

Abstract
At the Product Quality Research Institute (PQRI) Workshop held last January 14–15, 2014, participants from academia, industry, and governmental agencies involved in the development and regulation of nanomedicines discussed the current state of characterization, formulation development, manufacturing, and nonclinical safety evaluation of nanomaterial-containing drug products for human use. The workshop discussions identified areas where additional understanding of material attributes, absorption, biodistribution, cellular and tissue uptake, and disposition of nanosized particles would continue to inform their safe use in drug products. Analytical techniques and methods used for in vitro characterization and stability testing of formulations containing nanomaterials were discussed, along with their advantages and limitations. Areas where additional regulatory guidance and material characterization standards would help in the development and approval of nanomedicines were explored. Representatives from the US Food and Drug Administration (USFDA), Health Canada, and European Medicines Agency (EMA) presented information about the diversity of nanomaterials in approved and newly developed drug products. USFDA, Health Canada, and EMA regulators discussed the applicability of current regulatory policies in presentations and open discussion. Information contained in several of the recent EMA reflection papers was discussed in detail, along with their scope and intent to enhance scientific understanding about disposition, efficacy, and safety of nanomaterials introduced in vivo and regulatory requirements for testing and market authorization. Opportunities for interaction with regulatory agencies during the lifecycle of nanomedicines were also addressed at the meeting. This is a summary of the workshop presentations and discussions, including considerations for future regulatory guidance on drug products containing nanomaterials.
KEY WORDS: nanomaterials, nanomedicine, nanotechnology, PQRI, risk management, USFDA
INTRODUCTION
The Product Quality Research Institute (PQRI) Workshop for Nanomaterials in Drug Products (1) was designed as a collaborative effort among industry, academia, and the Center for Drug Evaluation and Research (CDER) Nanotechnology Working Group to discuss the current experience and risk management of potential risks from nanomaterials in drug products and to provide direction to future guidance efforts (2). The goals of the workshop were to address key areas for quality, safety, and efficacy of nanomaterial drug products, more specifically to:
Review analytical science and methods for characterizing nanomaterials. Discuss their application to the characterization and quality control of drug products;
Share experience and results using multiple formulation platforms for the same active pharmaceutical ingredient (API): effects on quality, safety, and efficacy of drug products containing nanosize constituents;
Discuss approaches to the management of potential risks of nanomaterials in drug products starting from early drug development and throughout product lifecycle. Implications for maintaining quality, safety, and efficacy were highlighted;
Gather input regarding the considerations for utilizing nanotechnology in pharmaceutical products;
Present experience and perspectives from international regulatory agencies and standard setting organizations on the use of nanotechnology in pharmaceutical products; and
Discuss areas where additional research on the effects of nanosize API on absorption, distribution, metabolism, excretion, and toxicity (ADME and toxicology) may be needed.
The following is a summary of the presentations, ensuing discussions, and key conclusions from the workshop.
SESSION I: NANOMATERIALS IN DRUG PRODUCTS: REGULATORY EXPERIENCE AND STANDARDS PERSPECTIVE
United States Food and Drug Administration’s Approach to Regulation of Nanotechnology Products: R. Nalubola, N. Sadrieh, and C. Cruz
The United States Food and Drug Administration (USFDA) recognizes that nanotechnology is an emerging technology that has the potential to be used across the full spectrum of USFDA-regulated products, including drugs, biological products, and medical devices. Over the past several years, USFDA has taken multiple steps to help ensure the responsible development of nanotechnology products.
In a presentation titled, “USFDA’s Approach to the Regulation of Nanotechnology Products,” Ritu Nalubola, Ph.D., a Senior Policy Advisor in USFDA’s Office of the Commissioner, provided an overview of the Agency’s overarching policy framework, regulatory science activities, and related regulatory cooperation efforts.
In a policy statement articulating its approach (3), USFDA noted that it does not categorically judge all products involving the application of nanotechnology to be either inherently benign or harmful. USFDA continues to regulate nanotechnology products under its existing statutory authorities in accordance with the specific legal standards applicable to each type of product under its jurisdiction. USFDA believes that this regulatory policy allows for tailored approaches that adhere to applicable legal frameworks and that reflect the characteristics of specific products or product classes and the evolving technology and scientific understanding. USFDA intends to ensure transparent and predictable regulatory pathways grounded in the best available science. USFDA’s approach is consistent with the broader US government principles for the regulation and oversight of emerging technologies (4) and nanotechnology (5).
In June 2014, after the PQRI workshop for Nanomaterials in Drug Products, USFDA issued a final guidance for industry, “Considering Whether an USFDA-Regulated Product Involves the Application of Nanotechnology,” to present its current thinking on nanotechnology (6), after taking into account public comment received on the corresponding draft guidance. As noted in that final guidance, USFDA has not adopted a regulatory definition of nanotechnology or related terms. In determining whether an USFDA-regulated product involves the use of nanotechnology, USFDA intends to ask:
-
Whether a material or end product is engineered to have at least one external dimension, or an internal or surface structure, in the nanoscale range (approximately 1 to 100 nm);
In addition, because materials or end products can also exhibit related properties or phenomena attributable to a dimension(s) outside the nanoscale range of approximately 1 to 100 nm that are relevant to evaluations of safety, effectiveness, performance, quality, public health impact, or regulatory status of products, USFDA will also ask:
Whether a material or end product is engineered to exhibit properties or phenomena, including physical or chemical properties or biological effects, that are attributable to its dimension(s), even if these dimensions fall outside the nanoscale range, up to 1 μm (1000 nm).
Table I provides a comparison of USFDA’s points to consider and the definitions proposed by Health Canada and the European Commission. The contents of Table I were updated based on the USFDA final guidance in June 2014, which was published after the PQRI Workshop. In 2014, USFDA also issued additional guidances for the industry to address product-specific technical issues related to the use of nanotechnology in cosmetic products in food substances and in food for animals (7). USFDA also established policies for animal drug submissions and continues to evaluate regulatory gaps for the review of drug products containing nanomaterials in order to formulate future guidance.
Table I.
Comparison of Nanomaterial-Related Descriptions
| USFDA’s points to consider | Health Canada working definition of nanomaterial (9) | European Commission recommendation on the definition of nanomaterial (97) |
|---|---|---|
| 1. Whether a material or end product is engineered to have at least one external dimension, or an internal or surface structure, in the nanoscale range (approximately 1 to 100 nm); and 2. Whether a material or end product is engineered to exhibit properties or phenomena, including physical or chemical properties or biological effects, that are attributable to its dimension(s), even if these dimensions fall outside the nanoscale range, up to 1 μm (1000 nm). |
Any manufactured substance or product and any component material, ingredient, device, or structure if: a. It is at or within the nanoscale in at least one external dimension, or has internal or surface structure at the nanoscale, or; b. It is smaller or larger than the nanoscale in all dimensions and exhibits one or more nanoscale properties/phenomena i. The term “nanoscale” means 1 to 100 nm, inclusive; ii. The term “nanoscale properties/phenomena” means properties which are attributable to size and their effects; these properties are distinguishable from the chemical or physical properties of individual atoms, individual molecules and bulk material; and, iii. The term “manufactured” includes engineering processes and the control of matter. |
A natural, incidental, or manufactured material containing particles, in an unbound state or as an aggregate or as an agglomerate and where, for 50% or more of the particles in the number size distribution, one or more external dimensions is in the size range 1–100 nm. In specific cases and where warranted by concerns for the environment, health, safety, or competitiveness, the number size distribution threshold of 50% may be replaced by a threshold between 1 and 50%. |
Dr. Nakissa Sadrieh, currently a Director in the Office of Cosmetics and Colors at CFSAN, a nanotechnology expert, and the previous CDER lead for nanotechnology, presented a comprehensive view of the types of nanotechnology-based products in CDER. Dr. Sadrieh summarized the technical profile of drug products in CDER based on nanomaterial type, route of administration, indication, and stage in product lifecycle. Overall, the trends show an increasing number of investigational new drugs using nanotechnology such as “nanoparticles” to deliver drugs. Liposomal injections for cancer indications are one of the most common classes of nanotechnology drug products. Nanomaterials can have unique physical or chemical properties that potentially offer great promise, but these same properties may also merit further examination to determine if they affect product safety or other product attributes. USFDA continues to invest in a nanotechnology regulatory science program to help address key scientific gaps in knowledge, methods, and tools necessary for regulatory assessments of nanotechnology products (8). Dr. Sadrieh presented some key topics currently covered under the USFDA research initiatives for nanotechnology-based products.
Dr. Celia N. Cruz, a Senior Quality Reviewer with the Office of Pharmaceutical Science and current lead of the CDER Nanotechnology Working Group, presented the results from the CDER nanotechnology risk assessment (2). The risk assessment identified the potential risks due to nanomaterials in drug product and any regulatory gaps for the risk management of these drug products, considering routes of administration and stage of the product lifecycle. The risk assessment also addressed the use of nanotechnology-based excipients in drug products. The working group concluded that the current framework for safety, quality, and efficacy assessment is sufficiently robust and flexible to address the potential risks for novel materials, including nanomaterials. Regardless of whether a product contains nanomaterials, USFDA asks relevant questions concerning product safety in order to ensure that the product meets statutory and regulatory requirements for safety. Industry remains responsible for ensuring that its products meet all applicable requirements, and the USFDA continues to offer technical advice and guidance, as needed to help the industry meet its obligations. However, the risk assessment exercise identified that increased reviewer training, targeted research on nanomaterial characterization methods, and the understanding of the impact of nanomaterial attributes on exposure and safety are key areas for continued improvement.
As noted in its draft guidance documents, USFDA encourages the industry to consult with the agency early in the drug development process to address questions related to the safety, effectiveness, or regulatory status of nanotechnology products. These early consultations afford an opportunity to clarify manufacturer’s obligations and discuss methodologies and data needed to meet those obligations.
During the subsequent discussion, USFDA clarified that the points to consider elaborated in the 2011 draft guidance focus on “engineered” substances because USFDA is particularly interested in the deliberate manipulation and control of particle size to produce specific properties. This is distinct from the more familiar use of biological or chemical substances that may naturally exist at small scales, including at the nanoscale, such as microorganisms or proteins. In addition, the points to consider would apply to different types of articles that are regulated by USFDA, including finished products as well as materials that are intended for use in a finished product.
With respect to collaboration with other Federal government agencies, USFDA noted that the Agency continues to collaborate and leverage its resources, as appropriate, with relevant domestic and international counterparts on both regulatory science activities and regulatory policy issues. For example, USFDA actively participates at the US government level in the National Nanotechnology Initiative and its related committees, the Emerging Technologies Interagency Policy Coordination Committee, as well as international regulatory cooperation forums. In addition, USFDA has an ongoing dialogue at the agency level with regulatory partners, including the European Commission, European Medicines Agency, and Health Canada.
Health Canada’s Approach to Regulation of Nanotechnology Products: H. Shahbazian
Hripsime Shahbazian, M. Sc., a Senior Science Advisor in the Office of Sciences and current chair of the Health Products and Food Branch Working Group on Nanotechnology, presented Health Canada’s current perspective on nanotechnology.
Nanomaterials are increasingly being used in the marketplace in a wide range of products and substances that Health Canada is responsible for regulating. It is recognized that nanomaterials exhibit unique physical and chemical properties which can be exploited for improved therapeutic benefits; however, these unique properties may lead to unanticipated behaviors. Health Canada acknowledges that new approaches may be necessary for risk assessment and risk management of nano-based health products to keep pace with advances in this area as there is inadequate information on risks associated with nanomaterials at this time. A number of working groups have been established at Health Canada to raise awareness and address nanotechnology-related issues.
Currently, Health Canada has no specific regulation for nanomaterial-containing regulated products. Health Canada relies on authorities within existing legislative and regulatory frameworks, which require the assessment of potential risks and benefits of products to the health and safety of Canadians before they can be authorized for sale. All health products, including those that contain nanomaterials, are regulated by the Food and Drugs Act and associated regulations. Nanomaterial-containing products are subject to the same rigorous health and safety regulations that apply to conventional health products.
To identify regulated products and substances that may contain nanomaterials and to ensure a consistent approach across diverse regulatory programs, Health Canada developed a general working definition which is described in the Policy Statement on Health Canada’s Working Definition for Nanomaterial (9). The “Working Definition” was adopted on October 6, 2011. It is intended to be used as a tool to help the Department gather safety information about nanomaterials to improve the understanding of nanomaterials in its risk assessment and risk management activities.
New health products can be sold in Canada once they have successfully passed a review process to assess their safety, efficacy, and quality. Responsibility for this review process rests with Health Canada’s Health Products and Food Branch (HPFB). To add visibility and transparency, the HPFB created a nanotechnology webpage entitled Nanotechnology-Based Health Products and Food (10). The webpage outlines applications of nanotechnology and provides general guidance to stakeholders regarding health products containing nanomaterial. The document advises sponsors and other stakeholders to communicate with responsible regulatory areas early in the development process if their products contain or make use of nanomaterial. It provides examples of the type of information that may be required for a nanotechnology-based product’s safety assessment.
To facilitate identification and tracking of nanomaterial-containing drug submissions, Health Canada released a revised Drug Submission Application Form for Human, Veterinary, Disinfectant Drugs and Clinical Trial Application/Attestation (HC/SC 3011) (11). The form asks the sponsor to self-identify when their application concerns a nanomaterial or “nanoproduct.” To capture this information in electronic form, a “Nanomaterial” subclass code was added to the Drug Submission Tracking System (DSTS) to allow queries by subclass code “Nanomaterial.”
Currently, Health Canada does not have any guidance documents specific to nano-based health products. Health Canada believes that, in general, its current risk assessment methodologies are applicable to nanomaterials as they allow for sufficient flexibility. To address unique physical, chemical, and biological properties of nanomaterials, each product is assessed on a case-by-case basis.
The state of science around nanomaterials is evolving. Joint efforts are needed to accelerate the achievements promised by nanotechnology. Strong relations and dialogue with international counterparts are important in achieving program objectives in an increasingly complex regulatory world. Health Canada is involved in various key international initiatives, including:
International Organization for Standardization (ISO) Technical Committee (TC) 229 on Nanotechnologies
Organization for Economic Co-operation and Development (OECD) Working Party on Manufactured Nanomaterials (WPMN)
Canada-US Regulatory Cooperation Council (RCC)
International Cooperation on Cosmetic Regulation (ICCR)
International Regulators Nanotechnology Working Group
European Medicines Agency Regulatory Approach to Nanotechnology Products: S. Haubenreisser
Dr. Sabine Haubenreisser, European Medicines Agency (EMA) liaison official at the USFDA, presented the European Medicines Agency’s perspectives on development of nanomedicines. She gave an overview of nanomedicines reviewed by the EMA and guidance on nanomedicines provided by the EMA to developers, and discussed future challenges.
The EMA has adopted a working definition for “nanomedicines,” as being purposely designed systems for clinical applications, with at least one component at nanoscale, and resulting in definable specific properties and characteristics. These are related to the specific nanotechnology application and intended use and the clinical advantages of the nanoengineering. Also, a nanomedicine needs to meet the definition of a medicinal product according to European legislation (needs to have a pharmacological, immunological, or metabolic mechanism of action or to be a diagnostic in vivo).
To date, the EMA has reviewed some 20 Marketing Authorisation Applications for nanomedicines, covering mainly anti-infectives, antineoplastic, and immunomodulating agents, and various types of nanomedicines systems, including liposomes and nanoparticles. EMA has given scientific advice or protocol assistance in over 50 instances. The Orphan Drug Committee has recommended orphan status for a number of nanomedicines under development.
Nanomedicines present scientific challenges, as they are an innovative and evolving scientific field. The EMA has to align with state-of-the-art knowledge and to evolve their methods for evaluation (12,13). From the regulatory standpoint, the EMA is faced with the evaluation of “nanosimilars” and the “next generation of nanomedicines.” Nanosimilars are follow-on nanomedicines as first-generation products come off patent and are claimed to be similar to a reference nanomedicine. In order to demonstrate similarity, in terms of quality, safety, and efficacy, there is a need for stepwise comparability studies. The EMA is also looking at “next-generation” nanomedicines, where recent advances in nanoscience are leading to the creation of even more complex, hybrid structures, paving the way for a wave of new pharmaceuticals, imaging agents, and combination products. Development and evaluation of such products will require special regulatory considerations.
In view of the scientific and regulatory challenges presented by nanomedicines, the EMA has established a pool of expertise for evaluation of both the scientific and regulatory aspects to ensure consistency and collaboration across the European Union (EU). The EMA identified the need for multidisciplinary expertise and, in 2009, established the Expert Group on nanomedicines, bringing together academia and regulators to provide scientific input to Scientific Advice, collate regulatory reflection for evaluation and approval of nanomedicines, monitor the uptake of technical advances in development and evaluation, and formulate guidelines. The EMA also established close cooperation with other EU scientific committees and international cooperation through the International Regulators Expert Group, chaired by the EMA and involving international partners from the USA (USFDA), Japan (Ministry of Health, Labour and Welfare), Canada (Health Canada), and Australia (TGA). Furthermore, the EMA is seeking a transparent dialogue with stakeholders and organized the first International Workshop on Nanomedicines in London in 2010.
The EU has a highly evolved system for the evaluation of the benefit/risk of medicines that has, so far, effectively accommodated new technologies. However, in view of the particular challenges of nanomedicines, EMA has to consider the need for additional guidance, for example, on quality, toxicology, and clinical development and monitoring aspects. In 2011, the Committee for Medicinal Products for Human Use (CHMP) commissioned the multidisciplinary Drafting Group to develop a series of four reflection papers on current scientific and regulatory thinking for nanomedicines. These documents cover the development both of new nanomedicines and of nanosimilars.
The reflection paper on the data requirements for intravenous liposomal products developed with reference to an innovator liposomal product (14)
The draft reflection paper on data requirements for intravenous iron-based nanocolloidal products (15)
The joint EMA/MHLW reflection paper on block copolymer micelle medicinal products (16)
The reflection paper on surface coating (17)
The EMA addresses needs for regulator training. In 2011 and 2012, the EMA held international webinar training sessions on block copolymer micelles and, in 2013, a training session for European regulators on liposomal formulations. An international training session on this topic is planned for this year and further sessions are planned for iron oxide nanoparticles and nanomedicines coating.
The EMA supports developers of nanomedicines in different ways. The Expert Group and Drafting Group on Nanomedicines have been mentioned above. The EMA also offers support through the Innovation Task Force, provides Scientific Advice with option of Parallel Scientific Advice with the USFDA, and has a specialized small- and medium-sized enterprises office (SME).
The Innovation Task Force is a multidisciplinary group that includes scientific, regulatory, and legal competences (18). The Innovation Task Force (ITF) provides a forum for early dialogue with applicants. It holds briefing meetings with applicants on emerging science and technologies with potential regulatory impact. These meetings are free of charge and are intended to facilitate the informal exchange of information and the provision of guidance early in the development process. A dedicated group has been established within ITF, focusing on nanotechnology scientific and regulatory aspects.
The EMA also provides Scientific Advice and protocol assistance to applicants developing nanomedicines (19,20). The Scientific Advice Working Party of the CHMP thus provides an EU view on scientific issues not covered by or deviating from existing guidance. Since experience with nanomedicines is limited, applicants are encouraged to seek early Scientific Advice on the specific data requirements. Applicants can also seek advice on the qualification of biomarkers (21) and can request parallel Scientific Advice from the EMA and USFDA with the aim of streamlining global development plans.
The EMA recognizes the significance of SMEs in the development of nanomedicines, which are often start-up companies or academic spin-offs. The EMA’s SME Office, launched in 2005, is dedicated to addressing the particular needs of smaller companies and aims at promoting innovation and the development of new medicines (22).
In summary, the EMA views nanotechnology as an emerging science which presents new opportunities for medicines in the fields of drug delivery, diagnostics, theranostics, and regenerative medicine and which will yield innovative products contributing to a more proactive paradigm for the diagnosis and therapy of diseases. The focus of the EMA is to facilitate the development of such products. The existing EU regulatory framework does accommodate nanomedicines and adapts constantly to address new challenges. Experience has allowed the EMA to assess the need for development of guidance specific to nanomedicines, which have been addressed so far through the four reflection papers. Applicants are encouraged to contact the EMA at the early stage of development. EMA can provide support through Scientific Advice or through informal briefing meetings with the ITF. Particular regulatory challenges are presented by the evaluation of “nanosimilars” and by advances in nanoscience giving rise to a new generation of complex, hybrid structures.
NANOMATERIALS IN DRUG PRODUCTS—INDUSTRY PERSPECTIVE
Nanomedicines Alliance: F. Malinoski
Dr. Frank Malinoski presented the perspective of the Nanomedicines Alliance on risk assessment and management of nanomedicines. The Nanomedicines Alliance (http://www.nanomedicines-alliance.org/), currently composed of ten pharmaceutical and biotechnology companies, is a unique industry group that promotes and facilitates scientific advancement, regulatory approval, safe use, and public appreciation of nanotechnology-based medicines for the diagnosis, treatment, and prevention of disease. The Alliance provides a forum for member companies to exchange information and discuss emerging issues in nanomedicine development. The Alliance also works with government partners and other stakeholders to define and address priority issues of science, regulation, and policies related to nanomedicine.
Since its formation in 2010, the Alliance has reported on key legislative, regulatory, and scientific developments related to nanomedicines via monthly newsletters. The Alliance has also developed unified industry comments to draft guidances issued by USFDA, National Institute of Occupational Safety and Health, and National Nanotechnology Initiative and established strong working relationships with government entities such as USFDA, National Cancer Institute, National Cancer Institute Alliance for Nanotechnology in Cancer, and Nanotechnology Characterization Laboratory. In 2013, the Alliance sponsored a well-received symposium where government agencies, academics, and industry scientists shared experiences, discussed challenges, and debated best practices in five distinct areas (i.e., design; preclinical pharmacology; toxicology; chemistry, manufacturing, and controls (CMC); and clinical studies) related to nanomedicine development. One white paper highlighting the symposium output was recently published in the AAPS Journal and another will be published soon. In 2014, the Alliance will continue to educate key stakeholders on the promise of nanotechnology for future medicines via publications, sponsorship and organization of conferences and roundtables, and other advocacy efforts.
Nanomedicines have the potential to improve drug targeting and effectiveness, while reducing toxicity and environmental burden. Nanotechnology-based diagnostic tools may be able to diagnose pathological conditions at an earlier stage than conventional diagnostics. There are striking differences (including intended usage, physiochemical properties, human exposure, risk assessment, and regulatory oversight) between nanomedicines and nonmedical nanoparticles, as summarized in Table II. Most importantly, nanomedicines are intended for human exposure and are subject to rigorous regulatory scrutiny before they can be used by patients. Any risk management strategy must weigh risks against benefits for a particular nanomedicinal product, as for other medicines (i.e., a case-by-case approach).
Table II.
Comparison of Nonmedical Nanoparticles and Nanomedicine
| Aspect | Nonmedical nanoparticles | Nanomedicines |
|---|---|---|
| Design goal | Engineering/technical performance | Medical benefit |
| Physicochemical properties | Generally uniform, homogeneous; relatively insoluble | Generally heterogeneous; soluble components |
| Human exposure | Unintended | Intended |
| Risk assessment | Hazard identification; appropriate controls | Safety characterized in development; aim for favorable benefit/risk |
| Regulatory authority oversight (US) | Occupational Health and Safety Administration, Environmental Protection Agency | USFDA |
The Nanomedicines Alliance believes that the current US regulatory framework is sufficiently comprehensive to accommodate nanomedicinal products and that this framework also allows for additional specific considerations on a case-by-case basis. The Alliance will continue to embrace and pragmatically apply, as appropriate, ongoing and emerging advances in bionanotechnology that may stimulate the development of new tools and approaches in the future.
SESSION II: CONSIDERATIONS FOR THE CHARACTERIZATION, DEVELOPMENT, MANUFACTURING, AND POST-COMMERCIALIZATION OF NANOMATERIALS IN DRUG PRODUCTS
Considerations Regarding the Impact of Nanomaterials on Drug Products: K. Tyner
Increased bioavailability, targeted delivery, and increased drug action have all been reported as direct outcomes when incorporating nanotechnology into drug products (23,24). Just as there are multiple reasons to develop a nanodrug, there is a wide diversity of the nanodrugs themselves, with size, shape, material, and route of administration all impacting the overall drug performance (25). Milled nanocrystals, liposomes, and metal colloids are just some of the examples of nanodrugs that are on the market today with even more complex nanodrug formulations on the horizon (26). This diversity provides a nanodrug landscape that is complex from a characterization, manufacturing, and regulatory standpoint. Since regulations and law do not separate nanotechnology products, applicable regulations such as Good Laboratory Practices for Non-Clinical Laboratory Studies (21 CFR 58) and cGMP (21 CFR 210 and 211) apply. However, applying those regulations to nanodrugs may be difficult. For example, fully describing a nanodrug formulation means characterizing it, but there is still much debate on what and how to characterize nanomaterials. In addition, terminology for nanodrugs may be different than for small molecule drugs (e.g., particle stability versus chemical stability). The goal of this session was to introduce the audience to characterization considerations for nanodrugs and to hear from industry representatives on manufacturing considerations for these complex products.
Analytical Considerations for the Characterization of Nanomaterial Drug Products: C. Sayes
The session focusing on the analytical techniques used to characterize nanomaterial drug products began with a platform presentation by Dr. Christie M. Sayes. In the research and development phases of nanomaterial drug product design, there are generally three stages: synthesis and formulation, physical and chemical characterization, and safety/efficacy evaluation. These stages build upon each other to ensure advantageous drug products enabled by nanotechnology. Understanding the biological effects as they relate to the drug’s characteristics enhances the potential for successful implementation of each experimental product, therefore resulting in a more streamlined approach to nanomaterial drug product development (27).
Throughout the R&D phases, choices are made based on tissue target and preferred delivery method. For example, during chemical synthesis, there is a choice between liquid or aerosol phase synthesis that will determine the stability of the product and state of aggregation over time. Other choices may include types of formulation (e.g., emulsification), physiochemical features (e.g., size, amount (weight or number per unit volume), composition (organic, polymeric, metallic), morphology (amorphous or crystalline), and surface functionality (charge, coating, conjugation)).
Synthesis and Formulation
There are two approaches to the synthesis of drug products, the top-down strategy and the bottom-up strategy (28). The top-down method focuses on physical techniques to break down larger particles into nanomaterials, i.e., mechanical techniques such as grinding, milling, or shredding. The bottom-up strategy involves chemical methods to construct nanometer-scale products from molecules (29). Such methods include organic synthesis, chemical reduction, self-assembly, and aerosol growth.
Physical and Chemical Characterization
Properties of nanomaterial drug products greatly impact their ability to be used in clinical applications. Such properties (Fig. 1) include size, shape, purity, and stability (28). Factors impacting these properties include the chemical precursors, reducing and capping agents, the molar ratio of precursors to reducing agents or capping agents, reaction temperature, reaction time, and cleanliness of the reactor vessels during synthesis, and separations to remove reagents from the final drug product (30).
Fig. 1.
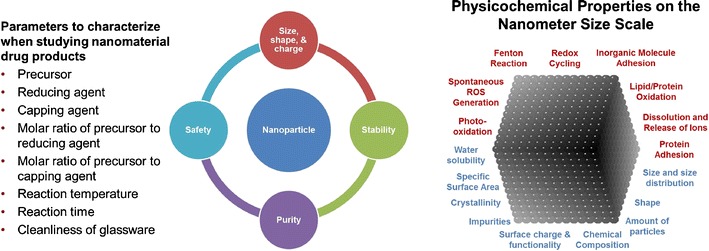
a Parameters and b physicochemical attributes to consider in the characterization of nanomaterial drug products
When designing nanomaterial drug products, it is important to consider the chemical properties that may impact the material’s interactions and function in a biological system. One such chemical property to consider is redox cycling. Free radical formation has been implicated in the toxicity of a wide range of xenobiotics. Other properties to measure include lipid/protein oxidation, protein adhesion, inorganic molecule adhesion, dissolution and release of ions, Fenton reaction, photo-oxidation, and spontaneous reactive oxygen species generation. Although short-term and long-term cytotoxicity was mentioned, a more elaborate discussion is summarized below in the Effect of Nanosized Excipients on Absorption, Distribution, Metabolism and Excretion (ADME) (31).
Similar to the consideration of chemical properties of nanomaterials engineered as drug products, the physical properties of such nanomaterials must also be designed and characterized carefully. Along with chemical composition, physical properties that impact the interaction of a nanomaterial with a biological system include size and shape, surface charge, and functionality (32–36). These properties can be measured using a variety of techniques including dynamic light scattering (DLS), electron microscopy (EM), nanoparticle tracking analysis (NTA), diffraction patterns, isoelectric point (IEP), water solubility, zeta potential, specific surface area, energy dispersive X-ray spectroscopy (EDS), and atomic emission spectroscopy (AES). The characterization of the nanomaterial in vitro needs to be complemented by its characterization in vivo, since the surface properties and fate of the nanomaterial in vivo are critically dependent on the coatings arising from nanoparticle-serum interactions (37–39).
Product Lifecycle: Transformation of Nanodrugs
There is a need to understand the analytical considerations for characterizing nanomaterial drug products. This need is best accomplished through an integrated product lifecycle approach. A lifecycle approach would include assessment of the pristine, nano-enabled intermediates and products, as well as product end-of-life stages (i.e., shelf life determination and disposal of nanomaterials into the environment). Pristine nanoparticles are generally well-characterized, studied, reported, and understood engineered nanomaterials. Nano-enabled products, however, include pharmaceuticals, devices, and packaging, and their physicochemical properties are not well understood. Product end-of-life scenarios also need additional attention. With this lifecycle approach, pristine and end-of-life particles must be characterized and assessed for safety. As we continue to compare the pristine and end-of-life nanoparticles against the nanomaterial drug product, a more relevant picture of nanotoxicology will be drawn. This enhanced picture will allow for “safer by design” nanomaterial pharmaceuticals.
BREAKOUT SESSION: ANALYTICAL METHODS USED FOR THE CHARACTERIZATION OF NANOMATERIALS: LIMITATIONS AND NEED FOR ADDITIONAL RESEARCH—MODERATORS: N SUBBRAO, K. TYNER, AND C. SAYES
Four major themes arose during the characterization breakout sessions—the first two centered on basic characterization of the drug product and the second two focused on impurities and related quality controls.
Characterization and Measurands
The approval of nanodrugs, like all drug products, is covered under the Code of Federal Regulations (CFR). The requirement to fully characterize a drug product may be found in 21 CFR 314.50(d)(1)(i), which requires a full description of the physical and chemical characteristics for the drug substance, including the identity, strength, and quality. When the product contains a nanomaterial, the tests and metrics that fully describe the drug substance may differ from those of traditional small molecules. There have been multiple groups and organizations that are involved in identifying the minimum requirements to adequately characterize a nanomaterial. These lists range from basic lists of size, shape, and composition to more extensive lists with 20+ measurands identified. In general, an exhaustive list of measurands is not useful to developers and regulators alike, as time and resources will be spent on tests that have no actual bearing on the quality, safety, or efficacy of the end product. As with most aspects of the drug development process, the specific critical quality attributes (CQAs) for a material will be product specific and will most likely include measurands that are both nano- and non-nanospecific. Risk assessment is oftentimes beneficial to determine what attributes need to be examined during development as well as when changes are made to the approved drug product. From a development perspective, standards for how information such as size and size distribution is reported would be beneficial and should be addressed early in the drug review process.
Characterization Tools
In addition to specific CQAs that emerge with the incorporation of nanomaterials, the tools and methods used to characterize these attributes may also be different from those used with small molecules. Techniques for the characterization of many aspects of nanodrugs are available, with more being developed to address specific attributes unique to nanomaterials. As with the identification of CQAs of the nanomaterial in the drug product, the techniques chosen to measure the attribute should be appropriate and validated. For example, if a particle size is supposed to be less than 100 nm, the instrument used for measuring the particle size should be validated to analyze at dimensions less than 100 nm. It is widely known that different instruments may provide different results when measuring nanomaterial attributes (such as size). To address this variability, it is generally recommended throughout the field that orthogonal/complementary methods be used when measuring the CQAs. Finally, the tests and tools used for development may not be the same as the ones used for release testing or additional quality checks. Knowledge sharing to keep the USFDA informed about technology used for measuring nano-attributes is recommended, especially when instrumentation and technology are new.
Impurities
In addition to the general considerations for impurities and impurity controls used for small molecules, nanodrugs may have additional impurity considerations. It has been reported previously how the size, shape, and structure of a nanomaterial may all be related to the function of a nanodrug. However, size and shape are rarely homogenous in a nanomaterial preparation and may itself be a source of drug performance variability. The extent and effect of observed variability should be studied and appropriately controlled. For example, a different shape or size of a nanomaterial could be considered an impurity if it is impacting the quality, safety, or efficacy of the product, even if it is the same composition. In addition, as with small molecule drugs, impurities may be introduced externally from processing conditions (e.g., milling media appearing in the final formulation), or internally (e.g., agglomeration occurring during processing, packaging, or storage). Using risk assessment and understanding if and when a change may be impactful to the quality, safety, and efficacy of the product will be critical in evaluating these impurities. Additional guidance exists for determining chemical impurity testing levels (40).
Quality Controls
The previous three sections highlight the need to understand the CQAs of a material and to choose the correct methods and tools to track these attributes throughout the lifecycle of the product. There is a general need to understand what type of testing is needed initially in the development of the drug versus release and quality testing later in the lifecycle of the product. Understanding the CQA will help determine the tolerable levels of heterogeneity lot to lot and process to process for the drug product. As with any drug, sponsors will need to demonstrate and justify what attributes are relevant and that the instruments used to characterize those attributes are robust enough to provide those data. In general, it is the outcome of the quality, safety, and efficacy of the product that will dictate the required tests.
INDUSTRIAL EXAMPLES OF NANOMATERIALS
Milled Nanomaterial Drug Intermediates and Products: M. Brewster
The pharmaceutical preparation of nanomaterial-based dosage forms is encouraged by a number of API, pharmaceutical, and biopharmaceutical drivers. For compounds whose water solubility or dissolution rate limits their oral bioavailability (e.g., BCS class II or IV materials), size reduction into the nanodomain can, via the Noyes-Whitney/Nernst-Bruner relationship, increase in vivo dissolution rate and fraction absorbed (41–44). These principles have led to a number of marketed products wherein a nanosuspension is either prepared as a liquid formulation (i.e., Megace ES®, megesterol acetate (Par, 2005)) or converted into a tablet or capsule (e.g., Rapamune®, sirolimus (Wyeth, 1999), Tricor®, fenofibrate (Abbott, 2004), Triglide®, fenofibrate (SkyPharma, 2005)) (45–48). Furthermore, nanosizing can also be of use in the design of parenteral dosage forms wherein poorly soluble APIs are milled to a specified size and size range resulting in not only useful bioavailability but also sustained release features (49). The antischizophrenic agent, Invega® Sustenna®, paliperidone palmitate (Janssen, 2009) provides 1 month of drug coverage after a single intramuscular injection, with a 3 month dosing interval project being developed along similar design. Other areas attracting interest include nano-aggregated inhalation platforms.
The development of nanosized formulations generally involved top-down approaches in which the API is milled or otherwise subjected to particle reduction strategies in an aqueous environment in the presence of a stabilizer or a set of stabilizers (50). Top-down strategies were considered more controllable and more robust as a function of process and design space for this type of manipulation. If the API is milled, it is often attrited under high energy in the presence of a milling media like highly reticulated polystyrene or zirconium milling beads. In some cases, piston gap high pressure homogenization can be used to generate the desired nanosuspension (51). Milling is generally monitored by an appropriate particle sizing technique and design and process spaces defined by milling curves first at small scale and then at production scale.
The production scale development of nanosized formulations when completed using ball milling usually involves high-energy mills which can recirculate the material, remilling it until a consistent product is generated (52,53). As suggested, the milling media can also abrade under the conditions of milling, and care should be taken that significant contamination of the nanosuspension by the milling media does not occur. The principle of high-energy milling can be carried across scales from 1 mL to more than 1000 L, and while scaling is a function of the product and the equipment, useful results are often obtained (45,54). Sterile nanosuspensions are also possible using high-energy ball mills by implementing isolator technology and other sterility-assuring components. Likewise, large-scale production using homogenizers is possible and this equipment type can offer advantages under various situations. Finally, several approaches allow for the conversion of the liquid nanosuspensions to be converted into resuspendable solid dosage forms (46,47).
Liposomes: D. Cabral-Lilly
Liposomes are perhaps the most established of carrier nanomedicines with several available on the commercial market and many more in clinical and preclinical development. A liposome carrier is used to increase a drug substance’s therapeutic index, often by multiple mechanisms including (1) favorably changing drug biodistribution and pharmacokinetics, (2) increasing drug substance stability in plasma, (3) decreasing drug toxicity, and/or (4) to deliver optimal ratios of drug combinations to the site of action with the goal of markedly increasing efficacy.
Considerations for manufacturing are similar for all liposome technologies, for example, microfluidization, ethanol injection, emulsion/extrusion, and remote loading. First, the manufacturing process must be reproducible and scalable from a few milliliters at the proof of principle stage to more than 200 L for commercial supplies. The process should minimize hazardous conditions where possible through choice of solvent mixture, equipment rating, and methods for isolating operators from the hazards. It is recommended to develop a scalable manufacturing process early to minimize the number of modifications that are required as the batch size is increased or product dosage form is changed. To date, liposome nanomedicines have been administered by routes that require the product to be sterile (inhalation, intravenous, intrathecal, intramuscular). For products with a relatively large mean particle size (>400 nm), manufacturing must be done as an aseptic process. Sterile filtration can be used for products with a mean particle size of approximately 100 nm or less, making compounding in class B or class C suites possible. This option is much more amenable to technology transfer to contract manufacturing sites, minimizes difficulties for the operator(s), and reduces costs significantly.
Characterization is an important component of the development and manufacturing of liposome nanomedicines. A set of core physicochemical attributes apply to all liposomal products: membrane phase transition temperature, morphology and lamellarity, trapped volume, and zeta potential. Product-specific characterization focuses on drug-excipient interaction, drug loading mechanisms, drug-drug interactions, and physical disposition of the drug in the carrier. An example was presented where data from two characterization techniques were used to help determine the final drug product formulation (48). CPX-1 (irinotecan/floxuridine) liposome injection is a clinical stage liposomal nanomedicine intended for the treatment of advanced colorectal cancer. The manufacturing process co-encapsulates irinotecan HCl and floxuridine at a 1:1 molar ratio into preformed liposomes. The aromatic region of the 1H NMR spectrum of the drug product shows three peaks from irinotecan. When the buffer inside the liposomes was sodium gluconate-triethanolamine (NaGluc-TEA), the 1H peaks are quite broad. When this entrapped buffer was copper gluconate-triethanolamine (CuGluc-TEA), the 1H peaks were much sharper suggesting an interaction between irinotecan and copper. Correspondingly, the drug release profiles from an in vitro release (IVR) assay also showed a difference. In the liposomes with NaGluc-TEA, the floxuridine and irinotecan were released from the liposomes at different rates, whereas with CuGlu-TEA as the entrapped buffer, the two drug substances released from the liposomes at the same rate under the conditions of the IVR assay.
The numerous components of liposome products (multiple membrane lipids, additional excipients, possibly multiple active agents), as well as the multistep/day manufacturing process pose challenges to performing development and validation studies using the standard quality by design (QbD) approach; the design space would be extremely complex. It should be possible, however, to obtain a good understanding by studying each step in the manufacturing campaign separately using science-based and process-based protocol designs. The CQAs of the product would be assured by determining the critical process parameters for liposome preparation, drug substance encapsulation, lyophilization, and the feasibility of reworking. An example was presented of a phase 3 clinical stage product, CPX-351 (cytarabine/daunorubicin) liposome injection, being developed for acute myelogenous leukemia (55). Here the two drug substances are encapsulated sequentially into preformed liposomes to achieve a 5:1 molar ratio in the final product. The batch size started at 0.5 L, progressed to 14, 21, and then to 200 L using a manufacturing process for the bulk product that is essentially unchanged. The drug product for phase 1 and 2 trials was a liposome suspension stored frozen at −20°C due to instability at refrigerated temperatures. The drug product was converted to a lyophilized form for phase 3 and comparability to the frozen product demonstrated using in vitro physicochemical characterization that included core parameters, product-specific characteristics, and in vitro release. Upon reconstitution with water, more than 95% of both cytarabine and daunorubicin remain encapsulated. The lyophilized product is stable for at least 3 years when stored refrigerated and for at least 2 years when stored at room temperature.
Gold Particles: L. Tamarkin
To be considered as a potential pharmaceutical product ready for the marketplace, nanomedicine manufacturing processes need to be robust, reproducible, and cost-effective. CytImmune has developed a simple method for manufacturing a family of anticancer nanomedicines that harness the therapeutic potential of the naturally occurring, anticancer hormone, tumor necrosis factor alpha (TNF) and are readily characterized (56).
Colloidal gold is the core of CytImmune’s proprietary nanotechnology. The mean size of the gold nanoparticles used as the core of CytImmune’s nanomedicine is 27 nm, where 95% of the particles range in size from 15 to 35 nm. The quality of the resultant gold sol is then confirmed by transmission electron microscopy and the quantity of gold is measured by inductively coupled plasma-atomic emissions spectroscopy (ICP-AES).
The next step in manufacturing is to independently bind two molecules to the gold nanoparticles, TNF and an analog of polyethylene glycol (PEG), using thiol binding to form dative covalent bonds with the gold nanoparticles. Each molecule serves a different purpose. TNF serves two functions, one as the tumor-targeting entity and a second as the API. The PEG analog, on the other hand, shields the gold particles from immune detection.
To ensure that both TNF and PEG-thiol bind to individual gold nanoparticles, the binding needs to occur in a small volume. This is accomplished by simultaneously drawing the colloidal gold sol and a mixture of TNF and PEG-thiol into a Y-connector using a single peristaltic pump. This dilute form of the final drug product is ultrafiltered and then vialed and lyophilized. To date, this lyophilized product has been shown to be stable for 3 years, and the process is scalable.
This nanomedicine, termed CYT-6091 (57,58), has a number of unique features that facilitate its characterization. First, TNF is bound to the surface of the nanocarrier, so that the API on the final drug product may be interrogated without destroying the final drug product. Second, the reagents bound to the gold nanoparticles may be stripped off the gold nanoparticles using a strong reducing agent, such as dithiothreitol or β-mercaptoethanol. And third, the gold nanoparticles may be centrifuged, enabling a simple process for separating gold-bound reagents from unbound (or free) reagents.
By measuring bound versus free TNF, the product specifications were established wherein 80% or more of the initial amount of TNF is in the final drug product that contains less than 5% free TNF. The range of APIs that this nanomedicine platform may carry is not limited to biologic molecules. CytImmune has developed thiolated linkers that release parent small molecule therapeutics either through hydrolysis or self-immolation (58). Now, using the same manufacturing process described above, CytImmune is able to bind a biologic (TNF), an analog of a small molecule therapeutic (e.g., paclitaxel) and PEG-thiol simultaneously to individual gold nanoparticles. Thus, this nanomedicine platform has the versatility to deliver multiple agents to tumors, each of which acts independently and potentially synergistically to target and destroy both the tumor vasculature and its cancer cells.
BREAKOUT SESSION: CURRENT AND EMERGING TECHNOLOGIES FOR MANUFACTURING STABLE NANOMATERIAL-CONTAINING DRUG PRODUCTS: MODERATORS: J. BARTLETT AND W.M. EICKHOFF
The breakout sessions stimulated a lot of very good discussion about the challenges and opportunities around nanomaterial-containing drug products. The following is an account of the major conclusions from the discussion during the breakout session. Some common themes that emerged from the different breakout sessions are summarized below.
Why Are There Not More Nanomaterial-Containing Drug Products on the Market?
From presentations given earlier in the day, it was shown that a major part of approved nanomedicines may be classified as the more “mature” technologies, for example, oral nanocrystals and liposomes. This topic of “why not more nanomedicines” was discussed and a few themes emerged. It is perceived that the development of a nanomedicine comes at a higher regulatory and technical risk. The perception that nanomedicines would require “enhanced drug development” which translates to greater costs and longer development timelines was also noted. This added development barrier comes at a cost to the innovator; therefore, a clear definition by the innovator companies around the medical benefit of the nanomedicine approach is key. For example, if a nanomedicine is being used to advance a candidate which displays poor aqueous solubility intended for oral delivery, the question should be addressed as to what advantage is the nanomedicine bringing compared with alternative oral solubilization approaches? There were a number of examples given over the course of the meeting where nanomedicines have the potential to offer significant medical benefits. Parenteral drug delivery employing nanomedicines was seen as the emerging technology where the greatest medical benefits may be observed. Oral nanomedicines were seen as more mature technologies, primarily employed to overcome poor dissolution and solubility of drug candidates.
Balance of a Deeper Understanding of the Technology Versus Development Timeline
Several points were captured around the perception that developing nanomedicines, particularly for the parenteral route of administration, will require a deeper understanding of the critical attributes of the formulation on efficacy and safety. There was discussion around the need to develop more and/or better in vitro or preclinical in vivo models that can accurately evaluate key performance endpoints (i.e., safety and efficacy) of the novel nanomedicines. For example, when a nanosystem is being used to increase the dissolution rate/bioavailability of a compound, predictive in vitro dissolution screens have been developed to assess the performance of the nanocrystals systems. It was noted that the challenges to developing these tests should not be underestimated. One of the key challenges in the development of more predictive models is that you need to know what to measure (biologically or in vitro) that is relevant to a clinical endpoint (i.e., safety and efficacy). Advancements are being made in this area, and several examples were presented with regard to the development of ADME screens to look at uptake/distribution of different nanomaterials. In some cases, several prototype nanomaterial-containing formulations are put directly into exploratory human clinical trials in order to explore the impact of nanoproperties, for example particle size distribution or surface properties, on pharmacokinetics (PK) or pharmacodynamics (PD) responses very early in the development program. From these results, correlations can be developed which can be used to develop predictive in vitro/in vivo models and to optimize future formulation efforts.
What Are the Major Barriers for Entry of Novel Nanomaterial-Containing Medicines: Technical, Regulatory, Funding?
This question was discussed at some length with varying opinions because “one size does not fit all.” There are multiple different nanomaterial-containing nanomedicine approaches (e.g., top-down versus bottom-up approaches) and multiple routes of administration (e.g., oral versus parenteral), and therefore, broadly defining the major barriers to entry for nanomedicines is a challenging topic. There was some discussion around classifying the different nanomedicines based on various attributes to help assign different levels of risk and provide more focused regulatory guidance, for example, attributes such as particle size, route of administration, longevity of the particles in the body, whether the particles are water soluble or water insoluble, extent of distribution within the body, etc. It was noted that there have been efforts in the literature regarding this approach (59) and by the EMA through the publication of focused reflection papers on the development of different nanomedicines (14–17).
There was a general understanding that if the medical benefit is clearly defined, and a market driver identified, technical hurdles with regard to the science, development, and manufacturing would be overcome. There was a general concern around the lack of regulatory guidance and acceptable level of manufacturing changes that may occur during the development of a new medicine. There was consensus that a specific SUPAC-like guidance for each type of nanomaterial was not practical and that a Quality Risk Management based on principles outlined in International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Q9 and ICH Q10 (40) would be more appropriate.1 The question was rooted in a concern that the development of a novel nanomaterial-containing drug product may carry additional development costs, timeline, and a higher level of regulatory risk that upon commercialization additional preclinical/clinical studies may be requested. To reduce this risk, it was noted that a more enhanced drug development program may be needed much earlier in a product development which comes at an added cost. Since many novel nanomedicines are being developed by small companies, the additional development costs could be a barrier. By more clearly defining what the key barriers to entry of new nanomedicines are, solutions could be identified to help overcome the obstacles.
SESSION III: SAFETY AND ADME CASE STUDIES OF NANOTECHNOLOGY-DERIVED DRUG PRODUCTS
Introduction: Safety and Toxicological Effects: A. Jacobs
Nanosize: What can we learn about nonclinical evaluations of nanomaterial drug products? There is an interplay between characterization of the nanomaterial and its toxicology. It is important to know that the material can be made reproducibly and is representative of what humans will be exposed to. There are many questions asked about nanomaterial drugs/formulations/carriers. For insoluble nanomaterials, it would be helpful to know to which part of the size range should any biologic effects be attributed, and which changes in which properties could affect biologic properties? This is not a simple task, but the developer should understand the critical variables.
There are two general types of regulatory submissions for nanomaterials. In one case, the product and active ingredient are completely de novo, and the standard ADME and toxicology studies used to evaluate non-nanomaterials during product development would apply. In other cases, there is a previously approved active pharmaceutical ingredient, which has been milled into the nanorange for better oral bioavailability. In this case, a bridging toxicity study and ADME study should generally suffice. However, there may be route-specific issues for nonoral routes of administration that may warrant evaluation.
Which in vitro tests are appropriate and in vivo safety concerns depend on whether the nanoparticle or carrier is not water soluble or biodegradable. If the product is biodegradable and water soluble then in vitro assays can have utility. These statements include some generalizations, but the final decision on what is needed will be based on a case-by-case approach.
Case Study: Safety and ADME and Toxicology of Nanotechnology in Parenteral Drug Products: W. Zamboni
Nanoparticle Anticancer Agents
Major advances in the use of carrier vehicles delivering pharmacologic agents to sites of disease, such as tumors, have occurred in the past 15 years (60,61). The primary types of carrier-mediated anticancer agents are liposomes, nanoparticles (NPs), and conjugated agents. Liposomes can be subdivided into stabilized (e.g., PEGylated) and nonstabilized (non-PEGylated) liposomes (62,63). The theoretical advantages of carrier-mediated drugs include increased solubility, prolonged duration of exposure, selective delivery of entrapped drug to the tumor, and improved therapeutic index (60). PEGylated liposomal doxorubicin (Doxil®, Lipodox®, PEGylated liposomal doxorubicin (PLD)), liposomal daunorubicin (DaunoXome®), liposomal vincristine (Marqibo®), and paclitaxel albumin-bound particles (Abraxane®) are the only USFDA-approved members of this relatively new class of drugs (61). However, there are hundreds of NP anticancer agents in development that may be improved based on the results and methods in this study. Thus, our proposed studies may have a far-reaching impact.
Pharmacokinetics and Pharmacodynamics of Nanoparticle Agents
NP and liposomal agents are cleared via the mononuclear phagocyte system (MPS), also called the reticuloendothelial system (RES), which is located primarily in the liver, spleen, and blood, as well as in the lung and bone marrow (64) (Fig. 2). Non-PEGylated or nonstabilized NPs are cleared relatively quickly via the MPS. PEGylated or stabilized NPs are also cleared via the MPS but at a much slower rate than nonstabilized carriers. We have reported that the slower clearance of PEGylated liposomal agents is associated with greater interpatient PK variability than non-PEGylated liposomal agents (65). The factors affecting the PK and PD variability of NPs remain unclear but most likely include the MPS (64). NPs can alter both the tissue distribution and the clearance of drugs because the drug takes on the PK characteristics of the carrier. Compared to small molecule formulations, the primary sites of accumulation of NPs are the tumor, liver and spleen. The delivery of NP agents to the tumor may also be affected by the MPS (66). Elucidating the mechanism(s) of interaction between NPs and the MPS and understanding the factors that alter MPS function are fundamental to the optimal clinical development of NPs.
Fig. 2.
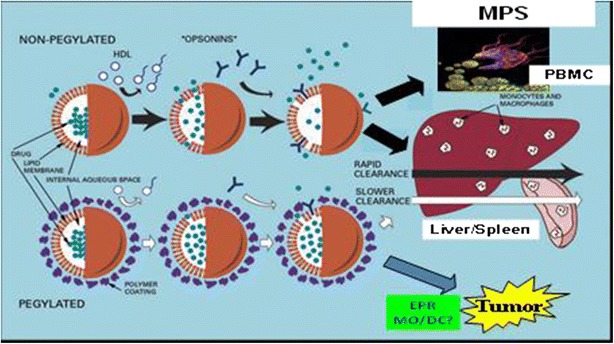
The clearance (CL) of non-PEGylated and PEGylated liposomal nanoparticle (NP) (e.g., liposomes) agents via the mononuclear phagocyte system (MPS) in the blood, liver, and spleen. NP are taken up and cleared via MPS cells (e.g., monocytes, macrophages, dendritic cells) in the blood, liver, and spleen. The MPS has been shown to be involved in the PK and PD of a wide variety of NP agents
The PK of liposomes and NPs are dependent upon the carrier and not the encapsulated drug until the drug is released from the carrier (64). The drug that remains encapsulated within liposomes or NPs is an inactive prodrug; thus, the drug must be released from the carrier to be active. After the drug is released from the carrier, the PK of the drug will be the same as if one administered the noncarrier form of the drug; however, the overall exposure of released drug is dependent on the variable clearance of the carrier (67). The PK of NPs is complex, and thus, detailed studies must be performed to evaluate the disposition of the NP encapsulated and the released forms.
PK and PD variability of NPs and liposomal anticancer agents administered intravenously (IV) is several-fold higher than small molecule drugs administered IV or oral (65). A meta-analysis comparing the interpatient PK variability of liposomal and small molecule formulations of the same anticancer agents (n = 9) reported significantly higher PK variability of the liposomal agents (65,68). The PK variability depicted as the area under the curve (AUC) CV% (P < 0.001) and ratio of max AUC to min AUC (P = 0.04) was significantly higher for the liposomal agents compared to the small molecule agents. In a phase I study of PEGylated liposomal CKD-602 (S-CKD602), the interpatient variability in the exposure of encapsulated and released CKD-602 ranged from 20- to 100-fold (69). Significant interpatient variability in PK and PD of PLD and other liposomal agents has also been observed.
In addition to the high interpatient variability in the PK of NPs, there also may be high intrapatient variability in the PK of NPs. Gabizon and colleagues reported that the clearance of sum total (encapsulated + released doxorubicin) decreased by approximately 25 to 50% from cycles 1 to 3 in patients with ovarian cancer (70). In addition, La and colleagues reported that this reduction in clearance of Doxil from cycle 1 to cycle 3 was associated with a reduction in precycle monocyte count (71). These studies suggest that there is a reduction in the clearance of liposomes over time that is associated with a reduction in MPS function. Thus, dose reductions may be needed in subsequent cycles to minimize the risk of toxicity. These results and prior results reporting a relationship between the reduction in monocyte counts in blood and the clearance of PEGylated liposomal agents in patients where greater monocyte reductions were associated with greater clearance of the liposomal agents suggest that there is a bidirectional interaction between NPs and the MPS, where the MPS cells take up NPs and the NPs have pharmacologic and/or toxicologic effects on the MPS cells (72,73). Interestingly, repeat dose studies of PEGylated liposomal doxorubicin in mice and rats did not report accumulation of drug in plasma suggesting that these preclinical models may not accurately reflect the disposition of PEGylated liposomal agents after repeated dosing (74,75). Thus, there is a need to develop better preclinical animal models for pharmacology and toxicology studies of liposomal and nanoparticle agents.
The PK variability contributes in part to variability in a drug’s PD effects, making it difficult to predict how a particular patient will respond in terms of efficacy and/or toxicity (64). This issue raises concern about the translational development and clinical utility of NP agents due to the high interpatient PK variability of NPs. Thus, there is a need to identify the factors associated with the PK and PD variability of NPs as methods to improve response and reduce toxicity.
MPS Effects on NP PK and PD
A fundamental and highly referenced principle of NP PK is the predominant effect of the MPS on the distribution and clearance of a wide variety, if not all, NP agents (64,66). New publications by Khanbeigi et al., Shah et al., Skoczen et al., Moghimi et al., Andersen et al., Crist et al. from NCI’s Nanotechnology Characterization Lab, and many others highlight the interaction of the MPS with a wide variety of NP agents (65,76,77). Specifically, studies by Shah et al. and Khanbeigi et al. reported that NPs with various surface properties (e.g., PEG and ligands) and sizes (i.e., 50 to 1000 nm), respectively, are all identified and taken up by the MPS. Our prior studies in animal models and in patients have reported that the MPS is highly involved in the clearance of liposomal agents, such as PLD (67). Moreover, there are no publications suggesting the MPS is not a predominant factor involved in the PK of NP agents, especially NP agents >50 nm.
Our previous studies suggest that the significantly high and clinically relevant variability in the PK and PD of liposomal and other NP anticancer agents is related to variability in the function of the MPS, which serves as the clearance pathway for NP agents (68). The patient factors that affect MPS function and NP PK and PD variability include age, gender, body habitus, sex hormone levels, and chemokines (78–80). However, these patient-related factors do not account for all of the PK and PD variability, and thus, the development of phenotypic probes of the MPS that incorporates or accounts for all of these factors may be a more appropriate approach to predicting the PK and PD of NP in animal models and in patients (Fig. 3). These probes could also be used to individualize the dose of NPs on cycle 1 and subsequent cycles as there is high inter- and intrapatient variability in MPS function and NP PK. Studies in preclinical animal models and in patients using phenotypic probes of MPS function to predict the clearance of PEGylated liposomal agents support this plan (67).
Fig. 3.

Summary of the process to use a phenotypic probe of MPS function in blood to measure MPS function in patients which would predict NP PK, efficacy, and toxicity. This type of probe could be used as a test that could retrospectively be used to explain patients with highly variable PK and PD. This type of probe could also be used as a method to individualize the dose of NPs as needed
Case Study: Safety and ADME Aspects of Nanotechnology in Topical Products (Sun Screens/Wound Healing/Topical/Ophthalmic Products): S. Ciotti and S. Gracon
Dr. Susan Ciotti and Dr. Stephen Gracon from NanoBio Corporation presented a case study of “soft nanoparticle” nanoemulsion-based therapies. A nanoemulsion is a high-energy emulsification of an aqueous phase, oil phase, nonionic surfactant, and a cationic surfactant. The nanoemulsion particles presented the range between approximately 180 and 500 nm. This “soft nanoparticle” formulation allows for permeation of the nanoemulsion (with drug) into the skin and mucosal surfaces.
Nanoemulsion droplets function in opposition to other soft formulations such as detergent micelles; nanoemulsions do not cross tight junctions or epithelial tissues, whereas detergent micelles can cross the tight junctions and disrupt cells and tissues, which may ultimately lead to irritation. This versatile platform allows for a broad spectrum of products, including ones incorporating antiviral and antimicrobial activity. The presented nanoemulsions are being probed for intranasal vaccines. A seasonal influenza vaccine based on these nanoemulsions is currently in phase I clinical trials.
BREAKOUT SESSION: THE SAFETY AND ADME EFFECTS OF NANOTECHNOLOGY ON DRUG PRODUCTS—MODERATORS: A. JACOBS AND E. MOREFIELD
In this breakout session, the following topics were discussed: the need to agree on specific terminology in the nanoscience and technology field, such as defining a “particle” or a “colloid”; addressing the gaps in current safety assessments and identifying testing needs specific for nanomaterial drug products; developing methods to characterize a nanodrug product and include tests for characterizing the stability of the product over time; develop methods for manufacturing reproducible products; understand the limitations of animal models when testing efficacy and safety; gain an appreciation of the physicochemical properties that are most important in efficacy and safety testing, such as crystal structure, formulations, and surface coatings; and develop methods for testing safety for dermal routes of exposures. The importance of only asking for studies that are “need to know” versus “nice to know” was emphasized. Equally important is understanding the minimum requisite analytical tools needed.
The following points were made during this breakout session:
Nanomaterial drug products are currently evaluated on a case-by-case basis. There is a desire to have some generally applicable tests to help minimize differences arising from a case-by-case approach. A suggestion was made to make a general nanochapter in the USP.
Another suggestion to the USFDA was that the training developed for agency reviewers be shared with nanoproduct developers.
- Other topics for a potential guidance were discussed and included:
- Whether there are enough nanorelated items to write a guidance.
- Whether a database should be developed to gather information on nanoproducts.
- Whether any potential guidance should be safety guidance or a quality guidance?
- How much variability is allowed for the physicochemical specifications and how variability should be addressed?
- If PK/distribution does not change with a change in physicochemical properties of a nanoproduct, what if any, analyses of the product or toxicology studies need to be repeated.
- Whether any change in a nanoproduct means that many analyses or toxicology tests need to be repeated.
- Guidance could include blinded examples of what changes in nanoproperties were acceptable, and what data were required for such changes.
Effect of Nanosized Excipients on Absorption, Distribution, Metabolism, and Excretion: R. David
Dr. R. David presented on the effect of nanosized excipient on ADME. Excipients can modify the bioavailability of the active ingredient by enhancing absorption and distribution or by decreasing metabolism and excretion. Nanomaterials as excipients also act to improve bioavailability. Understanding the properties of nanomaterials can aid in the development of new nanoexcipients. Liposomes have been used for many years and the history of liposomes as successful carriers can provide insight into how to craft new drug carrier excipients.
Any active ingredient has biological barriers to overcome to be effective. In their review, Alexis and coworkers (36) list those barriers as absorption, i.e., getting into the blood (assuming a nonparenteral route of administration); distribution limitations, i.e., getting to the target tissue and target cell; and avoiding metabolism and excretion mostly via scavenging into the reticuloendothelial system or filtration in the kidneys. Alexis et al. identify properties such as size (smaller particles are more easily absorbed into cells than are larger ones) and surface charge (negatively charged or neutral particles are retained in the circulation longer than are positively charged ones) as two properties of importance. Maeda and coworkers (81) agree with the biological barriers, but add that there are practical ones: will the active ingredient be efficacious; what are the regulatory and safety issues for the excipient; and what will be the cost/benefit of the new formulation? Indeed, the USFDA has already provided some guidance on the regulatory and safety issues (3) with encouragement to applicants for engaging in a presubmission consultation that will outline the specific issues for each new material or formulation.
Lessons on the impact of nanoexcipients can be learned from studying how liposomes have been used. Gregoriadis (82) seems to have been the first to describe drug “entrapped” in liposomes. He reported that liposomes reduced excretion of penicillin by a factor of 10 and reduced actinomycin excretion by a factor of 100. Furthermore, tissue levels of “entrapped” antibiotic were much higher than that of “free” antibiotic. Birrenbach and Speiser (83) describe the use of micelles as adjuvants for slow release of antigens to enhance immunization. They describe “nanoparts” of solidified micelles that contain drug; these nanoparts were used to encapsulate human IgG or tetanus toxoid, which were then more effective in eliciting antibody response than nonencapsulated forms. What is of greatest interest is the use of the term “nanoparts”—long before the term “nanotechnology” or prefixes of “nano” became popular. In 1978, Juliano and Stamp (84) described using liposomes to encapsulate antitumor agents. They found that the percentage of encapsulation differed with the drug and amount. For example, 66% of 1 μg actinomycin D and 71% of 50 μg were incorporated versus only 28% of 500 μg. Further, only 11% of 25 μg of vinblastine was incorporated even though it has an equivalent Kow as actinomycin D. Efflux from liposome also differed with drug with greater percentages of actinomycin being released compared with vinblastine. Thus, by the end of the 1970s, much was understood about how liposome could enhance delivery and bioavailability of active ingredients.
Over the years, liposomes had to be modified to improve “delivery.” Drug loading and release were improved by switching from liquid phase to solid phase. Also, selecting drugs with the right characteristics amenable to liposomes was important—one size did not fit all. The surface was modified to reduce opsonization with serum protein, which reduced excretion; and intracellular delivery was increased by using lipids that could fuse to cell membrane. Such strategies of selecting the right excipient for the active ingredient, modifying the surface chemistry, and utilizing the natural properties of the excipient can be applied to other nanoexcipients. The following will describe the properties of nanomaterials and how they influence ADME.
The biological effects of small particles are related to surface chemistry/properties, which are influenced by surface area and maybe shape. Movement/distribution of nanoparticles in the body is influenced by size, shape, and surface chemistry, which can be modified by surface coatings. In general, absorption into the body is influenced by size (smaller absorbed to a greater extent than larger), shape (sphere > ellipse), and surface charge (neutral/anionic > cationic), which can be modified by intentional coatings (or protein corona). Internalization into cells is accomplished primarily by endocytic processes or receptor-based internalization. For distribution in the body water, neutral or negatively charged surface nanoparticles have a reduced plasma protein adsorption and low rate of nonspecific cellular uptake; thus, the same principles that govern absorption govern distribution. Regarding metabolism/biotransformation, none is expected, although little is known about the fate of coatings other than lipid. Excretion occurs primarily into urine or feces, although little may be excreted and excretion may be particle dependent.
BREAKOUT SESSION: THE EFFECT OF NANOSIZED EXCIPIENTS ON ADME—MODERATORS: P. BROWN AND N. SUBBRAO
How are nanoexcipients being developed? Most novel excipient development is “bottom-up” development. This means that for most novel nanosized excipients, the intent of the manufacturer is to create an excipient with nanosized particles by assembling the particles and not by milling larger particles down to nanosize. This probably reflects the reasons that a developer would have for using a nanosized excipient. If the excipient is to be used to alter the delivery of the API or to have other specific physiochemical interactions with the API, then the qualities of the excipient needed for these interactions will often have to be intentionally engineered into the nanomaterials.
What are appropriate ways to characterize ADME and toxicology of nanoexcipients? In many cases, the nanoexcipient will be included in a drug product in order to alter the ADME and toxicology of the API. Consequently, assessment of ADME and toxicology for the entire product is likely to be necessary to understand the overall effects of the combination of API and excipients. An understanding of the stability of any nanoexcipient-API interactions may help when deciding the extent of testing needed with either component alone. In those cases where the nanoexcipient and API are readily dissociated in vivo, an assessment of the fate of the free nanoexcipient may be recommended.
It is recognized that “traditional” metabolism through phase I and II biotransformation pathways may not always apply to nanoexcipients. Some of the excipients may be inorganic or polymer based and therefore unlikely to be metabolized by these pathways. Depending on the molecular structure of the nanoexcipient, some breakdown of these molecules may occur through either less specific mechanisms (e.g., direct oxidation) or through enzymes such as lipases, peptidases, or nucleases. An understanding of the specific nanoexcipient structure can help guide decisions about how the in vivo fate of the nanoexcipient is determined and whether special studies are recommended.
If a quality attribute of an excipient such as particle size is critical to product performance, it is important and generally feasible to consistently ensure and control that quality. Some understanding of excipient to API ratio seems desirable but it is not clear that a full understanding of this at a molecular level is always achievable or necessary. Some variability may be acceptable as long as the critical attributes for safety and efficacy are adequately identified and controlled.
Some aspects of the safety of new nanoexcipients may be addressed through previous use of non-nanosize forms of the same excipient. However, as the change to a nanosize can alter a number of particle characteristics, it is likely that additional information will be needed to support switching from a non-nanosize to a nanosize. Use of a material in foods and its recognition as generally recognized as safe (GRAS) in that case does not necessarily translate to its safe use in a pharmaceutical product.
As with most considerations around the use of nanomaterials in drug products, the ADME and toxicology of nanoexcipients needs to consider the particular properties of each new material. A one-size-fits-all approach is not appropriate. A risk-based approach considering how the product will be used and worst case scenarios should be considered.
SESSION IV: REGULATORY PANEL DISCUSSION SUMMARY: R. UPPOOR, C. CRUZ, R. NALUBOLA, S. HAUBENREISSER, H. SHAHBAZIAN, AND A. JACOBS
ICH Common Technical Documents and ICH Guidelines (Series: Safety, Quality, Multidisciplinary) and Other Regional Guidelines Are Followed for Conventional Drug Products. In Addition, What Other Policy and Scientific Standards Do You Recommend the Sponsors and Applicants to Review and Follow for the Submission of Nanomaterial-Containing Drug Products?
The USFDA noted that ICH and USFDA guidance that applies to drug products generally also applies to nanotechnology-based drug products. This includes ICH M3 (R2) (40), which gives information on the timeline for nonclinical safety studies, and all relevant Pharm/Tox guidances. Another example is the Final Guidance for evaluation of excipients (85), in the event a novel excipient is introduced or used in a novel drug dosage form. There is more specific guidance relevant to nanomaterials or nanotechnology-based products, for example, the Draft Guidance for Industry on liposomes (86), and product-specific guidance regarding the bioequivalence of drug products that are nanotechnology based, e.g. (87–89). The USFDA Draft Guidance on considering whether a product involves nanotechnology (6) can be used to make an early assessment whether additional considerations or communication with the USFDA will be recommended.
Development of voluntary consensus standards (e.g., ISO and ASTM) can be helpful, when developing methods for characterization of nanomaterials, where regulatory standards are not available. All Federal agencies are directed to use voluntary consensus standards in lieu of government-unique standards in their procurement and regulatory practices, except where inconsistent with law or otherwise impractical. As with all drug products, justification and validation of the methods demonstrating that they work for their intended purpose is expected. If a drug developer is using uncommon methods for characterization and release of the nanomaterial or drug product, it is always recommended to discuss the scientific basis and qualification of the method early with the USFDA.
The EMA has implemented all ICH guidelines, provides complementary guidance documents on submission of applications and drafting of protocols, and has published dedicated reflection papers on nanomedicines (14–17). The principles laid down in these reflection papers can also be applied to other nanomedicines. The EMA website provides information on specific platforms applicants can use (13). There is also relevant guidance from the European Directorate for the Quality of Medicines & Healthcare (EDQM), ISO, Council for International Organizations of Medical Sciences (CIOMS), and OECD.
Health Canada noted that the Policy Statement on Health Canada’s Working Definition for Nanomaterial is applied in specific regulatory contexts across the Department to support the assessment of nanomaterials and to provide assistance to manufacturers and other stakeholders in meeting their respective statutory obligations for the health and safety of Canadians, pursuant to applicable Acts and Regulations. Health Canada has published numerous guidelines and policies to assist sponsors in the preparation and filing of drug submissions. ICH, as well as other guidance documents, forms, and policies needed to submit all the different types of applications are available at the Health Canada website (90). Sponsors of pharmaceutical or biological drug submissions should refer regularly to the Health Canada website for those guidelines and policies relating to a particular submission type of interest.
What About Combination Products?
Another area of considerable discussion involved combination products. Citing recent advances in complex nanostructures, which would enable the development of products with multiple functional attributes, stakeholders expressed the need for the agencies to clarify the distinction between a drug and a device as it relates to nanotechnology products. USFDA experts explained the existing review procedures (that apply to nanotechnology and conventional products, alike), which involve consultations among relevant Centers within the Agency, as needed, to make a determination regarding the classification of a product, when such questions arise. USFDA previously issued draft guidance, intended for both industry and USFDA staff, to provide information on when a product may be classified as a drug or a device (91).
Dr. Haubenreisser clarified that as soon as a device is combined with a nanomedicine and is overall defined as a medicine, it would be handled within the EU as a medicinal product and the same rules and regulations apply. For the part of the device, a national notified body will perform the conformity assessment to EU standards and provide the Conformité Européenne (CE, European conformity) mark.
Health Canada noted that classification of products is the first step in any regulatory review process at Health Canada. When the classification of a product is not clear, members of the Therapeutic Products Classification Committee (TPCC) may be consulted. The TPCC has set out the factors that influence the classification of a health product as a drug or medical device. These factors are described in the guidance document “Factors Influencing the Classification of Products at the Device-Drug Interface” (92). This guidance applies to therapeutic products at the device/drug interface when the appropriate regulatory framework is not immediately apparent. Health Canada also has a policy document on combination products (93). According to this policy, drug/device combination product classification decisions consider the principal mechanism of action by which the claimed effect or purpose of the product is achieved. The entire product then is regulated under either the Food and Drug Regulations or the Medical Devices Regulations.
What Regulatory Mechanisms Exist for the Protection of Intellectual Property of Materials and Equipment (Raw Materials, Excipients, Instruments, Algorithms Used, etc.) to the Suppliers of These Products, in Your Regions?
At USFDA, most chemistry, manufacturing, and controls data are kept confidential, and the confidentiality in a drug master file (DMF) is the same as in a New Drug Application (NDA). However, the applicant should understand and have quality control over all ingredients and components of their product and is ultimately responsible for assuring the quality of that product.
In the EU, the active substance masterfile (ASMF) does not apply to excipients. However, if a sponsor uses a new technology, the sponsor needs to share this information with the applicant. However, this information will not be available in the public domain since it is considered commercial confidential.
At Health Canada, the DMF files are considered confidential as well. In legal terms, this confidential business information belongs to the suppliers that generate it and it has traditionally been protected by intellectual property law. In Canada, intellectual property includes, in part, patents, trademarks, copyright, and trade secrets. The information in the DMF will be used to support a drug submission only if the DMF owner or agent provides Health Canada with a signed original letter of authorization to access the DMF (94).
What Are the Mechanisms and Pathways for Submitting Information to Various Regulatory Authorities and What Kind of Responses Can the Sponsors Accept? What Are Typical Timelines and Response Time?
Within USFDA, a request can be made to the division director of the appropriate review division for your product’s indication. You can get answers in writing or request a formal meeting. There are no fees for this process. Formal and informal mechanisms are available. You can also request a CMC-only meeting. Depending on the urgency of the meeting and the impact on the product development timeline, a type A, B, or C meeting may be requested and the response time is based on the meeting type (95). The best feedback is obtained when the meeting package contains high-quality information (e.g., specific questions, supporting data). Meeting packages are preferred which contain all the topics to be discussed. It is also good to state whether you would like to receive the answers in writing. Pre-IND meetings or type C meetings are an appropriate setting for industry to discuss and request feedback on alternative scenarios, new analytical approaches, when the developmental plans have not been finalized. In early stages in development, sponsors can submit questions, but USFDA may respond saying there is insufficient information for review depending on the quality of the submission.
In EMA, a sponsor developing a nanomedicine can request formal scientific advice or protocol assistance (18,20) relating to any question in relation to the development of the nanomedicine at anytime during development, prior to submission of a marketing authorization application and during the post-authorization phase. The sponsor can also apply for qualification advice on the use of a proposed method (21). The formal advice is given by the EMA’s Scientific Advice Working Party within 60–70 days to the sponsor. Scientific Advice requires payment of a fee, with fee reductions up to 90 or 100% for SMEs or orphan medicines. Sponsors can request for parallel scientific advice with USFDA, if seeking approval at the same time (20). The EMA’s ITF offers informal advice free of charge (19). Upon request, meetings are being organized within 60 days. The aim is to facilitate an informal exchange of information and provision of guidance early in the development. It can cover uncertainties, borderline products, and regulatory or scientific implications of emerging therapies. Any information can be presented or asked about. The ITF has a specific subgroup on nanotechnology.
At Health Canada, the manufacturer can discuss the application before filing a submission. There are no fees for this process. Health Canada encourages sponsors and other stakeholders to communicate with the responsible regulatory authority early in the development process, especially for combination products that are, contain, or make use of nanomaterials. In order to identify and assess potential risks and benefits of nanotechnology-based health and food products, the Department encourages manufacturers to request a presubmission meeting with the responsible regulatory authority to discuss the type of information that may be required for their product’s safety assessment. Guidance for Industry: Management of Drug Submissions provides clarification to sponsors of how Health Canada manages information and material submitted by sponsors in accordance with the Food and Drugs Act and Regulations (96).
KEY OUTCOMES AND NEXT STEPS
Overall conclusions from the Workshop can be summarized as follows:
The current regulatory framework involving review of safety, quality, and efficacy is adequate for the regulation of drug products containing nanomaterials. Agencies have successfully implemented subject matter expert groups that can provide advice to industry through guidance or meetings, train reviewers, prioritize research, and monitor the use of nanomaterials in drug products.
The diversity of nanomaterials and their intended use in drug products, as well as the combination of emerging and mature nanotechnologies, make a “one-size-fits-all” guidance challenging. However, there was consensus that focused understanding of nanomaterial attributes, its intended use (function of nanotechnology), and impact on efficacy (e.g., for drug delivery) and safety (e.g., reduction in toxicity) can be foundations for a risk management framework throughout the product lifecycle. USFDA/CDER will include this feedback in future guidance writing efforts for nanomaterials in drug products.
Clarity on the applicability of current drug product guidance to nanomaterials, on the general classification of nanomaterials based on potential risk, and on additional considerations for the evaluation and development of nanomaterials is sought by industry, regulators, and academics.
Current safety evaluation approaches are generally adequate, provided the materials being characterized are representative of the material to be used in clinical trials and the eventual marketed product. Attention to specialized methods for characterization of the nanomaterial attributes (e.g., size, surface properties, and structure) is important in most cases, in order to evaluate the impact on safety and efficacy of the nanomedicines and to maintain quality.
It was recognized that the characterization of a nanomaterial will be more involved and may require more specialized analytical methods. For novel methods, an early dialogue with the applicant is highly encouraged (via EMA ITF, Scientific Advice or EMA/USFDA parallel Scientific Advice, Pre-IND/IND/Pre-NDA discussions with USFDA).
The development of joint guidance as well as harmonization of guidance in the various regions will facilitate the development and approval of novel drug delivery systems. This has already been partly achieved through the continuous international collaboration led by the EMA, Health Canada, and USFDA. It is also important to reinforce the collaboration between regulatory authorities and the nongovernmental organizations (NGOs) involved in the development and characterization of novel methods of drug delivery for products containing nanomaterials.
Regulatory agencies recognize the value of training employees in the area of nanotechnologies. Such training will help assure that employees are aware of the areas in which existing policies and practice are appropriate for nanotechnologies and where new methods or approaches may be warranted. Agencies have already conducted such training and plan to continue to do so.
There are constant advances in the state of the science involving nanomaterial drug products. Additional research, however, is still needed across the field (including academia, industry, and regulatory entities) to further develop meaningful methods to appropriately characterize nanomaterials. Such research includes techniques to appropriately measure critical quality attributes as well as methodology (both in vitro and in vivo) to establish a nanomaterial’s effect on quality, safety, and efficacy.
Continuation of communication efforts between regulatory agencies and nongovernment organization through workshops and technical and regulatory forums is strongly encouraged.
Acknowledgments
The authors would like to thank Don Henry from USFDA and Vicky Penn from PQRI for their organization efforts on this workshop. The authors would also like to thank Drs. Susan Ciotti and Stephen Gracon, NanoBio Corporation, for their presentation on nanoemulsions, which is the basis for the topical case study discussed in this paper. The authors would also like to thank Professor Marisa Papaluca-Amati and Dr. Falk Ehmann, both from the EMA, Scientific Support, for their remote participation in the Q&A session on the “EMA Perspective on the Development of Nanomedicines.”
Abbreviations
- AAPS
American Association of Pharmaceutical Scientists
- ADME
absorption, distribution, metabolism, and excretion
- AES
atomic emission spectroscopy
- API
active pharmaceutical ingredient
- ASMF
active substance masterfile
- ASTM
American Society for Testing and Materials
- AUC
area under the curve
- BCS
Biopharmaceutical Classification System
- CDER
Center for Drug Evaluation and Research (at USFDA)
- CE
Conformité Européenne
- CFR
Code of Federal Regulations (United States)
- CFSAN
Center for Food Safety and Applied Nutrition (at USFDA)
- cGMP
current good manufacturing practices
- CHMP
Committee for Medicinal Products for Human Use (at EMA)
- CIOMS
Council for International Organizations of Medicinal Sciences
- CMC
chemistry, manufacturing, and controls
- CQAs
critical quality attributes
- CV
coefficient of variance
- DLS
dynamic light scattering
- DMF
drug master file
- DSTS
Drug Submission Tracking System (Health Canada)
- EDQM
European Directorate for the Quality of Medicines & Healthcare
- EDS
energy dispersive X-ray spectroscopy
- EM
electron microscope
- EMA
European Medicines Agency
- EU
European Union
- GRAS
generally recognized as safe
- HPFB
Health Canada’s Health Products and Food Branch
- ICCR
International Cooperation on Cosmetic Regulation
- ICH
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use
- ICP
inductively coupled plasma
- IEP
isoelectric point
- IND
Investigational New Drug application
- ISO
International Organization for Standardization
- ITF
Innovation Task Force (at EMA)
- IV
intravenous
- L
liter
- μg
micrograms
- MPS
mononuclear phagocyte system
- NCI
National Cancer Institute
- NDA
New Drug Application
- NGO
nongovernmental organization
- nm
nanometer
- NP
nanoparticle
- NTA
nanoparticle tracking analysis
- OECD
Organization for Economic Co-operation and Development
- PD
pharmacodynamics
- PEG
polyethylene glycol
- PK
pharmacokinetics
- PLD
PEGylated liposomal doxorubicin
- PQRI
Product Quality Research Institute
- QbD
quality by design
- R&D
research and development
- RCC
Canada-US Regulatory Cooperation Council
- RES
reticuloendothelial system
- SME
small- or medium-sized enterprise (EMA)
- SUPAC
scale-up and post-approval changes
- TGA
Therapeutic Goods Administration (Australia)
- TNF
tumor necrosis factor alpha
- TPCC
Therapeutic Products Classification Committee (at Health Canada)
- USFDA
United States Food and Drug Administration
- USP
United States Pharmacopeia
- WPMN
Working Party on Manufactured Nanomaterials (at the OECD)
Footnotes
A recently published PQRI White Paper (98) on this same topic outlines the best practices assessing scale-up and post-approval changes for drug products developed and approved using enhanced approaches.
The workshop was cosponsored by USFDA, USP, and AAPS and endorsed by the Society of Toxicology.
Dr. Marcus Brewster passed away before the completion of this report.
References
- 1.PQRI Workshop: Nanomaterial in Drug Products Current Experience and Risk Management. January 14–15, 2014, USP Meeting Center, Rockville, MD. Final Program Agenda at: http://www.pqri.org/workshops/Nanomaterials2013/Final.Program.Agenda.pdf. Accessed 21 Aug 2014.
- 2.Cruz CN, Tyner KM, Velezquez L, Hyams K, Jacobs A, Shaw A, et al. CDER risk assessment exercise to evaluate potential risks from the use of nanomaterials in drug products. AAPS J. 2013;15(3):623–8. doi: 10.1208/s12248-013-9466-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.FDA’s approach to regulation of nanotechnology products. Available online at: http://www.fda.gov/ScienceResearch/SpecialTopics/Nanotechnology/ucm301114.htm; see also Hamburg, 2012. Science 336:299–300. Accessed 21 Aug 2014.
- 4.Office of Science and Technology Policy, Office of Management and Budget, and the United States Trade Representative. Principles for regulation and oversight of emerging technologies, March 11, 2011; available online at: http://www.whitehouse.gov/sites/default/files/omb/inforeg/for-agencies/Principles-for-Regulation-and-Oversight-of-Emerging-Technologies-new.pdf. Accessed 21 Aug 2014.
- 5.Office of Science and Technology Policy, Office of Management and Budget, and the United States Trade Representative. Policy principles for the U.S. decision-making concerning regulation and oversight of applications of nanotechnology and nanomaterials, June 9, 2011; available online at: http://www.whitehouse.gov/sites/default/files/omb/inforeg/for-agencies/nanotechnology-regulation-and-oversight-principles.pdf. Accessed 21 Aug 2014.
- 6.Final guidance for industry: considering whether an FDA-regulated product involves the application of nanotechnology, June 2014. Available online at: http://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM401695.pdf. Accessed 21 Aug 2014.
- 7.FDA guidance on nanotechnology, issued June 2014; Available online at: http://www.fda.gov/ScienceResearch/SpecialTopics/Nanotechnology/default.htm. Accessed 05 Nov 2014.
- 8.FDA’s nanotechnology regulatory science research plan (accessible at: http://www.fda.gov/ScienceResearch/SpecialTopics/Nanotechnology/ucm273325.htm. Accessed 21 Aug 2014.
- 9.Policy Statement on Health Canada’s working definition for nanomaterial—available online at: http://www.hc-sc.gc.ca/sr-sr/pubs/nano/pol-eng.php. Accessed 21 Aug 2014.
- 10.Nanotechnology-based health products and food page—available online at: http://www.hc-sc.gc.ca/dhp-mps/nano-eng.php. Accessed 21 Aug 2014.
- 11.Drug Submission Application Form for Human, Veterinary, Disinfectant Drugs and Clinical Trial Application/Attestation (HC/SC 3011)—available online at: http://www.hc-sc.gc.ca/dhp-mps/prodpharma/applic-demande/form/hc3011_sc3011-eng.php. Accessed 05 Nov 2014.
- 12.Next-generation nanomedicines and nanosimilars: EU regulators’ initiatives relating to the development and evaluation of nanomedicines. Available online at: http://www.futuremedicine.com/doi/abs/10.2217/nnm.13.68. Accessed 21 Aug 2014 and published in Nanomedicine (2013) 8(5), 849–856, doi:10.2217/nnm.13.68. [DOI] [PubMed]
- 13.EMA website (nanotechnology page): http://www.ema.europa.eu/ema/index.jsp?curl=pages/special_topics/general/general_content_000345.jsp&murl=menus/special_topics/special_topics.jsp&mid=WC0b01ac05800baed9. Accessed 21 Aug 2014.
- 14.EMA draft reflection paper on the data requirements for intravenous liposomal products developed with reference to an innovator liposomal product. Available online at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/03/WC500140351.pdf. Accessed 21 Aug 2014.
- 15.The draft reflection paper on Data requirements for intravenous iron-based nano-colloidal products. Available online at: http://www.ema.europa.eu/ema/doc_index.jsp?curl=pages/includes/document/document_detail.jsp?webContentId=WC500149496&murl=menus/document_library/document_library.jsp&mid=0b01ac058009a3dc. Accessed 21 Aug 2014.
- 16.The joint EMA/MHLW reflection paper on block copolymer micelle medicinal products. Available online at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/01/WC500159411.pdf. Accessed 21 Aug 2014.
- 17.The EMA reflection paper on surface coating. Available online at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/08/WC500147874.pdf. Accessed 21 Aug 2014.
- 18.Innovation Task Force (ITF) itfsecretariat@ema.europa.eu: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000334.jsp&mid=WC0b01ac05800ba1d9. Accessed 21 Aug 2014.
- 19.EMA guidance for companies requesting scientific advice or protocol assistance. Available online at: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000049.jsp&mid=WC0b01ac05800229b9. Accessed 21 Aug 2014.
- 20.CHMP Scientific Advice and Novel methods qualification (e.g., biomarker): scientificadvice@ema.europa.eu http://www.ema.europa.eu/ema/index.jsp?curl=pages/special_topics/general/general_content_000349.jsp&mid=WC0b01ac05800baedb. Accessed 21 Aug 2014.
- 21.Qualification of novel methodologies for drug developments: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/document_listing/document_listing_000319.jsp&mid=WC0b01ac0580022bb0. Accessed 21 Aug 2014.
- 22.EMA SME Office: smeoffice@ema.europa.eu http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000059.jsp&mid=WC0b01ac05800240cc. Accessed 21 Aug 2014.
- 23.Liversidge GG, Cundy KC. Particle size reduction for improvement of oral bioavailability of hydrophobic drugs: I. Absolute oral bioavailability of nanocrystalline danazol in beagle dogs. Int J Pharm. 1995;25:91–7. doi: 10.1016/0378-5173(95)00122-Y. [DOI] [Google Scholar]
- 24.Petrelli F, Borgonova K, Barni S. Targeted delivery for breast cancer therapy: the history of nano-article-albumin-bound paclitaxel. Expert Opin Pharmacother. 2010;11:1413–32. doi: 10.1517/14656561003796562. [DOI] [PubMed] [Google Scholar]
- 25.Zolnik BS, Sadrieh N. Regulatory perspective on the importance of ADME assessment of nanoscale material containing drugs. Adv Drug Deliv Rev. 2009;61:422–7. doi: 10.1016/j.addr.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 26.Sultana S, Khan MR, Kumar M, Kumar S, Ali M. Nanoparticles-mediated drug delivery approaches for cancer targeting: a review. J Drug Target. 2013;21:107–25. doi: 10.3109/1061186X.2012.712130. [DOI] [PubMed] [Google Scholar]
- 27.Sayes CM. The relationships among structure, activity, and toxicity of engineered nanoparticles. KONA Powder Part J. 2014;31:10. doi: 10.14356/kona.2014002. [DOI] [Google Scholar]
- 28.D’Addio SM, Prud’homme RK. Controlling drug nanoparticle formation by rapid precipitation. Adv Drug Deliv Rev. 2011;63(6):417–26. doi: 10.1016/j.addr.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 29.Yu WW, Chang E, Falkner JC, Zhang J, Al-Somali AM, Sayes CM, et al. Forming biocompatible and nonaggregated nanocrystals in water using amphiphilic polymers. J Am Chem Soc. 2007;129(10):2871–9. doi: 10.1021/ja067184n. [DOI] [PubMed] [Google Scholar]
- 30.Sayes CM, Fortner JD, Guo W, Lyon D, Boyd D, Ausman K, et al. The differential cytotoxicity of water-soluble fullerenes. Nano Lett. 2004;4(10):1881–7. doi: 10.1021/nl0489586. [DOI] [Google Scholar]
- 31.Sayes CM, Reed KL, Warheit DB. Assessing toxicity of fine and nanoparticles: comparing in vitro measurements to in vivo pulmonary toxicity profiles. Toxicol Sci. 2007;97(1):163–80. doi: 10.1093/toxsci/kfm018. [DOI] [PubMed] [Google Scholar]
- 32.Boyd BJ. Past and future evolution in colloidal drug delivery systems. Expert Opin Drug Deliv. 2008;5(1):69–85. doi: 10.1517/17425247.5.1.69. [DOI] [PubMed] [Google Scholar]
- 33.Leroux JC, Allemann E, Jaeghere FD, Doelker E, Gurny R. Biodegradable nanoparticles—from sustained release formulations to improved site specific drug delivery. J Control Release. 1996;39(2–3):339–50. doi: 10.1016/0168-3659(95)00164-6. [DOI] [Google Scholar]
- 34.Owens DE, 3rd, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307(1):93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 35.Vonarbourg A, Passirani C, Saulnier P, Benoit JP. Parameters influencing the stealthiness of colloidal drug delivery systems. Biomaterials. 2006;27(24):4356–73. doi: 10.1016/j.biomaterials.2006.03.039. [DOI] [PubMed] [Google Scholar]
- 36.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–15. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi SW, Kim WS, Kim JH. Surface modification of functional nanoparticles for controlled drug delivery. J Dispers Sci Technol. 2003;24(3–4):475–87. doi: 10.1081/DIS-120021803. [DOI] [Google Scholar]
- 38.Dawson KA, Salvati A, Lynch I. Nanotoxicology: nanoparticles reconstruct lipids. Nat Nanotechnol. 2009;4:84–5. doi: 10.1038/nnano.2008.426. [DOI] [PubMed] [Google Scholar]
- 39.Monopoli MP, Bombelli FB, Dawson KA. Nanobiotechnology: nanoparticle coronas take shape. Nat Nanotechnol. 2010;6(1):11–2. doi: 10.1038/nnano.2010.267. [DOI] [PubMed] [Google Scholar]
- 40.International Harmonization Conference guidelines: http://www.ich.org/products/guidelines.html. Accessed 21 Aug 2014.
- 41.Kesisoglou F, Panmai S, Wu Y. Nanosizing—oral formulation development and biopharmaceutical evaluation. Adv Drug Deliv Rev. 2007;59:631–44. doi: 10.1016/j.addr.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 42.Chen H, Khemtong C, Yang X, Chsang X, Gao J. Nanonozation strategies for poorly water-soluble drugs. Drug Discov Today. 2011;16:354–60. doi: 10.1016/j.drudis.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 43.Gao L, Liu G, Ma J, Wang X, Zhou L, Li X, et al. Application of drug nanocrystal technologies on oral drug delivery of poorly soluble drugs. Pharm Res. 2012;30:307–24. doi: 10.1007/s11095-012-0889-z. [DOI] [PubMed] [Google Scholar]
- 44.Narang A, Chang R, Hussain M. Pharmaceutical development and regulatory considerations for nanoparticles and nanoparticulate drug delivery systems. J Pharm Sci. 2013;102(11):3867–82. doi: 10.1002/jps.23691. [DOI] [PubMed] [Google Scholar]
- 45.Moschwitzer J. Drug nanocrystals in the commercial pharmaceutical development process. Int J Pharm. 2013;453:142–56. doi: 10.1016/j.ijpharm.2012.09.034. [DOI] [PubMed] [Google Scholar]
- 46.Kayaert P, Anne M, Van den Mooter G. Bead layering as a process to stabilize nanosuspensions: influence of drug hydrophobicity on nanocrystal reagglomeration following in vitro release from sugar sphere. J Pharm Pharmacol. 2011;63:1446–53. doi: 10.1111/j.2042-7158.2011.01351.x. [DOI] [PubMed] [Google Scholar]
- 47.Chaubal M, Popescu C. Conversions of nanosuspensions in to into dry powders by spray drying: a case study. Pharm Res. 2008;25:2302–8. doi: 10.1007/s11095-008-9625-0. [DOI] [PubMed] [Google Scholar]
- 48.Dicko A, Frazier AA, Liboiron BD, Hindeliter A, et al. Intra- and inter-molecular interactions dictate the aggregation state of irinotecan co-encapsulated with floxuridine inside liposomes. Pharm Res. 2008;25:1702–13. doi: 10.1007/s11095-008-9561-z. [DOI] [PubMed] [Google Scholar]
- 49.Gilday E, Nasrallah H. Clinical pharmacology of paliperidone palmitate—a parenteral long acting formulation for the treatment of schizophrenia. Rev Recent Clin Trials. 2011;7(1):2–9. doi: 10.2174/157488712799363307. [DOI] [PubMed] [Google Scholar]
- 50.Shegokar R, Muller RH. Nanocrystals: industrially feasible multifunctional formulation technology for poorly soluble actives. Int J Pharm. 2010;399(1–2):129–39. doi: 10.1016/j.ijpharm.2010.07.044. [DOI] [PubMed] [Google Scholar]
- 51.Muller R, Keck C. Challenges and solutions for the delivery of biotech drugs—a review of drug nanocrystal technology and lipid nanoparticles. J Biotechnol. 2004;113:151–70. doi: 10.1016/j.jbiotec.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 52.Merisko-Liversidge E, Liversidge G. Nanosizing oral and parenteral drug delivery: a perspective on formulating poorly-water soluble compounds using wet media milling technology. Adv Drug Deliv Rev. 2011;63:427–40. doi: 10.1016/j.addr.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 53.Peltonen L, Hirvonen J. Pharmaceutical nanocrystals by nanomilling: critical process parameters, particle fracturing and stabilization methods. J Pharm Pharmacol. 2010;62:1569–79. doi: 10.1111/j.2042-7158.2010.01022.x. [DOI] [PubMed] [Google Scholar]
- 54.Singare D, Marella S, Gowthamarjan K, Kulkarni G, Vooturi R, Rao P. Optimization of formulation and process variables of nanosuspensions: an industrial perspective. Int J Pharm. 2010;402:213–20. doi: 10.1016/j.ijpharm.2010.09.041. [DOI] [PubMed] [Google Scholar]
- 55.Feldman EJ, Lancet JE, Kolitz JE, Ritchie EK, Roboz GJ, List AF, et al. First-in-man study of CPX-351: a liposomal carrier containing cytarabine and daunorubicin in a fixed 5:1 molar ratio for the treatment of relapsed and refractory acute myeloid leukemia. J Clin Oncol. 2011;29:979–85. doi: 10.1200/JCO.2010.30.5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paciotti GF, Myer L, Weinreich D, Goia D, Pavel N, McLaughlin RE, et al. Colloidal gold: a novel nanoparticle vector for tumor directed drug delivery. Drug Deliv. 2004;11:169–83. doi: 10.1080/10717540490433895. [DOI] [PubMed] [Google Scholar]
- 57.Libutti SK, Paciotti GF, Byrnes AA, Alexander HR, Jr, Gannon WE, Walker M, et al. Phase I and pharmacokinetic studies of CYT-6091, a novel PEGylated colloidal gold-rhTNF nanomedicine. Clin Cancer Res. 2010;16(24):6139–49. doi: 10.1158/1078-0432.CCR-10-0978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kingston DGI, Tamarkin L, Paciotti GF. Conformationally constrained and nanoparticle-targeted paclitaxels. Pure Appl Chem. 2012;10:1361–74. doi: 10.1351/PAC-CON-11-09-02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Muller RH, Gohla S, Keck CM. State of the art of nanocrystals—special features, production, nanotoxicology aspects and intracellular deliver. Eur J Pharm Biopharm. 2011;78:1–9. doi: 10.1016/j.ejpb.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 60.Zamboni WC, Torchilin V, Patri AK, Hrkach J, Stern S, Lee R, et al. Best practice s in cancer nanotechnology: perspective from NCI nanotechnology alliance. Clin Cancer Res. 2012;18(12):3229–41. doi: 10.1158/1078-0432.CCR-11-2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Duncan R, Gaspar R. Nanomedicine(s) under the microscope. Mol Pharm. 2011;8(6):2101–41. doi: 10.1021/mp200394t. [DOI] [PubMed] [Google Scholar]
- 62.Park JW, Benz CC, Martin FJ. Future directions of liposome- and immunoliposome-based cancer therapeutics. Semin Oncol. 2004;31(6 Suppl 13):196–205. doi: 10.1053/j.seminoncol.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 63.Zamboni WC, Yoshino K. Formulation and physiologic factors affecting the pharmacokinetics and pharmacodynamics of liposomal agents. Drug Deliv Syst. 2010;25(1):58–70. [Google Scholar]
- 64.Caron WP, Song G, Kumar P, Rawal S, Zamboni WC. Pharmacokinetic and pharmacodynamic disposition of carrier-mediated agents. Clin Pharmacol Ther. 2012;91(5):802–12. doi: 10.1038/clpt.2012.12. [DOI] [PubMed] [Google Scholar]
- 65.Moghimi SM, Parhamifar L, Ahmadvand D, Wibroe PP, Andresen TL, Farhangrazi ZS, et al. Particulate systems for targeting of macrophages: basic and therapeutic concepts. J Innate Immunol. 2012;4(5–6):509–28. doi: 10.1159/000339153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kumar P, Caron WP, Song G, Rawal S, Zamboni WC. Nanoparticle effects on the interaction with cells of the mononuclear phagocytic system. In: Dobrovolskaia M, editor. Immunological properties of engineered nanomaterials. First edition. World Scientific, in press. 2013.
- 67.Caron WP, Lay JC, Fong AM, La-Beck NM, Kumar P, Newman SE, et al. Translational studies of phenotypic probes for the mononuclear phagocyte system and liposomal pharmacology. J Pharmacol Exp Ther. 2013;347(3):599–606. doi: 10.1124/jpet.113.208801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schell RF, Sidone BJ, Caron WP, Walsh MD, White TF, Zamboni BA, et al. Meta-analysis of inter-patient pharmacokinetic variability of liposomal and non-liposomal anticancer agents. Nanomedicine. 2014;10:109–17. doi: 10.1016/j.nano.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zamboni WC, Strychor S, Maruca L, Ramalingam S, Zamboni BA, Wu H, et al. Pharmacokinetic study of pegylated liposomal CKD-602 (S-CKD602) in patients with advanced malignancies. Clin Pharmacol Ther. 2009;86(5):519–26. doi: 10.1038/clpt.2009.141. [DOI] [PubMed] [Google Scholar]
- 70.Gabizon A, Isacson R, Rosengarten O, Tzemach D, Shmeeda H, Sapir R. An open-label study to evaluate dose and cycle dependence of the pharmacokinetics of pegylated liposomal doxorubicin. Cancer Chemother Pharmacol. 2008;61(4):695–702. doi: 10.1007/s00280-007-0525-5. [DOI] [PubMed] [Google Scholar]
- 71.La-Beck NM, Zamboni BA, Gabizon A, Schmeeda H, Amantea M, Gehrig PA, et al. Factors affecting the pharmacokinetics of pegylated liposomal doxorubicin in patients. Cancer Chemother Pharmacol. 2012;69(1):43–50. doi: 10.1007/s00280-011-1664-2. [DOI] [PubMed] [Google Scholar]
- 72.Wu H, Ramanathan RK, Zamboni BA, Strychor S, Ramalingam S, Edwards RP, et al. Mechanism-based model characterizing bidirectional interaction between PEGylated liposomal CKD-602 (S-CKD602) and monocytes in cancer patients. Int J Nanomedicine. 2012;7:5555–64. doi: 10.2147/IJN.S35751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zamboni WC, Maruca LJ, Strychor S, Zamboni BA, Ramalingam S, Edwards RP, et al. Bidirectional pharmacodynamic interaction between pegylated liposomal CKD-602 (S-CKD602) and monocytes in patients with refractory solid tumors. J Liposome Res. 2011;21(2):158–65. doi: 10.3109/08982104.2010.496085. [DOI] [PubMed] [Google Scholar]
- 74.Charrois GJ, Allen TM. Multiple injections of pegylated liposomal doxorubicin: pharmacokinetics and therapeutic activity. J Pharmacol Exp Ther. 2003;306(3):1058–67. doi: 10.1124/jpet.103.053413. [DOI] [PubMed] [Google Scholar]
- 75.Suzuki T, Ichihara M, Hyodo K, Yamamoto E, Ishida T, Kiwada H, et al. Accelerated blood clearance of PEGylated liposomes containing doxorubicin upon repeated administration to dogs. Int J Pharm. 2012;436(1–2):636–43. doi: 10.1016/j.ijpharm.2012.07.049. [DOI] [PubMed] [Google Scholar]
- 76.Crist RM, et al. Common pitfalls in nanotechnology: lessons learned from NCI’s Nanotechnology Characterization Laboratory. Integr Biol (Camb) 2013;5:66–73. doi: 10.1039/C2IB20117H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Skoczen SL, Potter TM, Dobrovolskaia MA. In vitro analysis of nanoparticle uptake by macrophages using chemiluminescence. Methods Mol Biol. 2011;697:255–61. doi: 10.1007/978-1-60327-198-1_27. [DOI] [PubMed] [Google Scholar]
- 78.Song G, Moore S, Tarrant T, Dobrovolskaia M, et al. 126: Relationship between complement factors and CC chemokines and the pharmacokinetics (PK) and pharmacodynamics (PD) of PEGylated liposomal doxorubicin (PLD) in patients with refractory epithelial ovarian cancer (EOC) Eur J Cancer. 2012;48(SUPPL 6):39–40. doi: 10.1016/S0959-8049(12)71924-X. [DOI] [Google Scholar]
- 79.Kumar P, Caron WP, Song G, Gallagher K, et al. Relationship between serum hormone levels and pharmacokinetics (PK) of PEGylated liposomal doxorubicin (PLD) in patients with refractory ovarian cancer. Cancer Res. 2012;72(8 SUPPL 1). doi: 10.1158/1538-7445.AM2012-2673
- 80.Caron WP, Song G, Kumar P, Rawal S, Zamboni WC. Interpatient pharmacokinetic and pharmacodynamic variability of carrier-mediated anticancer agents. Clin Pharmacol Ther. 2012;91:802–12. doi: 10.1038/clpt.2012.12. [DOI] [PubMed] [Google Scholar]
- 81.Maeda H, Nakamura N, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev. 2013;65:71–9. doi: 10.1016/j.addr.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 82.Gregoriadis G. Drug entrapment in liposomes. FEBS Lett. 1973;36:292–6. doi: 10.1016/0014-5793(73)80394-1. [DOI] [PubMed] [Google Scholar]
- 83.Birrenbach G, Speiser PP. Polymerized micelles and their use as adjuvants in immunology. J Pharm Sci. 1976;65:1763–6. doi: 10.1002/jps.2600651217. [DOI] [PubMed] [Google Scholar]
- 84.Juliano RL, Stamp D. Pharmacokinetics of liposome-encapsulated anti-tumor drugs: studies with vinblastine, actinomycin D, cytosine, arabinoside, and daunomycin. Biochem Pharmacol. 1978;27:21–7. doi: 10.1016/0006-2952(78)90252-6. [DOI] [PubMed] [Google Scholar]
- 85.FDA guidance for industry: nonclinical studies for the safety evaluation of pharmaceutical excipients: May 2005: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm079250.pdf. Accessed 21 Aug 2014.
- 86.FDA Draft Guidance for Industry: liposome drug products: CMC, human pharmacokinetic and bioavailability; and labeling documentation: August 2002: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070570.pdf. Accessed 21 Aug 2014.
- 87.FDA draft guidance on doxorubicin hydrochloride: November 2013. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm199635.pdf. Accessed 21 Aug 2014.
- 88.FDA draft guidance on iron sucrose: November 2013: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM297630.pdf. Accessed 21 Aug 2014.
- 89.FDA draft guidance on aprepitant: November 2008 http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm082548.pdf. Accessed 21 Aug 2014.
- 90.Health Canada website http://hc-sc.gc.ca/dhp-mps/prodpharma/applic-demande/guide-ld/index-eng.php. Accessed 21 Aug 2014.
- 91.Draft Guidance for Industry and FDA Staff: Classification of products as drugs and devices & additional product classification issues, June 2011. Available from Office of Combination Products: http://www.fda.gov/aboutfda/centersoffices/officeofmedicalproductsandtobacco/officeofscienceandhealthcoordination/ucm2018184.htm Accessed 21 Aug 2014. At: http://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM258957.pdf. Accessed 21 Aug 2014.
- 92.Guidance document: factors influencing the classification of products at the device-drug interface available online at: http://www.hc-sc.gc.ca/dhp-mps/dev-drug-instr-drogue/class_prod_gd_ld-eng.php. Accessed 21 Aug 2014.
- 93.Drug/medical device combination products: http://hc-sc.gc.ca/dhp-mps/prodpharma/applic-demande/pol/combo_mixte_pol_2006-eng.php. Accessed 21 Aug 2014.
- 94.Health Canada draft guidance document—drug master files (DMFs) is available at: http://hc-sc.gc.ca/dhp-mps/prodpharma/applic-demande/guide-ld/chem/draft_ebauche_dmf_fmm_guide_ld-eng.php. Accessed 21 Aug 2014.
- 95.FDA guidance for industry: formal meetings between the FDA and sponsors or applicants: May 2009: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm153222.pdf. Accessed 21 Aug 2014.
- 96.Health Canada guidance for industry: food and drugs act and regulations: http://www.hc-sc.gc.ca/dhp-mps/prodpharma/applic-demande/guide-ld/mgmt-gest/mands_gespd-eng.php. Accessed 21 Aug 2014.
- 97.Commission Recommendation 2011/696/EU, OJ L 275, 20.10.2011. Available online at: http://ec.europa.eu/environment/chemicals/nanotech/pdf/commission_recommendation.pdf.
- 98.Van Buskirk GA, Asotra S, Balducci C, Basu P, DiDonato G, Dorantes A, et al. Best practices for the development, scale-up, and post-approval change control of IR and MR dosage forms in the current quality-by-design paradigm. AAPS Pharm Sci Technol. 2014;15:665–93. doi: 10.1208/s12249-014-0087-x. [DOI] [PMC free article] [PubMed] [Google Scholar]