

Abstract
Maternal food restriction (MFR) causes intrauterine growth restriction, a known risk factor for developing chronic lung disease. However, it is unknown whether this negative outcome is gender specific or preventable by blocking the MFR-induced hyperglucocorticoidism. Using a well-established rat model, we used metyrapone (MTP), an inhibitor of glucocorticoid synthesis, to study the MFR-induced lung changes on postnatal day (p) 21 in a gender-specific manner. From embryonic day 10 until delivery, pregnant dams were fed either an ad libitum diet or a 50% caloric restricted diet with or without MTP supplementation. Postnatally, the offspring were fed ad libitum from healthy dams until p21. Morphometric, Western blot, and immunohistochemical analysis of the lungs demonstrated that MTP mitigated the MFR-mediated decrease in alveolar count, decrease in adipogenic protein peroxisome proliferator-activated receptor γ, increase in myogenic proteins (fibronectin, α-smooth muscle actin, and calponin), increase in Wnt signaling intermediates (lymphoid enhancer-binding factor 1 and β-catenin), and increase in glucocorticoid receptor (GR) levels. The MFR-induced lung phenotype and the effects of MTP were similar in both genders. To elucidate the mechanism of MFR-induced shift of the adipogenic-to-myogenic phenotype, lung fibroblasts were used to independently study the effects of (1) nutrient restriction and (2) excess steroid exposure. Nutrient deprivation increased myogenic proteins, Wnt signaling intermediates, and GR, all changes blocked by protein supplementation. MTP also blocked, likely by normalizing nicotinamide adenine dinucleotide phosphate levels, the corticosterone-induced increase in myogenic proteins, but had no effect on GR levels. In summary, protein restriction and increased glucocorticoid levels appear to be the key players in MFR-induced lung disease, affecting both genders.
Keywords: bronchopulmonary dysplasia, intrauterine growth restriction, maternal food restriction, metyrapone, glucocorticoid, lung development
Introduction
Intrauterine growth restriction (IUGR) is a known risk factor for bronchopulmonary dysplasia (BPD),1 a negative outcome of prematurity that increases the risk of oxygen requirement,2 pulmonary infections,3 reactive airway disease,4 and chronic obstructive pulmonary disease.5 Despite extensive research, current preventive options such as postnatal glucocorticoids,6 exogenous surfactant,7 and caffeine8–10 may ameliorate acute respiratory symptoms but do not significantly impact the development of BPD.11 Avoiding invasive ventilation appears to be the primary method that may possibly reduce the incidence of BPD,12 yet this approach is overly simplistic since the oxygen requirement of the infant is ultimately constrained.13 Therefore, further work is required to discover other management strategies.
The Barker hypothesis for “fetal programming” suggests that the intrauterine environment may regulate genomic expression over the course of the individual's lifetime.14 Physiologic fetal glucocorticoid levels are essential for normal lung maturation,15 and a short therapeutic course may accelerate lung maturity16; however, chronically elevated glucocorticoid levels inhibit normal lung maturation.17 A rat model of maternal food restriction (MFR) causes IUGR and increased circulating corticosterone levels in the offspring, resulting in altered lung structure with reduced alveolar number, increased alveolar septal thickness, and reduced surfactant content, both at birth and in the adults.18,19 Thus, we reasoned that in the IUGR offspring resulting from MFR, chronically elevated glucocorticoid levels would be the unifying mechanism for BPD, and thus, limiting excessive glucocorticoid production would alleviate this negative effect. To inhibit elevated circulating glucocorticoid levels in IUGR offspring, we used metyrapone (MTP), an inhibitor of 11β-hydroxylase, which is essential for glucocorticoid biological activity.
Using a well-established rat model of MFR to induce IUGR,18,20 this study analyzes the effect of MTP on the lungs of MFR offspring at postnatal day (p) 21 separately in males and females. To gain mechanistic insight to the effects of MFR on the developing lung and its normalization with MTP, in vitro studies were conducted using cultured fetal lung fibroblasts. Based on the premise that MFR results in excess glucocorticoid levels that ultimately dysregulate normal pulmonary maturation, we hypothesize that “normalization” of the glucocorticoid levels with MTP would mitigate these abnormal changes.
Materials and Methods
Antenatal Food Restriction Animal Model
The Animal Research Committee of the Los Angeles Biomedical Research Institute at Harbor-UCLA Medical Center approved all studies, in compliance with the American Association for the Accreditation of Laboratory Animal Care and National Institutes of Health guidelines. First-time pregnant Sprague-Dawley rat dams (Charles River Laboratories, Inc, Hollister, California) were housed in a facility with a 12-hour light–12-hour dark cycle and constant temperature and humidity. From gestational day 10 until delivery (embryonic day 22), the dams were provided an ad libitum diet (AA, control) of standard laboratory rat chow: protein 23%, fat 4.5%, and metabolizable energy = 3030 kcal/kg; LabDiet 5001; Brentwood, Missouri) or a 50% food-restricted diet (FA), which was calculated based on the normal food intake of the ad libitum group every 24 hours. From gestational day 10 until delivery, the food-restricted dams were water supplemented either without (FA) or with MTP (FA + M; 0.5 mg/mL 2-methyl-1,2-di-3-pyridyl-1-propanone; Sigma-Aldrich, St Louis, Missouri).
After delivery, all pups were provided an ad libitum diet. Pups that were food-restricted in utero were cross-fostered to dams fed ad libitum during pregnancy. At day 1 after birth, the litters were separated by gender and the body weights of individual pups were recorded. To minimize bias toward selecting either heavier or lighter pups, on p1, all pups from each litter were weighed and 6 pups (3 females and 3 males) closest to the median body weight (according to gender) were included in the study, and the other pups were culled to keep the number of pups/litter for experimentation purposes at 6. All studies maintained a similar number of pups per dam between the control and experimental groups.
Pups were killed with Euthasol (pentobarbitol and phenytoin; Virbac, Fort Worth, Texas) on p21, and lungs were harvested for further analysis.
Western Analysis
Liquid nitrogen flash-frozen lung tissue samples were suspended in radioimmunoprecipitation assay buffer containing 1 mmol/L EDTA and ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA; Boston BioProducts, Ashland, Massachusetts) supplemented with 1 mmol/L phenylmethylsulfonyl fluoride (PMSF) and a complete protease inhibitor mixture (Roche Diagnostics, Indianapolis, Indiana), homogenized with a tissue grinder, sonicated, and centrifuged at 4°C for 10 minutes at 14 000 rpm. The concentration of protein was determined using the BCA Protein Assay Kit (Thermo Scientific Pierce, Rockford, Illinois). Equal aliquots (50 μg) of total protein for each sample were denatured with sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis sample buffer and separated by electrophoresis on an SDS–polyacrylamide gel. After transferring the samples to a nitrocellulose membrane, the membrane was blocked with Tris-buffered saline (TBS)-Tween + 5% milk and probed with the following primary antibodies (peroxisome proliferator-activated receptor γ [PPARγ], 1:500; fibronectin, 1:500; glucocorticoid receptor [GR], 1:200; β-catenin, 1:500; all from Santa Cruz Biotechnology, Santa Cruz, California; α smooth muscle actin (α-SMA), 1:50 000; calponin, 1:3000; both from Sigma-Aldrich; glyceraldehyde 3-phosphate dehydrogenase [GAPDH], 1:5000; from Millipore, New Bedford, Massachusetts] and the corresponding secondary antibodies (antirabbit antibody and antimouse antibody; both from GE Healthcare, United Kingdom). The membranes were washed with TBS containing 0.1% Tween-20 wash buffer after each antibody incubation cycle. SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific Pierce) was used for detection, and photographic emulsion was used to identify the protein bands, which were subsequently quantified by densitometry. Protein band intensities were normalized for loading using the corresponding GAPDH and expressed as arbitrary units. The densities of the specific protein bands were quantified with a scanning densitometer (Bio-Rad GS-800, Hercules, California), and the results are expressed as means ± standard error normalized to GAPDH.
Triglyceride Uptake Assay
Triolein uptake, a key marker for alveolar lipofibroblast (LIF) function, was used to quantitate the rate of triglyceride uptake by fetal rat lung explants using a previously described method.21 Briefly, culture medium was replaced with Dulbecco modified Eagle medium containing 20% adult rat serum mixed with [3H]triolein (5 μCi/mL). The explants were incubated at 37°C in 5% CO2-air balance for 4 hours. At the termination of the incubation, the medium was decanted, the explants were rinsed twice with 1 mL of ice-cold phosphate-buffered saline (PBS), and the tissue thoroughly homogenized. An aliquot of the tissue homogenate was taken for protein assay, and the remaining tissue homogenate was extracted to determine its radiolabeled neutral lipid content.
Preparation of Lung for Histological Analysis
After euthanasia, the lungs were fixed in situ by perfusing them with 4% (wt/vol) paraformaldehyde (PFA) in PBS solution. During the perfusion, a tracheal catheter was used to maintain a standard inflation pressure of 20 cm H2O. An Ethicon surgical suture (Ethicon, Inc, Somerville, New Jersey) was used to ligate the trachea, and fresh 4% PFA-PBS solution was used to incubate the lungs at 4°C for about 4 hours. Next, the PFA-PBS solution was rapidly replaced with 2 changes of cold PBS to eliminate luminal debris. Finally, the lungs were transferred to a filter-sterilized 30% sucrose-PBS solution and fully equilibrated at 4°C. The lungs were subsequently paraffin embedded. Cross-sections of 5 µm were prepared, transferred to Superfrost microscope glass slides (Fisher Scientific, Pittsburgh, PA), and processed using standard procedures.18 Processed paraffin sections were stained with either hematoxylin–eosin to assess tissue morphology and measure morphometric parameters or treat with antibodies for immunohistochemical analysis.
Lung Morphometry
Lung morphometric analysis was conducted by an investigator who was unaware of the treatment group assignment for each animal sample. Fifty randomly selected nonoverlapping fields from sections obtained from 12 blocks from each treatment group were included for measurement. Using a microscope at ×5 magnification, each field was digitally visualized with an AxioVision microscope (Carl Zeiss, Thornwood, New York), and the number of alveoli were counted, as described previously.18
Lung Immunohistochemistry
Immunohistochemistry (IHC) was conducted to assess in situ protein expression. Paraffin sections of 5 µm were deparaffinized in xylene and rehydrated with a graded series of ethanol solutions. Nonspecific binding was blocked with 5% normal goat serum (NGS). The sections were incubated with the specified monoclonal primary antibodies at room temperature for 1 hour as follows: for β-catenin staining, mouse monoclonal anti-β-catenin antibody (1:500; Santa Cruz Biotechnology); for calponin, mouse anti-calponin antibody (1:500; Sigma-Aldrich); and for α-SMA, mouse anti-α-SMA antibody (1:500; Sigma-Aldrich) were used as the primary antibodies. After several PBS washes at room temperature, the tissues were incubated sequentially with appropriate Alexa Fluor secondary antibodies (Alexa Fluor 568 Red, 1:100; Alexa Fluor 488 Green, 1:300; Invitrogen, Eugene, Oregon) in a humidified, darkened chamber. Sections were gently rinsed with PBS and mounted with ProLong Gold antifade reagent with 4′-6-diamidino-2-phenylindole (DAPI; Invitrogen) for fluorescent microscopic analysis.
Lipofibroblast Culture and In Vitro Studies
Embryonic day 19 (e19) pulmonary LIFs were isolated according to previously described methods.22 At 80% to 90% confluence, cells were cultured in Dulbecco minimal essential medium containing graded concentrations of either fetal bovine serum (FBS; 10%, 5%, 1%, or 0%) or bovine serum albumin (BSA; 0%, 1%, 2.5%, or 5%) for 24 hours, as previously described.22 At the termination of the experimental conditions, cell extracts were prepared in lysis buffer (20 mmol/L HEPES, 2 mol/L EGTA, 50 mmol/L β-glycerophosphate, 10% glycerol, 1% Triton X-100, 1 mmol/L dithiothreitol, 1 mmol/L vanadate, and 0.04 mmol/L PMSF) containing a mixture of protease inhibitors (Sigma-Aldrich) and subjected to Western analysis as described previously, using the appropriate primary antibodies (GR, 1:200; 11β-hydroxysteroid dehydrogenase 1 [11β-HSD1], 1:200; β-catenin, 1:500; lymphoid enhancer-binding factor 1 [LEF-1], 1:200; fibronectin, 1:500; calponin, 1:3000; all from Santa Cruz Biotechnology; GAPDH, 1:5000; from Millipore) and the secondary antibodies (antirabbit antibody and antimouse antibody; both from GE Healthcare).
These cell experiments were also performed to assess the effect of corticosterone exposure with and without MTP. At 80% to 90% confluence, cells were cultured in Dulbecco minimal essential medium + 0.1% FBS containing either corticosterone (CORT) 10−9 mol/L alone or with MTP 10−8 mol/L for 24 hours. The specific protein levels were determined by Western analysis with the antibodies as described previously with the exception of normalization to mouse β-actin (1:5000; Santa Cruz Biotechnology).
Determination of Nicotinamide Adenine Dinucleotide Phosphate/Reduced Form of Nicotinamide Adenine Dinucleotide Phosphate Levels
The nicotinamide adenine dinucleotide phosphate/reduced form of nicotinamide adenine dinucleotide phosphate (NADP/NADPH) assay kit was utilized to determine the levels of NADP and NADPH in the cell cultures (ab65349; Abcam, Cambridge, Massachusetts) following manufacturers’ instructions.
Immunocytochemistry
Embryonic day 19 LIFs were grown under different culture conditions on slide-backed chambers (Lab-Tek; Electron Microscopy Sciences, Hatfield, Pennsylvania). These cells were blocked with 5% NGS and subsequently incubated with primary and secondary antibodies. For β-catenin staining, mouse monoclonal anti-β-catenin antibody (1:50; Santa Cruz Biotechnology) was used as the primary antibody. For double-staining, rabbit polyclonal anti-LEF-1 antibody (1:50; Santa Cruz Biotechnology) and boron-dipyrromethene (BODIPY) 3922 (Invitrogen) were used to visualize changes in cellular fat content. After several PBS washes at room temperature, the cells were incubated with the appropriate Alexa Fluor secondary antibody (antimouse Alexa Fluor 488 Green and antirabbit Alexa Fluor 568 Red; both from Invitrogen) in a humidified, darkened chamber. Slides were gently rinsed with PBS and mounted with ProLong Gold antifade reagent with DAPI (Invitrogen) for fluorescent microscopic analysis.
Statistical Analysis
Experimental data were analyzed using 2-way analysis of variance, and as indicated, followed by Tukey post hoc test. A P value of <.05 was considered to indicate statistically significant differences between the experimental and control groups. Results are expressed as mean ± standard error of the mean. A minimum of 4 animals per group taken from 4 separate litters were used for data analysis.
Results
Weight and Growth of Pups
At p1, FA pups (males = 4.8 ± 0.4 g and females = 5.3 ± 0.5 g) demonstrated significantly (P < .05) lower birth weights than that of AA pups (males = 7.7 ± 0.6 g and females = 7.6 ± 0.7 g) for both genders (Figure 1A [males] and B [females]). Metyrapone (males = 6.6 ± 0.9 g and females = 6.0 ± 0.0 g, P < .05 vs FA pups) alleviated this weight discrepancy due to MFR. At p21, FA pups (males = 46.5 ± 1.1 g and females = 46.5 ± 1.3 g) still had lower weights than that of AA pups (males = 54.0 ± 3.0 g and females = 52.3 ± 2.5 g, P < .05 for both groups). With MTP therapy, FA + M pups (males = 58.0 ± 4.0 g and females = 51.7 ± 0.6) had weights greater than FA pups (P < .05 FA + M vs FA) and similar to the control group (P > .05 FA + M vs AA).
Figure 1.
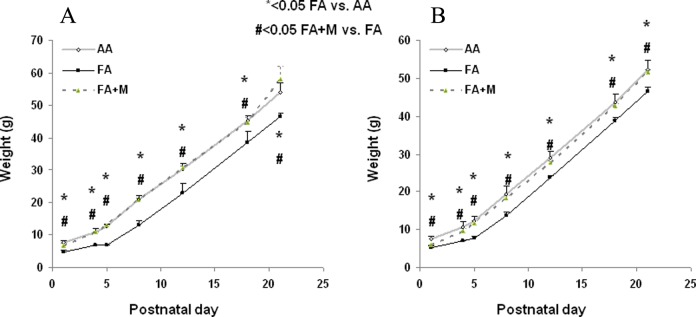
Metyrapone (MTP) ameliorates the effect of maternal food restriction (MFR) on offspring body weight from birth to postnatal day 21 (p21). In both males (A) and females (B), antenatal MTP treatment blocked the effect of MFR-induced intrauterine growth restriction (IUGR) at birth and on subsequent growth up to p21. * = P < .05 FA versus AA and # = P < .05 FA + M versus FA; N = 6. AA indicates ad libitum diet; FA + M, food-restricted diet with MTP.
Effect of MTP on MFR-induced changes in lung morphometry
At p21, for both genders (Figure 2A [males] and B [females]), MTP decreased the MFR-induced significant reduction in alveolar number (males AA = 358 ± 47 vs FA = 183 ± 14 vs FA + M = 299 ± 20, females AA = 320 ± 39 vs FA = 184 ± 12 vs FA + M = 275 ± 17; all expressed as /mm2, and for both genders P < .05 for FA vs AA and FA + M vs FA) and blocked the resultant simplification of the alveolar cytoarchitecture.
Figure 2.

Metyrapone (MTP) ameliorates the effect of maternal food restriction (MFR) on lung cytoarchitecture. At postnatal day 21 (p21), MTP blocked the MFR-induced decrease in alveolar number and simplification of the lung structure in both males (A) and females (B). * = P < .05 FA versus AA and # = P < .05 FA + M versus FA; N = 10. AA indicates ad libitum diet; FA+ M, food-restricted diet with MTP.
Effect of MTP on MFR-Induced Changes in Key Proteins of Alveolar Development and Injury/Repair
In the lungs of both genders at p21, MFR decreased the expression of PPARγ (Figure 3A), and increased the expression of fibronectin (Figure 3B), α-SMA (Figure 3C), calponin (Figure 3D), GR (Figure 3E), and β-catenin (Figure 3F; a key mediator of the Wnt signaling pathway). Metyrapone therapy blocked these changes caused by MFR in both males and females. Similar to the Western blot data, IHC studies of male and female p21 lungs demonstrated that MFR increased the expression of α-SMA (Figure 4A), calponin (Figure 4B), β-catenin (Figure 4C), and LEF-1 (Figure 4D; for brevity only male data are shown). Blockage of these changes by MTP was also corroborated by IHC.
Figure 3.


Metyrapone (MTP) blocks the effects of maternal food restriction (MFR) on key pulmonary lipogenic and myogenic differentiation markers. At postnatal day 21 (p21), in both males and females, MTP normalized the MFR-induced decrease in peroxisome proliferator-activated receptor γ (PPARγ; A) and MFR-induced increase in fibronectin (B), α smooth muscle actin (α-SMA; C), calponin (D), GR (E), and β-catenin (F) protein levels, as determined by Western blotting. * = P < .05 FA versus AA and # = P < .05 FA + M versus FA; N = 6. AA indicates ad libitum diet; FA+ M, food-restricted diet with MTP.
Figure 4.

Metyrapone (MTP) blocks the effect of maternal food restriction (MFR)-induced increase in pulmonary myogenic proteins and Wnt signaling intermediates. Immunohistochemistry of postnatal day 21 (p21) lungs shows that MFR-induced increase in protein levels of α smooth muscle actin (a-SMA; A), calponin (B), β-catenin (C), and lymphoid enhancer-binding factor 1 (LEF-1; D) is blocked by MTP.
Metyrapone Blocks the MFR-Induced Functional Decrease in Lung Mesenchymal Differentiation
In both genders, adverse effects of MFR were reflected by a significant decrease in the rate of triolein uptake, a marker of LIF function, by cultured lung explants of p21 lungs; MTP treatment not only blocked the MFR-induced decrease in triolein uptake but also in fact increased it (Figure 5; males AA = 11 951 ± 305, FA = 7810 ± 1708, and FA + M = 16 438 ± 1673; females AA = 4450 ± 784, FA = 1801 ± 803, and FA + M = 9569 ± 91; all values are expressed as cpm/mg protein).
Figure 5.
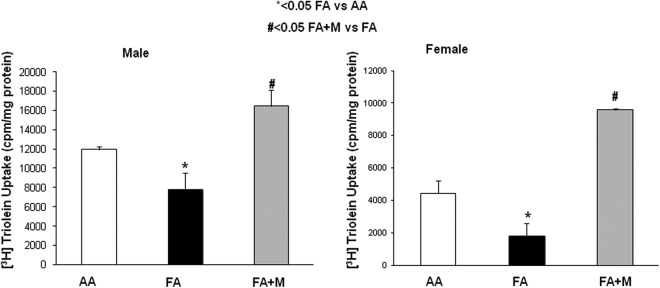
Maternal food restriction (MFR)-induced decrease in triolein uptake, a functional marker for lipofibroblasts, is blocked by metyrapone (MTP). Both male (A) and female (B) postnatal day 21 (p21) lung explants showed significant decrease in triolein uptake; MTP treatment not only blocked the MFR-induced decrease in triolein uptake but also in fact increased it. * = P < .05 FA versus AA and # = P < .05 FA + M versus FA; N = 6. AA indicates ad libitum diet; FA + M, food-restricted diet with MTP.
Effect of Protein Supplementation on Nutrient Restriction in Fetal Lung Fibroblast Differentiation
Lung fibroblast differentiation is the key to determining the alveolar phenotype. In order to mimic the effect of MFR on lung fibroblasts, mixed e19 male and female fetal rat lung fibroblasts were grown in the presence or absence of FBS supplementation. Analogous to the MFR in vivo data for p21 lungs, the absence of FBS resulted in increased protein levels of fibronectin, calponin, β-catenin, and LEF-1 (Figure 6A) indicating Wnt activation and differentiation of these fibroblasts to a myogenic phenotype. Bovine serum albumin supplementation blocked all these effects of FBS-restriction on the fibroblast phenotype (Figure 6B-E). Fluorescence staining of cultured e19 fibroblasts showed that FBS restriction decreased lipid (BODIPY) staining but increased the expression of the Wnt signaling intermediates LEF-1 and β-catenin, indicating lipo-to-myofibroblast trandifferentiation (Figure 6F). Translocation of β-catenin from the cytoplasm to the nucleus upon FBS restriction further supports Wnt activation. Importantly, all these FBS-restriction-induced changes were blocked by BSA supplementation. In line with our in vivo data, FBS restriction for cultured e19 fibroblasts resulted in increased GR protein levels (Figure 7A), which was accompanied by decreased 11β-HSD1 levels (Figure 7B). Bovine serum albumin supplementation reversed the effect of FBS restriction on GR levels but had no effect on 11β-HSD1 levels (Figure 7C and D).
Figure 6.



Fetal bovine serum (FBS) restriction and bovine serum albumin (BSA) supplementation affect the expression of key myogenic proteins by cultured e19 fetal rat lung lipofibroblasts (LIFs). In e19 LIFs, FBS restriction increased the expression of fibronectin, calponin, β-catenin, and LEF-1 (A); BSA supplementation blocked these effects (B-E). After a 24-hour incubation period, FBS-restricted cells demonstrated increased β-catenin levels along with its increased nuclear translocation, increased LEF-1 levels, and decreased lipid content; all of these effects were blocked by BSA supplementation (F). * = P < .05 versus 10% FBS and # = P < .05 versus 0% FBS; N = 4. e19 indicates embryonic day 19; LEF-1, lymphoid enhancer-binding factor 1.
Figure 7.

The effect of fetal bovine serum (FBS) restriction and bovine serum albumin (BSA) supplementation on the expression of glucocorticoid receptor (GR) and 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) levels in embryonic day 19 (e19) fetal rat lung lipofibroblasts (LIFs). The FBS restriction increased GR levels (A) but decreased HSD1 levels (B) dose dependently. The BSA supplementation blocked the effect of FBS restriction on GR levels (C) but not on HSD1 levels (D). * = P < .05 versus 10% FBS and # = P < .05 versus 0% FBS; N = 4.
Effect of Glucocorticoids on e19 Fibroblasts and its Inhibition by MTP
To study the effect of glucocorticoids directly on fetal lung fibroblasts, mixed e19 male and female fetal rat lung fibroblasts were grown in the presence of CORT. Analogous to the in vivo MFR and in vitro FBS restriction studies, CORT treatment resulted in increased protein levels of fibronectin, calponin, and LEF-1, with these changes being blocked by MTP (Figure 8A-C). In contrast to the nutrient restriction studies, CORT had no effect on the level of GR but increased the level of HSD1, an effect that was blocked by MTP (Figure 8D and E). Further studies demonstrated that CORT resulted in an increased NADPH level and an increased NADPH–total NADP ratio, but MTP reversed these changes induced by CORT (Figure 9A and B).
Figure 8.

The effect of corticosterone (CORT) and metyrapone (MTP) treatment of e19 fetal rat lung lipofibroblasts (LIFs). In e19 LIFs, CORT treatment increased the expression of fibronectin (A), calponin (B), lymphoid enhancer-binding factor 1 (LEF-1; C), and hydroxysteroid dehydrogenase type 1 (HSD1; E) but had no effect on GR levels (D). Metyrapone blocked these effects of corticosterone. * = P < .05 versus control and # = P < .05 versus CORT; N = 4. e19 indicates embryonic day 19.
Figure 9.
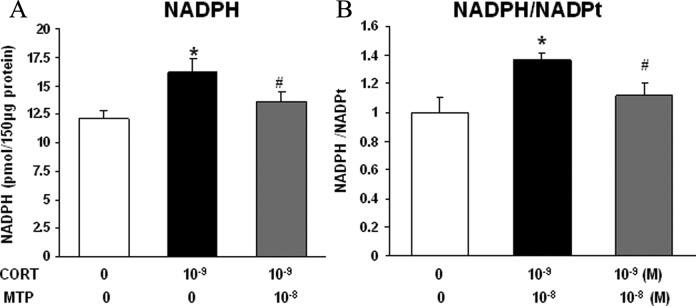
The effect of corticosterone (CORT) and metyrapone (MTP) treatment on NADPH/NADPt levels in e19 fetal rat lung lipofibroblasts (LIFs). In e19 LIFs, CORT treatment increased the levels of (A) NADPH and (B) NADPH/NADPt. Metyrapone blocked this effect of corticosterone. * = P < .05 versus control and # = P < .05 versus CORT; N = 4. e19 indicates embryonic day 19; NADPt, total NADP; NADPH, nicotinamide adenine dinucleotide phosphate.
Discussion
Chronic lung disease of prematurity is a complex, multifactorial condition whose definition and pathological features have evolved since its initial description in the 1960s23,24 as a result of the advancements in neonatal management. Although the precise mechanisms are still being elucidated, the primary risk factors include prematurity and exposure to insults such as hyperoxia, volutrauma, infection, and IUGR.
Intrauterine growth restriction can result from any condition where the nutritional needs of the growing fetus are not being met by the supply from the mother (eg, nutrient restriction, uteroplacental insufficiency from any cause). Our study used a well-established model of MFR-induced IUGR in rats.18,20 To elucidate the role of increased corticosterone levels in association with IUGR in the developing offspring lung, we examined how MTP, a glucocorticoid synthesis inhibitor, affects the lung phenotype in IUGR offspring. We chose to study offspring lungs at p21, the stage by which alveolarization has by and large been completed.
We hypothesized that prolonged elevated circulating glucocorticoids would be the principal factor causing abnormal lung development in the IUGR offspring. To test this hypothesis, one can either inhibit glucocorticoid synthesis, or its local bioactivation, or its binding site, or its downstream targets. Metyrapone inhibits the final enzyme in the pathway for corticosterone synthesis25 and thus was a logical option to blunt the effects of elevated glucocorticoid levels seen in MFR-induced IUGR pups.
Consistent with our prior data,18 MFR caused IUGR (Figure 1) and inhibited lung development, with a decreased number of alveoli at p21 in both males and females (Figure 2). As hypothesized, MTP therapy blunted the MFR-induced IUGR effect based on the birth weights (Figure 1) and restored the normal lung cytoarchitecture in both males and females (Figure 2). In addition to the direct receptor-mediated cellular effects of elevated glucocorticoids on the developing lung, these effects, at least in part, can also be explained by protein restriction since exposure to prolonged elevated glucocorticoids is known to increase protein catabolism and decrease protein synthesis.26 This is supported by our data, since both MTP and protein supplementation blocked nutrient restriction-induced changes in the lung and fibroblast phenotypes, respectively. We also analyzed the expression of proteins that reflect adipogenic (homeostatic) versus myogenic (dyshomeostatic) lung development.27–30 Consistent with our previous studies, MFR decreased the amount of the adipogenic protein PPARγ18; however, MTP therapy blocked this effect (Figure 3A). In contrast, MFR increased the amount of various myogenic proteins—fibronectin (Figure 3B), α-SMA (Figures 3C and 4A), and calponin (Figures 3D and 4B). Metyrapone therapy also blocked these MFR-induced changes. Because the myogenic phenotype is determined by stimulation of the Wnt signaling pathway, we also examined the expression of key Wnt signaling pathway intermediates in the MFR offspring, and how MTP supplementation affects this expression. Maternal food restriction increased both β-catenin and LEF-1,28 2 important intermediates in the Wnt signaling pathway, both of which were normalized by MTP administration (Figures 3F and 4C and D).
We also found that both MFR-offspring lungs (Figure 3E) and nutrient-restricted cultured LIFs (Figure 7A) demonstrated elevated GR protein levels. Metyrapone administration to the pups (Figure 3E) or protein (BSA) supplementation of the fetal lung fibroblast (Figure 7C) blocked these changes. This is in line with our previous studies where we demonstrated increased circulating corticosterone levels in MFR dams, in association with increased GR, HSD1, and HSD2 levels in the lungs of the adult offspring of MFR pregnancies.31 In contrast to these in vivo data, in response to nutrient restriction to cultured fibroblasts, we observed decreased HSD1 levels despite increased GR levels. This is likely due to differences in these 2 models despite nutritional restriction being a common denominator. For example, the in vivo response is likely to be driven by increased circulating corticosterone levels, as well as by paracrine influences. In line with the findings of other systems, both chronically elevated glucocorticoids32–34 and elevated GR35,36 are likely to stimulate the Wnt signaling pathway, as also evidenced by increased expression of LEF-1 (Figure 6E and F) and β-catenin (Figure 6D and F), and its translocation to the nucleus (Figure 6F), increasing the expression of myogenic proteins such as fibronectin (Figure 6A and B) and calponin (Figure 6A and C). Nutrient-restriction stress also led to lipid depletion from LIFs (BODIPY staining, Figure 6F), associated with activated Wnt signaling, which is known to result in increased myogenic differentiation37,38 under both in vivo and in vitro conditions.
Since MTP corrected both nutrition restriction and corticosterone-induced changes in fibroblast differentiation, it is logical that in addition to inhibition of systemic corticosterone synthesis, MTP’s effects might be mediated via its local cellular action. Prior studies have shown that MTP inhibits the local synthesis of glucocorticoids by depleting NADPH in the endoplasmic reticulum, affecting adipocyte differentiation.39 Likewise, we observed that CORT-treated fetal rat lung fibroblasts demonstrated elevated NADPH levels, while MTP blocked this CORT-induced effect (Figure 9A and B). Therefore, it is logical that MTP inhibits corticosterone-induced activation of Wnt signaling and fibroblast differentiation to a myogenic phenotype (Figure 8A-C). However, since both maternal nutrient restriction and MTP administration during pregnancy can affect fetal androgens and estrogen levels,40,41 the precise mediators of increased Wnt signaling in alveolar interstitial fibroblasts observed by us can only be speculated. Additionally, although maternal Adrenocorticotropic hormone at physiological concentrations does not cross the placenta,42 at pathologically higher levels, it has been shown to cross placenta and affect fetal hypothalamic pituitary adrenal axis and fetal androgens and estrogen levels.43 Unfortunately we didn’t measure these hormones; and therefore, the contributions of alterations in these hormones in our model can’t be ascertained. Another important limitation of our study is the noninclusion of an ad libitum fed and MTP-treated group, which would have provided information on MTP’s effect on the developing lung in the absence of hyperglucocorticoidism. It is important to point out that we initially did conduct a pilot study with MTP supplementation without MFR, but didn’t see any significant differences in lung protein levels of a limited number of markers examined (fibronectin and α-SMA) in comparison to the control (fed ad libitum, untreated) group; therefore, we didn’t formally include this group in our study. Of note, parenteral administration MTP at 45 mg/kg/dose twice daily during the last 3 days of rat gestation has been shown delay fetal lung maturation and decrease postnatal survival.25 Finally, as with any model, the in vitro model cannot and does not account for all the variables (complex interactions between different organ systems) that exist in the in vivo model. Our in vitro models of pulmonary LIFs were utilized to individually assess the effects of nutrient (FBS) restriction and the resultant stress response (ie, elevated corticosteroid levels). We used BSA to counteract the effects of FBS restriction in these lung fibroblasts because our prior data had shown the effective blockage of FBS restriction-induced increased extracellular matrix elastin levels with BSA supplementation.19
Regarding the effect of MTP on increased triolein uptake (Figure 5), hypocorticoidism may have caused a net increase in 11β-HSD1 activity since it has been shown that corticosteroids inhibit 11β-HSD2 activity44 causing net increase in reductase within the lung, increasing local production of corticosterone and increased triolein uptake. Alternatively, it has been shown that during the pseudoglandular period of lung development, cortisol stimulates lung fibroblast growth, potentially increasing net 11β-HSD1 activity due to the presence of more lung fibroblasts.45 It is also important to point out that though both exogenous46 and endogenous47 glucocorticoids are known to affect the gender difference in lung development, this effect is only known to occur antenatally.48 Therefore, although our study was not designed to compare gender differences, it is not surprising that at p21 we didn’t observe any gender differences in lung phenotype either as a result of MFR or in response to MTP.
In summary, both in vivo and in vitro studies show that nutrient restriction to the developing lung leads to elevated GR levels, upregulating the Wnt signaling pathway, resulting in the increased expression of myogenic proteins, and decreased expression of the adipogenic nuclear transcription factor PPARγ, consistent with LIF-to-myofibroblast transdifferentiation, an effect blocked by MTP (Figure 10A and B). Our data suggest that the development of chronic lung disease in the IUGR offspring of MFR pregnancies is likely to result from at least 2 mechanisms—first, protein/nutrient restriction stress not only limits the supply of essential materials for lung development but also activates Wnt signaling and second, increased circulating glucocorticoid levels impair the process of lung development, also by activating Wnt signaling. The ability of MTP to block the myogenic pulmonary phenotype secondary to nutrient restriction highlights the importance of chronically elevated systemic corticosteroid and local NADPH levels in the development of chronic lung disease in the IUGR offspring and offers novel treatment targets.
Figure 10.
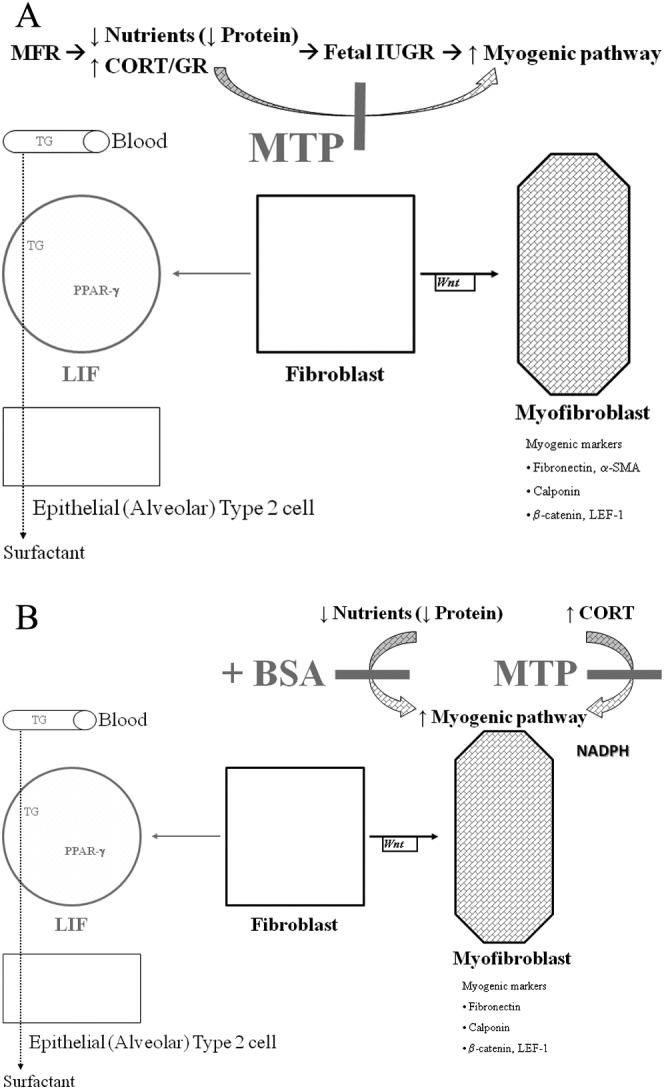
In vivo (A) and in vitro models to show the effect of nutrient restriction on fibroblast differentiation and how metyrapone (MTP) might affect this process. Adepithelial alveolar fibroblasts can either differentiate into the lipofibroblasts (LIF) by the adipogenic pathway or myofibroblast by the myogenic pathway, under both in vivo and in vitro conditions. Triglyceride (TG) uptake by LIFs and transfer to alveolar type II cells are the hallmarks of alveolar lipofibroblasts, an essential step for surfactant synthesis and alveolar homeostasis. Chronically elevated CORT levels (as seen in intrauterine growth restriction [IUGR] fetuses) shift the balance toward the myofibroblast phenotype. Protein supplementation and MTP normalize the CORT levels and allow the differentiation of fibroblasts to a lipofibroblastic phenotype, normalizing the MFR-induced lung phenotype. CORT indicates corticosterone.
Footnotes
Authors’ Note: This study was presented in part at the Pediatric Academic Societies Meeting, Boston, MA, USA, May 2012.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: NIH Grants HL75405, HD51857, HL107118, HD071731, HD058948, and HL55268.
References
- 1. Aucott SW, Donohue PK, Northington FJ. Increased morbidity in severe early intrauterine growth restriction. J Perinatol. 2004;24 (7):435–440. [DOI] [PubMed] [Google Scholar]
- 2. Jobe AH, Bancalari E. Bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2001;163 (7):1723–1729. [DOI] [PubMed] [Google Scholar]
- 3. Carpenter TC, Stenmark KR. Predisposition of infants with chronic lung disease to respiratory syncytial virus-induced respiratory failure: a vascular hypothesis. Pediatr Infect Dis J. 2004;23 (1 suppl):S33–S40. [DOI] [PubMed] [Google Scholar]
- 4. Northway WH, Jr, Moss RB, Carlisle KB, et al. Late pulmonary sequelae of bronchopulmonary dysplasia. N Engl J Med. 1990;323 (26):1793–1799. [DOI] [PubMed] [Google Scholar]
- 5. Narang I. Review series: what goes around, comes around: childhood influences on later lung health? Long-term follow-up of infants with lung disease of prematurity. Chron Respir Dis. 2010;7 (4):259–269. [DOI] [PubMed] [Google Scholar]
- 6. Van Marter LJ, Allred EN, Leviton A, Pagano M, Parad R, Moore M. Antenatal glucocorticoid treatment does not reduce chronic lung disease among surviving preterm infants. J Pediatr. 2001;138 (2):198–204. [DOI] [PubMed] [Google Scholar]
- 7. Bancalari E, del Moral T. Bronchopulmonary dysplasia and surfactant. Biol Neonate. 2001;80 (suppl 1):7–13. [DOI] [PubMed] [Google Scholar]
- 8. Schmidt B, Roberts RS, Davis P, et al. Caffeine therapy for apnea of prematurity. N Engl J Med. 2006;354 (20):2112–2121. [DOI] [PubMed] [Google Scholar]
- 9. Schmidt B, Roberts RS, Davis P, et al. Long-term effects of caffeine therapy for apnea of prematurity. N Engl J Med. 2007;357 (19):1893–1902. [DOI] [PubMed] [Google Scholar]
- 10. Spitzer AR. Evidence-based methylxanthine use in the NICU. Clin Perinatol. 2012;39 (1):137–148. [DOI] [PubMed] [Google Scholar]
- 11. Schulzke SM, Pillow JJ. The management of evolving bronchopulmonary dysplasia. Paediatr Respir Rev. 2010;11 (3):143–148. [DOI] [PubMed] [Google Scholar]
- 12. Birenbaum HJ, Dentry A, Cirelli J, et al. Reduction in the incidence of chronic lung disease in very low birth weight infants: results of a quality improvement process in a tertiary level neonatal intensive care unit. Pediatrics. 2009;123 (1):44–50. [DOI] [PubMed] [Google Scholar]
- 13. Trevisanuto D, Gizzi C, Martano C, et al. Oxygen administration for the resuscitation of term and preterm infants. J Matern Fetal Neonatal Med. 2012;25 (suppl 3):26–31. [DOI] [PubMed] [Google Scholar]
- 14. Barker DJ. Fetal origins of coronary heart disease. BMJ. 1995;311 (6998):171–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cole TJ, Blendy JA, Monaghan AP, et al. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. 1995;9 (13):1608–1621. [DOI] [PubMed] [Google Scholar]
- 16. Bunton TE, Plopper CG. Triamcinolone-induced structural alterations in the development of the lung of the fetal rhesus macaque. Am J Obstet Gynecol. 1984;148 (2):203–215. [DOI] [PubMed] [Google Scholar]
- 17. Massaro GD, Massaro D. Formation of pulmonary alveoli and gas-exchange surface area: quantitation and regulation. Annu Rev Physiol. 1996;58:73–92. [DOI] [PubMed] [Google Scholar]
- 18. Karadag A, Sakurai R, Wang Y, et al. Effect of maternal food restriction on fetal rat lung lipid differentiation program. Pediatr Pulmonol. 2009;44 (7):635–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rehan VK, Sakurai R, Li Y, et al. Effects of maternal food restriction on offspring lung extracellular matrix deposition and long term pulmonary function in an experimental rat model. Pediatr Pulmonol. 2012;47 (2):162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Desai M, Gayle D, Babu J, Ross MG. Programmed obesity in intrauterine growth-restricted newborns: modulation by newborn nutrition. Am J Physiol Regul Integr Comp Physiol. 2005;288 (1):R91–R96. [DOI] [PubMed] [Google Scholar]
- 21. Rehan VK, Wang Y, Sugano S, et al. In utero nicotine exposure alters fetal rat lung alveolar type II cell proliferation, differentiation, and metabolism. Am J Physiol Lung Cell Mol Physiol. 2007;292(1):L323–L333. [DOI] [PubMed] [Google Scholar]
- 22. Rehan VK, Sugano S, Wang Y, et al. Evidence for the presence of lipofibroblasts in human lung. Exp Lung Res. 2006;32 (8):379–393. [DOI] [PubMed] [Google Scholar]
- 23. Stahlman MT, Young WC, Gray J, Shepard FM. The management of respiratory failure in the idiopathic respiratory distress syndrome of prematurity. Ann N Y Acad Sci. 1965;121:930–941. [DOI] [PubMed] [Google Scholar]
- 24. Northway WH, Jr, Rosan RC, Porter DY. Pulmonary disease following respirator therapy of hyaline-membrane disease. Bronchopulmonary dysplasia. N Engl J Med. 1967;276 (7):357–368. [DOI] [PubMed] [Google Scholar]
- 25. Sosenko IR, Lewis PL, Frank L. Metyrapone delays surfactant and antioxidant enzyme maturation in developing rat lung. Pediatr Res. 1986;20(7):672–675. [DOI] [PubMed] [Google Scholar]
- 26. Kim YS, Kim Y. Glucocorticoid inhibition of protein synthesis in vivo and in vitro. J Biol Chem. 1975;250 (6):2293–2298. [PubMed] [Google Scholar]
- 27. Rehan VK, Torday JS. PPARgamma signaling mediates the evolution, development, homeostasis, and repair of the lung. PPAR Res. 2012;2012:289867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sakurai R, Cerny LM, Torday JS, Rehan VK. Mechanism for nicotine-induced up-regulation of Wnt signaling in human alveolar interstitial fibroblasts. Exp Lung Res. 2011;37 (3):144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Torday JS, Rehan VK. Developmental cell/molecular biologic approach to the etiology and treatment of bronchopulmonary dysplasia. Pediatr Res. 2007;62 (1):2–7. [DOI] [PubMed] [Google Scholar]
- 30. Torday JS, Rehan VK. The evolutionary continuum from lung development to homeostasis and repair. Am J Physiol Lung Cell Mol Physiol. 2007;292 (3):L608–L611. [DOI] [PubMed] [Google Scholar]
- 31. Rehan VK, Li Y, Corral J, et al. Metyrapone blocks maternal food restriction-induced changes in female rat offspring lung development. Reprod Sci. 2014;21 (4):517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang Q, Zhao DY. Effects of dexamethasone on lung morphogenesis in rats and the expression of Wnt signal transduction pathway in the lung of offspring [in Chinese]. Zhonghua Er Ke Za Zhi. 2008;46 (4):286–290. [PubMed] [Google Scholar]
- 33. Guan Y, Rubenstein NM, Failor KL, Woo PL, Firestone GL. Glucocorticoids control beta-catenin protein expression and localization through distinct pathways that can be uncoupled by disruption of signaling events required for tight junction formation in rat mammary epithelial tumor cells. Mol Endocrinol. 2004;18 (1):214–227. [DOI] [PubMed] [Google Scholar]
- 34. Miyata S, Koyama Y, Takemoto K, et al. Plasma corticosterone activates SGK1 and induces morphological changes in oligodendrocytes in corpus callosum. PLoS One. 2011;6 (5):e19859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fonte C, Grenier J, Trousson A, et al. Involvement of {beta}-catenin and unusual behavior of CBP and p300 in glucocorticosteroid signaling in Schwann cells. Proc Natl Acad Sci USA. 2005;102 (40):14260–14265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhou H, Mak W, Kalak R, et al. Glucocorticoid-dependent Wnt signaling by mature osteoblasts is a key regulator of cranial skeletal development in mice. Development. 2009;136 (3):427–436. [DOI] [PubMed] [Google Scholar]
- 37. Portilho DM, Martins ER, Costa ML, Mermelstein CS. A soluble and active form of Wnt-3a protein is involved in myogenic differentiation after cholesterol depletion. FEBS Lett. 2007;581 (30):5787–5795. [DOI] [PubMed] [Google Scholar]
- 38. Mermelstein CS, Portilho DM, Mendes FA, Costa ML, Abreu JG. Wnt/beta-catenin pathway activation and myogenic differentiation are induced by cholesterol depletion. Differentiation. 2007;75 (3):184–192. [DOI] [PubMed] [Google Scholar]
- 39. Marcolongo P, Senesi S, Gava B, et al. Metyrapone prevents cortisone-induced preadipocyte differentiation by depleting luminal NADPH of the endoplasmic reticulum. Biochem Pharmacol. 2008;76 (3):382–390. [DOI] [PubMed] [Google Scholar]
- 40. Oakey RE, Heys RF. Regulation of the production of oestrogen precursors in the foetus. Acta Endocrinol (Copenh). 1970;65 (3):502–508. [DOI] [PubMed] [Google Scholar]
- 41. Maeyama M, Nakagawa T, Tuchida Y, Matuoka H. The role of human fetal adrenals in steroidogenesis: effect of adrenocorticosterone on urinary excretion of estrogens in pregnancy. Steroids. 1969;13 (1):59–67. [DOI] [PubMed] [Google Scholar]
- 42. Dupouy JP, Chatelain A, Allaume P. Absence of transplacental passage of ACTH in the rat: direct experimental proof. Biol Neonate. 1980;37 (1-2):96–102. [DOI] [PubMed] [Google Scholar]
- 43. Dassler CG. Influence of corticotropin on estrogen excretion in pregnancy and in intrauterine fetal death [in German]. Acta Endocrinol (Copenh). 1966;53 (3):401–406. [PubMed] [Google Scholar]
- 44. Zallocchi ML, Matkovic L, Calvo JC, Damasco MC. Adrenal gland involvement in the regulation of renal 11beta-hydroxysteroid dehydrogenase 2. J Cell Biochem. 2004;92 (3):591–602. [DOI] [PubMed] [Google Scholar]
- 45. Smith BT, Torday JS, Giroud CJ. Evidence for different gestation-dependent effects of cortisol on cultured fetal lung cells. J Clin Invest. 1974;53 (6):1518–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Effect of antenatal dexamethasone administration on the prevention of respiratory distress syndrome. Am J Obstet Gynecol. 1981;141 (3):276–287. [PubMed] [Google Scholar]
- 47. Nielsen HC, Torday JS. Sex differences in avian embryo pulmonary surfactant production: evidence for sex chromosome involvement. Endocrinology. 1985;117 (1):31–37. [DOI] [PubMed] [Google Scholar]
- 48. Torday J. Cellular timing of fetal lung development. Semin Perinatol. 1992;16 (2):130–139. [PubMed] [Google Scholar]