

Abstract
The prevalence of metabolic syndrome (MS) has been rising alarmingly worldwide, including in the United States, but knowledge on specific genetic determinants of MS is very limited. Therefore, we planned to identify the genetic determinants of MS as defined by National Cholesterol Education Program/Adult Treatment Panel III (NCEP/ATPIII) criteria. We performed linkage screen for MS using data from 692 Mexican Americans, who participated in the San Antonio Family Diabetes/Gallbladder Study (SAFDGS). We found strong evidence for linkage of MS on chromosome 7q (LOD = 3.6, empirical P = 6.0 × 10−5), between markers D7S2212 and D7S821. In addition, six chromosomal regions exhibited potential evidence for linkage (LOD ≥ 1.2) with MS. Further, we examined 29 single nucleotide polymorphisms (SNPs) from the fatty acid translocase (FAT or CD36, 18 SNPs) gene and guanine nucleotide binding protein, alpha transducing 3 (GNAT3, 11 SNPs) gene, located within the 1-LOD support interval region for their association with MS and its related traits. Several SNPs were associated with MS and its related traits. Remarkably, rs11760281 in GNAT3 and rs1194197 near CD36 exhibited the strongest associations with MS (P = 0.0003, relative risk [RR] = 1.6 and P = 0.004, RR = 1.7 respectively) and several other related traits. These two variants explained about 18% of the MS linkage evidence on chromosome 7q21, and together conferred approximately 3-fold increase in MS risk (RR = 2.7). In conclusion, our linkage and subsequent association studies implicate a region on chromosome 7q21 to influence MS in Mexican Americans.
Keywords: Metabolic syndrome, NCEP/ATPIII, linkage/genome scan, variance components linkage analysis, single nucleotide polymorphisms, association analysis, Mexican Americans, CD36/FAT, GNAT3
Metabolic abnormalities such as obesity, insulin resistance and hyperinsulinemia, impaired glucose tolerance, dyslipidemia, and hypertension often cluster to constitute the Metabolic Syndrome [MS]. The MS is a predictor of both type 2 diabetes (T2DM) and Coronary Heart Disease (CHD), and its prevalence has been rising alarmingly worldwide. In the United States (US), more than 47 million people are affected with MS. Specifically, its prevalence is high in minority populations such as Mexican Americans, who had the highest age-adjusted prevalence of MS [∼32%] when compared to the non-Hispanic whites, African Americans and other ethnic groups (1). Added to this burden, over the past 20 years, the prevalence of MS among children and adolescents has also been increasing at epidemic rates in several populations including Mexican Americans and African Americans (2-4).
The MS and its components are complex disease conditions that are influenced by genetic and environmental factors and their interactions MS can be defined as a single, composite phenotype for genetic analysis using criteria such as the National Cholesterol Education Program [NCEP]/Adult Treatment Panel III [ATPIII] (NCEP/ATPIII) recommendations, the most widely used definition in the US (5,6). In addition to the NCEP/ATPIII criteria, several organizations have proposed different definitions for MS. While the various definitions use similar core components of MS, primarily including the measurements of obesity, glucose, lipids, and blood pressure, they differ in the number of traits and the cut-off points required for the diagnosis. Despite certain caveats, several studies have used the NCEP/ATPIII definition for its simplicity, in the context of clinical practice and epidemiological investigations (7); most importantly, as noted by Grundy et al. (6), it avoids emphasis on a single cause.
Several genome-wide linkage and association studies have been performed to understand the genetic basis of the components of MS by using a number of analytical tools such as multivariate genetic analysis and factor analysis (8-12). Such studies have identified several genetic regions linked to MS related traits, but showing limited concordance in their results (11). In contrast to the above approaches, a single, composite MS phenotype using the NCEP/ATPIII or other criteria can be used to identify genetic regions with potential common genetic influences on MS; however, there have been a few such studies (13, 14).
Therefore, the aim of our study was to identify the susceptibility loci for MS using the NCEP/ATPIII definition in a Mexican American population who are at high risk for the development of MS. Here, we report the results of our genome-wide linkage screen for MS performed by using the multipoint variance components linkage analysis and the phenotypic and genotypic (∼10 cM map) data collected from the Mexican American individuals who participated in our San Antonio Family Diabetes/Gallbladder Study (SAFDGS). In addition, we report the results of our preliminary MS association study performed by examining the genetic variants in two of the positional candidate genes, which are physically close to each other, located within the 1-LOD support interval region of our MS linkage signal on chromosome 7q.
Materials and Methods
San Antonio Family Diabetes/Gallbladder Study (SAFDGS)
Phenotypic and genotypic data for the present study were obtained from about 720 individuals from 39 low-income Mexican American families, who were recruited during the years 1998 through 2001 for the San Antonio Family Gallbladder Study [SAFGS], a follow-up and extension of the San Antonio Family Diabetes Study [SAFDS]. These studies are collectively referred to as the San Antonio Family Diabetes/Gallbladder Study (SAFDGS) and the particulars and procedures of the SAFDGS participant's recruitment and data collection have been detailed elsewhere (15,16).
Phenotype Data
Blood samples from the participants were obtained after a 12-h fast, and measured for various metabolic parameters including glucose concentrations, triglycerides, high density lipoprotein cholesterol (HDL-C) levels at the Frederic C. Bartter General Clinical Research Center (GCRC) laboratory, South Texas Veterans Healthcare System, Audie L. Murphy Division, San Antonio, as described elsewhere (16). Blood samples were collected again 2-h after a standardized oral glucose load for plasma glucose measurements. In addition to the collection of demographic and medical history information, the anthropometric (height, weight, and waist circumference) and blood pressure (systolic and diastolic blood pressure) data were collected using standardized protocols (16). T2DM was diagnosed in accordance with the 1999 criteria of the World Health Organization. Also, individuals who did not meet these criteria but reported that they were under treatment with either oral antidiabetic agents or insulin and gave a history of diabetes were considered to have T2DM (16).
The MS was defined in accordance with NCEP/ATPIII criteria which use the presence of at least 3 or more of the following 5 risk factors: increased waist circumference [>102 cm in men and >88cm in women], hypertriglyceredemia [≥150 mg/dl], low HDL-C levels [< 40 mg/dl in men and < 50 mg/dl in women], hypertension [≥ 130/85 mm Hg or individuals who were on hypertensive medication with normal blood pressure values], and high fasting glucose concentrations [≥ 110 mg/dl or a diagnosis of T2DM, as defined above] (5). Subsequently, the NCEP/ATPIII definition has changed the threshold for high fasting glucose values to ≥ 100 mg/dl, in accordance with the ADA recommendation for impaired fasting glucose (17, 6). In the present study, although we are aware of this change, we have used the earlier definition which uses the threshold for high fasting glucose values ≥ 110 mg/dl (NCEP/ATPIII, 2001), for the purpose of comparison with other studies. However, we have also reanalyzed the data with the newer threshold for impaired fasting glucose (i.e., ≥ 100 mg/dl).
Genotype Data
The genotypic data and genetic maps used for this study were described in detail previously (15,16). Briefly, we used the genotypic data (i.e., ∼10 cM map) generated by the Center for Inherited Disease Research (CIDR) that contained information from 382 highly polymorphic autosomal markers. The marker genotyping errors and the pedigree discrepancies were resolved using analytical techniques as implemented in programs Simwalk2 and PREST (16). The Marshfield genetic maps were used, and the multipoint identical-by-descent (IBD) matrices given a number of genetic markers (map distance in Haldane cM) were calculated using Markov chain Monte Carlo methods implemented in the program Loki (16).
Positional Candidate Genes, SNP identification, selection and genotyping
As part of our preliminary screening of positional candidate genes for identifying the potential functional variants within the 1 LOD-unit support interval surrounding the MS linkage peak on chromosome 7q21, the fatty acid translocase (FAT; also called leukocyte differentiation antigen CD36; MIM 173510) gene with potential functional relevance to MS and its flanking gene guanine nucleotide binding protein, alpha transducing 3 (GNAT3; MIM 139395) were chosen for initial association studies. We performed partial resequencing (coding exons, including their flanking regions, 5′-UTRs, 3′-UTRs, and approximately 1 kb of the putative promoter regions of the reported alternatively spliced transcripts; 18) of the CD36 gene. Sequencing was performed by Polymorphic DNA Technologies (Alameda, CA) from 30 SAFDGS subjects, who contributed positively to the evidence for linkage of MS on chromosome 7q21. Briefly, primers were designed for the above mentioned regions based on the genomic sequences (NCBI Build36) of the alternative transcripts NM_001001547; AK129899; NM _001001548; AK096858; NM_0072. NA10791. Using conventional PCR amplification these regions were amplified and sequenced in both directions using Big Dye Terminator system on ABI 3730xl automated sequencers. DNA from chromosome 7 specific somatic cell hybrid was used in all the sequencing reactions as a “hemizygous” nonpolymorphic control representing a single allelic version of all variants on this chromosome. The sequencing results of all these 30 subjects, sent as chromatograms by the company were aligned, visualized and compared against the reference sequence, to identify the sequence variations between individuals, using software Sequencher version 3 (Gene Codes Corporation, Ann Arbor, MI). As a quality control one encrypted duplicate was included in all the sequencing reactions which showed 100% concordance in the sequences. All the identified sequence variants were confirmed with the chromatogram sequences derived from the other direction.
Partial resequencing of CD36 gene and single nucleotide polymorphism (SNP) identification
We identified 22 SNPs, of which one was unique to our study population. Among the 22 SNPs, 14 SNPs which were polymorphic and for whom probes were designable were chosen for genotyping. In addition, 5 SNPs in the CD36 gene and 11 SNPs in the GNAT3 gene available in the public SNP database (dbSNP) were chosen for genotyping to assure adequate coverage of the locus. All together, 30 single nucleotide polymorphisms (SNPs) were examined.
Genotyping was performed in ∼720 SAFDGS subjects by the PCR based, 5′ nuclease Taqman allelic discrimination method, employed by Applied Biosystems's (ABI) TaqMan platform per the company's protocol (Foster City, CA). Following PCR amplification, end-point fluorescence was read in the ABI's Primer 7900HT sequence detection system and genotypes were assigned using SDS2.1 Allelic Discrimination Software. Each analysis included negative controls with no DNA and 10% duplicated samples (randomly selected and genotyped) for reproducibility.
Variance-Components Linkage Analysis
The variance-components (VC) approach uses information from all possible biological relationships simultaneously in an attempt to disentangle the genetic architecture of a quantitative trait. The VC method was extended to analyze the dichotomous trait MS using a liability or threshold model. The variance components (i.e., heritability attributed to the susceptibility locus and heritability attributed to the residual additive genetic effects), and covariates for MS (i.e., age) were estimated simultaneously in likelihood terms. Since sex information was used for the definition of certain risk factors of MS, it was not considered as a covariate for the genetic analysis. The hypothesis of no linkage (i.e., h2q = 0) was tested by comparing the likelihood of this restricted model with that of a model in which the parameter h2q was estimated. Twice the difference in likelihoods of these two models yields a test statistic that is asymptotically distributed as a ½:½ mixture of a χ21 and a point mass at zero. To obtain lod scores, the ln likelihood values were converted into values of log10. A LOD score of 3.0 and above was considered as strong or significant evidence in support of linkage (19). For the purpose of discussion, other genetic regions across the genome with LOD ≥ 1.2 were considered as evidence for potential linkage.
Because the SAFDGS families were ascertained on T2DM probands, as a conservative approach, all our genetic analyses incorporated correction for ascertainment bias by computing the likelihood of a pedigree conditional on the phenotype (i.e., MS) of the proband. To verify our major MS linkage finding on chromosome 7q, we performed simulation analysis to determine the empirical P value, using information obtained from 100,000 replicates. All analytical procedures are incorporated in the program SOLAR as described elsewhere (16).
Association Analysis
We performed association analysis of the SNP data with MS and various MS related traits, using the measured genotype analysis (MGA) and Bayesian quantitative trait nucleotide (BQTN) analysis. The genotypic data were checked for Mendelian consistency using the program SimWalk2 (20). This program employs Markov Chain Monte Carlo and simulated annealing algorithms to assign probabilities of mistyping to each genotype that will be used to make decisions about the appropriate genotypes to blank. This approach also takes into account the low likelihood of recombination events within the gene; thus, can be used to identify spurious recombinations due to typing error. Using SOLAR, the allele frequencies were calculated, SNPs were tested for Hardy–Weinberg Equilibrium (HWE), and linkage disequilibrium (LD) between SNP pairs was estimated using r2 values.
Measured genotype analysis (MGA)
In the variance components (VC) approach described above, variance components are modeled as random effects, whereas the effects of measured covariates are modeled as fixed effects on the trait mean. In the MGA, the marker genotypes are generally incorporated in the mean effects model as a measured covariate assuming additivity of allelic effects (19). The maximum likelihood techniques were used to estimate the VCs, the association parameters, and the other covariate effects (e.g., age and sex). The hypothesis of no association was tested by comparing likelihood of a model in which the effect of measured genotype was estimated with a model where the effect of measured genotype was fixed at zero. Prior to performing MGA, the presence of population stratification was tested using quantitative transmission disequilibrium test [QTDT], as implemented in SOLAR. With regard to the dichotomous traits, the threshold-based liability model was projected onto the dichotomous outcome in a manner directly analogous to logistic regression to obtain genotypic relative risks. These calculations of genotype-specific prevalence were obtained from the estimated liability model using a standard normal distribution function. In reference to the quantitative traits, we estimated mean trait values by genotype category. The program SOLAR was used to carry out these analyses.
Bayesian quantitative trait nucleotide (BQTN) analysis
The BQTN technique was used to analyze SNPs simultaneously to find the nucleotide variants responsible for influencing MS, using a measured genotype approach (22). This technique can be used with a set of SNP data from a candidate gene to identify the sequence variants that are either functional or that exhibit the highest disequilibria with the true functional sites and is detailed elsewhere (22, 23). Given that a candidate gene may contain a number of SNPs that could generate several possible competing models of QTN action, a Bayesian model averaging approach was used to analyze the SNP data simultaneously in order to estimate the probability that each SNP has an effect on the MS phenotype. The BQTN approach is described in detail elsewhere (23).
Conditional Linkage/Association Analysis
To determine whether the associated SNP(s) account for the MS linkage signal, we combined the QTN analysis with our identity by descent (IBD)-based variance component linkage analysis (21). If a variant or set of variants is/are responsible for the observed linkage signal, linkage analysis conditional on a fixed-effect measured genotype analysis of the polymorphism will yield an expected LOD score near zero. Alternatively, if the associated polymorphism is in less than complete linkage disequilibrium with the true functional site, linkage analysis will generally yield a non-zero LOD score. The analytical procedures described above are implemented in the program SOLAR.
Results
The clinical characteristics of the 692 individuals, for whom information on MS (NCEP/ATPIII) were available, are shown in Table 1. The mean age (± SD) was 44.5 (± 16.3) years; and ∼61% of the study sample were females. The prevelence of MS in our family data was 43.5% and the prevalence rates of its components ranged from 33.0% (high fasting glucose) to 60.7% (waist circumference or abdominal obesity) [Table 2].
Table 1. Characteristics of the 692 SAFDGS participants.
| Quantitative MS components* | Mean ± SD or % | ||
|---|---|---|---|
| Total (N = 692) | Males (N = 270) | Females (N = 422) | |
| Mean age (years) | 44.5 ± 16.3 | 44.2 ± 16.7 | 44.8 ± 16.1 |
| Waist circumference (mm) | 1003.0 ± 161.7 | 1029.7 ± 146.0 | 976.2 ± 177.4 |
| Fasting glucose (mg/dl) | 115.9 ± 49.2 | 116.2 ± 47.4 | 115.5 ± 51.0 |
| Systolic blood pressure (mm Hg) | 127.7 ± 17.4 | 130.4 ± 15.4 | 126.0 ± 18.4 |
| Diastolic blood pressure (mm Hg) | 70.3 ± 9.7 | 72.1 ± 9.6 | 68.8 ± 9.4 |
| Triglycerides (mg/dl) | 163.8 ± 170.7 | 184.4 ± 241.6 | 143.2 ± 99.8 |
| High density lipoprotein cholesterol (mg/dl) | 45.5 ± 11.9 | 43.0 ± 11.2 | 47.9 ± 12.6 |
| Dichotomous MS components* and MS | |||
| Abdominal Obesity | 37.1 | 45.6 | 69.4 |
| High fasting glucose | 53.8 | 35.2 | 32.2 |
| High blood pressure | 60.7 | 55.9 | 42.0 |
| High Triglycerides | 33.0 | 44.5 | 32.5 |
| Low HDL-C | 46.5 | 43.4 | 60.3 |
| Metabolic syndrome** | 43.5 | 43.3 | 43.6 |
the sample size used for the lipid traits were slightly different from the 692 as shown below: Triglycerides/High density lipoprotein cholesterol = Total: 665/663; Males: 256/256; Females: 409/407.
The MS was defined in accordance with NCEP/ATPIII criteria (2001). The criteria uses the presence of at least 3 or more of the following 5 risk factors: increased waist circumference [>102 cm in men and >88cm in women], hypertriglyceredemia [≥150 mg/dl], low HDL-C levels [< 40 mg/dl in men and < 50 mg/dl in women], hypertension [≥ 130/85 mm Hg or individuals who were on hypertensive medication with normal blood pressure values], and high fasting glucose concentrations [≥ 110 mg/dl or a diagnosis of T2DM as described in the text].
Table 2. a. Chromosomal regions linked to MS with LOD scores ≥ 1.2 based on multipoint linkage analyses.
| Marker region | Distance from p-ter (cM)a | Chromosomal location | Maximum LODb |
|---|---|---|---|
| D2S1360 | 38 | 2p24.2 | 1.2 |
| D2S1328 | 133 | 2q14.3 | 1.3 |
| D5S2845 | 36 | 5p14.3 | 1.5 |
| D7S2212-D7S821/ GATA5D08 | 95-109 | 7q21.11-7q21.3 | 3.6 |
| D8S1136/GATA41A01 | 82 | 8q13.1 | 2.6 |
| D11S1999 | 17 | 11p15.4 | 2.3 |
| D13S1265-D13S285 | 99-111 | 13q33.3-13q34 | 1.6 |
| aMarshfield data (Kosambi cM) | |||
| bMS adjusted for covariate effects of age | |||
| b. Evidence for linkage of the five dichotomous MS risk factors as defined by NCEP/ATPIII criteria and the corresponding quantitative traits on chromosome 7q | ||
|---|---|---|
| Traits (Dichotomous; Quantitative) | Distance from p-ter (cM)a | Maximum LODb |
| Abdominal Obesity; WC | 95-109 | 1.45; 1.65 |
| High Triglycerides; TG | 95-109 | 0.76; 0.19 |
| Low HDL-C; HDL-C | 95-109 | 1.12; 1.36 |
| High fasting glucose; FG | 95-109 | 0.85; 0.22 |
| High blood pressure; SBP and DBP | 95-109 | 0.43; 0.05 and 0.54 |
Marshfield data (Kosambi cM)
all dichotomous traits adjusted for covariate effects of age as in the case of original linkage analysis (Table 2a); quantitative traits were adjusted for age and sex terms as well as for medication status for TG, HDL-C, FG, SBP and DBP
Multipoint linkage analysis
Prior to conducting linkage analysis, after accounting for the age effects on MS, we detected a high heritability for MS (h2 = 0.51 ± 0.10). The results of our genome-wide linkage screen are provided in Table 2a and Figure 1. The strongest evidence for linkage of MS (LOD = 3.6; empirical P = 6.0 × 10−5) occurred on chromosome 7q, between markers D7S2212 and D7S821, after accounting for age influences (Figures 1 & 2). Of the examined 39 families, 22 families (56.4%) positively contributed to this linkage signal, and the evidence for linkage in this subset of 22 families is much stronger (LOD = 7.8). As shown in Table 2b, all of the five MS risk factors as defined by NCEP/ATPIII criteria in the total data set, exhibited some evidence of linkage at this location. For the purpose of comparison, evidence for linkage of the quantitative measures of these dichotomized risk factors was also reported in Table 2b. In addition, six genetic locations representing 5 chromosomes across the genome exhibited potential evidence for linkage (i.e., LOD ≥ 1.2) with MS (Table 3, Figure 1). The 1 LOD-unit support interval surrounding the MS linkage peak on chromosome 7q is flanked by the markers D7S2490 and D7S2539. This chromosomal region is ∼21 cM long and harbors about 123 genes (NCBI Build 36.2). Of the positional candidate genes most relevant to MS in this region, the CD36 gene (MIM 173510) and its adjacent gene GNAT3 were chosen for our preliminary association studies in order to identify the potential functional variant(s) that could relate to our MS linkage signal. The reanalysis of our data using the newer threshold for impaired fasting glucose (i.e., ≥ 100 mg/dl) was found to be moderately heritable (h2 = 0.43 ± 0.10). The strongest evidence for linkage (LOD = 3.1) again occurred at the 7q chromosomal region, between markers D7S2212 and D7S821, although it was attenuated slightly. Such an observation suggests that the threshold for impaired fasting glucose (i.e., ≥ 110 mg/dl) used for the original analysis was more informative for linkage.
Figure 1. Multipoint linkage findings of Metabolic Syndrome in Mexican Americans.

Figure 2. Linkage* of MS on chromosome 7q21 without (M1) and with (M2) associations involving rs11760281 (GNAT3) and rs1194197 (CD36).
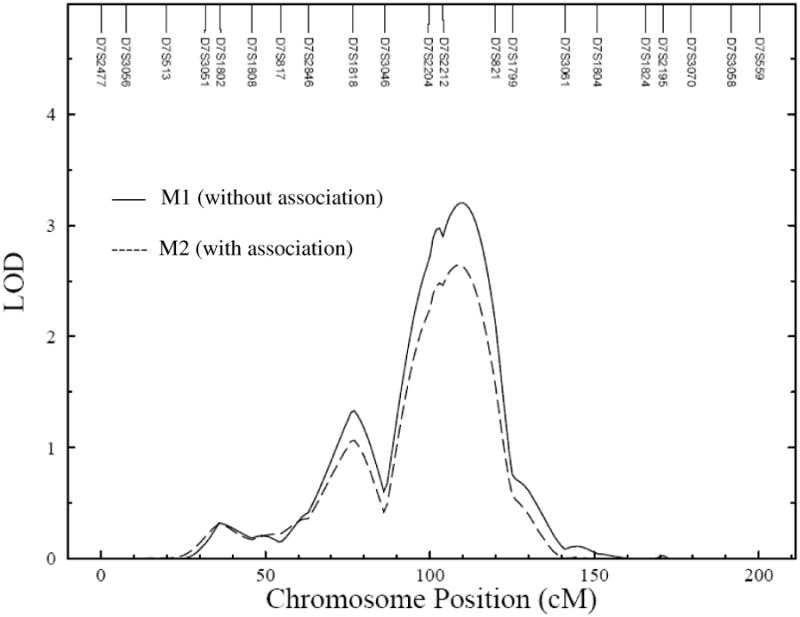
*Reanalyzed data (N = 648), given the availability of rs11760281 and rs1194197 marker data.
Table 3. Characteristics of the SNPs near or in CD36 and GNAT3 genes and their association with MS (NCEP/ATPIII).
| Gene | SNP | Major/minor alleles* | Physical distance (bp)** | Location** | Minor allele frequency (MAF) | P-value |
|---|---|---|---|---|---|---|
| GNAT3 | SNP1 (rs799975) | G/C | 79920950 | downstream of 3′-UTR | 0.228 | 0.0414 |
| SNP2 (rs2944398)a | C/T | 79924797 | near 3′-UTR | 0.425 | 0.0326 | |
| SNP3 (rs6942728) | T/C | 79932864 | intron | 0.048 | 0.0437 | |
| SNP4 (rs1473122)a | A/T | 79933295 | intron | 0.380 | 0.0271 | |
| SNP5 (rs799929)a | A/G | 79935512 | intron | 0.403 | 0.0737 | |
| SNP6 (rs6961082) | C/A | 79938905 | intron | 0.012 | 0.4313 | |
| SNP7 (rs11760281) | G/A | 79942485 | intron | 0.442 | 0.0003 | |
| SNP8 (rs10280807) | T/C | 79946464 | intron | 0.093 | 0.5679 | |
| SNP9 (rs6467212)b | A/G | 79955940 | intron | 0.060 | 0.4319 | |
| SNP10 (rs10234980) | C/T | 79958990 | intron | 0.035 | 0.8846 | |
| SNP11 (rs1524600)b | C/T | 79976239 | intron | 0.067 | 0.5536 | |
| CD36 | SNP1 (rs1194197) | T/C | 80013542 | upstream of 5′-UTR1 | 0.476 | 0.0044 |
| SNP2 (rs1194182)c | G/C | 80069440 | 5′-UTR1 | 0.375 | 0.0164 | |
| SNP3 (rs1984112) | A/G | 80080856 | Intron1 | 0.266 | 0.2819 | |
| SNP4 (rs1761667)c | A/G | 80082875 | intron1 | 0.378 | 0.0431 | |
| SNP5 (rs57312550)d | C/A | 80090631 | intron1 | 0.070 | 0.4929 | |
| SNP6 (rs59637606)d | A/G | 80090743 | intron1 | 0.070 | 0.5638 | |
| SNP7 (rs62461694) | T/C | 80090989 | intron1 | 0.043 | 0.6636 | |
| SNP8 (rs2151916)e | T/C | 80091319 | intron1 | 0.272 | 0.4414 | |
| SNP9 (rs2366855)f | A/T | 80091391 | intron1 | 0.445 | 0.0898 | |
| SNP10 (new) | G/T | 80105623 | near 5′-UTR2 | 0.022 | 0.5163 | |
| SNP11 (rs1049654)f | C/A | 80113391 | 5′-UTR2 | 0.413 | 0.2377 | |
| SNP12 (rs3211805)g | G/T | 80113512 | intron1 & 2 | 0.004 | 0.4243 | |
| SNP13 (rs997906)e | A/T | 80117771 | intron1 & 2 | 0.244 | 0.8259 | |
| SNP14 (rs3173798) | T/C | 80123786 | intron1 & 2 | 0.145 | 0.0481 | |
| SNP15 (rs3211891)g | T/C | 80128284 | intron1 & 2 | 0.004 | 0.4248 | |
| SNP16 (rs3211956) | T/G | 80141698 | near 3′-UTR1; intron2 | 0.056 | 0.2740 | |
| SNP17 (rs7755)h | G/A | 80144207 | 3′-UTR2 | 0.324 | 0.5031 | |
| SNP18 (rs1049673)h | C/G | 80144286 | 3′-UTR2 | 0.318 | 0.3493 |
risk allele in bold font
(NCBI build36; dbSNP BUILD127);
Transscript NM_001001547.1,
Transscript NM_001001548.2;
Three markers are in strong LD with each other (r2 ≥ 0.84);
Two markers are in strong LD with each other (r2 = 0.90);
Two markers are in strong LD with each other (r2 = 0.97);
Two markers are in strong LD with each other (r2 = 1.00);
Two markers are in strong LD with each other (r2 = 0.86);
Two markers are in strong LD with each other (r2 = 0.88);
Two markers are in strong LD with each other (r2 = 1.00);
Two markers are in strong LD with each other (r2 = 0.99)
Association Analysis and Conditional Linkage/Association Analysis
As described earlier, 19 SNPs in or near the CD36 gene and 11 SNPs in the GNAT3 gene were genotyped in our entire sample of approximately 720 individuals. We tested all the 30 SNP data for Hardy-Weinberg Equilibrium (HWE), in turn finding that only one SNP in the CD36 gene (rs13230419) deviated from the HWE expectation. This SNP was thus excluded from further analysis. The particulars of the 29 SNPs (i.e., CD36 = 18 and GNAT3 = 11) including their physical location and minor allele frequencies (MAFs) considered for further analysis are reported in Table 4. The LD patterns between 18 SNPs in CD36 are depicted in Supplementary Figure 1 S1 online. The pair wise LD (r2) for the 18 SNPs examined in CD36 ranged between 0 and 1, and the following SNP pairs exhibited strong LD (r2 > 0.80): rs57312550 and rs59637606 [r2 = 1.0], rs7755-rs1049673 (r2 = 0.99), rs1194182-rs1761667 (r2 = 0.97), rs2366855-rs1049654 (r2 = 0.88), and rs2151916-rs997906 (r2 = 0.86). The LD patterns between 11 SNPs in GNAT3 are shown in Supplementary Figure 2 S2 online. The range of pair wise LD observed for the 11 SNPs in GNAT3 was between 0 and 0.93. The following SNP pairs showed strong pair wise LD (r2 > 0.80): rs2944398-rs799929 (r2 = 0.93), rs1473122-rs799929 (r2 = 0.90), rs6467212-rs1524600 (r2 = 0.90), and rs2944398- rs1473122 (r2 = 0.84).
Table 4. Characteristics of the SNPs near or in CD36 and GNAT3 genes and their association with MS (NCEP/ATPIII).
| Gene | SNP | Major/minor alleles* | Physical distance (bp)* | Location* | Minor allele frequency (MAF) | P-value |
|---|---|---|---|---|---|---|
| GNAT3 | SNP1 (rs799975) | G/C | 79920950 | downstream of 3′-UTR | 0.228 | 0.0414 |
| SNP2 (rs2944398)a | C/T | 79924797 | near 3′-UTR | 0.425 | 0.0326 | |
| SNP3 (rs6942728) | T/C | 79932864 | intron | 0.048 | 0.0437 | |
| SNP4 (rs1473122)a | A/T | 79933295 | intron | 0.380 | 0.0271 | |
| SNP5 (rs799929)a | A/G | 79935512 | intron | 0.403 | 0.0737 | |
| SNP6 (rs6961082) | C/A | 79938905 | intron | 0.012 | 0.4313 | |
| SNP7 (rs11760281) | G/A | 79942485 | intron | 0.442 | 0.0003 | |
| SNP8 (rs10280807) | T/C | 79946464 | intron | 0.093 | 0.5679 | |
| SNP9 (rs6467212)b | A/G | 79955940 | intron | 0.060 | 0.4319 | |
| SNP10 (rs10234980) | C/T | 79958990 | intron | 0.035 | 0.8846 | |
| SNP11 (rs1524600)b | C/T | 79976239 | intron | 0.067 | 0.5536 | |
| CD36 | SNP1 (rs1194197) | T/C | 80013542 | upstream of 5′-UTR1 | 0.476 | 0.0044 |
| SNP2 (rs1194182)c | G/C | 80069440 | 5′-UTR1 | 0.375 | 0.0164 | |
| SNP3 (rs1984112) | A/G | 80080856 | Intron1 | 0.266 | 0.2819 | |
| SNP4 (rs1761667)c | A/G | 80082875 | intron1 | 0.378 | 0.0431 | |
| SNP5 (rs57312550)d | C/A | 80090631 | intron1 | 0.070 | 0.4929 | |
| SNP6 (rs59637606)d | A/G | 80090743 | intron1 | 0.070 | 0.5638 | |
| SNP7 (rs62461694) | T/C | 80090989 | intron1 | 0.043 | 0.6636 | |
| SNP8 (rs2151916)e | T/C | 80091319 | intron1 | 0.272 | 0.4414 | |
| SNP9 (rs2366855)f | A/T | 80091391 | intron1 | 0.445 | 0.0898 | |
| SNP10 (new) | G/T | 80105623 | near 5′-UTR2 | 0.022 | 0.5163 | |
| SNP11 (rs1049654)f | C/A | 80113391 | 5′-UTR2 | 0.413 | 0.2377 | |
| SNP12 (rs3211805)g | G/T | 80113512 | intron1 & 2 | 0.004 | 0.4243 | |
| SNP13 (rs997906)e | A/T | 80117771 | intron1 & 2 | 0.244 | 0.8259 | |
| SNP14 (rs3173798) | T/C | 80123786 | intron1 & 2 | 0.145 | 0.0481 | |
| SNP15 (rs3211891)g | T/C | 80128284 | intron1 & 2 | 0.004 | 0.4248 | |
| SNP16 (rs3211956) | T/G | 80141698 | near 3′-UTR1; intron2 | 0.056 | 0.2740 | |
| SNP17 (rs7755)h | G/A | 80144207 | 3′-UTR2 | 0.324 | 0.5031 | |
| SNP18 (rs1049673)h | C/G | 80144286 | 3′-UTR2 | 0.318 | 0.3493 |
(NCBI build36; dbSNP BUILD127);
Transscript NM_001001547.1,
Transscript NM_001001548.2;
Three markers are in strong LD with each other (r2 ≥ 0.84);
Two markers are in strong LD with each other (r2 = 0.90);
Two markers are in strong LD with each other (r2 = 0.97);
Two markers are in strong LD with each other (r2 = 1.00);
Two markers are in strong LD with each other (r2 = 0.86);
Two markers are in strong LD with each other (r2 = 0.88);
Two markers are in strong LD with each other (r2 = 1.00);
Two markers are in strong LD with each other (r2 = 0.99)
Association between MS and SNPs in CD36 and GNAT3 was examined using MGA, and the presence of population stratification was tested by performing QTDT prior to MGA. There was no evidence for the presence of population stratification. The nominally significant results (P ≤ 0.05) of our association analysis are reported in Table 4. Briefly, among the 18 SNPs examined in CD36, 4 were associated with MS (rs1194197: P = 0.0044; rs1194182: P = 0.0164; rs1761667: P = 0.0431; and rs3173798: P = 0.0481), although the markers rs1194182 and rs1761667 were found to be in strong LD (r2 = 0.97). Of the 11 SNPs examined in GNAT3, 5 exhibited nominally significant associations with MS (rs799975: P = 0.0414; rs2944398: P = 0.0326; rs6942728: P = 0.0437; rs1473122: P = 0.0271; and rs11760281: P = 0.0003). As shown in Table 4, the markers rs2944398, rs1473122, and rs799929 were in strong LD (r2 ≥ 0.84). To address the issue of multiple testing, given the LD patterns among the examined SNPs, an effective number of SNPs was obtained using the method of Li and Ji (22) to derive a P value adjusted for multiple testing. The effective number of SNPs was 17, and the required significance threshold was 0.0030. Thus, the observed association between MS and rs11760281 remained significant after correction for multiple testing and the association between MS and rs1194197 appeared to exhibit a similar trend, but failed to meet the required statistical significance threshold.
Subsequently, we performed BQTN analysis on our SNP data from both CD36 and GNAT3 genes simultaneously. Models which used rs1194197 and rs11760281 provided the strongest evidence for associations with the estimated probability of 0.78 and 0.77 respectively. As stated above, these were the two markers which exhibited relatively strong associations with MS. The minor allele A of marker rs11760281 and the major allele T of marker rs1194197 were associated with increased risk for MS. The relative risks (RR) associated with these markers were estimated to be 1.6 (rs11760281) and 1.7 (rs1194197), respectively. These two variants together conferred an approximately 3-fold increase in MS risk (RR = 2.7). Subsequently, we reanalyzed our data by performing linkage analysis conditional on the associations involving these two variants; and, determined that these two sequence variants explained about 18% of the MS linkage evidence on chromosome 7q (Figure 2).
Secondary data analyses of quantitative traits related to MS
After determining the correlations between the 29 SNPs and MS, association analyses of the same markers with various MS-related quantitative traits, including the MS individual components (Table 1) treated as continuous traits, were performed. As can be seen in Supplementary Table S1 online, several of the traits were associated with various SNPs examined in this study. On multiple occasions, more than one MS-related quantitative trait was associated with a given SNP. For example, rs11760281, the SNP that showed the strongest association with MS, also was found to be significantly associated with waist circumference (P = 0.0090), BMI (P = 0.0028), fasting glucose (P = 0.0171), and fasting insulin (nondiabetic individuals only) (P = 0.0037). As detailed in Supplementary Table 1, the carriers of the minor allele were found to be at increased risk for all of these four traits as indicated by the mean trait value by genotype class. In contrast, the marker rs1194197 that exhibited the second strongest association with MS, also showed significant associations with waist circumference (P = 0.0087), BMI (P = 0.0507), fasting glucose (P = 0.0054). The carriers of the major allele were at increased risk for all of the three traits as indicated by the mean trait value by genotype class. Since the traits/models were correlated (thus, not completely independent), no attempt was made to adjust the P values reported in Supplementary Table 1 S1 online for multiple comparisons.
Discussion
Metabolic syndrome is a complex phenotype, representings a constellation of several traits related to obesity, T2DM and CHD. Despite the debate on its clinical utility (25, 7), as stated earlier, previous studies including our own have shown that the MS-related traits are under potentially strong common genetic (or pleiotropic) influences, using multivariate genetic analyses and/or factor analytical techniques. In this study, we aimed to determine the heritability of MS and to localize its susceptibility loci, using the NCEP/ATPIII definition in a Mexican American population at high risk for the development of MS. The prevalence of MS in our family data was 43.5%. This study suggests that additive genetic effects (h2 = 51%) significantly influence variation in susceptibility to MS. Given that the heritability estimates are population sample specific, some previous studies determined the heritability for MS defined by NCEP/ATPIII criteria to be ∼13-60% (26, 27, 11), which are comparable to our finding in Mexican Americans.
Importantly, we found the strongest evidence (LOD = 3.6) for linkage of MS on chromosome 7q21 between the markers D7S2212 (7q21.11) and D7S821/GATA5D08 (7q21.3). This region has been implicated by several studies, including our previous studies, to contain susceptibility loci that influence MS or its related traits (see Supplementary Table S2 online). These findings consistent with ours suggest that the relatively broad 7q chromosomal region of interest in this study could harbor one or more MS susceptibility genes with potential common genetic influences.
As a preliminary susceptibility gene identification effort, we screened two positional candidate genes in the 1-LOD support interval region on chromosome 7q, CD36 and GNAT3, which are physically very close to each other (Table 4); and, found two sequence variants, one each from these genes (GNAT3, rs11760281 and CD36, rs1194197), that were relatively strongly associated with MS and together explained together about 18% of the observed MS linkage evidence. Of these two genes, CD36 has been the focus of various investigations in humans with regard to MS and/or its correlated disease conditions. A unique feature of the CD36 gene is alternative splicing of its exons and their upstream promoters, which appear to be tissue specific (18, 26). Indeed, as remarked by Andersen et al. (28), the CD36 gene is regulated by an unusually complex molecular mechanism, mirroring the multifunctional role of CD36 in different tissues and conditions. Given that insulin resistance, obesity and their associated inflammatory processes are the major underlying mechanisms of MS, experimental evidence suggests that CD36 could play a significant role in such interrelated complex mechanisms (29). Thus, CD36 is a strong candidate gene for susceptibility to various components of MS including obesity, insulin resistance, dyslipidemia, inflammation and atherosclerosis.
In the spontaneous hypertensive rat (SHR), an animal model for MS, a region on chromosome 4 provided evidence for linkage of various MS related traits. Subsequently, a deletion in the CD36 gene located in the linked chromosome 4 region was found to be responsible for the underlying insulin resistance, defective fatty acid metabolism and hypertriglyceridemia (30). The insulin insensitivity was restored in these rats by transgenically rescuing the CD36 gene (31). In humans, deficiency in CD36 was found to be associated with insulin resistance in the Japanese population (32). In addition, following their previous findings of association between CD36 sequence variants and HDL-C levels (33), Love-Gregory et al. (34) have recently reported the influence of such HDL-associated genetic polymorphisms on CD36 expression, revealing the causality of the link between expression and HDL-C. In consideration of its potential functional significance and location on chromosome 7q in humans, it is possible that the genetic variants in CD36 by themselves or together with other variants in the neighboring gene(s) could contribute to the MS linkage signal found in this study. Of the 29 SNPs examined, 11 from CD36 and 9 from GNAT3 were significantly associated with MS and/or MS-related traits, although some of the SNPs were found to be in LD (Table 4 and Supplementary Table S1 online).
Numerous studies have reported findings in support of the association between genetic variation at CD36 and MS and its related traits (Supplementary Table S3 online). Although there is a certain degree of overlap of the SNP sets used among the studies, some of the associations reported by such studies were not always between a specific variant and a MS related trait; instead, with a different, but correlated MS related trait(s). Two studies used MS diagnostic criteria similar to the one used in this study involving African Americans (33) and Boston Puerto Ricans (35). The best associated CD36 SNP (rs1194197) in our study was not examined in these studies, instead they found other SNPs in this gene to be associated with MS. Of the remaining three SNPs nominally associated with MS in our study, rs3173798 was evaluated in the African American data, and rs1761667 was examined in the Puerto Rican sample. Although rs3173798 was not correlated with MS in African Americans, it exhibited association with HDL-C levels (33). In the Puerto Rican data, rs1761667 exhibited suggestive association with MS (P = 0.016); but, another SNP (rs7807607) in strong LD with it was part of one of the haplotypes consisting of three SNPs (rs1049673, rs3211931, and rs7807607) significantly associated with MS (35). It is worth noting that rs1049673 is associated with fasting glucose in both the Puerto Rican and our data sets.
Several studies have reported associations between CD36 genetic variants and MS correlates (Supplementary Table S3 online). Some SNPs found to be associated with MS related traits in our study were also shown to be correlated with other related phenotypes by several investigations as discussed below. Ma et al. (36) reported the association of some common genetic variants in CD36 with increased risk of cardiovascular disease and serum lipid levels in a Caucasian population. A common haplotype derived from five tagging SNPs was found to be associated with free fatty acids (FFA) and triglycerides in men. Interestingly, two of these tagging polymorphisms (i.e. rs1761667 and rs1049673) which were associated with FFA, were also found to be associated in our study, with MS (rs1761667), fasting glucose (rs1761667 and 1049673), and diastolic blood pressure (rs1049673). In a North Indian population, T2DM was strongly associated with rs1761667 (37). This polymorphism (i.e., rs1761667) was one of the four CD36 SNPs recently found to be associated with decreased CD36 expression in monocytes (34). It should be noted that the observation of more of the SNPs associated with glucose traits in our study compared to others may be due to the high background prevalence of diabetes in our data. However, a thorough evaluation of the genetic region of interest to identify potential functional variants with the use of deep sequencing could reveal the true patterns of correlation between a full spectrum of sequence variants, both common and rare, and correlated MS-related traits.
To our knowledge, sequence variants in the GNAT3 (or α-gustducin) gene, which flanks CD36, have not been examined for their correlation with MS or its components. Gustducin is a G-protein originally found to be expressed in taste receptor cells and is considered to be an essential component of the mammalian sweet taste transduction pathway (38, 39). The GNAT3 gene encodes α-Gustducin, one of the three subunits of Gustducin. Fushan et al. (39) have recently reported significant associations between 11 SNPs in GNAT3 and sweet taste perception, using data from individuals with Caucasian, African-American and Asian population backgrounds. Of these variants, two were examined in our study (rs1524600 and rs6961082), but only rs6961082 exhibited significant associations with BMI, fasting glucose and 2-hr glucose levels. It is worthwhile noting that, Laugerette et al. (40), based on studies in animal models, have shown that CD36 is involved in oral long-chain fatty acid detection; thus, highlighting its role in “fat taste” perception (38). Toguri examined whether CD36 genetic variants (rs1984112, rs1761667, rs1527483, and rs1049673) were associated with habitual fat intake in Caucasian and Asian populations, in turn finding associations of rs1984112 and rs1761667 with %energy from fat in Caucasians and rs1049673 with %energy from carbohydrate in Asians, respectively (41; Ph.D dissertation; http://proquest.umi.com/pqdlink?Ver=1&Exp=11-07-2016&FMT=7&DID=1671905181&RQT=309&attempt=1&cfc=1). All of these three associated SNPs were examined in our study, and rs1761667 and rs1049673 exhibited associations with MS or its components (i.e., fasting glucose and diastolic blood pressure).
It is well known that glucagon-like-peptide-1 (GLP-1) is a regulator of appetite, insulin secretion and gut motility which is released in the enteroendocrine cells of the gut in response to glucose. Still the mechanism of how the glucose taken orally can signal the release of GLP in the gut is not well understood. Interestingly, recent studies have shown that the taste G protein gustducin (α-gustducin, GNAT3) along with other taste signaling elements could play a very important role in this link (42-45). Recently, in a sample of 97 Japanese individuals at high risk for cardiovascular disease with abdominal obesity, fasting serum GLP-1 levels were found to be on average 28% higher in individuals with MS compared to those with pre-MS, and the GLP-1 levels were elevated with increasing numbers of MS components or risk factors (46). In the present study, 5 of 11 GNAT3 SNPs examined were associated with MS (Table 4), and several SNPs exhibited correlations with various MS-related quantitative traits including obesity measures; fasting lipids, insulin and glucose concentrations; and 2-hr glucose concentrations (Supplementary Table S1 online).
We also tested rs11760281 for association with MS-related quantitative traits in an independent sample of Mexican American families ascertained on probands with or without a previous diagnosis of gestational diabetes mellitus; the BetaGene Study (47). Data were available on a maximum of 1,703 non-diabetic individuals (687 males, 1016 females) with average age of 34.8 ± 8.4 years (mean ± SD) and BMI of 29.5 ± 5.9 (mean ± SD) kg/m2. The A allele of rs11760281 was associated with increased 30-minute OGTT insulin levels, both absolute (P = 0.006) and the increment above fasting (P = 0.007), increased insulinogenic index (P = 0.004), and with increased acute insulin response to intravenous glucose (P = 0.036) adjusting for age and sex. The additional adjustment for body fat did not substantially alter the results. These results are consistent with the hypothesis that gut-expressed proteins related to taste signaling may regulate glucose-stimulated secretion of GLP-1 and its effects as an insulin secretagogue.
In conclusion, we performed a genome-wide linkage screen to localize MS susceptibility loci in Mexican Americans, a population at high risk for the development of MS, and found the strongest evidence for MS linkage on chromosome 7q21. Subsequent screening and examination of sequence variants in positional candidate genes CD36 and its flanking gene GNAT3 provided strong evidence for their association with MS and their related traits. Importantly, two variants (one from each gene) that exhibited strongest associations partially contributed to the linkage signal on chromosome 7q21. On one hand, the current observations shed light on the role of genetic variation at CD36 and GNAT3 genes, which are associated with complex pathways related to insulin action, insulin secretion and food taste perception, in determining susceptibility to MS and/or its risk factors including obesity, insulin resistance/hyperinsulinemia, hyperglycemia, dyslipidemia, and elevated blood pressure. On other hand, these findings strongly support an immediate need for a systematic targeted sequencing of not only CD36 and GNAT3 genes but also other genes within the 1-LOD support interval in the 7q chromosomal region of interest to identify the causal genes and the functional relevance of the implicated genetic variants, both common and rare. Such activities may ultimately help to develop effective strategies to prevent disease or treat individuals that are at high risk for MS and its co-morbid disease conditions.
Supplementary Material
Supplementary Figure 1 S1 online: Linkage Disequilibrium (LD) between the SNP pairs in CD36
Supplementary Figure 2 S2 online: Linkage Disequilibrium (LD) between the SNP pairs in GNAT3
Supplementary Table S1. Association of the SNPs in CD36 and GNAT3 with MS related phenotypes
Supplementary Table S2. Linkage reports of MS-related phenotypes by various studies on chromosome 7q
Supplementary Table S3. Association of SNPs in CD36 with MS related traits reported in previous studies that are not discussed in the text.
Acknowledgments
This study was supported by grants from the National Institute of Diabetes, Digestive and Kidney Diseases (DK53889, DK42273, and DK47482), Kronkosky Foundation, and Texas Biomedical Research Institute (formerly, Southwest Foundation for Biomedical Research) Forum Research Support Funds. The nursing and dietetic care provided by the staff of the Frederic C. Bartter General Clinical Research Center at the South Texas Veterans Health Care Systems - Audie Murphy Division is appreciated. We wish to acknowledge Richard Granato, Irma Jean Gomez, and Roy G. Resendez for excellent technical support. We thank the Center for Inherited Disease Research (CIDR) for providing a genome scan using the SAFDGS data. We warmly thank the families from SAFDGS for their enthusiasm and cooperation.
Footnotes
Disclosure: No conflict of interest.
References
- 1.Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287:356–359. doi: 10.1001/jama.287.3.356. [DOI] [PubMed] [Google Scholar]
- 2.Halley-Castillo E, Borges G, Talavera JO, et al. Body mass index and the prevalence of metabolic syndrome among children and adolescents in two Mexican populations. J Adolesc Health. 2007;40:521–526. doi: 10.1016/j.jadohealth.2006.12.015. [DOI] [PubMed] [Google Scholar]
- 3.Chen W, Berenson GS. Metabolic syndrome: definition and prevalence in children. J Pediatr (Rio J) 2007;83:1–2. doi: 10.2223/JPED.1584. [DOI] [PubMed] [Google Scholar]
- 4.Steinberger J, Daniels SR, Eckel RH, et al. Progress and challenges in metabolic syndrome in children and adolescents: a scientific statement from the American Heart Association Atherosclerosis, Hypertension, and Obesity in the Young Committee of the Council on Cardiovascular Disease in the Young; Council on Cardiovascular Nursing; and Council on Nutrition, Physical Activity, and Metabolism. Circulation. 2009;119:628–647. doi: 10.1161/CIRCULATIONAHA.108.191394. [DOI] [PubMed] [Google Scholar]
- 5.NCEP/ATPIII Expert panel on detection, evaluation and treatment of High Blood Cholesterol in adults. Executive summary of the Third report of National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High blood cholesterol in adults (ATP III) JAMA. 2001;285:2486–2497. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 6.Grundy SM, Cleeman JI, Daniels SR, et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112:2735–2752. doi: 10.1161/CIRCULATIONAHA.105.169404. [DOI] [PubMed] [Google Scholar]
- 7.Simmons RK, Alberti KG, Gale EA, et al. The metabolic syndrome: useful concept or clinical tool? Report of a WHO Expert Consultation. Diabetologia. 2010;53:600–605. doi: 10.1007/s00125-009-1620-4. [DOI] [PubMed] [Google Scholar]
- 8.Duggirala R, Blangero J, Almasy L, et al. A major locus for fasting insulin concentrations and insulin resistance on chromosome 6q with strong pleiotropic effects on obesity-related phenotypes in nondiabetic Mexican Americans. Am J Hum Genet. 2001;68:1149–1164. doi: 10.1086/320100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arya R, Blangero J, Williams K, et al. Factors of insulin resistance syndrome--related phenotypes are linked to genetic locations on chromosomes 6 and 7 in nondiabetic Mexican-Americans. Diabetes. 2002;51:841–847. doi: 10.2337/diabetes.51.3.841. [DOI] [PubMed] [Google Scholar]
- 10.Lehman DM, Arya R, Blangero J, et al. Bivariate linkage analysis of the insulin resistance syndrome phenotypes on chromosome 7q. Hum Biol. 2005;77:231–246. doi: 10.1353/hub.2005.0040. [DOI] [PubMed] [Google Scholar]
- 11.Teran-Garcia M, Bouchard C. Genetics of the metabolic syndrome. Appl Physiol Nutr Metab. 2007;32:89–114. doi: 10.1139/h06-102. [DOI] [PubMed] [Google Scholar]
- 12.Kraja AT, Vaidya D, Pankow JS, et al. A Bivariate Genome-Wide Approach to Metabolic Syndrome: STAMPEED Consortium. Diabetes. 2011;60:1329–1339. doi: 10.2337/db10-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ng MC, So WY, Lam VK, et al. Genome-wide scan for metabolic syndrome and related quantitative traits in Hong Kong Chinese and confirmation of a susceptibility locus on chromosome 1q21-q25. Diabetes. 2004;53:2676–2683. doi: 10.2337/diabetes.53.10.2676. [DOI] [PubMed] [Google Scholar]
- 14.Langefeld CD, Wagenknecht LE, Rotter JI, et al. Insulin Resistance Atherosclerosis Study Family Study. Linkage of the metabolic syndrome to 1q23-q31 in Hispanic families: the Insulin Resistance Atherosclerosis Study Family Study. Diabetes. 2004;53:1170–1174. doi: 10.2337/diabetes.53.4.1170. [DOI] [PubMed] [Google Scholar]
- 15.Hunt KJ, Lehman DM, Arya R, et al. Genome-wide linkage analyses of type 2 diabetes in Mexican Americans: the San Antonio Family Diabetes/Gallbladder Study. Diabetes. 2005;54:2655–2662. doi: 10.2337/diabetes.54.9.2655. [DOI] [PubMed] [Google Scholar]
- 16.Puppala S, Dodd GD, Fowler S, et al. A Genomewide Search Finds Major Susceptibility Loci for Gallbladder Disease on Chromosome 1 in Mexican Americans. Am J Hum Genet. 2006;78:377–392. doi: 10.1086/500274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Genuth S, Alberti KG, Bennett P, et al. Follow-up report on the diagnosis of diabetes mellitus. Diabetes Care. 2003;26:3160–3167. doi: 10.2337/diacare.26.11.3160. [DOI] [PubMed] [Google Scholar]
- 18.Noushmehr H, D'Amico E, Farilla L, et al. Fatty acid translocase (FAT/CD36) is localized on insulin-containing granules in human pancreatic beta-cells and mediates fatty acid effects on insulin secretion. Diabetes. 2005;54:472–481. doi: 10.2337/diabetes.54.2.472. [DOI] [PubMed] [Google Scholar]
- 19.Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Gene. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 20.Sobel E, Lange K. Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet. 1996;58:1323–1337. [PMC free article] [PubMed] [Google Scholar]
- 21.Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet. 1998;62:1198–1211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blangero J, Goring HH, Kent JW, Jr, et al. Quantitative trait nucleotide analysis using Bayesian model selection. Hum Biol. 2005;77:541–559. doi: 10.1353/hub.2006.0003. [DOI] [PubMed] [Google Scholar]
- 23.Curran JE, Jowett JBM, Elliott KS, et al. Genetic variation in selenoprotein S influences inflammatory response. Nat Genet. 2005;37:1234–1241. doi: 10.1038/ng1655. [DOI] [PubMed] [Google Scholar]
- 24.Li J, Ji L. Adjusting multiple testing in multilocus analyses using the eigenvalues of a correlation matrix. Heredity. 2005;95:221–227. doi: 10.1038/sj.hdy.6800717. [DOI] [PubMed] [Google Scholar]
- 25.Tenenbaum A, Fisman EZ. “The metabolic syndrome… is dead”: These reports are an exaggeration. Cardiovasc Diabetol. 2011;10:11. doi: 10.1186/1475-2840-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henneman P, Aulchenko YS, Frants RR, van Dijk KW, Oostra BA, van Duijn CM. Prevalence and heritability of the metabolic syndrome and its individual components in a Dutch isolate: the Erasmus Rucphen Family study. J Med Genet. 2008;45:572–577. doi: 10.1136/jmg.2008.058388. [DOI] [PubMed] [Google Scholar]
- 27.Sung J, Lee K, Song YM. Heritabilities of the metabolic syndrome phenotypes and related factors in Korean twins. J Clin Endocrinol Metab. 2009;94:4946–52. doi: 10.1210/jc.2009-1268. [DOI] [PubMed] [Google Scholar]
- 28.Andersen M, Lenhard B, Whatling C, Eriksson P, Odeberg J. Alternative promoter usage of the membrane glycoprotein CD36. BMC Mol Biol. 2006;7:8. doi: 10.1186/1471-2199-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang Z, Ming XF. CD36: the common soil for inflammation in obesity and atherosclerosis? Cardiovasc Res. 2011;89:485–486. doi: 10.1093/cvr/cvq406. [DOI] [PubMed] [Google Scholar]
- 30.Aitman TJ, Glazier AM, Wallace CA, et al. Identification of Cd36 (Fat) as an insulin-resistance gene causing defective fatty acid and glucose metabolism in hypertensive rats. Nat Genet. 1999;21:76–83. doi: 10.1038/5013. [DOI] [PubMed] [Google Scholar]
- 31.Pravenec M, Landa V, Zidek V, et al. Transgenic rescue of defective Cd36 ameliorates insulin resistance in spontaneously hypertensive rats. Nat Genet. 2001;27:156–158. doi: 10.1038/84777. [DOI] [PubMed] [Google Scholar]
- 32.Miyaoka K, Kuwasako T, Hirano K, Nozaki S, Yamashita S, Matsuzawa Y. CD36 deficiency associated with insulin resistance. Lancet. 2001;357:686–687. doi: 10.1016/s0140-6736(00)04138-6. [DOI] [PubMed] [Google Scholar]
- 33.Love-Gregory L, Sherva R, Sun L, et al. Variants in the CD36 gene associate with the metabolic syndrome and high-density lipoprotein cholesterol. Hum Mol Genet. 2008;17:1695–1704. doi: 10.1093/hmg/ddn060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Love-Gregory L, Sherva R, Schappe T, et al. Common CD36 SNPs reduce protein expression and may contribute to a protective atherogenic profile. Hum Mol Genet. 2011;20:193–201. doi: 10.1093/hmg/ddq449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Noel SE, Lai CQ, Mattei J, Parnell LD, Ordovas JM, Tucker KL. Variants of the CD36 gene and metabolic syndrome in Boston Puerto Rican adults. Atherosclerosis. 2010;211:210–215. doi: 10.1016/j.atherosclerosis.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma X, Bacci S, Mlynarski W, Gottardo L, et al. A Common haplotype at the CD36 locus is associated with high free fatty acid levels and increased cardiovascular risk in Caucasians. Hum Mol Genet. 2004;13:2197–2205. doi: 10.1093/hmg/ddh233. [DOI] [PubMed] [Google Scholar]
- 37.Banerjee M, Gautam S, Saxena M, Bid HM, Agrawal CG. Association of CD36 gene variants rs1761667 (G > A) and rs1527483 (C > T) with Type 2 diabetes in North Indian population. In J Diabetes Mellitus. 2010;2:179–183. [Google Scholar]
- 38.Garcia-Bailo B, Toguri C, Eny KM, El-Sohemy A. Genetic variation in taste and its influence on food selection. OMICS. 2009;13:69–80. doi: 10.1089/omi.2008.0031. [DOI] [PubMed] [Google Scholar]
- 39.Fushan AA, Simons CT, Slack JP, Drayna D. Association between common variation in genes encoding sweet taste signaling components and human sucrose perception. Chem Senses. 2010;35:579–592. doi: 10.1093/chemse/bjq063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laugerette F, Passilly-Degrace P, Patris B, et al. CD36 involvement in orosensory detection of dietary lipids, spontaneous fat preference, and digestive secretions. J Clin Invest. 2005;115:3177–3184. doi: 10.1172/JCI25299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Toguri C. M Sc Dissertation. University of Toronto; 2008. Genetic variation in CD36 and dietary fat intake. MR45176. [Google Scholar]
- 42.Jang HJ, Kokrashvili Z, Theodorakis MJ, et al. Gut-expressed gustducin and taste receptors regulate secretion of glucagon-like peptide-1. Proc Natl Acad Sci U S A. 2007;104:15069–15074. doi: 10.1073/pnas.0706890104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kokrashvili Z, Mosinger B, Margolskee RF. T1r3 and alpha-gustducin in gut regulate secretion of glucagon-like peptide-1. Ann N Y Acad Sci. 2009;1170:91–94. doi: 10.1111/j.1749-6632.2009.04485.x. [DOI] [PubMed] [Google Scholar]
- 44.Kokrashvili Z, Mosinger B, Margolskee RF. Taste signaling elements expressed in gut enteroendocrine cells regulate nutrient-responsive secretion of gut hormones. Am J Clin Nutr. 2009;90:822S–825S. doi: 10.3945/ajcn.2009.27462T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steinert RE, Gerspach AC, Gutmann H, Asarian L, Drewe J, Beglinger C. (2011). The functional involvement of gut-expressed sweet taste receptors in glucose-stimulated secretion of glucagon-like peptide-1 (GLP-1) and peptide YY (PYY) Clin Nutr. 2011 doi: 10.1016/j.clnu.2011.01.007. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 46.Yamaoka-Tojo M, Tojo T, Takahira N, et al. Elevated circulating levels of an incretin hormone, glucagon-like peptide-1, are associated with metabolic components in high-risk patients with cardiovascular disease. Cardiovasc Diabetol. 2010;9:17. doi: 10.1186/1475-2840-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Watanabe RM, Allayee H, Xiang AH, et al. Transcription factor 7-like 2 (TCF7L2) is associated with gestational diabetes mellitus and interacts with adiposity to alter insulin secretion in Mexican Americans. Diabetes. 2007;56:1481–1485. doi: 10.2337/db06-1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 S1 online: Linkage Disequilibrium (LD) between the SNP pairs in CD36
Supplementary Figure 2 S2 online: Linkage Disequilibrium (LD) between the SNP pairs in GNAT3
Supplementary Table S1. Association of the SNPs in CD36 and GNAT3 with MS related phenotypes
Supplementary Table S2. Linkage reports of MS-related phenotypes by various studies on chromosome 7q
Supplementary Table S3. Association of SNPs in CD36 with MS related traits reported in previous studies that are not discussed in the text.