect are unclear, since it has multiple pharmacological activities (summarized in 21). Its actions are highly concentration-dependent; the targets that are inhibited by >50 μM extracellular suramin include IL-2, insulin growth factor-1, tumor necrosis factor β, and topoisomerase II; the targets that are inhibited by <50 μM extracellular suramin include fibroblast growth factors, reverse transcriptase, protein kinase C, and RNA polymerase. With respect to telomerase, two earlier studies show that inhibition by suramin occurs at high cytotoxic concentrations of ≥200 μM in intact C6 rat glioma cells and human osteosarcoma cells (24–96 h treatment) [36, 37]. As these concentrations are several times higher compared with the levels required for chemosensitization, it is unclear if telomerase inhibition contributes to suramin chemosensitization. The present study investigated the pharmacodynamics of noncytotoxic suramin on telomerase activity and telomere maintenance in vitro and in vivo. The results show noncytotoxic suramin (10–50 μM) was effective in inhibiting telomerase, shortening telomeres, and inducing cell senescence in multiple human cancer cells that depend on telomerase for telomere maintenance. The onset of senescence was slow, observed after continuous suramin treatment for 6–11 weeks. The clinical implication of the delayed onset senescence is discussed.
MATERIALS AND METHODS
Chemicals and reagents
Suramin, sulforhodamine B, p-phenylenediamine, and BCA kit for protein determination were purchased from Sigma Co. (St. Louis, MO); polynucleotide kinase and DNA molecular weight marker V from Roche Molecular Biochemicals (Indianapolis, IN); cefotaxime sodium from Hoechst-Roussel (Somerville, NJ); gentamicin from Solo Pak Laboratories (Franklin Park, IL); minimum essential medium (MEM), RPMI 1640 medium, fetal bovine serum (FBS), and colcemid from Life Technologies, Inc. (Grand Island, NY); γ-32P-adenosine-5′-triphosphate (ATP) from ICN (Costa Mesa, CA); 3H-suramin sodium from Moravek Biochemicals (Brea, CA); and Advantage cDNA Polymerase Mix from Clontech (Palo Alto, CA). All chemicals and reagents were used as received.
Pharmacodynamics of suramin cytotoxicity
Human cancer cells (FaDu, prostate PC3, breast MCF7, and osteosarcoma Saos-2) were obtained from the American Type Culture Collection (Rockville, MD). Culture media (RPMI1640 for PC3 cells, MEM for FaDu, MCF7, and Saos-2 cells) were supplemented with 9% heat-inactivated FBS, 0.1% 10 mM non-essential amino acids, 2 mM l-glutamine, 90 μg/ml gentamicin, and 90 μg/ml cefotaxime sodium. Cells in exponential growth phase were harvested at ~70% confluence.
The antiproliferative effect of suramin was measured as inhibition of 5-bromodeoxyuridine incorporation using Cell Proliferation ELISA (Roche), and the apoptotic effect was measured by monitoring the cytoplasmic histone-associated DNA fragments (mono- and oligonucleosomes) using Cell Death Detection ELISA (Roche, absorbance measured at 405 nm). The overall cytotoxicity was determined using the sulforhodamine B assay that measures total cellular protein content [14]. The drug concentration causing 50% growth inhibition (IC50) or 50% cells death (DC50) was determined as previously described [38].
Suramin uptake in cells
This was studied in FaDu cells. Briefly, cells were cultured in six-well plates and dosed with a mixture of 3H-suramin (125 nM) and non-radiolabeled suramin (1, 10, or 100 μM). Cells were harvested and washed twice with MEM. The radioactivity in cells and culture media was measured using liquid scintillation and converted to drug concentrations using the corresponding specific activity and the cell volume of 1.7 × 10−6 μl per cell [39, 40].
Telomerase activity in cell lysates
Telomerase activity in cell lysate was measured using a modified nonradioactive telomeric repeat amplification protocol (TRAP), as we previously described [41]. Briefly, cells were lysed in sodium dodecyl sulfate-based lysis buffer and CHAPS-based buffer (1:3 ratio). The resulting cell lysate was added to the reaction mixture (10 mM Tris–HCl pH 8.3, 1.5 mM MgCl2, 50 mM KCl, 0.005% Tween-20, 1 mM EGTA, 50 μM dNTPs, 0.05 μg of TS primer, 0.1 mg/ml bovine serum albumin, and suramin) and incubated at 30°C for 30 min for primer extension, then at 90°C for 2 min to terminate the reaction. The extended TS primer was purified by phenol/chloroform extraction and ethanol precipitation and used as template for the subsequent polymerase chain reaction (PCR) amplification; this purification step eliminates potential inhibitors (including suramin) of Tag DNA polymerase that would introduce artifacts in the PCR results. For example, a pilot study indicated that suramin, at high concentrations (e.g., ten times the telomerase inhibitory concentrations) inhibited Taq DNA polymerase. Negative controls used lysis buffer in place of cell lysates to correct for the background signal. The PCR products were electrophoresed on a 10% polyacrylamide gel and stained with ethidium bromide, and the image was captured by a gel documentation system and analyzed by GPTools software (Gel Print 2000i, Biophotonics, Ann Arbor, MI).
Telomerase activity in intact cells
The telomerase activity in intact or whole cells was evaluated using our previously described intracellular TRAP method [38]. Briefly, suramin-treated and control cells (about 1 × 105) were harvested and washed twice with phosphate-buffered saline (PBS). The cell pellet was resuspended and incubated with reaction mixture (100 μl of serum-free RPMI 1640 containing 5 U/ml of streptolysin O, 2 μM of TS primer, 50 μM of dNTP) at room temperature for 5 min, followed by the addition of 200 μl of RPMI 1640 containing 10% FBS to stop the permeating process. The mixture was incubated at 30°C for 30 min to allow the extension of intracellular TS primer by telomerase. The extended TS primer was then extracted by CHAPS-based lysis buffer and used as template for PCR amplification as described for cell lysate. Negative controls used lysis buffer in place of cells.
Telomere length of cultured cells
The effect of suramin on telomere length in cultured cells was studied in FaDu and PC3 cells that depend on telomerase for telomere maintenance and the telomerase-negative Saos-2 cells that do not depend on telomerase for telomere maintenance [42]. Cells were continuously treated with suramin for 6–11 weeks, with weekly medium change and cell harvesting. We measured both the average telomere length in individual cells and all cells.
Fluorescence in situ hybridization (FISH) was used to measure the telomere signals in individual cells as we previously described [43]. Briefly, cells were treated with colcemid (0.1 μg/ml for 4 h), harvested, treated with hypotonic solution, and fixed with acetic acid and methanol, dropped onto slides, air-dried and stored at −20°C. Cells were denatured at 80°C for 2 min and hybridized to fluorescein-labeled peptide nucleic acid probe (CCCTAA)3 (PerSeptiveBiosystems, Framingham, MA) at room temperature for 2 h. The slides were washed at room temperature with 70% formamide and PBS and the chromosomes counterstained with propidium iodide and examined under a fluorescence microscopy. The digital images were analyzed by Scion Image software (NIH Image for PC).
Two methods were used to measure the mean telomere length in total cells. The first method was the previously described solution hybridization-based method that measures the telomere amount and length (TALA) [43]. Briefly, genomic DNA was isolated and digested at 37°C overnight with HinfI/CfoI/HeaIII. The oligonucleotide probe (TTAGGG)4 was labeled by γ-32P-ATP with polynucleotide T4 kinase and added to DNA solution (3 ng of probe in 2.5 μg DNA). After denaturation at 98°C for 5 min, hybridization was performed at 55°C overnight. The samples were electrophoresed on 0.7% agarose gel. After drying under vacuum without heating, the gel was exposed to phosphor-image screen, and the result was analyzed using the area-under-curve method of the ImageQuaNT software from Molecular Dynamics (Sunnyvale, CA). The point which equally divides the area-under-curve represents the mean telomere length. The second method was the modified monochrome multiplex quantitative PCR method [44]. Briefly, DNA was isolated using DNA isolation kit (Omega BioTek, Norcross, GA) according to the manufacturer’s protocol. Telomere length was measured using real time PCR; albumin was simultaneously amplified with the telomere template to normalize for the amount of DNA per sample. The primer sequences were: Forward 5′ACACTAAGGTTTGGGTTTGGGTTTGGGTTTGGGTTAGTGT-3′ and Reverse 5′- TGTTAGGTATCCCTATCCCTATCCCTATCCCTATCCCTAACA-3′ for telomere; and Forward 5′-CGGCGGCGGGCGGCGCGGGCTGGGCGGAAATGCTGCACAGAATCCTTG-3 and Reverse 5′-GCCCGGCCCGCCGCGCCCGTCCCGCCGGAAAAGCATGGTCGCCTGTT-3′ for albumin.
Telomere length of in vivo tumors
The in vivo suramin effect was evaluated in FaDu xenograft tumors. FaDu cells (0.5 ~ 1 × 106 cell) were implanted subcutaneously in male BALB/c nu/nu mice (National Cancer Institute, Bethesda, MD). Mice received intravenous tail vein injections of 10 mg/kg suramin or normal saline solution immediately after tumor implantation and twice a week thereafter for up to 6 weeks. This suramin dose yields a peak plasma concentration of about 50 μM and a steady-state concentration of 1 μM at 72 h [45] and has no antitumor activity or host toxicity on its own, but significantly enhances the activity of chemotherapy in multiple xenograft tumor models [21–25].
Tumors from suramin- and saline-treated groups were harvested weekly after 2 to 6 weeks of suramin treatments. Due to the presence of mouse stromal cells that have much longer telomere length compared with human tumor cells (>32 vs. 4.3 kbp), we used FISH to determine telomere length in individual cells (frozen tumor sections). About ten fields were randomly chosen from each tumor section, and the percentage of tumor cells with attenuated or lost telomere signals was quantified.
Cell senescence
The above studies measured the average telomere length in all cells. However, it has been shown that telomere dysfunction and senescence are triggered by the shortest telomeres and that a single critically short telomere is sufficient to cause senescence in individual cells [2, 4]. Hence, we measured cell senescence using two additional methods. First, senescent cells were identified by β-galactosidase staining as described previously [46]. Briefly, drug-treated cells were washed with PBS, fixed for 3 min with 2% formaldehyde and 0.2% glutaraldehyde, again washed with PBS, followed by staining at 37°C overnight in a solution containing 1 mg/ml of X-gal (5-bromo-4-chloro-3-indolyl P3-D-galactoside), 5 mM potassium ferricyanide, and 2 mM MgCl2 in PBS, pH 6.0.
The second method was to quantify the mRNA levels of p16 and p21 using quantitative real-time PCR. Total RNA was isolated from FaDu cells using Quick RNA MiniPrep (Zymo Research, Irvine, CA) and reverse-transcribed to cDNA using iScript cDNA Synthesis Kit (Quanta, Gaithersburg, MD). RT-PCR was performed in duplicates using iQ SyBR Green (Biorad) according to the manufacturer’s protocol. Data were standardized to GAPDH mRNA. The primer sequences were: Forward 5′-GGAGTTAATAGCACCTCCTCC-3′ and Reverse 5′-TTCAATCGGGGATGTCTGAGG-3′ for p16; and Forward 5′-GTCAGTTCCTTGTGGAGCCG-3′ and Reverse 5′-GAAGGTAGAGCTTGGGCAGG-3′ for p21.
Immunoblotting of hTERT protein
FaDu cell pellets were collected and lysed with M-PER Mammalian Protein Extraction Reagent with Halt Protease Inhibitor Cocktail (Thermo Scientific, Rockford, IL). Lysates were centrifuged at 14,000 rpm for 5 min, and protein concentrations in the supernatant were measured using the BCA kit (Thermo Scientific). Aliquots containing 15 μg of protein were run on a Bolt™ 4–12% Bis–Tris Plus Gel (Life technologies, Carlsbad, CA) and protein were transferred to a nitrocellulose membrane using the Trans-blot Turbo Transfer System (Biorad, Hercules, CA). Blots were probed with hTERT (1:500, Abcam, Cambridge, MA), and tubulin (1:1000, Cell Signaling, Danvers, MA) primary antibodies followed by horseradish peroxidase-conjugated anti-rabbit secondary antibodies (1:2000, Cell Signaling) using the iBind™ Western System (Life Technologies). Chemiluminescence was detected, and the band intensity was analyzed using the Molecular Imager ChemiDoc XRS (Biorad). Protein levels were normalized to tubulin loading control.
Statistical analysis
Differences in measurements among multiple treatment groups were analyzed by one-way ANOVA with post hoc Tukey testing. For mRNA results, the variances in mRNA expression were heterogeneous between groups and tests were performed on the number of amplification cycles of the real-time PCR determinations.
RESULTS
Cytotoxicity of suramin
Figure 1 shows the dose–response curves of suramin in FaDu, PC3, and MCF7 cells, obtained with three cytotoxicity assays, i.e., overall cytotoxicity (sulforhodamine B assay), antiproliferation effect (5-bromodeoxyuridine incorporation), and cell death (cytoplasmic histone–DNA complex). The results are summarized in Table I. Suramin (96 h treatment) produced concentration-dependent cytotoxicity in all three cells. The IC50 values for overall cytotoxicity in all three cells were comparable to the values for the antiproliferation effect (ranged from 60 to 165 μM) whereas >3 times higher concentrations were required for 50% apoptosis induction (from 190 to >1,000 μM). Short-term exposure for 3 weeks or less to suramin at 25 μM suramin did not result in cytotoxicity, whereas longer exposure resulted in reduction in cell number in all three cell lines (about 10% reduction after 4 weeks and about 30–50% reduction after 6 weeks).
Fig. 1.

Pharmacodynamics of suramin cytotoxicity. FaDu, PC3, and MCF7 cells were treated with suramin for 96 h. The overall cytotoxicity was measured using the sulforhodamine B assay (SRB). The antiproliferation effect was measured as inhibition of 5-bromodeoxyuridine incorporation (BrdU). The apoptotic effect was measured as release of DNA–histone complex to cytoplasm (cell death). Representative dose–response curves are shown. FaDu: solid lines. PC3: broken lines. MCF7: dotted lines. Note the different scales
Table I.
Pharmacodynamics of Suramin Cytotoxicity and Telomerase Inhibition
| IC50 or DC50, μM | CxT50, μM-h | |||||
|---|---|---|---|---|---|---|
| FaDu | MCF7 | PC3 | FaDu | MCF7 | PC3 | |
| Telomerase inhibition in cell lysates | 1.7 ± 1 | 2.8 ± 1.5 | 1.6 ± 0.7 | 42 ± 24 | 67 ± 35 | 38 ± 16 |
| Telomerase inhibition in intact cells | 1.5 ± 0.7 | 1.4 ± 1.1 | 2.3 ± 0.9 | 36 ± 1 | 34 ± 26 | 55 ± 21 |
| Antiproliferative effect | 140 ± 15 | 64 ± 18 | 114 ± 16 | (13.4 ± 1.4) × 103 | (61.4 ± 1.7) × 103 | (10.9 ± 1.5) × 103 |
| Apoptotic effect | 190 ± 27 | >1,000 | >1,000 | (18.2 ± 2.6) × 103 | >96 × 103 | >96 × 103 |
| Overall cytotoxicity | 132 ± 9 | 63 ± 6 | 165 ± 15 | (12.7 ± 0.9) × 103 | (6.1 ± 0.6) × 103 | (15.8 ± 1.4) × 103 |
Telomerase inhibition by suramin in cell lysates and intact cells was measured using the modified TRAP assay (24 h treatment). The antiproliferative and apoptotic effects were measured as inhibition of 5-bromodeoxyuridine incorporation and release of DNA–histone complex to cytoplasm, respectively. Overall cytotoxicity was measured using the sulforhodamine B assay. All three cytotoxicity assays were studied using 96 h treatment. The total drug exposure for 50% effect (CxT50) was calculated as the product of IC50/DC50 values and time. Data are mean ± SD of three experiments.
IC 50 50% inhibitory drug concentration, DC 50 drug concentration causing 50% cell death, CxT 50 concentration–time product (total drug exposure) for 50% effect
Uptake of suramin
Inhibition of intracellular telomerase requires suramin to enter cells whereas the physicochemical properties of suramin (highly charged anionic molecule [47] and high molecular weight of 1,429 Da) are not favorable for uptake into cells. Hence, we investigated the kinetics of suramin uptake in FaDu cells; the results are shown in Fig. 2. For the lower extracellular concentrations of 1 and 10 μM, the plateauing intracellular concentrations were identical to the extracellular concentrations and were reached rapidly at 3 and 6 h and maintained for over 96 h. The identical intracellular and extracellular drug concentrations and the rapid uptake at lower extracellular concentrations are consistent with passive diffusion. In contrast, the intracellular concentration at the higher extracellular concentration of 100 μM increased at a slower rate, reached 100 μM at about 36 h, followed by a continued increase that eventually exceeded the extracellular concentration (e.g., ~150 μM at 96 h). The difference in drug uptake kinetics at low and high extracellular concentrations suggests concentration-dependent uptake mechanisms.
Fig. 2.
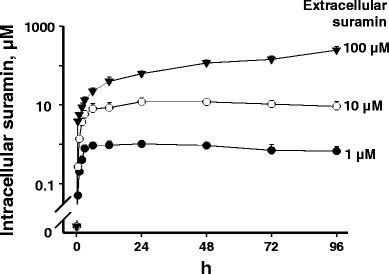
Kinetics of suramin uptake in FaDu cells. FaDu cells were treated with 3H-suramin plus nonradiolabeled suramin. Suramin concentrations in cells were determined as described in the section on “MATERIALS AND METHODS.” Data represent mean ± SD (n = 3)
Noncytotoxic suramin inhibits telomerase in cell lysates and intact cells
Figure 3 shows the results of modified TRAP assays of telomerase activity. Suramin (24 h treatment) yielded concentration-dependent telomerase inhibition in lysates of FaDu, MCF7, and PC3 cells, as well as in intact cells, with IC50 values of between 1 and 3 μM. The nearly identical IC50 values in cell lysates and intact cells are in agreement with the rapid equilibration of intracellular and extracellular drug concentrations. Table I compares the IC50 and the corresponding total drug exposure CxT50 (concentration–time product) for telomerase inhibition to those for suramin cytotoxicity (96 h treatment); the values for telomerase inhibition were, respectively, 120- to >715-fold and 500- to >3,000-fold lower. As shown below, the telomerase-mediated cytotoxicity of suramin occurred slowly and required drug exposure for several weeks.
Fig. 3.

Noncytotoxic suramin inhibits telomerase. Human pharynx cancer FaDu cells were treated with suramin for 24 h. Telomerase activity was detected in cell lysates (a) and intact cells (b). Data points are mean ± SD (n = 3). The concentration–response curves were generated by computer fitting using nonlinear least-squares regression. M: pBR322/HaeIII DNA marker, NC: negative control using lysis buffer in place of cells or cell lysates
We studied the effect of noncytotoxic suramin on hTERT, the protein/enzyme component of telomerase, in order to determine if suramin exerted its telomerase inhibition via hTERT perturbation. The results, shown in Fig. 4, indicate no significant changes in hTERT levels in intact FaDu cells, at up to 100 μM suramin.
Fig. 4.

Noncytotoxic suramin does not alter hTERT level. FaDu cells were treated with suramin (1–100 μM) for 24 h, extracted, and then analyzed for the level of hTERT as described in “MATERIALS AND METHODS.”. The hTERT levels were expressed relative to the untreated control. Data represent mean ± SD (n = 4)
Noncytotoxic suramin shortens telomere length in cultured cells
Effect of suramin on telomere length was measured qualitatively using FISH, semi-quantitatively using TALA, and quantitatively using monochrome multiplex quantitative PCR. The FISH results showed that treatment with suramin (25 μM for 6 weeks) reduced telomere signals in individual cells, compared to no changes in control cells (Fig. 5a). The TALA results showed a suramin concentration-dependent shortening of telomeres in the telomerase-positive FaDu and PC3 cells after 6-week continuous treatment with 25 μM suramin. In contrast, suramin had no effect on the telomerase-negative Saos-2 cells that do not depend on telomerase for telomere maintenance (Fig. 5b). The PCR results showed significant telomere shortening after treatments at both 10 and 25 μM for 11 weeks (Fig. 4c, p < 0.05).
Fig. 5.

Noncytotoxic suramin shortens telomeres in cultured cells. FaDu, PC3, and Saos-2 cells were continuously treated with suramin. a Fluorescence in situ hybridization results (continuous treatment with 25 μM suramin for 6 weeks). b Solution hybridization results (6-week treatments at 25 μM). M: DNA molecular weight markers (from top to bottom: 21, 5.1, 5.0, 4.3, 3.5, 2.0, 1.9, 1.6, and 1.4 kbp). The horizontal white bars depict the location of the average length. c Monochrome multiplex quantitative PCR results for FaDu cells treated with 25 μM suramin for 6 and 11 weeks (mean ± SD, n = 6–8 per group; *p < 0.05 compared with control)
Noncytotoxic suramin induces senescence in cultured cells
Cell senescence was measured semi-quantitatively using the β-galactosidase staining method and quantitatively by measuring the expression of p16 and p21. The β-galactosidase results in MCF7 and PC3 cells indicated between 10 and 35% cells were senescent after treatment with 25 μM suramin for 6 weeks (Fig. 6a). Figure 6b shows the time- and suramin concentration-dependent increases in the mRNA levels of p16 and p21; the increases were statistically significant at the higher 25 μM concentration after 6 weeks of treatment and at both 10 and 25 μM concentrations after 11 weeks (p < 0.05 or < 0.01). We did not study the Saos-2 cells because it uses the alternative telomere lengthening mechanism.
Fig. 6.

Noncytotoxic suramin induces cell senescence. FaDu, MCF7, and/or PC3 cells were treated with 25 μM suramin for 6 and 11 weeks. a β-gal staining (green) in MCF7 and PC3 cells. b Time- and concentration-dependent increases in mRNA levels of senescence markers p16 and p21, in FaDu cells (mean ± SD, n = 5–7 per group; *p < 0.05 and **p < 0.01 compared with control)
Effect of suramin on telomere length in animal tumors
Mice treated with saline or 10 mg/kg suramin twice weekly for 6 weeks showed similar tumor establishment rate (i.e., 100%), tumor growth rate, and body weight (data not shown), confirming that noncytotoxic suramin had no antitumor activity as we previously reported [23, 24, 48, 49]. On the other hand, the FISH results indicate gradual telomere shortening by suramin; the cell fraction that had attenuated or lost telomere signals were significantly higher after 3 weeks and reached over 95% at 6 weeks (Fig. 7, p < 0.05).
Fig. 7.

Noncytotoxic suramin shortens telomeres in animal tumors. FaDu tumor-bearing mice were treated with suramin at a dose of 10 mg/kg intravenously, twice a week, for 6 weeks. a Fluorescence in situ hybridization results showed progressively reduced telomere signals. b Fraction of tumor cells with eroded telomeres. *p < 0.05 compared with controls
DISCUSSION
Our results demonstrate that suramin is an effective telomerase inhibitor. Human telomerase comprises a protein component (hTERT which catalyzes the addition of nucleotides to the telomere) and a RNA component (hTR which serves as a template for the synthesis of the telomeric repeats), and several other proteins [50, 51]. Telomerase inhibitors usually target either hTR or hTERT [14, 52, 53]. As suramin is a potent reverse transcriptase inhibitor, with IC50 of <1 μg/ml for the RNA virus enzyme [54], the most likely target is hTERT. Our results show that suramin did not alter the hTERT protein level. A possibility is that suramin interacts with the template primer binding site of hTERT, as suggested for the inhibition of RNA virus reverse transcriptase [54]. However, as suramin also acts on other molecules that regulate telomerase, e.g., growth factors, protein kinase C, and Akt [38, 55–60], it is possible that other indirect mechanisms are involved.
The present study shows several noteworthy findings on the pharmacodynamics of suramin on telomerase and telomere. First, the IC50 or CxT50 values of suramin for telomerase inhibition are 120- to >3,000-fold lower compared with the values for short-term cytotoxicity. However, prolonged treatment with suramin at these lower, telomerase-inhibitory concentrations, for 6 weeks, resulted in reduced cell numbers (30–50% growth reduction). These findings indicate telomerase inhibition is not a major mechanism of the short-term suramin cytotoxicity but that this effect produces slow-onset cytotoxicity. This is likely because cell death due to telomere shortening occurs when the telomeres reach a critical minimum length. For example, when the telomerase in HeLa cells was inhibited by antisense constructs, cells continue to proliferate for 23–26 doublings while the average telomere length decreases by 25% from 3.3 to 2.4 kb, at which time cell death occurs [5]. Similarly, yeast cells with a fully eroded telomere continue to divide for several generations before undergoing senescence [4]. Second, the IC50 values for telomerase inhibition in three human epithelial tumor cell lines (MCF7, PC3, FaDu) observed in the present study are 100- to 200-fold lower compared with the previously reported values in other cell types (1–2 vs. 200 μM in rat glioma or human osteosarcoma cells; 37, 38), suggesting the telomerase inhibitory effect of suramin is cell type-dependent. Third, our findings show, for the first time, that sustained telomerase inhibition by noncytotoxic suramin (10–50 μM) for 6 to 11 weeks resulted in telomere shortening and cellular senescence, in vitro and in vivo. Three senescence biomarkers, i.e., β-galactosidase activation, and gene expression of p16 and p21, were observed. While the current study did not measure the p16 and p21 protein levels, other ongoing studies found three- to five-fold increases in these proteins in PC3 xenograft tumors treated with noncytotoxic suramin twice weekly for 3 weeks (unpublished results). The clinical implications of these findings are as follows.
The IC50 and CxT50 for the telomerase inhibition and telomere shortening effects of suramin are clinically achievable and are well within the ranges that have shown broad-spectrum chemosensitization in human xenograft tumors. This finding, together with the finding by us and others that telomerase inhibitors enhance chemosensitivity (e.g., [8, 15, 16]), suggest telomerase inhibition as a potential mechanism of in vivo chemosensitization by noncytotoxic suramin.
We have designed the earlier clinical trials of noncytotoxic suramin in part based on two preclinical findings: (a) noncytotoxic suramin enhances tumor sensitivity to multiple classes of conventional chemotherapeutics in mice bearing human epithelial tumor xenografts, and (b) suramin chemosensitization is achieved by co-administering suramin and chemotherapeutics [23, 24, 48, 49]. These findings, plus the clinical practice, led to the trial design whereby suramin is given immediately before the chemotherapeutics, and the treatment is terminated in the event of disease progression or intolerable toxicity [27–35]. The current findings suggest that noncytotoxic suramin may also be used as a single agent, without concurrent chemotherapeutics, to inhibit telomerase that is selectively expressed in tumor cells and thereby induce tumor senescence. As discussed above, cell senescence through telomerase inhibition is a slow process. Hence, one approach to utilize the telomerase inhibition effect will be to continue the noncytotoxic suramin treatment beyond the duration of cytotoxic chemotherapy. The concept of maintenance therapy using agents such as antiangiogenics has been gaining popularity in clinical practice [61, 62]. Another approach is to use noncytotoxic suramin in situations where the tumor burden is not life-threatening and/or the tumor is slow-growing, such that the telomerase inhibition-induced senescence may result in clinical benefits.
In summary, results of the present study indicate suramin, at concentrations with no appreciable acute cytotoxicity (both antiproliferation and cell death), inhibited telomerase, shortened telomeres, and induced cell senescence in multiple human epithelial cancer cell lines that depend on telomerase for telomere maintenance. These findings improve our understanding of the mechanisms of its in vivo chemosensitization effect and may be used to design future clinical trials to capture these pharmacodynamic properties.
Acknowledgments
This study was supported in part by a research grant RO1CA77091 from the National Cancer Institute, NIH, DHHS.
Abbreviations
- CxT50
Concentration-time product (total drug exposure) for 50% effect
- DC50
Drug concentration causing 50% cell death
- FBS
Fetal bovine serum
- FISH
Fluorescence in situ hybridization
- IC50
50% inhibitory drug concentration
- hTERT
Reverse transcriptase component of human telomerase
- hTR
RNA component of human telomerase
- MEM
Minimum essential medium
- PBS
Phosphate-buffered saline
- TALA
Solution hybridization-based telomere amount and length assay
- TRAP
Telomeric repeat amplification protocol
References
- 1.Zakian VA. Telomeres: beginning to understand the end. Science. 1995;270:1601–7. doi: 10.1126/science.270.5242.1601. [DOI] [PubMed] [Google Scholar]
- 2.Hemann MT, Strong MA, Hao LY, Greider CW. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell. 2001;107:67–77. doi: 10.1016/S0092-8674(01)00504-9. [DOI] [PubMed] [Google Scholar]
- 3.Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463–79. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abdallah P, Luciano P, Runge KW, Lisby M, Geli V, Gilson E, et al. A two-step model for senescence triggered by a single critically short telomere. Nat Cell Biol. 2009;11:988–93. doi: 10.1038/ncb1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feng J, Funk WD, Wang SS, Weinrich SL, Avilion AA, Chiu CP, et al. The RNA component of human telomerase. Science. 1995;269:1236–41. doi: 10.1126/science.7544491. [DOI] [PubMed] [Google Scholar]
- 6.Au JL, Kumar RR, Li D, Wientjes MG. Kinetics of hallmark biochemical changes in paclitaxel-induced apoptosis. AAPS Pharm Sci. 1999;1:E8. doi: 10.1208/ps010308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moos PJ, Fitzpatrick FA. Taxanes propagate apoptosis via two cell populations with distinctive cytological and molecular traits. Cell Growth Differ. 1998;9:687–97. [PubMed] [Google Scholar]
- 8.Stone AA, Chambers TC. Microtubule inhibitors elicit differential effects on MAP kinase (JNK, ERK, and p38) signaling pathways in human KB-3 carcinoma cells. Exp Cell Res. 2000;254:110–9. doi: 10.1006/excr.1999.4731. [DOI] [PubMed] [Google Scholar]
- 9.Strahl C, Blackburn EH. Effects of reverse transcriptase inhibitors on telomere length and telomerase activity in two immortalized human cell lines. Mol Cell Biol. 1996;16:53–65. doi: 10.1128/mcb.16.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gangemi RMR, Santamara B, Bargellesi A, Cosulich E, Fabbi M. Late apoptotic effects of taxanes on K562 erythroleukemia cells: apoptosis is delayed upstream of caspase-3 activation. Int J Cancer. 2000;85:527–33. doi: 10.1002/(SICI)1097-0215(20000215)85:4<527::AID-IJC14>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 11.Campisi J, d’Adda di FF. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–40. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 12.Kelland L. Targeting the limitless replicative potential of cancer: the telomerase/telomere pathway. Clin Cancer Res. 2007;13:4960–3. doi: 10.1158/1078-0432.CCR-07-0422. [DOI] [PubMed] [Google Scholar]
- 13.Kim NM, Piatzszek MS, Prowese KR, Harley CB, West MD, Ho PL, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–5. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 14.Harley CB. Telomerase and cancer therapeutics. Nat Rev Cancer. 2008;8:167–79. doi: 10.1038/nrc2275. [DOI] [PubMed] [Google Scholar]
- 15.Lee KH, Rudolph KL, Ju YJ, Greenberg RA, Cannizzaro L, Chin L, et al. Telomere dysfunction alters the chemotherapeutic profile of transformed cells. Proc Natl Acad Sci U S A. 2001;98:3381–6. doi: 10.1073/pnas.051629198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mo Y, Gan Y, Song S-H, Johnston J, Xiao X, Wientjes MG, et al. Simultaneous targeting of telomeres and telomerase as a cancer therapeutic approach. Cancer Res. 2003;63:579–85. [PubMed] [Google Scholar]
- 17.Stein CA. Suramin: a novel antineoplastic agent with multiple potential mechanisms of action. Cancer Res. 1993;54:2239–48. [PubMed] [Google Scholar]
- 18.Church D, Zhang Y, Rago R, Wilding G. Efficacy of suramin against human prostate carcinoma DU145 xenograft in nude mice. Cancer Chemother Pharmacol. 1999;43:198–204. doi: 10.1007/s002800050884. [DOI] [PubMed] [Google Scholar]
- 19.Vincent TS, Hazen-Martin DJ, Garvin AJ. Inhibition of insulin like growth factor II autocrine growth of Wilms’ tumor by suramin in vitro and in vivo. Cancer Lett. 1996;103:49–56. doi: 10.1016/0304-3835(96)04186-9. [DOI] [PubMed] [Google Scholar]
- 20.Yonega R, Williams P, Rhine C, Boyce BF, Dunstan C, Mundy GR. Suramin suppresses hypercalcemia and osteoclastic bone resorption in nude mice bearing a human squamous cancer. Cancer Res. 1995;55:1989–93. [PubMed] [Google Scholar]
- 21.Song S, Wientjes MG, Walsh C, Au JL. Nontoxic doses of suramin enhance activity of paclitaxel against lung metastases. Cancer Res. 2001;61:6145–50. [PubMed] [Google Scholar]
- 22.Song S, Wientjes MG, Gan Y, Au JL. Fibroblast growth factors: an epigenetic mechanism of broad spectrum resistance to anticancer drugs. Proc Natl Acad Sci U S A. 2000;97:8658–63. doi: 10.1073/pnas.140210697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Song S, Yang F, Au JL, Wientjes MG. Nontoxic doses of suramin enhance activity of doxorubicin in prostate tumors. J Pharmacol Exp Ther. 2001;299:426–33. [PubMed] [Google Scholar]
- 24.Lu Z, Wientjes TS, Au JL. Nontoxic suramin treatments enhance docetaxel activity in chemotherapy-pretreated non-small cell lung xenograft tumors. Pharm Res. 2005;22:1069–78. doi: 10.1007/s11095-005-6038-1. [DOI] [PubMed] [Google Scholar]
- 25.Yu B, Song S-H, Wientjes MG, Au JL. Suramin enhances activity of CPT-11 in human colorectal xenograft tumors. Proc Am Assoc Cancer Res. 2003;44.
- 26.ClinTrials.Gov. Available from: https://clinicaltrials.gov/ct2/show/NCT01671332.
- 27.Au JL, Olencki T, Wientjes MG, Otterson G, Saab T, Grainger A, et al. A phase I study of nontoxic suramin as a chemosensitizer in the pretreated/refractory non-small cell lung cancer (NSCLC) patients. J Thorac Oncol. 2007;2:S663–4. doi: 10.1097/01.JTO.0000283938.40281.52. [DOI] [Google Scholar]
- 28.George S, Dreicer R, Au JL, Shen T, Rini BI, Roman S, et al. Phase I/II trial of 5-fluorouracil and a noncytotoxic dose level of suramin in patients with metastatic renal cell carcinoma. Clin Genitourin Cancer. 2008;6:79–85. doi: 10.3816/CGC.2008.n.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lam ET, Au JL, Otterson GA, Wientjes MG, Chen L, Shen T, et al. Phase I trial of non-cytotoxic suramin as a modulator of docetaxel and gemcitabine therapy in previously treated patients with non-small cell lung cancer. Cancer Chemother Pharmacol. 2010;66:1019–29. doi: 10.1007/s00280-010-1252-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lustberg MB, Pant S, Ruppert AS, Shen T, Wei Y, Chen L, et al. Phase I/II trial of non-cytotoxic suramin in combination with weekly paclitaxel in metastatic breast cancer treated with prior taxanes. Cancer Chemother Pharmacol. 2012;70:49–56. doi: 10.1007/s00280-012-1887-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olencki T, Wientjes MG, Otterson G, Saab T, Grainger A, Yeh T, et al. Modulation of chemotherapy resistance with low dose suramin in refractory non-small cell lung cancer (NSCLC) patients: a phase I study of sequential non-cross resistant chemotherapy. J Clin Oncol. 2005;23:2104. [Google Scholar]
- 32.Shapiro CL, Sheils D, Barton L, Young D, Shen T, Chen L, et al. CTEP-sponsored phase I/II trial of paclitaxel and low dose suramin in metastatic breast cancer. Breast Cancer Res Treat. 2007;106:S270. [Google Scholar]
- 33.Villalona-Calero MA, Otterson GA, Kanter S, Young D, Fischer B, Straiko M, et al. A phase I, pharmacokinetic (PK), and biological study of FGF inhibition modulating paclitaxel/carboplatin (P/C) chemotherapy in non-small cell lung cancer (NSCLC) patients (pts) Clin Cancer Res. 2001;417:3738S. [Google Scholar]
- 34.Villalona-Calero MA, Otterson GA, Wientjes MG, Weber F, Bekaii-Saab T, Young D, et al. Noncytotoxic suramin as a chemosensitizer in patients with advanced non-small-cell lung cancer: a phase II study. Ann Oncol. 2008;19:1903–9. doi: 10.1093/annonc/mdn412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Villalona-Calero MA, Wientjes MG, Otterson GA, Kanter S, Young D, Murgo AJ, et al. Phase I study of low-dose suramin as a chemosensitizer in patients with advanced non-small cell lung cancer. Clin Cancer Res. 2003;9:3303–11. [PubMed] [Google Scholar]
- 36.Erguven M, Akev N, Ozdemir A, Karabulut E, Bilir A. The inhibitory effect of suramin on telomerase activity and spheroid growth of C6 glioma cells. Med Sci Monit. 2008;14:BR165–73. [PubMed] [Google Scholar]
- 37.Trieb K, Blahovec H. Suramin suppresses growth, alkaline-phosphatase and telomerase activity of human osteosarcoma cells in vitro. Int J Biochem Cell Biol. 2003;35:1066–70. doi: 10.1016/S1357-2725(02)00308-4. [DOI] [PubMed] [Google Scholar]
- 38.Gan Y, Mo Y, Johnston J, Lu J, Wientjes MG, Au JL. Telomere maintenance in telomerase-positive human ovarian SKOV-3 cells cannot be retarded by complete inhibition of telomerase. FEBS Lett. 2002;527:10–4. doi: 10.1016/S0014-5793(02)03141-1. [DOI] [PubMed] [Google Scholar]
- 39.Kuh HJ, Jang SH, Wientjes MG, Au JL. Computational model of intracellular pharmacokinetics of paclitaxel. J Pharmacol Exp Ther. 2000;293:761–70. [PubMed] [Google Scholar]
- 40.Gao Y, Li M, Chen B, Shen Z, Guo P, Wientjes MG, et al. Predictive models of diffusive nanoparticle transport in 3-dimensional tumor cell spheroids. AAPS J. 2013;15:816–31. doi: 10.1208/s12248-013-9478-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gan Y, Lu J, Johnson A, Wientjes MG, Schuller DE, Au JL. A quantitative assay of telomerase activity. Pharm Res. 2001;18:488–93. doi: 10.1023/A:1011006427733. [DOI] [PubMed] [Google Scholar]
- 42.Bryan TM, Englezou A, Dalla-Pozza L, Dunham MA, Reddel RR. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat Med. 1997;3:1271–4. doi: 10.1038/nm1197-1271. [DOI] [PubMed] [Google Scholar]
- 43.Gan Y, Engelke KJ, Brown CA, Au JL. Telomere amount and length assay. Pharm Res. 2001;18:1655–9. doi: 10.1023/A:1013306109801. [DOI] [PubMed] [Google Scholar]
- 44.Cawthon RM. Telomere length measurement by a novel monochrome multiplex quantitative PCR method. Nucleic Acids Res. 2009;37:e21. doi: 10.1093/nar/gkn1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu L. Suramin pharmacokinetics after regional or systemic administration. Dissertation, The Ohio State University, 2005
- 46.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eisenberger MA, Reyno LM. Suramin. Cancer Treat Rev. 1994;20:259–73. doi: 10.1016/0305-7372(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 48.Song S, Yu B, Wei Y, Wientjes MG, Au JL. Low-dose suramin enhanced paclitaxel activity in chemotherapy-naive and paclitaxel-pretreated human breast xenograft tumors. Clin Cancer Res. 2004;10:6058–65. doi: 10.1158/1078-0432.CCR-04-0595. [DOI] [PubMed] [Google Scholar]
- 49.Xin Y, Lyness G, Chen D, Song S, Wientjes MG, Au JL. Low dose suramin as a chemosensitizer of bladder cancer to mitomycin C. J Urol. 2005;174:322–7. doi: 10.1097/01.ju.0000161594.86931.ea. [DOI] [PubMed] [Google Scholar]
- 50.Artandi SE, DePinho RA. Telomeres and telomerase in cancer. Carcinogenesis. 2010;31:9–18. doi: 10.1093/carcin/bgp268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wojtyla A, Gladych M, Rubis B. Human telomerase activity regulation. Mol Biol Rep. 2011;38:3339–49. doi: 10.1007/s11033-010-0439-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raymond E, Soria JC, Izbicka E, Boussin F, Hurley L, Von Hoff DD. DNA G-quadruplexes, telomere-specific proteins and telomere-associated enzymes as potential targets for new anticancer drugs. Invest New Drugs. 2000;18:123–37. doi: 10.1023/A:1006373812586. [DOI] [PubMed] [Google Scholar]
- 53.White LK, Wright WE, Shay JW. Telomerase inhibitors. Trends Biotechnol. 2001;19:114–20. doi: 10.1016/S0167-7799(00)01541-9. [DOI] [PubMed] [Google Scholar]
- 54.De Clercq E. Suramin: a potent inhibitor of the reverse transcriptase of RNA tumor viruses. Cancer Lett. 1979;8:9–22. doi: 10.1016/0304-3835(79)90017-X. [DOI] [PubMed] [Google Scholar]
- 55.La Rocca RV, Danesi R, Cooper MR, Jamis-Dow CA, Ewing MW, Linehan WM, et al. Effect of suramin on human prostate cancer cells in vitro. J Urol. 1991;145:393–8. doi: 10.1016/s0022-5347(17)38351-9. [DOI] [PubMed] [Google Scholar]
- 56.Haïk S, Gauthier LR, Granotier C, Peyrin J-M, Lages CS, Dormont D, et al. Fibroblast growth factor 2 up regulates telomerase activity in neural precursor cells. Oncogene. 2000;19:2957–66. doi: 10.1038/sj.onc.1203596. [DOI] [PubMed] [Google Scholar]
- 57.Tsumuki H, Hasunuma T, Kobata T, Kato T, Uchida A, Nishioka K. Basic FGF-induced activation of telomerase in rheumatoid synoviocytes. Rheumatol Int. 2000;19:123–8. doi: 10.1007/s002960050115. [DOI] [PubMed] [Google Scholar]
- 58.Li H, Zhao L, Yang Z, Funder JW, Liu JP. Telomerase is controlled by protein kinase C-alpha in human breast cancer cells. J Biol Chem. 1998;273:33436–42. doi: 10.1074/jbc.273.50.33436. [DOI] [PubMed] [Google Scholar]
- 59.Yu CC, Lo SC, Wang TC. Telomerase is regulated by protein kinase C-zeta in human nasopharyngeal cancer cells. Biochem J. 2001;355:459–64. doi: 10.1042/0264-6021:3550459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kang SS, Kwon T, Kwon DY, Do SI. Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. J. Biol Chem. 1999;274:13085–90. [DOI] [PubMed]
- 61.Marshall JL. Maintenance therapy for colorectal cancer. Oncology (Williston Park). 2014;28:322–4 [PubMed]
- 62.Khalique S, Hook JM, Ledermann JA. Maintenance therapy in ovarian cancer. Curr Opin Oncol. 2014;26:521–8 [DOI] [PubMed]