

Abstract
There is rapidly mounting evidence that intracellular proteases in bacteria are compelling targets for antibacterial drugs. Multiple reports suggest that the human pathogen Mycobacterium tuberculosis and other actinobacteria may be particularly sensitive to small molecules that perturb the activities of self-compartmentalized peptidases, which catalyze intracellular protein turnover as components of ATP-dependent proteolytic machines. Here, we report chemical syntheses and evaluations of structurally diverse β-lactones, which have a privileged structure for selective, suicide inhibition of the self-compartmentalized ClpP peptidase. β-lactones with certain substituents on the α- and β-carbons were found to be toxic to M. tuberculosis. Using an affinity-labeled analog of a bioactive β-lactone in a series of chemical proteomic experiments, we selectively captured the ClpP1P2 peptidase from live cultures of two different actinobacteria that are related to M. tuberculosis. Importantly, we found that the growth inhibitory β-lactones also inactivate the M. tuberculosis ClpP1P2 peptidase in vitro via formation of a covalent adduct at the ClpP2 catalytic serine. Given the potent antibacterial activity of these compounds and their medicinal potential, we sought to identify innate mechanisms of resistance. Using a genome mining strategy, we identified a genetic determinant of β-lactone resistance in Streptomyces coelicolor, a non-pathogenic relative of M. tuberculosis. Collectively, these findings validate the potential of ClpP inhibition as a strategy in antibacterial drug development and define a mechanism by which bacteria could resist the toxic effects of ClpP inhibitors.
Keywords: ClpP, Mycobacterium, β-lactone, inhibitor, chemical proteomics, genome mining, Streptomyces
The rising number of deaths resulting from infections with multi-drug resistant pathogens is a public health crisis (1). The drug-resistance problem is evident in the case of Mycobacterium tuberculosis, the causative agent of tuberculosis (2). Efforts are underway for the discovery and development of new antibacterial agents that are effective against multi-drug resistant strains of M. tuberculosis (1,2,5). Drug development has been aided by a renewed focus on the physiology of this pathogen (2,7,9–12). Through screens for conditionally or absolutely essential genes, it has been established that enzymes associated with intracellular protein turnover are particularly important in mycobacterial physiology. For example, genes encoding the ClpP peptidase and its AAA+ partners, ClpX and ClpC1, are absolutely essential for the viability of the bacterium (7, 10). The ClpP peptidase is a self-compartmentalized barrel-shaped tetradecamer, composed of two heptameric rings that enclose a degradation chamber (7,8,18). By itself, ClpP degrades small peptides, but its small axial pores exclude large peptides and folded proteins from the proteolytic chamber. The AAA+ partners of ClpP are ring hexamers that recognize, unfold, and translocate substrates into the ClpP chamber in ATP-dependent reactions (8,13,14). Mycobacterial ClpP plays critical roles in protein turnover, but is distinct in many ways from its orthologs in other bacteria. For instance, ClpP is essential in mycobacteria and in other actinobacteria, but is only required for virulence and stress tolerance in other pathogenic bacteria (12,15–17). Another unusual feature of mycobacteria and other actinobacteria is that they often harbor two clpP genes (clpP1 and clpP2), typically in a dicistronic operon (7,12,18); whereas most bacteria have a single clpP gene. Biochemical studies suggest that mycobacterial ClpP1 and ClpP2 form distinct homo-heptameric rings, which assemble into an active ClpP17•ClpP27 heterotetradecameric complex (7, 18), which we will call ClpP1P2.
Because the genes encoding ClpP1 and ClpP2 are essential for the viability of M. tuberculosis, there have been efforts to discover and develop small molecules that perturb the activities of these enzymes (12,19–29). Parish and co-workers reported that acyldepsipeptides (ADEPs), which dysregulate ClpP function by opening the axial pore and causing unregulated protein degradation, are toxic to M. tuberculosis (10,29). Although ADEPs are attractive drug leads, they must be used in conjunction with efflux-pump inhibitors to realize low MICs in M. tuberculosis (10,29). Cyclomarin A1, a molecule that activates ClpP by binding to its AAA+ ClpC1 partner, has also been reported to kill M. tuberculosis (27,29). These drug leads validate the strategy of killing M. tuberculosis via small molecule-mediated activation of ClpP1P2 (10,27,29).
An appealing but unvalidated strategy for killing M. tuberculosis is small molecule-mediated inhibition of ClpP1P2. Insights into how the mycobacterial ClpP might be inhibited can be gleaned from molecules that inhibit its counterparts in other bacteria. For example, the Keiler group identified cyclic peptides that inhibit ClpXP in Caulobacter crescentus by an unknown mechanism (28). Consistent with the requirement of the clpP and clpX genes for cell-cycle progression in C. crescentus, these inhibitory peptides are toxic to this bacterium. Analogously, Sieber and co-workers have reported that ClpP-dependent production of virulence factors in Staphylococcus aureus and Listeria monocytogenes is suppressed by β-lactones that selectively inhibit ClpP (20–23,25). These molecules are “suicide inhibitors” because the active-site serine of ClpP attacks the electrophilic carbonyl of the β-lactone ring, resulting in the formation of an inactive O-acyl-enzyme product (25). Here, we report syntheses of novel β-lactones and show that some of them are toxic to Mycobacterium smegmatis and M. tuberculosis. Chemical proteomic experiments indicate that the β-lactones react with actinobacterial ClpP in vivo. We present evidence that these compounds inhibit the peptidase activity of M. tuberculosis ClpP1P2 in vitro by modifying the active-site serine of ClpP2. Furthermore, we successfully used a genome mining strategy to identify an innate mechanism of β-lactone resistance in a Streptomyces coelicolor, a non-pathogenic relative of mycobacteria (30). Collectively, these results have important implications for the development of ClpP inhibitors as anti-tuberculosis drugs.
RESULTS AND DISCUSSION
β-lactones are toxic to Mycobacteria
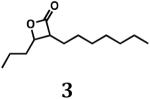
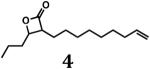
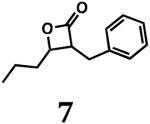
Based on the essentiality of the clpP locus in M. tuberculosis, we hypothesized that the bacterium would be susceptible to molecules possessing the β-lactone core structure that is common to suicide inhibitors of ClpP peptidases in a variety of bacterial species. Because there was insufficient information to predict which structural features an inhibitor of mycobacterial ClpP might possess, we used methodology developed by either Danheiser or Romo (31,32) to synthesize compounds with a common β-lactone core but with structurally disparate substituents attached to the α- and β-carbons, including hydrogen atoms, straight and branched aliphatic groups, or an aromatic moiety. Our collection also included β-lactones with two substituents at the β-carbon or with fused rings. In total, we synthesized 12 different β-lactones (compounds 2–13; Fig. 1), typically in moderate to poor yields because of chemical instability and/or difficulties in separating the desired products from starting materials.
Figure 1.

β-lactone structures
For biological testing, we included two commercially available compounds (β-lactones 1 and 14; Fig. 1) together with our newly synthesized β-lactones. Of these 14 molecules, four prevented growth of M. smegmatis MC2155 (Table 1), a non-pathogenic surrogate of the human pathogen M. tuberculosis. Three of these inhibitory compounds were trans-disubstituted β-lactones with unbranched alkyl substituents on both the α- and β-carbons (β-lactones 2–4; Fig. 1). For these molecules, there was a positive correlation between the length of the alkyl group on the α-carbon and antibacterial activity. However, the most potent compound (β-lactone 7; Fig. 1; MIC = 10 μg/mL) had an aromatic benzyl substituent on the α-carbon and an unbranched n-propyl group on the β-carbon. Compounds with only one substituent, with branching or disubstitution at the β-carbon, or with the β-lactone fused to another ring (β-lactones 1, 5, 6, 8–14; Fig. 1) did not prevent growth of M. smegmatis at the concentrations tested. Clearly, the simple β-lactone core structure is not sufficient for inhibition of M. smegmatis growth, and the chemical identity of groups attached to the α- and β-carbons dictates antibacterial activity against M. smegmatis and potentially other actinobacteria.
Table 1.
Minimal Inhibitory Concentrations of β-lactones for Mycobacteria
| Compound | MIC (μg/mL) | |
|---|---|---|
| M. smegmatis MC2115 | M. tuberculosis H37Rv | |
| 2 | 250 | Not tested |
| 3 | 150 | Not tested |
| 4 | 45 | 47 |
| 7 | 10 | 28 |
β-lactone minimal inhibitory concentrations (MICs) for M. smegmatis and M. tuberculosis. MICs of representative antibacterial drugs against M. tuberculosis H37Rv are as follows: moxifloxacin - 8 μg/mL, isoniazid - 2 μg/mL, kanamycin - 1.2 μg/mL, streptomycin - 16 μg/mL, ethambutol - 4 μg/mL (49).
The two most active compounds (β-lactone 4 and 7) from the activity assays with M. smegmatis were subsequently tested for their ability to inhibit the growth of the human pathogen, M. tuberculosis H37Rv. Strikingly, both molecules also inhibited growth of M. tuberculosis H37Rv (Table 1), with β-lactone 7 being the most active against M. smegmatis and M. tuberculosis. These findings are noteworthy because β-lactones were not previously recognized as a family of privileged structures that inhibit the growth of M. tuberculosis.
β-lactones target actinobacterial ClpP1P2 in vivo
Disubstituted β-lactones are thought to be privileged structures for the selective inhibition of the ClpP peptidase. We designed a chemical proteomic experiment in which we could capture and analyze cytoplasmic proteins with which the β-lactones had reacted in vivo. Specifically, we synthesized a bioactive analog of β-lactone 7 (the most potent compound) that has a n-pentynl substituent on the β-carbon in place of the parent's n-propyl substituent. The alkyne moiety was purposefully included because it could be used as a reactant in a “click” reaction for capture of proteins with which the β-lactone formed adducts. For the experiments, shaken liquid cultures of both S. coelicolor and M. smegmatis were treated with the alkynyl β-lactone. These organisms were chosen because they represent two different genera of actinobacteria that require a heterotetradecameric ClpP for viability (7,43,44). After mild cell lyses, the lysates were sequentially treated with azido-biotin, a copper (II) catalyst for the click reaction, and an avidin-functionalized agarose resin. In parallel, control experiments were performed in which the cell lysates were not treated with the Cu (II)-catalyst or with azido-biotin or were treated only with the avidin-functionalized agarose resin. In all cases, the protein-enriched resins were treated with trypsin and the resulting peptides were identified by a combination of peptide mass-fingerprinting and bioinformatic analysis via MASCOT software. After the elimination of all proteins that were identified in the negative control experiments, we identified 8 and 13 proteins that were captured from the cell lysates of S. coelicolor and M. smegmatis, respectively (Tables 2, S4 and S5). Presuming that growth inhibitory β-lactones would have the same targets in both S. coelicolor and M. smegmatis, we generated a list of the proteins that were captured from the lysates of both species via the click reaction. We were gratified to find ClpP1 and ClpP2 among the three proteins on the list (Table 2). The only other protein that was affinity captured from the cell lysates was the α-subunit of the proteasome. While it is not clear whether or not the β-lactone inhibits the actinobacterial proteasome, inhibition of this non-essential enzyme could not explain the growth inhibitory effects of the β-lactones. These results provide strong evidence that the bioactivity of the β-lactones can be ascribed to inhibition of the essential ClpP peptidase in actinobacteria.
Table 2.
Proteins identified in chemical proteomic analyses
| Streptomyces coelicolor | Mycobactehum smegmatis |
|---|---|
| Aconitate hydratase | Beta-lactamase |
| Aminolevulinic acid dehydratase | ClpB |
| ClpP1 | ClpP1 |
| ClpP2 | ClpP2 |
| DNA binding protein, EngD | D-alanyl-D-alanine carboxylpeptidase |
| Molybdenum cofactor protein, MoaC | Leucyl-tRNA synthetase |
| Proteasome α-subunit | Malate dehydrogenase |
| Pyridoxal lyase, PdxS | Purine biosynthesis protein, PurH |
| Proteasome β-subunit | |
| Prokaryotic Ubiquitin-like Protein, PUP | |
| Thiosulfate sulfurtransferase | |
| Tryptophan synthase | |
| Tryptophan-tRNA synthetase |
Proteins identified in both S. coelicolor and M. smegmatis are shown in bold text. In the bioinformatic identifications of the peptide fragments, a trypsin specificity with two missed cleavage sites was allowed and the MS mass tolerance was 7 ppm while the MS-MS tolerance was 0.5 Daltons. Identifications were contingent on at least one unique peptide spectrum match (psm) in the molecular weight search (MOWSE) with a protein score cut-off of 32.8. An additional information on the proteins in this list can be formed in Tables S4and S5.
β-lactones Inhibit the Activity of M. tuberculosis ClpP in vitro
To determine if M. tuberculosis ClpP1P2 peptidase activity can be inhibited by β-lactones, we performed enzymatic assays in the presence and absence of the compounds. For these experiments, we cloned the clpP1 and clpP2 gene products from M. tuberculosis with C-terminal His6 tags, over-expressed the proteins in a clpP null strain of Escherichia coli, and purified ClpP1 and ClpP2 by metal-affinity, anion-exchange, and size-exclusion chromatography (13,33,34). Formation and activation of the M. tuberculosis ClpP1P2 complex has only been observed in vitro in the presence of short N-protected hydrophobic dipeptides, such as Z-Leu-Leu-CHO (18), which presumably serve as agonists by binding to the peptidase active sites. For our inhibition assays, we activated ClpP1P2 using a combination of Z-Leu-Leu-Nva-CHO (an N-protected tripeptide aldehyde, which activated to higher levels and at lower concentrations than dipeptides), E. coli ClpXΔN (which activated independently and in combination with agonists) and ATPγS (33). In the presence of ATPγS, ClpXΔN presumably enhances catalytic activity by binding to and stabilizing catalytically active ClpP1P2 (33). The reactions were initiated by the addition of the fluorogenic tripeptide substrate, Z-Gly-Gly-Leu-AMC, and monitored by following the increase in fluorescence resulting from hydrolytic release of the aminomethylcoumarin (AMC) fluorophore.
β-lactones 4 and 7, which were most toxic to M. tuberculosis, displayed dose-dependent inhibition of ClpP1P2 peptidase activity (Figs. 2A, 2B), supporting the hypothesis that inhibition in vivo occurs via modification of this target. β-lactone 7, the most potent inhibitor identified in this study, is structurally distinct from previously reported β-lactone inhibitors of ClpP (20–23). It contains a benzylic substituent on the α-carbon, rather than an unbranched aliphatic chain, and an alkyl chain on the β-carbon. Interestingly, we note that significantly lower concentrations of β-lactones 4 and 7 were required to suppress mycobacterial growth than for inhibition of ClpP activity in vitro and that the inhibition was not quantitative. Sieber and co-workers reported similar observations wherein β-lactones that suppress virulence factor production in S. aureus in the nanomolar range only partially inhibit the ClpP peptidase in vitro in the micromolar range (20, 25). The differences in the observations in vivo and in vitro could be explained by the possibility that a modest degree of ClpP inhibition may be sufficient for suppression of mycobacterial growth. While we cannot rule out the possibility that the compounds have other essential targets, these observations could also be explained by the poor solubilities of the compounds in the enzymatic assays or by incomplete reconstitution of ClpP1P2 activity in vitro. Indeed, the reconstitution of a catalytically active tetradecamer of M. tuberculosis ClpP1P2 is not trivial and requires the inclusion of agonists (18) and/or a heterologous AAA+ partner. Although the mechanistic basis of the agonists is ill-defined (18), their requirement in assays in vitro suggests that an unidentified activator or regulatory partner may stimulate M. tuberculosis ClpP1P2 activity in vivo.
Figure 2.

Evidence for in vitro inhibition of ClpP1P2 by β-lactones
Following inhibition of ClpP1P2 with β-lactone 7 in vitro, we searched for sites of modification by mass spectrometry following digestion with trypsin and Asp-N. As evidenced by a 204 Dalton increase in the peptide masses, these experiments revealed β-lactone modification of the active-site serine of ClpP2 (Fig. 2C). In contrast, we did not recover modified peptides from ClpP1 active site. Thus, under the conditions of these assays, β-lactone 7 selectively inhibits ClpP2. We found that β-lactone 4 also selectively inhibits ClpP2 (data not shown). Similar observations have recently been reported with the ClpP1P2 peptidase in L. moncytogenes (35). These results are consistent with the observation that the peptidase activities of ClpP1 and ClpP2 have somewhat different specificities (18,45). They also provide an explanation for the partial inhibition of ClpP peptidase activity shown in Figures 2A and 2B. Interestingly, both ClpP1 and ClpP2 in M. smegmatis and in S. coelicolor (Table 2) were captured in the aforementioned chemical proteomic experiments. Based on the in vitro analyses, it is likely that ClpP1 is affinity captured in vivo not by a reaction, but by its association with ClpP2. These results are noteworthy because they provide clear evidence that the β-lactones that suppress the growth of M. tuberculosis also inhibit its ClpP peptidase in vitro.
An Auxiliary clpP Locus in Streptomyces coelicolor Confers Resistance to β-lactones
Mining the genomes of antibiotic-producing bacteria has proven to be a powerful strategy for the identification of antibacterial drug resistance genes (46–49). To identify putative determinants of resistance to β-lactone inhibitors of ClpP, we searched publicly available genome sequences of antibiotic-producing Streptomyces bacteria and found that S. coelicolor A3(2), S. avermitilis, and S. lividans possess a clpP3clpP4 locus that is paralogous to clpP1clpP2 (see, www.streptomyces.org.uk). Paralogous genes are often associated with antibiotic resistance (36, 38–42). We chose to study the role of the paralogous clpP genes in β-lactone resistance in S. coelicolor because it is the model organism of the genus and readily amenable to genetic manipulation. In S. coelicolor, the clpP1clpP2 locus is comprised by genes SCO2618 and SCO2619, while the clpP3P4 locus includes genes SCO7280 and SCO7281 (30). Further, the S. coelicolor clpP1 and clpP3 gene products are 59% identical in sequence, whereas the products encoded by clpP2 and clpP4 share 56% identity. Consistent with our prediction about the role of the auxiliary clpP locus, we found that S. coelicolor grew in media supplemented with high concentrations of β-lactones 4 or 7, whereas S. griseus IFO13350, a strain lacking clpP3clpP4, did not grow (Table 3). These results suggested that the auxiliary clpP locus confers β-lactone resistance. To test this hypothesis more directly, we constructed clpP1clpP2 and clpP3clpP4 null strains of S. coelicolor and assayed their susceptibilities to β-lactones. In support of this model, the clpP3clpP4-null strain was killed by aliphatic β-lactones 3 and 4, whereas the clpP1clpP2-null strain was resistant (Fig. 3; Table 3). The clpP3clpP4-null strain, by contrast, was only slightly more susceptible to β-lactone 7 than the clpP1clpP2-null strain, suggesting that β-lactone 7 inhibits both ClpP1P2 and ClpP3P4, albeit to different degrees. Further evidence that the compounds preferentially inhibit ClpP1P2 was the observation that wild-type S. coelicolor exhibited delayed sporulation in a manner reminiscent of the clpP1clpP2-null strain when grown on media supplemented with inhibitory β-lactones (Fig. S1).
Table 3.
Minimal Inhibitory Concentrations of β-lactones for Streptomyces bacteria
| Compound | Strain | MIC (μg/mL) |
|---|---|---|
|
WT | >800 |
| Δ clpP 1/2::apr | >800 | |
| Δ clpP 3/4::apr | 600 | |
| S. griseus | 650 | |
|
WT | >800 |
| Δ clpP 1/2::apr | >800 | |
| Δ clpP 3/4:: apr | 550 | |
| S. griseus | 580 | |
|
WT | >800 |
| Δ clpP 1/2::apr | 300 | |
| Δ clpP 3/4::apr | 250 | |
| S. griseus | 250 |
β-lactone minimal inhibitory concentrations (MICs) for wild-type S. coelicolor, S. coelicolor clpP1clpP2 and clpP3clpP4 null strains, and S.griseus. The β-lactones are partially insoluble at concentrations above 800 μg/mL.
Figure 3.
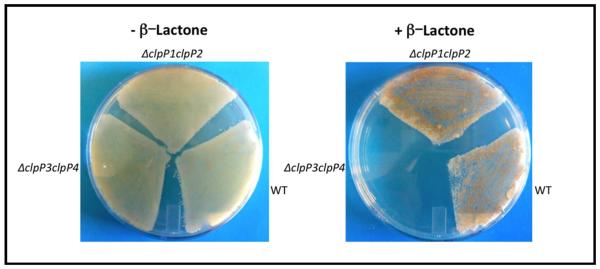
S. coelicolor null mutant susceptibly to β-lactones
In S. lividans, the clpP3clpP4 locus is only highly transcribed in a clpP1clpP2-null strain (40,43) because ClpP1P2 continuously degrades PopR, a regulatory protein that activates clpP3clpP4 transcription (40,43). Based on this model, we hypothesized that β-lactones might induce clpP3clpP4 transcription in S. coelicolor by inhibiting ClpP1P2. To test this prediction, we used semi-quantitative RT-PCR to assay clpP3clpP4 transcripts in wild-type S. coelicolor treated with either β-lactone or a DMSO control and found that clpP3clpP4 mRNA was only abundant after treatment with β-lactones 2,3,4,7 (Fig. 4A). In control experiments with the clpP1clpP2-null strain, we observed constitutive transcription of clpP3clpP4 (Fig. 4B), in a manner reminiscent of the analogous null strain of S. lividans (40, 43). Using 5′-RACE, we determined that the transcriptional-start sites of the clpP3clpP4 operon are located 80 and 82 base pairs upstream of the predicted translation start site. The positions of these start sites are similar to the one for clpP3clpP4 transcription in S. lividans and are close to an inverted-repeat sequence to which PopR is likely to bind. (43) Since the regulatory gene popR is divergently transcribed from clpP3clpP4 operon in S. coelicolor, S. lividans, and S. avermitilis, these genes may comprise a mobile, autoregulatory antibiotic-resistance locus.
Figure 4.
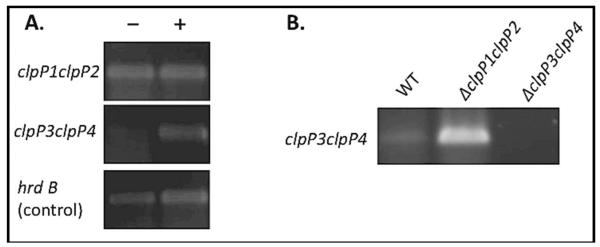
Transcriptional Analysis of clpP3clpP4
CONCLUSION
We report that disubstituted β-lactones inhibit the growth of wild-type strains of M. tuberculosis and M. smegmatis. The inhibition of growth by these molecules is markedly different from reports that compounds with related structures are not toxic to, but suppress virulence of pathogenic bacteria like Staphylococcus aureus and Listeria monocytogenes. The simple explanation for species-specific responses to the compounds is that ClpP is essential for viability in mycobacteria and other actinomycetes, but is only required for virulence in S. aureus and L. monocytogenes. While we cannot rule out other mechanisms by which these compounds could inhibit the growth of actinomycetes, observations reported here indicate that the toxic effects of these molecules can be ascribed to inhibition of essential ClpP peptidase activity. Specifically, we show that the toxic β-lactones inhibit M. tuberculosis ClpP peptidase activity in vitro. Consistent with the inhibition of the peptidolytic activity was the observation of a covalent adduct at the ClpP2 active site serine residue in a β-lactone treated preparation of the M. tuberculosis ClpP1P2 via mass spectrometry. The selective inhibition of ClpP2 by these compounds and their suppression of growth are consistent with reports that both clpP2 and clpP1 are essential for the viability of M. tuberculosis (7,11). Further, ClpP1 and ClpP2 were the only essential proteins identified in lysates of both S. coelicolor and M. smegmatis in in vivo chemical proteomic experiments, wherein proteins that reacted with an alkyne-substituted β-lactone were captured via a click reaction and analyzed. Additional evidence for ClpP inhibition by β-lactones is the observation that treatment of wild-type S. coelicolor with these compounds caused the same defect in morphological differentiation seen in the clpP1clpP2 null strain. The proposed mode of antibacterial activity by β-lactones is further validated by our findings that a S. coelicolor strain lacking one of the two paralogous clpP loci (i.e., clpP3clpP4) is susceptible to disubstituted β-lactones, whereas the wild-type strain is insensitive to these compounds. Collectively, these observations indicate that inhibition of essential ClpP peptidase activity by small molecules critically compromises bacterial growth.
Genome mining efforts led to the discovery that paralogous clpP genes mediate resistance to β-lactones. This finding is yet another example of a paralogous gene encoding an antibiotic resistance determinants (38–42). The connection of the auxiliary clpP3clpP4 locus in S. coelicolor with resistance to β-lactones is noteworthy because it has recently been associated with resistance to ADEP antibiotics that act by dysregulating ClpP (40). Accordingly, it is remarkable the peptidase encoded by clpP3clpP4 confers resistance to two distinct structural classes of antibiotics that affect ClpP activity by completely different mechanisms. Fortunately, the sensitivity of a S. coelicolor strain harboring only ClpP3P4 to benzyl-substituted β-lactone 7 indicates that the apparent resistance of this ClpP isoform can be circumvented. However, our findings regarding clpP3clpP4 do take on special relevance because there is evidence for the transfer of antibiotic resistance genes from Streptomyces bacteria to mycobacteria (52). In any case, the absence of a clpP3clpP4 locus in M. tuberculosis bodes well for the use of β-lactones and ADEPs as antibacterial drugs.
In summary, we report that β-lactones are a new privileged structure for inhibiting growth of mycobacteria and define a mechanism by which related actinobacteria can resist the toxic effects of these compounds. Our finding that the most potent β-lactone, which inhibits the growth of mycobacteria and both clpP null mutants of S. coelicolor, has a benzyl substituent on the α-carbon is novel and suggests that the potency of β-lactones can be enhanced by the inclusion of aromatic substituents at this carbon. Further investigations into this substituent effect as it relates to antibacterial activity and ClpP inhibition, as well as mechanistic studies of ClpP3P4, are underway in these laboratories. Collectively, our results lend further credence to predictions that ClpP and other intracellular proteases in bacteria are pharmacologically tractable drug targets (12, 29), like their human and viral counterparts. Continued research in this area will hasten the development of new chemical entities in the armamentarium used to treat tuberculosis and other bacterial infections.
METHODS
Strains, Media and Culture Conditions
The strains and plasmids used in this work are listed in Tables S1 and S2. E. coli strains DH5α and ET12567/pUZ8002 were grown in Luria-Bertani medium at 37 °C for routine subcloning, whereas E. coli strain BW25113/pIJ790 was grown at 30 °C to maintain selection of pIJ790 (50). Streptomyces strains were grown at 30 °C on mannitol soya flour medium (SFM), Difco nutrient agar medium (DNA), yeast extract-malt extract medium (YEME), or minimal liquid medium (NMMP) (37). For transcriptional analyses, S. coelicolor strains were grown in NMMP. M. smegmatis MC2155 was grown in Luria-Bertani medium supplemented with 0.2% (vol/vol) glycerol at 37 °C. M. tuberculosis H37Rv was maintained in Middlebrook 7H9 broth supplemented with 0.2% (vol/vol) glycerol, 0.05% Tween 80, and 10% (vol/vol) oleic acid-albumin-dextrose-catalase. For E. coli strains, ampicillin, apramycin, chloramphenicol, hygromycin, and kanamycin were employed at 100, 50, 25, 80, and 50 μg/mL, respectively. Nalidixic acid was used at 20 μg/mL for counter selections of E. coli in conjugations with S. coelicolor. For S. coelicolor, apramycin and hygromycin were used at 50 μg/mL.
Synthesis of β-lactones
With the exceptions of β-lactone 1 (purchased from Aldrich) and β-lactone 14 (salinosporamide, a gift from Prof. B. Moore), β-lactones were chemically synthesized by published methods (31, 32). Detailed synthetic procedures are available in the supporting information. All reagents were purchased from Sigma-Aldrich or VWR. Column chromatography was performed using 60 Å (230–400 mesh ASTM) silica gel. NMR analyses were performed on a Bruker Advance Ultrashield Spectrometer (400 or 300 MHz).
MIC assays
S. coelicolor and S. griseus MIC assays were performed on Difco nutrient agar medium supplemented with specific β-lactones. Growth was assessed after incubation at 30 °C for 48 h. The lowest drug concentration that inhibited ≥90% growth was considered to be the MIC. M. smegmatis MIC assays were performed on Difco nutrient agar medium supplemented with 0.2% glycerol and specific β-lactones. Growth was assessed after incubation at 30 °C for 72 h. The lowest drug concentration that inhibited ≥90% growth was considered to be the MIC. To determine the MICs of compounds for M. tuberculosis, the resazurin-based microplate assay was performed. Briefly, β-lactones were resuspended in DMSO and tested in a range from 10 to 0.08 mg/L following a 2-fold dilution scheme. After addition of the bacterial cells at 105 cfu/mL, the 96-well plates were incubated at 37 °C for 5 days. Resazurin was added (0.05 mL of a 0.1% solution), and cultures were incubated for 2 additional days at 37 °C. Fluorescence was measured using a Fluoroskan Ascent microplate fluorimeter (Thermo Scientific) with an excitation of 530 nm and emission of 590 nm. Wells containing only β-lactones were used to detect auto-fluorescence of compounds. The lowest drug concentration that inhibited ≥90% growth was considered to be the MIC. A 2-fold variation in MIC was considered to be within the error range of the assay. The final concentration of DMSO in all wells was 0.625%.
Affinity Capture of β-lactone Modified Proteins
Shaken liquid cultures (10 mL) of Streptomyces coelicolor M145 and Mycobacterium smegmatis MC2155 were grown to stationary phase and harvested by centrifugation at 4,000 rpm for 10 min. The resulting bacterial cell pellets were washed twice with 5 mL of phosphate buffer saline (PBS) to remove secreted proteins, and resuspended in 500 μL PBS. The alkynyl β-lactone was added to a final concentration of 200 μg/mL and incubated at room temperature on a nutating mixer for 2 hours. (The β-lactone probe was applied from a 20 mg/mL DMSO stock solution, such that DMSO concentration in the cell suspension did not exceed 1%.) Following incubation with the β-lactone probe, the cells were pelleted and washed three times with 1 mL PBS to remove excess probe. The cell suspensions were then lysed by treatment with lysozyme at a final concentration of 1 mg enzyme/mL of culture for 45 min at 37°C and clarified. To remove proteins that bound non-specifically to the affinity matrix, 50 μL of a commercially available solution containing avidin-agarose beads (Sigma) were added to the lysates and the resulting solution was incubated at room temperature for 30 minutes. After removal of the beads by centrifugation, 20 μM azido-PEG3-biotin (Alfa Aesar) and 1 mM Tris (2-carboxyethyl) phosphine hydrochloride (TCEP) were added to the lysates and they were gently mixed by brief vortexing. In turn, the click reaction (cycloaddition) was promoted by the addition of 1 mM CuSO4 catalyst and allowed to proceed for 1 hour at room temperature with gentle agitation. Once the CuSO4 was removed by centrifugation, the click reaction products were captured by incubation with 50 μL of the commercially available solution containing avidin-agarose beads for 1 hour. Control experiments in which the samples were not treated with either azido-biotin or with the Cu(II) catalyst were performed in parallel. To prepare for the protein-enriched beads for proteomic analysis, they were washed five times with 1 mL PBS and three times with 1 mL of a saturated NH4Cl solution.
Protein identification via mass spectrometry and bioinformatics
In all cases, the bead-bound proteins were tryptically digested and the resulting peptides were loaded onto a reverse phase column and separated by an Agilent 1200 HPLC. In addition to analysis of the experimental samples in this way, we also analyzed the protein-enriched beads used to remove non-specifically binding proteins, as well as those from all of the aforementioned negative controls. The peptides were identified by tandem, high-resolution MS/MS analyses using a coupled LTQ Orbitrap Velos Mass Spectrometer. The mass spectral data were searched using the MASCOT software algorithm against the corresponding bacterial protein databases. In the bioinformatic identifications of the peptide fragments, trypsin specificity with two missed cleavage sites was allowed and the MS mass tolerance was 7 ppm while the MS-MS tolerance was 0.5 Daltons. Identifications were contingent on at least one unique peptide spectrum match (PSM) in the molecular weight search (MOWSE) with a protein score cut-off of 32.8. A complete list of the proteins that were identified can be found in the supporting information.
Plasmids and primers
Standard cloning procedures were employed to generate the plasmids listed in Table S2. Genes encoding mature M. tuberculosis ClpP1 and ClpP2 (spanning residues 7–200 and 13–214, respectively (18) were amplified by PCR from M. tuberculosis H37Rv genomic DNA (ATCC) and cloned into the expression vector pET-22b (EMD) in frame with a C-terminal His6 tag. PCR was performed with Taq (Invitrogen) and Pfu (Stratagene, Agilent Technologies). All PCRs were performed with 5% (vol/vol) dimethylsulfoxide (DMSO).
Protein expression and purification
ClpP1 and ClpP2 were individually overexpressed in the clpP− E. coli strain JK10 (13) in LB broth at room temperature, following induction with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Protein was purified from clarified lysates by metal affinity (HisPur Ni-NTA agarose, Thermo), anion exchange (MonoQ, GE Healthcare), and size-exclusion chromatography (Superdex 200, GE Healthcare). Purified ClpP1 and ClpP2 were concentrated in Amicon centrifugal concentration devices (Millipore) to ≥80 μM tetradecamer in storage buffer (25 mM HEPES pH 7.5, 150 mM NaCl, 10% glycerol).
E. coli ClpXΔN (residues 62–425), bearing an N-terminal His6 tag followed by a tobacco etch virus (TEV) protease cleavage site, was cloned into pET-21a (EMD) and overexpressed in ER2566 (NEB) E. coli. ClpXΔN was purified from clarified lysates by metal affinity chromatography. The His6 tag was removed by overnight incubation at 4 °C with TEV protease (1 μg TEV per 50 μg ClpXΔN) in 25 mM HEPES pH 8, 50 mM NaCl, 1 mM EDTA, and 1 mM DTT. ClpXΔN was further purified by anion exchange (MonoQ, GE Healthcare) and size-exclusion chromatography (Superdex 200, GE Healthcare), and was concentrated to 30 M hexamer in storage buffer.
Peptidase assays
Peptidase assays were carried out at 30 °C in 25 mM HEPES (pH 7.0), 100 mM KCl, 5 mM MgCl2, 0.5 mM EDTA, 1 mM tris(2-carboxyethyl)phosphine (TCEP), and 20% (vol/vol) DMSO. M. tuberculosis ClpP1P2 (0.5 μM tetradecamer), Z-Leu-Leu-Nva-CHO (50 μM; Enzo Life Sciences), E. coli ClpXΔN (1 μM hexamer), and adenosine 5'-(γ-thiotriphosphate) (1 mM ATPγS; EMD) were incubated with the inhibitor for 30 min. Reactions (40 μL) were initiated by the addition of Z-Gly-Gly-Leu-AMC substrate (Enzo Life Sciences) to a final concentration of 100 μM. Substrate cleavage was monitored in a SpectraMax M5 microplate reader (Molecular Devices) by exciting at 380 nm and following the increase in 460 nm fluorescence.
LC-MS/MS
ClpP1P2 (3 μM tetradecamer), Z-Leu-Leu-Nva-CHO (50 μM), ClpXΔN (2 μM hexamer) and ATP-γ-S (1.5 mM) were incubated with β-lactone 7 (200 μM) for 1 h at 30 °C, and buffer exchanged into 50 mM ammonium bicarbonate using a SpinOUT desalting column (G-biosciences). Follow treatment with dithiothreitol, the protein was alkylated with iodoacetamide, and subjected to serial overnight digestion at 37 °C by trypsin (Sigma) and endopeptidase Asp-N (Sigma). LC-MS/MS analyses of the resulting peptides was performed on a LTQ Ion Trap mass spectrometer (Thermo Scientific) at the Koch Institute Proteomics Core Facility. Peptides were identified by the Mascot software package (Matrix Science) by searching against all possible peptides resulting from trypsin/Asp-N cleavage of bacterial protein sequences in the Swiss-Prot database. Data were visualized using the Scaffold software package (Proteome Software).
Construction of clpP1clpP2 and clpP3clpP4 null mutants
Using a published procedure for gene replacement in Streptomyces bacteria (50), strains of S. coelicolor were constructed in which either the clpP1clpP2 locus (SCO2618-19) or the clpP3clpP4 locus (SCO7280-81) was replaced with an apramycin (apr) resistance cassette. The loci replacements were confirmed by PCR analyses of genomic DNA from the null strains. In both, the null mutants' phenotypes were suppressed by complementation. Detailed procedures for the construction of the null strains are available in the supporting information.
Transcriptional Analyses
To analyze the transcription of the clpP operons, strains were grown for 21 hrs in NMMP media prior to treatment with 50 μg/ml of β-lactone or DMSO as a negative control. Cells were further incubated for 3 hrs. 1.5-mL culture aliquots were obtained and washed once with 10.3% (wt/vol) aqueous sucrose, followed by the addition of 100 μL of 10 mg/mL lysozyme solution (50 mM Tris-HCl, 1 mM EDTA, pH 8.0). The cells were incubated at 37 °C for 15 min. Total RNA was isolated using the RNeasy Mini Kit (Qiagen) following the manufacturer's protocol. RNA concentrations were measured using a NanoDrop ND-1000 spectrophotometer. An equal quantity of total RNA (1.0 μg) was employed in all reverse transcription-PCRs (RT-PCRs). RT-PCR was performed with the OneStep RT-PCR kit (Qiagen) according to the manufacturer's protocol for transcripts with high GC content. A 215-bp complementary DNA (cDNA) corresponding to the clpP1clpP2 (SCO2618-19) transcript and a 314-bp cDNA corresponding to the clpP3clpP4 (SCO7280-81) transcript were detected with clpP1clpP2 RT-PCR primers and clpP3clpP4 RT-PCR primers, respectively. In a positive control experiment, a 486-bp cDNA corresponding to the hrdB gene (SCO5820) transcript, which encodes a vegetative sigma factor, was detected with hrdB RT-PCR primers. All primer sequences are listed in Table S3 in the supporting information. The PCR program used for the detection of all transcripts was 50 °C for 30 min, 95 °C for 15 min, 30 cycles of 94 °C for 30 s, 57 °C for 30 s, and 72 °C for 60 s, and a final elongation at 72 °C for 10 min. No signals were detected in control experiments with Pfu polymerase, indicating that the RT-PCR signals correspond to the amplification of mRNA transcripts.
The transcriptional start site of the clpP3p4 locus was identified using the 5′-RACE System for Rapid Amplification of cDNA Ends, Version 2.0 (Invitrogen), using the manufacturer's protocol for high GC-content transcripts.
Supplementary Material
Acknowledgements
JKS and CLC gratefully acknowledge Brown University for funding. JKS is the recipient of a NSF CAREER Award. Supported in part by NIH grant GM-101988. Expert technical support on NMR and mass spectrometry were provided by R. Hopson, T. Shen, and J. Clifton (Brown University Department of Chemistry and Center for Genomics and Proteomics), and by R. Schiavoni (MIT Koch Institute Mass Spectrometry facility). We gratefully acknowledge S. Franzblau at the University of Illinois, Chicago Institute for Tuberculosis Research for determining minimal inhibitory concentrations of the compounds against M. tuberculosis.
Footnotes
Supporting information: This material is available free of charge via the internet at http://pubs.acs.org/.
Author Contributions. CLC synthesized the β-lactones, constructed the S. coelicolor clpP-null strains, and performed the proteomic and transcriptional analyses. KRS purified proteins for assays. CLC and KRS designed and performed the ClpP-inhibition assays. All authors contributed to designing experiments and writing the manuscript.
References
- 1.Cox G, Wright G. Intrinsic antibiotic resistance: Mechanisms, origins, challenges and solutions. Int. J. Med. Microbiol. 2013;303:287–292. doi: 10.1016/j.ijmm.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 2.Christian L, Philippe G, Mukund U, Knut L, Haileyesus G, Raviglione M. Global tuberculosis control: Lessons learnt and future prospects. Nat. Rev. Microbiol. 2012;10:407–416. doi: 10.1038/nrmicro2797. [DOI] [PubMed] [Google Scholar]
- 3.World Health Organization Global tuberculosis report. 2012 http://apps.who.int/iris/bitstream/10665/75938/1/9789241564502_eng.pdf.
- 4.Dorman S, Chaisson R. From magic bullets back to the magic mountain: the rise of extensively drug-resistant tuberculosis. Nat. Med. 2007;13:295–298. doi: 10.1038/nm0307-295. [DOI] [PubMed] [Google Scholar]
- 5.Koul A, Arnoult E, Lounis N, Guillemont J, Andries K. The challenge of new drug discovery for tuberculosis. Nature. 2011;469:483–490. doi: 10.1038/nature09657. [DOI] [PubMed] [Google Scholar]
- 6.Boucher H, Talbot G, Bradley J, Edwards J, Gilbert D, Rice L, Scheld M, Spellberg B, Bartlet J. Bad bugs, no drugs: No ESKAPE! an update from the infectious diseases society of America. Clin. Infect. Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 7.Raju R, Ennikrishnan M, Rubin D, Krishnamoorthy V, Kandror O, Akopian T, Goldberg A, Rubin E. Mycobacterium tuberculosis ClpP1 and ClpP2 function together in protein degradation and are required for viability in vitro and during infection. Plos Pathog. 2012;8 doi: 10.1371/journal.ppat.1002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baker T, Sauer R. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim. Biophys. Acta, Mol. Cell Res. 2012;1:15–28. doi: 10.1016/j.bbamcr.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Darwin K, Ehrt S, Gutierrez-Ramos J, Weich N, Nathan C. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science. 2003;302:1963–1966. doi: 10.1126/science.1091176. [DOI] [PubMed] [Google Scholar]
- 10.Ollinger J, O'Malley T, Kesicki E, Odingo J, Parish T. Validation of the essential ClpP protease in Mycobacterium tuberculosis as a novel drug target. J. Bacteriol. 2012;194:663–668. doi: 10.1128/JB.06142-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sassetti C, Rubin E. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. U. S. A. 2003;100:12989–12994. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raju R, Goldberg A, Rubin E. Bacterial proteolytic complexes as therapeutic targets. Nat. Rev. Drug Discovery. 2012;11:777–789. doi: 10.1038/nrd3846. [DOI] [PubMed] [Google Scholar]
- 13.Kenniston J, Baker T, Sauer R. Partitioning between unfolding and release of native domains during ClpXP degradation determines substrate selectivity and partial processing. Proc. Natl. Acad. USA. 2005;102:1390–1395. doi: 10.1073/pnas.0409634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Hartling J, Flanagan J. The structure of ClpP at 2.3 Å resolution suggests a model for ATP-dependent proteolysis. Cell. 1997;91:447–456. doi: 10.1016/s0092-8674(00)80431-6. [DOI] [PubMed] [Google Scholar]
- 15.Gaillot O, Pellegrini E, Bregenholt S, Nair S, Berche P. The ClpP serine protease is essential for the intracellular parasitism and virulence of Listeria monocytogenes. Mol. Microbiol. 2000;35:1286–1294. doi: 10.1046/j.1365-2958.2000.01773.x. [DOI] [PubMed] [Google Scholar]
- 16.Robertson G, Ng W, Foley J, Gilmour R, Winkler M. Global transcriptional analysis of clpP mutations of type 2 Streptococcus pneumoniae and their effects on physiology and virulence. J. Bacteriol. 2002;184:3508–3520. doi: 10.1128/JB.184.13.3508-3520.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie F, Zhang Y, Li G, Zhou L, Liu S, Wang C. The ClpP protease is required for the stress tolerance and biofilm formation in Actinobacillus pleuropneumoniae. PloS One. 2013;8 doi: 10.1371/journal.pone.0053600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akopian T, Kandror O, Raju R, UnniKrishnan M, Rubin E, Goldberg A. The active ClpP protease from M. tuberculosis is a complex composed of a heptameric ClpP1 and a ClpP2 ring. EMBO J. 2012;31:1529–1541. doi: 10.1038/emboj.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Socha A, Tan N, LaPlante K, Sello J. Diversity-oriented synthesis of cyclic acyldepsipeptides leads to the discovery of a potent antibacterial agent. Bioorg. Med. Chem. 2010;18:7193–7202. doi: 10.1016/j.bmc.2010.08.032. [DOI] [PubMed] [Google Scholar]
- 20.Zeiler E, Vadim S, Korotkov, Lorenz-Baath K, Böttcher T, Sieber S. Development and characterization of improved β-lactone-based anti-virulence drugs targeting ClpP. Bioorg. Med. Chem. 2012;20:583–591. doi: 10.1016/j.bmc.2011.07.047. [DOI] [PubMed] [Google Scholar]
- 21.Bottcher T, Sieber S. β-lactones as specific inhibitors of ClpP attenuate the production of extracellular virulence factors of Staphylococcus aureus. J. Am. Chem. Soc. 2008;130:14400–14401. doi: 10.1021/ja8051365. [DOI] [PubMed] [Google Scholar]
- 22.Böttcher T, Sieber S. β-lactones decrease the intracellular virulence of Listeria monocytogenes in macrophages. ChemMedChem. 2009;4:1260–1263. doi: 10.1002/cmdc.200900157. [DOI] [PubMed] [Google Scholar]
- 23.Böttcher T, Sieber S. Structurally refined B-lactones as potent inhibitors of devastating bacterial virulence factors. ChemBioChem. 2009;10:663–666. doi: 10.1002/cbic.200800743. [DOI] [PubMed] [Google Scholar]
- 24.Brotz-Oesterhelt H, Beyer D, Kroll H, Endermann R, Ladel C, Schroeder W, Hinzin B, Raddatz S, Paulson H, Henninger K, Bandow J, Sahl H, Labischinski H. Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat. Med. 2005;11:1082–1087. doi: 10.1038/nm1306. [DOI] [PubMed] [Google Scholar]
- 25.Gersch M, Gut F, Korotkov V, Lehmann J, Bottcher T, Rusch M, Hedberg C, Waldmann H, Klebe G, Sieber S. The mechanism of caseinolytic protease (ClpP) inhibition. Angew. Chem., Int. Ed. Engl. 2013;52:3009–3014. doi: 10.1002/anie.201204690. [DOI] [PubMed] [Google Scholar]
- 26.Kirstein J, Hoffmann A, Lille H, Schmidt R, Rubsamen-Walgmann H, BrotzOesterhelt H, Mogk A, Turgay K. The antibiotic ADEP reprogrammes ClpP, switching it from a regulated to an uncontrolled protease. EMBO Mol. Med. 2009;1:37–49. doi: 10.1002/emmm.200900002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmitt E, Riwanto M, Sambandamurthy V, Roggo S, Miault C, Zwingelstein C, Krastel P, Noble C, Beer D, Rao S, Au M, Niyomrattanakit P, Lim V, Zheng J, Jeffery D, Pethe K, Camacho L. The natural product cyclomarin kills Mycobacterium tuberculosis by targeting the ClpC1 subunit of the caseinolytic protease. Angew. Chem., Int. Ed. Engl. 2011;123:6011–6013. doi: 10.1002/anie.201101740. [DOI] [PubMed] [Google Scholar]
- 28.Cheng L, Naumann T, Horswill A, Hong S, Venters B, Tomsho J, Benkovic S, Keiler K. Discovery of antibacterial cyclic peptides that inhibit the ClpXP protease. Protein Sci. 2007;16:1535–1542. doi: 10.1110/ps.072933007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roberts D, Personne Y, Ollinger J, Parish T. Proteases in Mycobacterium tuberculosis pathogenesis: potential as drug targets. Future Microbiol. 2013;8:621–631. doi: 10.2217/fmb.13.25. [DOI] [PubMed] [Google Scholar]
- 30.Bentley S, Chater K, Cerdeno-Tarrage A, Challis G, Thomson N, James K, Harris D, Quail M, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen C, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S, Huang C, Kieser T, Larke L, Murphy L, Oliver K, O'Neil S, Rabbinowitsch E, Rajandream M, Rutherford K, Rutter S, Seeger K, Saunders D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorreck A, Woodward J, Barrell B, Parhill J, Hopwood D. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2) Nature. 2002;417:141–147. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- 31.Liu G, Shirley M, Romo D. A diastereoselective, nucleophile promoted aldollactonization of ketoacids leading to bicyclic-beta-lactones. J. Org. Chem. 2012;77:2496–2500. doi: 10.1021/jo202252y. [DOI] [PubMed] [Google Scholar]
- 32.Danheiser R, Nowick J. A practical and efficient method for the synthesis of β-lactones. J. Org. Chem. 1991;56:1176–1185. [Google Scholar]
- 33.Lee M, Baker T, Sauer R. Control of substrate gating and translocation into ClpP by channel residues and ClpX binding. J. Mol. Biol. 2010;399:707–718. doi: 10.1016/j.jmb.2010.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gottesman S, Roche E, Zhou Y, Sauer R. The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA-tagging system. Genes Dev. 1998;12:1338–1347. doi: 10.1101/gad.12.9.1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zeiller E, List A, Alte F, Gersch M, Wachtel R, Poreba M, Drag M, Groll M, Sieber S. Structural and functional insights into caseinolytic proteases reveal an unprecedented regulation principle of their catalytic triad. Proc. Natl. Acad. USA. 2013;110:11302–11307. doi: 10.1073/pnas.1219125110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walsh C. Molecular mechanisms that confer antibacterial drug resistance. Nature. 2000;406:775–781. doi: 10.1038/35021219. [DOI] [PubMed] [Google Scholar]
- 37.Kieser T, Bibb M, Buttner M, Chater K, Hopwood D. The John Innes Foundation. 2000. Practical Streptomyces genetics. [Google Scholar]
- 38.Vecchione J, Sello J. Characterization of an inducible, antibiotic-resistant aminoacyl-tRNA synthetase gene in Streptomyces coelicolor. J. Bacteriol. 2008;190:6253–6257. doi: 10.1128/JB.00737-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vecchione J, Sello J. A novel tryptophanyl-tRNA synthetase gene confers high-level resistance to indolmycin. Antimicrob. Agents Chemother. 2009;53:3972–3980. doi: 10.1128/AAC.00723-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gominet M, Seghezzi N, Mazodier P. Acyl depsipeptide (ADEP) resistance in Streptomyces. Microbiol. 2011;157:2226–2234. doi: 10.1099/mic.0.048454-0. [DOI] [PubMed] [Google Scholar]
- 41.Maurer K, Pfeiffer F, Zehender H, Mecke D. Characterization of two glyceraldehyde-3-phosphate dehydrogenase isoenzymes from the pentalenolactone producer Streptomyces arenae. J. Bacteriol. 1983;153:930–936. doi: 10.1128/jb.153.2.930-936.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thiara S, Cundliffe E. Cloning and characterization of a DNA gyrase B gene from Streptomyces sphaeroides that confers resistance to novobiocin. EMBO J. 1988;7:2255–2259. doi: 10.1002/j.1460-2075.1988.tb03065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Viala J, Mazodier P. ClpP-dependent degradation of PopR allows tightly regulated expression of the clpP3 clpP4 operon in Streptomyces lividans. Mol. Microbiol. 2002;44:633–643. doi: 10.1046/j.1365-2958.2002.02907.x. [DOI] [PubMed] [Google Scholar]
- 44.Viala J, Rapoport G, Mazodier P. The clpP multigenic family in Streptomyces lividans: Conditional expression of the clpP3 clpP4 operon is controlled by PopR, a novel transcriptional activator. Mol. Microbiol. 2000;38:602–612. doi: 10.1046/j.1365-2958.2000.02155.x. [DOI] [PubMed] [Google Scholar]
- 45.Personne Y, Brown AC, Schuessler DL, Parish T. Mycobacterium tuberculosis ClpP Proteases Are Co-transcribed but Exhibit Different Substrate Specificities. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0060228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sello J. Mining the antibiotic resistome. Chemistry and Biology. 2012;19(10):1220–1. doi: 10.1016/j.chembiol.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 47.D'Costa V, McGrann K, Hughes D, Wright G. Sampling the antibiotic resistome. Science. 2006;311:374–377. doi: 10.1126/science.1120800. [DOI] [PubMed] [Google Scholar]
- 48.Spanogiannopoulos P, Thaker M, Koteva K, Waglechner N, Wright GD. Characterization of a rifampin-inactivating glycosyltransferase from a screen of environmental atinomycetes. Antimicrob. Agents Chemother. 2012;56:5061–5069. doi: 10.1128/AAC.01166-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Forsberg K, Reyes A, Wang B, Selleck E, Sommer M, Dantas G. The shared antibiotic resistome of soil bacteria and human pathogens. Science. 2012;337:1107–1111. doi: 10.1126/science.1220761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gust B, Kieser T, Chater K. REDIRECT technology: PCR-targeting system in Streptomyces coelicolor. 2002 http://streptomyces.org.uk/redirect/protocol_V1_4.pdf.
- 51.Franzblau S, Witzig R, McLaughlin J, Torres P, Madico G, Hernandez A, Degnan M, Cook M, Quenzer V, Ferguson R, Gilman R. Rapid, Low-Technology MIC Determination with Clinical Mycobacterium tuberculosis Isolates by Using the Microplate Alamar Blue Assay. J. Clin. Microbiol. 1998;36:362–366. doi: 10.1128/jcm.36.2.362-366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pang Y, Brown B, Steingrube V, Wallace R, Roberts M. Tetracycline resistance determinants in Mycobacterium and Streptomyces species. Antimicrob. Agents Chemother. 1994;38:1408–1412. doi: 10.1128/aac.38.6.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.