

Abstract
Development of peptide drugs is challenging but also quite rewarding. Five blockbuster peptide drugs are currently on the market, and six new peptides received first marketing approval as new molecular entities in 2012. Although peptides only represent 2% of the drug market, the market is growing twice as quickly and might soon occupy a larger niche. Natural peptides typically have poor absorption, distribution, metabolism, and excretion (ADME) properties with rapid clearance, short half-life, low permeability, and sometimes low solubility. Strategies have been developed to improve peptide drugability through enhancing permeability, reducing proteolysis and renal clearance, and prolonging half-life. In vivo, in vitro, and in silico tools are available to evaluate ADME properties of peptides, and structural modification strategies are in place to improve peptide developability.
KEY WORDS: ADME, peptides, pharmacokinetics, proteolysis, renal clearance
INTRODUCTION
The human genome project indicated there are 30,000 human genes (1). Among them, ∼3,000 are disease-modifying genes and ∼3,000 are druggable genes (1). This suggests that only 2–5% of the human genome are small molecule drug targets (∼600–1,500), which presents a great opportunity to use peptides or large molecules to treat human diseases.
Development of peptides into drugs has long been recognized as an important opportunity in order to address certain disease targets that would otherwise be challenging using small molecules. In the 1980s, huge investments were channeled into the development of peptide-based drugs, only to realize the nondrug-like nature of this structure class (2). Subsequently, many drug developers and investors dismissed peptides as potential drugs.
With the advances in genomics, transcriptomics, and proteomics, many new disease targets feature protein–protein interactions with shallow binding pockets covering wide surface area. Peptides and proteins are more suitable for these types of disease targets than small molecules. Personalized medicine and greater emphasis on efficacy and safety in the current regulatory environment require therapeutic agents to have high versatility, specificity, and safety. Peptides have greater potential to meet the ever-increasing expectations of new drugs, as they are highly specific to individual protein targets, amenable to site-specific modification and highly selective. In addition, the advances in recombinant protein expression technologies, the development of more efficient and economic peptide synthesis, the improvement of peptide purification systems and new analytical tools have been essential for the revival of the peptide field in the recent decade. Peptides have the potential to offer the advantages of both small molecule drugs and proteins.
Unlike small molecule drugs, peptides represent a very small portion (2%) of the worldwide drug market (3). The annual sales of peptide drugs are about $20 billion (3). However, peptide drugs can be quite successful with several blockbuster peptide drugs on the market, e.g., Copaxone, Lupron, Zoladex, Sandostatin, and Velcade (4–6). Six new peptides received first marketing approval as new molecular entities in 2012 (6). Most peptide drugs (∼85%) are synthetically made, which is largely credited to the development of solid-state peptide synthesis, and a small number (∼15%) are prepared using recombinant technology. With the use of unnatural amino acids and pseudo-peptide bonds, chemical synthesis offers more diversity and patentability than peptides derived from recombinant technology (7). There are currently ∼70 approved peptide drugs on the market, ∼200 in clinical development, and ∼600 in the preclinical drug discovery stage (Fig. 1) (3). The peptide market is growing twice as quickly as the rest of the drug market, suggesting peptides might soon occupy a larger niche (7,8).
Fig. 1.
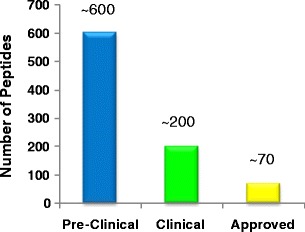
Stages of peptide drugs in discovery and development (3)
Peptide drugs cover a wide range of therapeutic areas, such as diabetics, cancer, osteoporosis, hormone therapy, cardiovascular diseases, anemia, bowel syndrome, Cushing’s disease, multiple sclerosis, HIV, and many more (6,9). The focus of peptide drugs is shifting from hormone therapy and diagnosis to cancer and infection.
Ranging in size between small molecules and proteins, peptides present a unique opportunity and challenge for the pharmaceutical industry compared to small molecules (Table I) (9,10). Small molecules represent ∼80% of the drug market and peptides only represent ∼2%. Small molecules are usually cheaper and relatively easier to synthesize. Peptides are typically more difficult to make and have synthetic and production cost about 10–100-fold higher than small molecules (7,11). Though some small molecules also face similar challenges, many of them are permeable through cell membranes, stable, and have good oral bioavailability. Peptides usually have low cell membrane permeability, limited stability, poor oral bioavailability, and are usually administrated by injection [e.g., subcutaneous (SC), intramuscular (IM), intravenous (IV)]. Unmodified peptides tend to have short half-lives and are typically limited to extracellular targets due to poor cell membrane penetration. With all these challenges, why are we interested in developing peptide drugs? Peptides have unique properties that are very appealing as therapeutic agents, such as high binding affinity toward therapeutic targets, easier to identify as the mechanisms of action are well-defined, excellent target specificity, broad coverage of disease targets, low toxicity and immunogenicity, minimal risk of drug-drug interaction potential, and low impact of generic erosions (3,5,6,12). Therefore, peptides are an area of high interest for many drug developers.
Table I.
Comparison Between Peptides and Small Molecules
| Small molecules | Peptides |
|---|---|
| • ∼80% drug market • Low cost • Permeable • Stable • Good oral bioavailability • Easier synthesis |
• ∼2% drug market • High cost • Low permeability • Limited stability • Poor oral bioavailability • More challenging synthesis • Short half-life • Limited to extracellular targets • High binding affinity • Easier to identify • Excellent target specificity • Broad disease targets • Mechanism of action well understood • Low toxicity and immunogenicity • Low risk of drug–drug interaction potential • Lower impact from generics |
ADME CHALLENGES OF PEPTIDES
It is often challenging for peptides to become successful drugs due to multiple absorption, distribution, metabolism, and excretion (ADME) issues such as low permeability, metabolic instability, short half-life, and limited residence time in tissues. Most peptides have low cell membrane permeability owing to high hydrogen bonding capacity and low lipophilicity (13). Low oral bioavailability of peptides is mostly caused by low absorption and high first-pass extraction due to enzymatic- and pH-mediated hydrolysis in the gastrointestinal (GI) tract and liver (14). Consequently, peptide drugs are frequently administered as injectables or through other alternative delivery routes, such as inhaled, buccal, intranasal, and transdermal. Unmodified peptides usually have very short half-lives (e.g., minutes) resulting from extensive proteolysis in blood, kidneys, or liver and/or rapid renal clearance (15). In silico, in vitro, and in vivo tools have been developed to address the ADME challenges of peptides. Structural modification strategies are in place to enhance peptide developability (5,12,16,17).
Absorption
With a few exceptions (e.g., cyclosporine A), most peptides have less than 1% oral bioavailability (18). They tend to show high inter-subject and inter-species variability in plasma exposure (19). In spite of the major hurdles to achieve oral bioavailability, there is still significant effort in developing oral peptides owing to their high market potential and patient compliance (20,21). Examples of oral peptides in development include calcitonin in phase III, glucagon-like peptide-1 (GLP-1), and parathyroid hormone in phase I (5,12,15).
Following SC or IM delivery routes, peptides enter systemic circulation either through blood capillaries (molecular weight (MW) <1 kDa) or lymphatic vessels (MW >16–22 kDa) (15,22). For most therapeutic peptides with typical MW of 1–10 kDa, combined absorption through both blood and lymphatic systems is expected with the diffusion-driven uptake into blood capillaries being the predominant pathway (15). “Flip-flop” pharmacokinetics (PK) (absorption constant is much slower than elimination constant) may occur when absorption is slower than elimination leading to prolonged half-life and pharmacodynamic (PD) effect (15). An example is the gonadotropin-releasing hormone (GnRH) agonist leuprolide, where a long-acting monthly IM depot injection releases the peptide slowly and continually into blood to provide long-term suppression of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) for the treatment of prostate cancer and endometriosis (23).
Permeability Methods for Peptides
Peptides can be absorbed by (a) passive diffusion through the lipid membrane, (b) paracellular pathway, and (c) transporter-mediated processes [e.g., peptide transporter 1 (PEPT1), sodium-dependent multivitamin transporter (SMVT), vitamin B12 transport system] (12). Small molecule in vitro permeability and transporter assay platforms are largely applicable to study peptide permeability and transporter characteristics, such as log D (16,24), parallel artificial membrane permeability assay (PAMPA) (13,25), Madin-Darby canine kidney (MDCK) (26,27), Caco-2 (28–30), PEPT1 (31–33), and SMVT (34). The Caco-2 cell line possesses many of the human intestinal transporters (e.g., PEPT1, SMVT) and can be used to identify peptides with high absorption potential not only by transcellular or paracellular passive diffusion mechanisms but also by active uptake processes (30). The challenges of these assays for measuring peptide permeability are as follows: (1) high nonspecific binding to assay plates, pipette tips, and transwell filter membranes and (2) degradation mediated by enzymes expressed in the cell systems or by pH-mediated hydrolysis. To minimize the impact of nonspecific binding, low binding tips and plates are typically used, which are commercially available (http://www.labonline.com.au/products/48460-Eppendorf-LoBind-Tubes-and-Plates-and-epT-I-P-S-LoRetention-pipette-tips). Serum proteins [e.g., bovine serum albumin (BSA)] are often added to the receiver wells to create a sink condition and minimize nonspecific binding. To reduce enzymatic degradation during the assay, protease inhibitors or cocktails [e.g., aprotinin, 4-(2-aminoethyl) benzenesulfonyl fluoride (AEBSF), bestatin] are usually added to the system to reduce peptide proteolysis.
A number of in silico models have also been developed to predict permeability of peptides (13,35,36). Stenberg et al. found that using dynamic molecular surface properties, Caco-2 permeability of peptides was successfully predicted (35). Rezai et al. reported an atomistic physical model to predict PAMPA passive membrane permeability of cyclic peptides that did not involve “training data” (13). Rafi et al. discussed an all-atom force field-based method to calculate changes in free energy associated with the transfer of the peptidic molecules from water to membrane (36). The method correctly predicted rank order experimental permeability trends from MDR1-MDCK cells within congeneric series and was much more predictive than methods that do not consider three-dimensional peptide conformation (36). It was found that the intentional introduction of hydrogen bond acceptor–donor pairs in peptides can improve membrane permeability (36). The key descriptors for peptide permeability are hydrogen-bonding capacity (especially intramolecular hydrogen bonding), hydrophobicity/lipophilicity, size, and polar surface area (13,35,36). In silico models are particularly useful prior to peptide synthesis to estimate permeability values and when it is technically challenging to measure permeability experimentally due to stability, nonspecific binding, sensitivity, and other issues.
In vivo animal models are frequently used to study peptide bioavailability and fraction absorbed in nonsurgical or portal vein cannulated (PVC) animals. In vitro data can be used in conjunction with in vivo studies to determine whether low oral bioavailability is due to poor absorption or rapid first-pass liver extraction. Transporter knockout animals (e.g., PEPT1 knockout mice) are useful to understand the contribution of uptake transporters in oral absorption (37). Bioanalysis of peptides can be challenging due to low sensitivity and selectivity, high nonspecific binding and protein binding, low recovery, carryover, solubility, and stability issues (38–40). For peptides with poor stability, blood/plasma samples need to be stabilized once they are removed from the in vivo systems (41). Protease inhibitors or cocktails (e.g., aprotinin to avoid proteolysis or oxidation of cysthiols) are usually added to the collection tubes on wet ice to cool the samples immediately after blood collection and to prevent further hydrolysis during sample preparation and analysis (42). Nonabsorptive collection tubes are used to minimize nonspecific binding (http://www.labonline.com.au/products/48460-Eppendorf-LoBind-Tubes-and-Plates-and-epT-I-P-S-LoRetention-pipette-tips). Displacement proteins (e.g., serum albumin) or peptides (structural analogs) are sometimes added to compete for the surface-binding sites (40). Organic solvents, acids, salts, or surfactants (e.g., Triton X-100 or Tween-20) can be added to overcome nonspecific binding by increasing peptide solubility (40,42).
Strategies to Enhance Peptide Permeability
Many strategies have been developed to enhance peptide permeability, including N-methylation to reduce hydrogen bonding potential, cyclization to increase rigidity, and introducing intramolecular hydrogen bonds to reduce intermolecular hydrogen bonds and flexibility (30,36,43–46). Cyclosporine A, an 11-residue peptide, comprises all of these strategies in its structure, i.e., cyclic backbone, seven N-methyl groups, and four intramolecular hydrogen bonds (5,47). Other approaches include stapled peptides (produced by connecting two amino acids to increase helicity, potency, stability, and permeability), prenylated peptides with farnesyl (C15) and geranylgeranyl (C20) chains, and pepducins (containing a short peptide derived from a GPCR intracellular loop tethered to a hydrophobic moiety) where the N-terminus of the peptide is lipidated with palmitoyl or other fatty acids (C12–C18) (48,49). Reversible lipidization (covalently attached long chain fatty acid to a peptide) has been shown to prolong plasma half-life and increase GI stability and oral bioavailability (50–52). It overcomes the limitations of a conventional lipidization approach (e.g., incompatible reaction media, insoluble in water, low biological activity) (51).
Transporter-mediated processes play an important role in peptide oral absorption. In the study of a library of 54 cyclic peptides with different N-methylation patterns, it was found that the similarity of the backbone conformation with the well-absorbed peptides (e.g., cyclosporine A and N-methylated somatostatin analog) was important for uptake by absorptive transporters in the GI lumen (30). SMVT has been shown to be a promising transporter to improve oral absorption of peptide–biotin conjugates (53). The strategy of using the vitamin B12 uptake system to deliver peptide orally has shown great potential (54,55). With the increasing knowledge and understanding of transporter–peptide structure relationships, it will increase the success of oral delivery of peptides.
Formulation can help improve oral absorption of peptides by using absorption enhancers. A large number of permeability enhancers have been reported to increase intestinal absorption of peptides, such as surfactants, bile salts, phospholipids, fatty acids, and glycerides (56–58). The mechanisms by which the enhancers increase permeability are (20,21): (1) opening epithelial tight junctions reversibly to increase paracellular transport (e.g., EDTA increases paracellular permeability by chelating the calcium that is required to form intercellular tight junctions) (59); (2) mildly perturbing the mucosal surface by altering membrane fluidity to enhance transcellular permeation (e.g., transient permeability enhancers with medium chain fatty acids) (60); and (3) forming noncovalent complex with payload to be absorbed (e.g., Eligen®) (61). Despite 50 years of research on oral permeability enhancers, clinical success has yet to be achieved (20). The major challenges of oral permeability enhancers are low and variable oral bioavailability and safety concerns. These hurdles are currently being addressed by both academia and industry (20). A number of peptide clinical trials with permeability enhancers are ongoing, and several of them have shown promising oral efficacy (20).
Proteolytic Stability
Peptides are susceptible to proteolysis by proteases or peptidases due to the amide bonds in their structures. Both luminally secreted enzymes (e.g., pepsins, elastase, trypsin, and chymotrypsin) and brush border membrane-bound enzymes (e.g., endopeptidases, aminopeptidases, and carboxypeptidase) play important roles in peptide proteolysis. More than 550 putative proteases are ubiquitously distributed throughout the body (62). Proteolysis is a major elimination pathway for most peptides, and clearance of peptides can exceed cardiac output due to blood degradation.
Structure–Proteolytic Stability Relationship
In general, the N-terminus residue of a peptide correlates to its half-life in plasma. Peptides with N-terminus containing Met, Ser, Ala, Thr, Val, or Gly typically have longer half-lives. Peptides with N-terminus containing Phe, Leu, Asp, Lys, or Arg usually have shorter half-lives. Peptide domains rich in Pro, Glu, Ser, and Thr are more prone to enzymatic degradation. Proteolytic enzymes, their substrates, and site specificities have been well documented (17,63). A number of software programs are available to predict peptide cleavage sites, such as PeptideCutter (http://web.expasy.org/peptide_cutter/), PROSPER (https://prosper.erc.monash.edu.au/), and CutDB (http://cutdb.burnham.org/login). In drug discovery, the proteolytic enzymes for a specific peptide are not always known. In practice, the sites of cleavage are typically identified using liquid chromatography–mass spectrometry (LC-MS/MS). Knowing the cleavage sites allows a well-directed modification of peptide structure to minimize enzymatic degradation (17). Hydrolytic products are sometimes tested for pharmacological activity as they might be active against the disease target.
Stability Assays
Peptides can be incubated with biological matrices to evaluate their stability (64–66). Both kinetic information (in vitro intrinsic clearance and half-life) and degradation products can be determined. The typical matrices of various species are as follows: (1) plasma/serum and blood to evaluate degradation in systemic circulation; (2) GI fluids [simulated gastric fluid (SGF), simulated intestinal fluid (SIF)], intestine brush border membrane vesicles (BBMV), and intestine microsomes or S9 to examine GI stability and predict oral bioavailability; (3) liver microsomes, S9, cytosol, and hepatocytes to study liver metabolism by the various liver enzymes; (4) kidney BBMV, microsomes, or homogenates to assess kidney degradation; (5) tissue homogenates to examine tissue stability; and (6) assay media and formulation vehicles to ensure acceptable stability. LC-MS/MS is used to monitor parent drug depletion and examine the structures of the degradation products (67,68). The information is used to guide structure modification to improve peptide stability.
Strategies to Stabilize Peptides from Proteolysis
Many approaches are available to enhance stability of peptides through structure modification (17,69). Some approaches not only improve stability, but also enhance other ADME properties, e.g., cyclization can increase stability and permeability; conjugation to macromolecules can improve stability and reduce renal clearance (70–75). It is important to maintain potency and avoid toxicity while improving stability and ADME properties of peptides.
-
Protecting N- and C-terminus
A number of proteolytic enzymes in blood/plasma, liver or kidney are exopeptidases, aminopeptidases and carboxypeptidases and they break down peptide sequences from the N- and C-termini. Modification of the N- or/and C-termini can often improve peptide stability. Many examples have reported that N-acetylation, and C-amidation increase resistance to proteolysis (8,76). For example, N-terminal acetylated somatostatin analogs were reported to be much more stable than the native peptide (69). The N-acetylated 7-34 form of glucagon-like peptide-1 (GLP-1 7-34) has been shown to be much more stable than the unprotected peptides (77). Even though N-acetylation and C-amidation are known to increase stability against exopeptidases, it was found to improve resistance against endopeptidases for EFK17 (EFKRIVQRIKDFLRNLV) peptide when applied in conjunction with amino acid substitutions (78). Tesamorelin has a hexenoyl group attached to the N-terminus tyrosine residue and has a much longer half-life (1 h) than the natural growth hormone-releasing hormone (GHRH, 6.8 min) (79).
-
Replacing l-amino acids with d-amino acids
Substituting natural l-amino acids with nonnatural d-amino acids decreases the substrate recognition and binding affinity of proteolytic enzymes and increases stability. One example is vasopressin, which contains an l-Arg and has a half-life of 10–35 min in humans (80). The d-Arg analog, desmopressin, has a half-life of 3.7 h in healthy human volunteers (81). Octreotide, a drug for the treatment of gastrointestinal tumors, has a shorter amino acid sequence than the endogenous hormone somatostatin (8 vs. 14 amino acids) and differs by the substitution of l-amino acids with d-amino acids. The human in vivo half-life of octreotide improved to 1.5 h from a few minutes for somatostatin (82). In the study of a bicyclic peptide inhibitor of the cancer-related protease urokinase-type plasminogen activator (uPA), replacement of a specific glycine with a d-serine not only improves potency by 1.8-fold but also increases stability by 4-fold in mouse plasma (83). In MUC2 epitope peptide, the partial d-amino acid-substituted peptide exhibited high resistance against proteolytic degradation in plasma and lysosomal preparation (84). However, exceptions do exist. Dermorphin analogs with additional d-amino acid substitutions were found to be more rapidly cleaved than the parent peptides, potentially due to remote secondary structural features that are important for enzyme recognition (85). In another case, d-amino acid-substituted analogs of growth hormone-releasing factor 1-29 amide did not show significant improvement of half-life in rats (86). These exceptions highlighted the importance of the understanding of key structural features and interactions to guide successful peptide modifications. Recently, the novel concept of d-peptide (minor image of natural all l-peptide) significantly improved stability and half-life. Rotigaptide (antiarrhythmic) and PIE12 (HIV) are two d-peptides in development (87,88). One major concern of using unnatural amino acids is the potential toxicity. Unnatural amino acid substitutions have been found to associate with adverse effects as they can accumulate in the liver and other organs (89,90).
-
Modification of amino acids
Modification of natural amino acids can improve the stability of peptides by introducing steric hindrance or disrupting enzyme recognition (17). For example, gonadotropin-releasing hormone has a very short half-life (minutes), while buserelin, in which one Gly is replaced with a t-butyl-d-Ser and another Gly is substituted by ethylamide, has a much longer half-life in humans (http://products.sanofi.ca/en/suprefact-depot.pdf). Ipamorelin, a pentapeptide, has 2′-naphthylalanine and phenylalanine in the D configuration and the C-terminal l-alanine replaced by 2-aminoisobutyric acid, resulting in improved terminal half-life of ~2 h in humans (91,92).
-
Cyclization
Cyclization introduces conformation constraint, reduces the flexibility of peptides, and increases stability and permeability. Depending on the functional groups, peptides can be cyclized head-to-tail, head/tail-to-side-chain, or side-chain-to-side-chain. Cyclization is commonly accomplished through lactamization, lactonization, and sulfide-based bridges (4). Cyclic enkephalin analog was found to be highly resistant to enzymatic degradation (93). A cyclic epitope peptide derived from herpes simplex virus glycoprotein was completely stable in 50% human serum, but the linear peptide was totally unstable (94). Stapled peptides have been shown to significantly enhance serum stability by reinforcing an α-helix to create a shield from proteolysis (95–97). ALRN-5281, a stapled peptide, is currently in a clinical trial for treating orphan endocrine disorders (98). Disulfide bridges create folding and conformational constraints that can improve potency, selectivity, and stability. A number of disulfide bond-rich peptides are on the market or in preclinical or clinical development, e.g., linaclotide, lepirudin, and ziconotide (15).
-
Conjugation to Macromolecules
Conjugation to macromolecules (e.g., polyethylene glycol (PEG), albumin) is an effective strategy to improve stability of peptides and reduce renal clearance. This will be discussed in the “Renal Clearance” section.
Renal Clearance
Many peptides exhibit promising in vitro pharmacological activity but fail to demonstrate in vivo efficacy due to very short in vivo half-life (minutes). The rapid clearance and short half-life of peptides hamper their development into successful drugs. The main causes of rapid clearance of peptides from systemic circulation are enzymatic proteolysis or/and renal clearance. The glomeruli have a pore size of ∼8 nm, and hydrophilic peptides with MW <2–25 kDa are susceptible to rapid filtration through the glomeruli of the kidney. Since peptides are not easily reabsorbed through the renal tubule, they frequently have high renal clearance and short half-life. Other minor routes of peptide clearance are endocytosis and degradation by proteasome and the liver. Comparison between systemic and renal clearance in animal models provides useful information on whether renal clearance is likely to be a major elimination pathway.
For renal-impaired patients, dose adjustment may be needed for peptide drugs to avoid accumulation and higher drug exposure, as inappropriate dosing in patients with renal dysfunction can cause toxicity or ineffective therapy (99,100). Several strategies have been developed to reduce peptide renal clearance and prolong half-life. These will be reviewed next.
-
Increase plasma protein binding
Renal clearance of peptides is reduced when they are bound to membrane proteins or serum proteins. An example is the cyclic peptide drug octreotide, a treatment for endocrine tumors, which has about 100 min half-life in humans due to binding to lipoproteins (fraction unbound 0.65) (101,102).
-
Covalent Linkage to Albumin-Binding Small Molecules
Covalently attaching albumin-binding small molecules to peptides can reduce glomerular filtration, improve proteolytic stability, and prolong half-life by indirectly interacting with albumin through the highly bound small molecules (74). Liraglutide is a GLP-1 analog that is linked via a γ-l-glutamyl spacer to a 16-carbon fatty acid residue (Fig. 2) (103). The lipopeptide binds to albumin, thus decreasing proteolysis and renal clearance (104–106). Half-life increased to 8 h after IV administration compared to a few minutes for native GLP-1. The SC half-life of liraglutide is 11–15 h, enabling QD dosing. Another example is a bicyclic peptide linked to an albumin-binding peptide (107). The conjugate was completely resistant to proteolysis and had a 50-fold longer half-life (107).
-
Conjugation to Large Polymers
Conjugation of peptides to large synthetic or natural polymers or carbohydrates can increase their molecular weight and hydrodynamic volume, thus reducing their renal clearance. The common polymers used for peptide conjugation are PEG, polysialic acid (PSA), and hydroxyethyl starch (HES). An example is peginesatide, a PEGylated synthetic peptide approved by the FDA recently for the treatment of anemia associated with chronic kidney disease but was withdrawn as a result of new post-marketing reports of serious hypersensitivity reactions (http://www.takeda.com/news/2013/20130701_5854.html). It has an elimination half-life of 18.9 h after IV administration in healthy volunteers (108).
-
Fusion to Long-Live Plasma Proteins
Plasma proteins, such as albumin and immunoglobulin (IgG) fragments, have long half-lives of 19–21 days in humans (74). Because of the high MW (67–150 kDa), these proteins have low renal clearance, and their binding to neonatal Fc receptor (FcRn) reduces the elimination through pinocytosis by the vascular epithelium. Covalent linkage of peptides to albumin or IgG fragments can reduce renal clearance and prolong half-life. An example is the albumin-exendin-4 conjugate (CJC-1134-PC). It has a half-life of ~8 days in humans and is currently in a phase II clinical trial for the treatment of type II diabetes mellitus (109). The recently FDA-approved drug, albiglutide, is a DPPIV-resistant GLP-1 dimer fused to human albumin and has a half-life of 6–7 days, which enables weekly dosing for the treatment of type 2 diabetics (110,111).
Fig. 2.
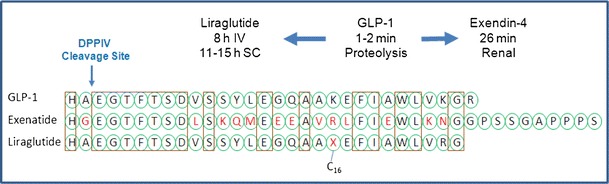
Strategies to enhance peptide stability: GLP-1 peptide has a half-life of 1–2 min. Modification of labile amino acids resulted in exenatide having enhanced serum stability, and clearance shifted from proteolysis to renal clearance, half-life 26 min. Addition of palmitoyl chain (C16 fatty acid) to GLP-1 formed liraglutide with decreased proteolysis and renal clearance, half-life 8 h IV, 11–15 h SC enabled QD dosing
Predicting PK Parameters of Peptides
Predictions of peptide PK and PK/PD are important for candidate selection and dose regimen design. Peptide clearance mechanisms can be similar to either small molecules or proteins depending on their structures and physicochemical properties. Certain small and lipophilic peptides (cyclosporine, bortezomib) have clearance mechanisms similar to small molecules (e.g., via P450-mediated metabolism) (112–114). Strategies that apply to small molecule drugs can be used to scale human in vivo clearance of peptides (115). If peptides behave more like proteins that are eliminated through proteolysis, renal filtration, catabolism, and endocytosis, allometric scaling appears to be successful in predicting human PK parameters from preclinical species (22,116). A number of studies have shown that allometric scaling is effective in predicting human volume of distribution and clearance with some exceptions (22,117–120).
Because of the ubiquitous distribution of proteases throughout the body, proteolytic degradation is not limited to classical clearance organs (e.g., liver, kidney). Allometric scaling has been shown to provide satisfying human clearance prediction in the absence of nonlinear PK and species-specific clearance mechanisms. In a comprehensive study of 34 therapeutic proteins, including 12 monoclonal antibodies and Fc fusion proteins, human clearance values were reasonably well predicted with simple allometric scaling and a fixed exponent of 0.8 (116). About 95% was within 2-fold of the observed values when using all available species (e.g., mouse, rat, dog, monkey, rabbit) or about 90% using monkey single species scaling (116). Evaluation of single species scaling with a fixed exponent of 0.75 using rat clearance of 20 peptides showed good prediction of human clearance with a correlation coefficient of 0.90 (Fig. 3). Applying a fixed exponent with one to two preclinical species is simple, resource saving, and minimizes systematic bias compared to a fitted exponent method (116). However, due to potentially unrecognized pitfalls of allometric scaling (e.g., species-dependent clearance mechanisms), special cautions (e.g., understanding of clearance mechanisms) need to be applied when using this approach (22).
Fig. 3.
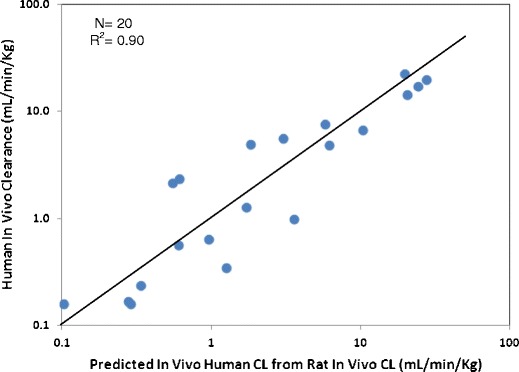
Correlation of observed human clearance and predicted values from single species scaling from rat clearance for 20 peptides. CLhuman (L/h) = CLrat (L/h) × (BWhuman / BWrat)0.75
Peptides cannot cross biomembranes easily and, therefore, are mostly confined in the extracellular space. Diffusion and convection are both involved in the distribution of peptides and the relative contribution is dependent on the size and structure of the peptides (22). Volume of distribution of peptides is typically small and not greater than the volume of the extracellular body fluid (Vss < 15 L or 0.2 L/Kg). Vss is reasonably well predicted with allometric scaling using animal data with an exponent near 1 (22).
Some peptides showed nonlinear PK caused by saturation of target-mediated drug disposition (TMDD). A combined model of TMDD and allometric scaling was able to simultaneously describe preclinical PK of exenatide from mouse, rat, and monkey following both IV and SC dosing (121). The model structure was successfully applied to predict human concentration–time profiles (121). The advantages of such mechanistic models compared to empirical models are their abilities to extrapolate from preclinical to clinical species and from healthy volunteers to disease state and special populations.
CONCLUSIONS
Development of peptide drugs is, no doubt, not only full of challenges and risks but also offers great potential and promise. Future enhancement of the ADME tools will help accelerate the development of peptides into successful drugs. Developing deeper understanding of physicochemical properties that govern peptide conformation is critical to assessing the impact on potency and ADME properties (e.g., permeability, stability, and PK). Predictive in silico or rule-based ADME tools are useful to guide peptide design with improved drug-like properties while maintaining target potency. Transporters can play a vital role in uptake of peptides for enhanced oral absorption and cell membrane penetration. Effective and physiologically relevant transporter assays will help define substrate specificity and guide peptide design with improved uptake and transport characteristics. Continued refinement of mechanistic PK and PD models will provide powerful insights in designing future generations of peptide drugs with the greatest safety and efficacy.
Acknowledgments
The author would like to thank Karen Atkinson for editing the manuscript; Angela Doran, Angela Wolford, and Amit Kalgutkar for the study on peptide PK prediction; and Larry Tremaine, Tess Wilson, and Charlotte Allerton for their leadership and support.
References
- 1.Hopkins AL, Groom CR. Opinion: the druggable genome. Nat Rev Drug Discov. 2002;1(9):727–30. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 2.Gongora-Benitez M, Tulla-Puche J, Albericio F. Multifaceted roles of disulfide bonds. Peptides as therapeutics. Chem Rev (Washington, DC, U S) 2014;114(2):901–26. doi: 10.1021/cr400031z. [DOI] [PubMed] [Google Scholar]
- 3.Sun L. Peptide-based drug development. Mod Chem Appl. 2013;1(1):1–2. [Google Scholar]
- 4.Goodwin D, Simerska P, Toth I. Peptides as therapeutics with enhanced bioactivity. Curr Med Chem. 2012;19(26):4451–61. doi: 10.2174/092986712803251548. [DOI] [PubMed] [Google Scholar]
- 5.Craik DJ, Fairlie DP, Liras S, Price D. The future of peptide-based drugs. Chem Biol Drug Des. 2013;81(1):136–47. doi: 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]
- 6.Kaspar AA, Reichert JM. Future directions for peptide therapeutics development. Drug Discov Today. 2013;18(17–18):807–17. doi: 10.1016/j.drudis.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 7.Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Synthetic therapeutic peptides: science and market. Drug Discov Today. 2010;15(1/2):40–56. doi: 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 8.Ladner RC, Sato AK, Gorzelany J, De Souza M. Phage display-derived peptides as therapeutic alternatives to antibodies. Drug Discov Today. 2004;9(12):525–9. doi: 10.1016/S1359-6446(04)03104-6. [DOI] [PubMed] [Google Scholar]
- 9.Lax R, Meenan C. Challenges for therapeutic peptides part 1: on the inside, looking out. Innovations Pharm Technol. 2012;42:54–6. [Google Scholar]
- 10.Lax R, Meenan C. Challenges for therapeutic peptides part 2: delivery systems. Innovations Pharm Technol. 2012;43:42–4. [Google Scholar]
- 11.Bray BL. Innovation: large-scale manufacture of peptide therapeutics by chemical synthesis. Nat Rev Drug Discov. 2003;2(7):587–93. doi: 10.1038/nrd1133. [DOI] [PubMed] [Google Scholar]
- 12.Edmonds DJ, Price DA. Oral GLP-1 modulators for the treatment of diabetes. Annu Rep Med Chem. 2013;48:119–30. [Google Scholar]
- 13.Rezai T, Bock JE, Zhou MV, Kalyanaraman C, Lokey RS, Jacobson MP. Conformational flexibility, internal hydrogen bonding, and passive membrane permeability: successful in silico prediction of the relative permeabilities of cyclic peptides. J Am Chem Soc. 2006;128(43):14073–80. doi: 10.1021/ja063076p. [DOI] [PubMed] [Google Scholar]
- 14.Mahato RI, Narang AS, Thoma L, Miller DD. Emerging trends in oral delivery of peptide and protein drugs. Crit Rev Ther Drug Carrier Syst. 2003;20(2–3):153–214. doi: 10.1615/critrevtherdrugcarriersyst.v20.i23.30. [DOI] [PubMed] [Google Scholar]
- 15.Diao L, Meibohm B. Pharmacokinetics and pharmacokinetic-pharmacodynamic correlations of therapeutic peptides. Clin Pharmacokinet. 2013;52(10):855–68. doi: 10.1007/s40262-013-0079-0. [DOI] [PubMed] [Google Scholar]
- 16.Rand AC, Leung SSF, Eng H, Rotter CJ, Sharma R, Kalgutkar AS, et al. Optimizing PK properties of cyclic peptides: the effect of side chain substitutions on permeability and clearance. Med Chem Comm. 2012;3(10):1282–9. doi: 10.1039/C2MD20203D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Werle M, Bernkop-Schnuerch A. Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids. 2006;30(4):351–67. doi: 10.1007/s00726-005-0289-3. [DOI] [PubMed] [Google Scholar]
- 18.Zhou XH, Li Wan Po A. Peptide and protein drugs: II. Non-parenteral routes of delivery. Int J Pharm. 1991;75(2–3):117–30. [Google Scholar]
- 19.Hellriegel ET, Bjornsson TD, Hauck WW. Interpatient variability in bioavailability is related to the extent of absorption: implications for bioavailability and bioequivalence studies. Clin Pharmacol Ther (St Louis) 1996;60(6):601–7. doi: 10.1016/S0009-9236(96)90208-8. [DOI] [PubMed] [Google Scholar]
- 20.Maher S, Brayden DJ. Overcoming poor permeability: translating permeation enhancers for oral peptide delivery. Drug Discov Today: Technol. 2012;9(2):e113–e9. doi: 10.1016/j.ddtec.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 21.Chin J, Foyez Mahmud KA, Kim SE, Park K, Byun Y. Insight of current technologies for oral delivery of proteins and peptides. Drug Discov Today: Technol. 2012;9(2):e105–e12. doi: 10.1016/j.ddtec.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 22.Lin JH. Pharmacokinetics of biotech drugs: peptides, proteins and monoclonal antibodies. Curr Drug Metab. 2009;10(7):661–91. doi: 10.2174/138920009789895499. [DOI] [PubMed] [Google Scholar]
- 23.Periti P, Mazzei T, Mini E. Clinical pharmacokinetics of depot leuprorelin. Clin Pharmacokinet. 2002;41(7):485–504. doi: 10.2165/00003088-200241070-00003. [DOI] [PubMed] [Google Scholar]
- 24.Munegumi T. Hydrophobicity of peptides containing D-amino acids. Chem Biodivers. 2010;7(6):1670–9. doi: 10.1002/cbdv.200900370. [DOI] [PubMed] [Google Scholar]
- 25.Ano R, Kimura Y, Shima M, Matsuno R, Ueno T, Akamatsu M. Relationships between structure and high-throughput screening permeability of peptide derivatives and related compounds with artificial membranes: application to prediction of Caco-2 cell permeability. Bioorg Med Chem. 2004;12(1):257–64. doi: 10.1016/j.bmc.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 26.Kramer SD, Wunderli-Allenspach H. No entry for TAT(44–57) into liposomes and intact MDCK cells: novel approach to study membrane permeation of cell-penetrating peptides. Biochim Biophys Acta Biomembr. 2003;1609(2):161–9. doi: 10.1016/s0005-2736(02)00683-1. [DOI] [PubMed] [Google Scholar]
- 27.Tang F, Borchardt RT. Characterization of the efflux transporter(s) responsible for restricting intestinal mucosa permeation of the coumarinic acid-based cyclic prodrug of the opioid peptide DADLE. Pharm Res. 2002;19(6):787–93. doi: 10.1023/a:1016196514217. [DOI] [PubMed] [Google Scholar]
- 28.Ano R, Kimura Y, Urakami M, Shima M, Matsuno R, Ueno T, et al. Relationship between structure and permeability of dipeptide derivatives containing tryptophan and related compounds across human intestinal epithelial (Caco-2) cells. Bioorg Med Chem. 2004;12(1):249–55. doi: 10.1016/j.bmc.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 29.Stevenson CL, Augustijns PF, Hendren RW. Use of Caco-2 cells and LC/MS/MS to screen a peptide combinatorial library for permeable structures. Int J Pharm. 1999;177(1):103–15. doi: 10.1016/s0378-5173(98)00331-7. [DOI] [PubMed] [Google Scholar]
- 30.Beck JG, Chatterjee J, Laufer B, Kiran MU, Frank AO, Neubauer S, et al. Intestinal permeability of cyclic peptides: common key backbone motifs identified. J Am Chem Soc. 2012;134(29):12125–33. doi: 10.1021/ja303200d. [DOI] [PubMed] [Google Scholar]
- 31.Bhardwaj RK, Herrera-Ruiz D, Sinko PJ, Gudmundsson OS, Knipp G. Delineation of human peptide transporter 1 (hPepT1)-mediated uptake and transport of substrates with varying transporter affinities utilizing stably transfected hPepT1/Madin-Darby canine kidney clones and Caco-2 cells. J Pharmacol Exp Ther. 2005;314(3):1093–100. doi: 10.1124/jpet.105.087148. [DOI] [PubMed] [Google Scholar]
- 32.Faria TN, Timoszyk JK, Stouch TR, Vig BS, Landowski CP, Amidon GL, et al. A novel high-throughput PepT1 transporter assay differentiates between substrates and antagonists. Mol Pharm. 2004;1(1):67–76. doi: 10.1021/mp034001k. [DOI] [PubMed] [Google Scholar]
- 33.Balimane PV, Chong S, Patel K, Quan Y, Timoszyk J, Han Y-H, et al. Peptide transporter substrate identification during permeability screening in drug discovery: comparison of transfected MDCK-hPepT1 cells to Caco-2 cells. Arch Pharmacal Res. 2007;30(4):507–18. doi: 10.1007/BF02980227. [DOI] [PubMed] [Google Scholar]
- 34.Vadlapudi AD, Vadlapatla RK, Mitra AK. Sodium dependent multivitamin transporter (SMVT): a potential target for drug delivery. Curr Drug Targets. 2012;13(7):994–1003. doi: 10.2174/138945012800675650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stenberg P, Luthman K, Artursson P. Prediction of membrane permeability to peptides from calculated dynamic molecular surface properties. Pharm Res. 1999;16(2):205–12. doi: 10.1023/a:1018816122458. [DOI] [PubMed] [Google Scholar]
- 36.Rafi SB, Hearn BR, Vedantham P, Jacobson MP, Renslo AR. Predicting and improving the membrane permeability of peptidic small molecules. J Med Chem. 2012;55(7):3163–9. doi: 10.1021/jm201634q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jappar D, Hu Y, Smith DE. Effect of dose escalation on the in vivo oral absorption and disposition of glycylsarcosine in wild-type and Pept1 knockout mice. Drug Metab Dispos. 2011;39(12):2250–7. doi: 10.1124/dmd.111.041087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li W, Zhang J, Tse FLS. Handbook of LC-MS bioanalysis: best practices, experimental protocols, and regulations 2013.
- 39.Letzel T, Editor. Protein and peptide analysis by LC-MS: experimental strategies. [In: RSC Chromatogr. Monogr., 2011; 15]2011. 172 pp.
- 40.van den Broek I, Sparidans RW, Schellens JHM, Beijnen JH. Quantitative bioanalysis of peptides by liquid chromatography coupled to (tandem) mass spectrometry. J Chromatogr B Anal Technol Biomed Life Sci. 2008;872(1–2):1–22. doi: 10.1016/j.jchromb.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 41.Li W, Zhang J, Tse FLS. Strategies in quantitative LC-MS/MS analysis of unstable small molecules in biological matrices. Biomed Chromatogr. 2011;25(1–2):258–77. doi: 10.1002/bmc.1572. [DOI] [PubMed] [Google Scholar]
- 42.Nowatzke WL, Rogers K, Wells E, Bowsher RR, Ray C, Unger S. Unique challenges of providing bioanalytical support for biological therapeutic pharmacokinetic programs. Bioanalysis. 2011;3(5):509–21. doi: 10.4155/bio.11.2. [DOI] [PubMed] [Google Scholar]
- 43.Kuhn B, Mohr P, Stahl M. Intramolecular hydrogen bonding in medicinal chemistry. J Med Chem. 2010;53(6):2601–11. doi: 10.1021/jm100087s. [DOI] [PubMed] [Google Scholar]
- 44.Lokey RS. Testing the conformational hypothesis of membrane permeability using cyclic peptide diastereomers. Abstracts of Papers, 232nd ACS National Meeting, San Francisco, CA, United States, Sept 10–14, 2006. 2006:BIOL-167.
- 45.Rezai T, Yu B, Millhauser GL, Jacobson MP, Lokey RS. Testing the conformational hypothesis of passive membrane permeability using synthetic cyclic peptide diastereomers. J Am Chem Soc. 2006;128(8):2510–1. doi: 10.1021/ja0563455. [DOI] [PubMed] [Google Scholar]
- 46.White TR, Renzelman CM, Rand AC, Rezai T, McEwen CM, Gelev VM, et al. On-resin N-methylation of cyclic peptides for discovery of orally bioavailable scaffolds. Nat Chem Biol. 2011;7(11):810–7. doi: 10.1038/nchembio.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alex A, Millan DS, Perez M, Wakenhut F, Whitlock GA. Intramolecular hydrogen bonding to improve membrane permeability and absorption in beyond rule of five chemical space. Med Chem Comm. 2011;2(7):669–74. [Google Scholar]
- 48.Milletti F. Cell-penetrating peptides: classes, origin, and current landscape. Drug Discov Today. 2012;17(15–16):850–60. doi: 10.1016/j.drudis.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 49.Tressel SL, Koukos G, Tchernychev B, Jacques SL, Covic L, Kuliopulos A. Pharmacology, biodistribution, and efficacy of GPCR-based pepducins in disease models. Methods Mol Biol (N Y, NY, U S) 2011;683:259–75. doi: 10.1007/978-1-60761-919-2_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang J, Shen D, Shen W-C. Preparation, purification, and characterization of a reversibly lipidized desmopressin with potentiated antidiuretic activity. Pharm Res. 1999;16(11):1674–9. doi: 10.1023/a:1018929312715. [DOI] [PubMed] [Google Scholar]
- 51.Wang J, Chow D, Heiati H, Shen W-C. Reversible lipidization for the oral delivery of salmon calcitonin. J Control Release. 2003;88(3):369–80. doi: 10.1016/s0168-3659(03)00008-7. [DOI] [PubMed] [Google Scholar]
- 52.Wang J, Shen W-C. Gastric retention and stability of lipidized Bowman-Birk protease inhibitor in mice. Int J Pharm. 2000;204(1–2):111–6. doi: 10.1016/s0378-5173(00)00489-0. [DOI] [PubMed] [Google Scholar]
- 53.Chae SY, Jin C-H, Shin HJ, Youn YS, Lee S, Lee KC. Preparation, characterization, and application of biotinylated and biotin-PEGylated glucagon-like peptide-1 analogues for enhanced oral delivery. Bioconjugate Chem. 2008;19(1):334–41. doi: 10.1021/bc700292v. [DOI] [PubMed] [Google Scholar]
- 54.Clardy-James S, Chepurny OG, Leech CA, Holz GG, Doyle RP. Synthesis, characterization and pharmacodynamics of vitamin-B12-conjugated glucagon-like peptide-1. ChemMedChem. 2013;8(4):582–6. doi: 10.1002/cmdc.201200461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clardy SM, Allis DG, Fairchild TJ, Doyle RP. Vitamin B12 in drug delivery: breaking through the barriers to a B12 bioconjugate pharmaceutical. Expert Opin Drug Deliv. 2011;8(1):127–40. doi: 10.1517/17425247.2011.539200. [DOI] [PubMed] [Google Scholar]
- 56.Shaji J, Patole V. Protein and peptide drug delivery: oral approaches. Indian J Pharm Sci. 2008;70(3):269–77. doi: 10.4103/0250-474X.42967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aungst BJ. Intestinal permeation enhancers. J Pharm Sci. 2000;89(4):429–42. doi: 10.1002/(SICI)1520-6017(200004)89:4<429::AID-JPS1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 58.Whitehead K, Karr N, Mitragotri S. Safe and effective permeation enhancers for oral drug delivery. Pharm Res. 2008;25(8):1782–8. doi: 10.1007/s11095-007-9488-9. [DOI] [PubMed] [Google Scholar]
- 59.LeCluyse EL, Sutton SC. In vitro models for selection of development candidates. Permeability studies to define mechanisms of absorption enhancement. Adv Drug Deliv Rev. 1997;23(1–3):163–83. [Google Scholar]
- 60.Wang X, Maher S, Brayden DJ. Restoration of rat colonic epithelium after in situ intestinal instillation of the absorption promoter, sodium caprate. Ther Deliv. 2010;1(1):75–82. doi: 10.4155/tde.10.5. [DOI] [PubMed] [Google Scholar]
- 61.Goldberg M, Gomez-Orellana I. Challenges for the oral delivery of macromolecules. Nat Rev Drug Discov. 2003;2(4):289–95. doi: 10.1038/nrd1067. [DOI] [PubMed] [Google Scholar]
- 62.Puente XS, Gutierrez-Fernandez A, Ordonez GR, Hillier LW, Lopez-Otin C. Comparative genomic analysis of human and chimpanzee proteases. Genomics. 2005;86(6):638–47. doi: 10.1016/j.ygeno.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 63.Woodley JF. Enzymatic barriers for GI peptide and protein delivery. Crit Rev Ther Drug Carrier Syst. 1994;11(2–3):61–95. [PubMed] [Google Scholar]
- 64.Powell MF, Grey H, Gaeta F, Sette A, Colon S. Peptide stability in drug development: a comparison of peptide reactivity in different biological media. J Pharm Sci. 1992;81(8):731–5. doi: 10.1002/jps.2600810802. [DOI] [PubMed] [Google Scholar]
- 65.Powell MF, Stewart T, Otvos L, Jr, Urge L, Gaeta FCA, Sette A, et al. Peptide stability in drug development. II. Effect of single amino acid substitution and glycosylation on peptide reactivity in human serum. Pharm Res. 1993;10(9):1268–73. doi: 10.1023/a:1018953309913. [DOI] [PubMed] [Google Scholar]
- 66.Noto PB, Abbadessa G, Cassone M, Mateo GD, Agelan A, Wade JD, et al. Alternative stabilities of a proline-rich antibacterial peptide in vitro and in vivo. Protein Sci. 2008;17(7):1249–55. doi: 10.1110/ps.034330.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eng H, Sharma R, McDonald TS, Landis MS, Stevens BD, Kalgutkar AS. Pharmacokinetics and metabolism studies on the glucagon-like peptide-1 (GLP-1)-derived metabolite GLP-1(9–36)amide in male Beagle dogs. Xenobiotica. 2014;44(9):842–8. doi: 10.3109/00498254.2014.897011. [DOI] [PubMed] [Google Scholar]
- 68.Sharma R, McDonald TS, Eng H, Limberakis C, Stevens BD, Patel S, et al. In vitro metabolism of the glucagon-like peptide-1 (GLP-1)-derived metabolites GLP-1(9–36)amide and GLP-1(28–36)amide in mouse and human hepatocytes. Drug Metab Dispos. 2013;41(12):2148–57. doi: 10.1124/dmd.113.054254. [DOI] [PubMed] [Google Scholar]
- 69.Adessi C, Soto C. Converting a peptide into a drug: strategies to improve stability and bioavailability. Curr Med Chem. 2002;9(9):963–78. doi: 10.2174/0929867024606731. [DOI] [PubMed] [Google Scholar]
- 70.Linde Y, Ovadia O, Safrai E, Xiang Z, Portillo FP, Shalev DE, et al. Structure-activity relationship and metabolic stability studies of backbone cyclization and N-methylation of melanocortin peptides. Biopolymers. 2008;90(5):671–82. doi: 10.1002/bip.21057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ovadia O, Linde Y, Haskell-Luevano C, Dirain ML, Sheynis T, Jelinek R, et al. The effect of backbone cyclization on PK/PD properties of bioactive peptide-peptoid hybrids: the melanocortin agonist paradigm. Bioorg Med Chem. 2010;18(2):580–9. doi: 10.1016/j.bmc.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 72.Hess S, Linde Y, Ovadia O, Safrai E, Shalev DE, Swed A, et al. Backbone cyclic peptidomimetic melanocortin-4 receptor agonist as a novel orally administrated drug lead for treating obesity. J Med Chem. 2008;51(4):1026–34. doi: 10.1021/jm701093y. [DOI] [PubMed] [Google Scholar]
- 73.Byk G, Halle D, Zeltser I, Bitan G, Selinger Z, Gilon C. Synthesis and biological activity of NK-1 selective, N-backbone cyclic analogs of the C-terminal hexapeptide of substance P. J Med Chem. 1996;39(16):3174–8. doi: 10.1021/jm960154i. [DOI] [PubMed] [Google Scholar]
- 74.Pollaro L, Heinis C. Strategies to prolong the plasma residence time of peptide drugs. Med Chem Comm. 2010;1(5):319–24. [Google Scholar]
- 75.Pisal DS, Kosloski MP, Balu-Iyer SV. Delivery of therapeutic proteins. J Pharm Sci. 2010;99(6):2557–75. doi: 10.1002/jps.22054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sato AK, Viswanathan M, Kent RB, Wood CR. Therapeutic peptides: technological advances driving peptides into development. Curr Opin Biotechnol. 2006;17(6):638–42. doi: 10.1016/j.copbio.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 77.John H, Maronde E, Forssmann W-G, Meyer M, Adermann K. N-terminal acetylation protects glucagon-like peptide GLP-1-(7–34)-amide from DPP-IV-mediated degradation retaining cAMP-and insulin releasing capacity. Eur J Med Res. 2008;13(2):73–8. [PubMed] [Google Scholar]
- 78.Stroemstedt AA, Pasupuleti M, Schmidtchen A, Malmsten M. Evaluation of strategies for improving proteolytic resistance of antimicrobial peptides by using variants of EFK17, an internal segment of LL-37. Antimicrob Agents Chemother. 2009;53(2):593–602. doi: 10.1128/AAC.00477-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ferdinandi ES, Brazeau P, High K, Procter B, Fennell S, Dubreuil P. Non-clinical pharmacology and safety evaluation of TH9507, a human growth hormone-releasing factor analogue. Basic Clin Pharmacol Toxicol. 2007;100(1):49–58. doi: 10.1111/j.1742-7843.2007.00008.x. [DOI] [PubMed] [Google Scholar]
- 80.Sharman A, Low J. Vasopressin and its role in critical care. Contin Educ Anaesth, Crit Care Pain. 2008;8(4):134–7. [Google Scholar]
- 81.Agerso H, Larsen LS, Riis A, Lovgren U, Karlsson MO, Senderovitz T. Pharmacokinetics and renal excretion of desmopressin after intravenous administration to healthy subjects and renally impaired patients. Br J Clin Pharmacol. 2004;58(4):352–8. doi: 10.1111/j.1365-2125.2004.02175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Harris AG. Somatostatin and somatostatin analogues: pharmacokinetics and pharmacodynamic effects. Gut. 1994;35(3 Suppl):S1–4. doi: 10.1136/gut.35.3_suppl.s1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen S, Gfeller D, Buth SA, Michielin O, Leiman PG, Heinis C. Improving binding affinity and stability of peptide ligands by substituting glycines with D-amino acids. Chem Bio Chem. 2013;14(11):1316–22. doi: 10.1002/cbic.201300228. [DOI] [PubMed] [Google Scholar]
- 84.Tugyi R, Uray K, Ivan D, Fellinger E, Perkins A, Hudecz F. Partial D-amino acid substitution: improved enzymatic stability and preserved Ab recognition of a MUC2 epitope peptide. Proc Natl Acad Sci U S A. 2005;102(2):413–8. doi: 10.1073/pnas.0407677102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Darlak K, Benovitz DE, Spatola AF, Grzonka Z. Dermorphin analogs: resistance to in vitro enzymatic degradation is not always increased by additional D-amino acid substitutions. Biochem Biophys Res Commun. 1988;156(1):125–30. doi: 10.1016/s0006-291x(88)80813-1. [DOI] [PubMed] [Google Scholar]
- 86.Rafferty B, Coy DH, Poole S. Pharmacokinetic evaluation of superactive analogues of growth hormone-releasing factor (1–29)-amide. Peptides. 1988;9(1):207–9. doi: 10.1016/0196-9781(88)90029-0. [DOI] [PubMed] [Google Scholar]
- 87.Nattel S, Carlsson L. Innovative approaches to anti-arrhythmic drug therapy. Nat Rev Drug Discov. 2006;5(12):1034–49. doi: 10.1038/nrd2112. [DOI] [PubMed] [Google Scholar]
- 88.Welch BD, Francis JN, Redman JS, Paul S, Weinstock MT, Reeves JD, et al. Design of a potent D-peptide HIV-1 entry inhibitor with a strong barrier to resistance. J Virol. 2010;84(21):11235–44. doi: 10.1128/JVI.01339-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Verschraegen CF, Westphalen S, Hu W, Loyer E, Kudelka A, Volker P, et al. Phase II study of cetrorelix, a luteinizing hormone-releasing hormone antagonist in patients with platinum-resistant ovarian cancer. Gynecol Oncol. 2003;90(3):552–9. doi: 10.1016/s0090-8258(03)00408-6. [DOI] [PubMed] [Google Scholar]
- 90.Heredi-Szabo K, Murphy RF, Lovas S. Is IGnRH-III the most potent GnRH analog containing only natural amino acids that specifically inhibits the growth of human breast cancer cells? J Pept Sci. 2006;12(11):714–20. doi: 10.1002/psc.783. [DOI] [PubMed] [Google Scholar]
- 91.Raun K, Hansen BS, Johansen NL, Thogersen H, Madsen K, Ankersen M, et al. Ipamorelin, the first selective growth hormone secretagogue. Eur J Endocrinol. 1998;139(5):552–61. doi: 10.1530/eje.0.1390552. [DOI] [PubMed] [Google Scholar]
- 92.Gobburu JVS, Agerso H, Jusko WJ, Ynddal L. Pharmacokinetic-pharmacodynamic modeling of ipamorelin, a growth hormone releasing peptide, in human volunteers. Pharm Res. 1999;16(9):1412–6. doi: 10.1023/a:1018955126402. [DOI] [PubMed] [Google Scholar]
- 93.Weber SJ, Greene DL, Hruby VJ, Yamamura HI, Porreca F, Davis TP. Whole body and brain distribution of [3H]cyclic [D-Pen2, D-Pen5]enkephalin after intraperitoneal, intravenous, oral and subcutaneous administration. J Pharmacol Exp Ther. 1992;263(3):1308–16. [PubMed] [Google Scholar]
- 94.Tugyi R, Mezo G, Fellinger E, Andreu D, Hudecz F. The effect of cyclization on the enzymatic degradation of herpes simplex virus glycoprotein D derived epitope peptide. J Pept Sci. 2005;11(10):642–9. doi: 10.1002/psc.669. [DOI] [PubMed] [Google Scholar]
- 95.Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, et al. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science (Washington, DC, U S) 2004;305(5689):1466–70. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL. Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide [Erratum to document cited in CA146:397011] J Am Chem Soc. 2007;129(16):5298. doi: 10.1021/ja0693587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bird GH, Madani N, Perry AF, Princiotto AM, Supko JG, He X, et al. Hydrocarbon double-stapling remedies the proteolytic instability of a lengthy peptide therapeutic. Proc Natl Acad Sci U S A. 2010;107(32):14093–8. doi: 10.1073/pnas.1002713107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Grigoryev Y. Stapled peptide to enter human testing, but affinity questions remain. Nat Med (N Y, NY, U S) 2013;19(2):120. doi: 10.1038/nm0213-120a. [DOI] [PubMed] [Google Scholar]
- 99.Czock D, Keller F, Seidling HM. Pharmacokinetic predictions for patients with renal impairment: focus on peptides and protein drugs. Br J Clin Pharmacol. 2012;74(1):66–74. doi: 10.1111/j.1365-2125.2012.04172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Verbeeck RK, Musuamba FT. Pharmacokinetics and dosage adjustment in patients with renal dysfunction. Eur J Clin Pharmacol. 2009;65(8):757–73. doi: 10.1007/s00228-009-0678-8. [DOI] [PubMed] [Google Scholar]
- 101.Chanson P, Timsit J, Harris AG. Clinical pharmacokinetics of octreotide. Therapeutic applications in patients with pituitary tumours. Clin Pharmacokinet. 1993;25(5):375–91. doi: 10.2165/00003088-199325050-00004. [DOI] [PubMed] [Google Scholar]
- 102.Kutz K, Nuesch E, Rosenthaler J. Pharmacokinetics of SMS 201–995 in healthy subjects. Scand J Gastroenterol Suppl. 1986;119:65–72. doi: 10.3109/00365528609087433. [DOI] [PubMed] [Google Scholar]
- 103.Malm-Erjefalt M, Bjoernsdottir I, Vanggaard J, Helleberg H, Larsen U, Oosterhuis B, et al. Metabolism and excretion of the once-daily human glucagon-like peptide-1 analog liraglutide in healthy male subjects and its in vitro degradation by dipeptidyl peptidase IV and neutral endopeptidase. Drug Metab Dispos. 2010;38(11):1944–53. doi: 10.1124/dmd.110.034066. [DOI] [PubMed] [Google Scholar]
- 104.Hou J, Manaenko A, Hakon J, Hansen-Schwartz J, Tang J, Zhang JH. Liraglutide, a long-acting GLP-1 mimetic, and its metabolite attenuate inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2012;32(12):2201–10. doi: 10.1038/jcbfm.2012.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Levy Odile E, Jodka Carolyn M, Ren Shijun S, Mamedova L, Sharma A, Samant M, et al. Novel exenatide analogs with peptidic albumin binding domains: potent anti-diabetic agents with extended duration of action. PLoS One. 2014;9(2):e87704. doi: 10.1371/journal.pone.0087704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lindgren J, Refai E, Zaitsev Sergei V, Abrahmsen L, Berggren P-O, Karlstrom AE. A GLP-1 receptor agonist conjugated to an albumin-binding domain for extended half-life. Biopolymers. 2014;102(3):252–9. doi: 10.1002/bip.22474. [DOI] [PubMed] [Google Scholar]
- 107.Angelini A, Morales-Sanfrutos J, Diderich P, Chen S, Heinis C. Bicyclization and tethering to albumin yields long-acting peptide antagonists. J Med Chem. 2012;55(22):10187–97. doi: 10.1021/jm301276e. [DOI] [PubMed] [Google Scholar]
- 108.Bronson J, Black A, Dhar TGM, Ellsworth BA, Merritt JR. To market, to market—2012. Annu Rep Med Chem. 2013;48:471–546. [Google Scholar]
- 109.Baggio LL, Huang Q, Cao X, Drucker DJ. An albumin-exendin-4 conjugate engages central and peripheral circuits regulating murine energy and glucose homeostasis. Gastroenterology. 2008;134(4):1137–47. doi: 10.1053/j.gastro.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 110.Poole RM, Nowlan ML. Albiglutide: first global approval. Drugs. 2014:Ahead of Print. [DOI] [PubMed]
- 111.Pratley RE, Nauck MA, Barnett AH, Feinglos MN, Ovalle F, Harman-Boehm I, et al. Once-weekly albiglutide versus once-daily liraglutide in patients with type 2 diabetes inadequately controlled on oral drugs (HARMONY 7): a randomised, open-label, multicentre, non-inferiority phase 3 study. Lancet Diabetes Endocrinol. 2014;2(4):289–97. doi: 10.1016/S2213-8587(13)70214-6. [DOI] [PubMed] [Google Scholar]
- 112.Delaforgea M, Bouille G, Jaouen M, Jankowski CK, Lamouroux C, Bensoussan C. Recognition and oxidative metabolism of cyclodipeptides by hepatic cytochrome P450. Peptides (N Y, NY, U S) 2001;22(4):557–65. doi: 10.1016/s0196-9781(01)00364-3. [DOI] [PubMed] [Google Scholar]
- 113.Wacher VJ, Silverman JA, Zhang Y, Benet LZ. Role of P-glycoprotein and cytochrome P450 3A in limiting oral absorption of peptides and peptidomimetics. J Pharm Sci. 1998;87(11):1322–30. doi: 10.1021/js980082d. [DOI] [PubMed] [Google Scholar]
- 114.Pekol T, Daniels JS, Labutti J, Parsons I, Nix D, Baronas E, et al. Human metabolism of the proteasome inhibitor bortezomib: identification of circulating metabolites. Drug Metab Dispos. 2005;33(6):771–7. doi: 10.1124/dmd.104.002956. [DOI] [PubMed] [Google Scholar]
- 115.Di L, Feng B, Goosen TC, Lai Y, Steyn SJ, Varma MV, et al. A perspective on the prediction of drug pharmacokinetics and disposition in drug research and development. Drug Metab Dispos. 2013;41(12):1975–93. doi: 10.1124/dmd.113.054031. [DOI] [PubMed] [Google Scholar]
- 116.Wang W, Prueksaritanont T. Prediction of human clearance of therapeutic proteins: simple allometric scaling method revisited. Biopharm Drug Dispos. 2010;31(4):253–63. doi: 10.1002/bdd.708. [DOI] [PubMed] [Google Scholar]
- 117.Mordenti J, Chen SA, Moore JA, Ferraiolo BL, Green JD. Interspecies scaling of clearance and volume of distribution data for five therapeutic proteins. Pharm Res. 1991;8(11):1351–9. doi: 10.1023/a:1015836720294. [DOI] [PubMed] [Google Scholar]
- 118.Richter WF, Gallati H, Schiller C-D. Animal pharmacokinetics of the tumor necrosis factor receptor-immunoglobulin fusion protein lenercept and their extrapolation to humans. Drug Metab Dispos. 1999;27(1):21–5. [PubMed] [Google Scholar]
- 119.Grene-Lerouge NAM, Bazin-Redureau MI, Debray M, Scherrmann JMG. Interspecies scaling of clearance and volume of distribution for digoxin-specific Fab. Toxicol Appl Pharmacol. 1996;138(1):84–9. doi: 10.1006/taap.1996.0101. [DOI] [PubMed] [Google Scholar]
- 120.Mahmood I. Interspecies scaling of protein drugs: prediction of clearance from animals to humans. J Pharm Sci. 2004;93(1):177–85. doi: 10.1002/jps.10531. [DOI] [PubMed] [Google Scholar]
- 121.Chen T, Mager DE, Kagan L. Interspecies modeling and prediction of human exenatide pharmacokinetics. Pharm Res. 2013;30(3):751–60. doi: 10.1007/s11095-012-0917-z. [DOI] [PMC free article] [PubMed] [Google Scholar]