

Abstract
This study aimed to gain new insights into both systemic and placental leptin and its receptors, with reference to the maternal prepregnancy body mass index (BMI). Thus, 84 women (29 lean, 24 overweight, and 31 obese) were recruited and maternal, cord blood, and placental tissues collected prior to term labor. Plasma levels were measured by enzyme-linked immunosorbent assay and for placenta, immunohistochemistry and messenger RNAs (mRNAs) were quantitated. We confirmed that maternal leptin increased linearly as the soluble receptor decreased with BMI (P = .001). Fetal leptin increased with maternal BMI (P = .02) and birth weight (P = .006) and was higher in female infants (P < .001). Placental mRNA levels of leptin and its receptors showed no change in BMI. However, we show a significant (P = .043) linear increase in leptin in the placental vascular endothelial cells with maternal obesity, while leptin in syncytiotrophoblast showed no statistical change. Leptin receptors localized to syncytiotrophoblast and intravillous macrophages and were unchanged with BMI.
Keywords: leptin, leptin receptor, pregnancy, obesity
Introduction
Obesity affects more than 30% of reproductive age women and continues to be a major public health problem in the United States.1 The rate of morbid obesity in reproductive age women exceeds 7%, which is 50% higher compared to men in the same age group.1 Obesity in pregnancy has been associated with increased pregnancy complications. Recent studies have shown that when compared to normal weight women, obese women are at increased risk of having stillbirths,2 cesarean section, infections, preeclampsia, and gestational diabetes.3,4 Their neonates are at increased risk of having congenital malformations,5–7 being large for gestational age, and having low Apgar scores at birth.3,4 In addition, the mother’s obesity has been linked to childhood obesity and metabolic syndrome.8,9
Several possible mechanisms for the association between obesity and adverse pregnancy outcomes have been proposed. One explanation is that obesity predisposes women to a proinflammatory environment from excess adipose tissue, which causes these adverse outcomes. Adipocytes have been demonstrated to synthesize and secrete numerous cytokines, such as leptin, visfatin, interleukin 6, and tumor necrosis factor α.10 More recently in pregnancy, the placenta has also been shown to be a major source of several adipocytokines, and their increased levels have been linked to gestational diabetes, hypertension, and intrauterine growth restriction.11
Leptin, the protein product of the ob gene, is a 16-kDa hormone widely secreted by white adipose tissue and important in maintaining energy homeostasis in the body. It functions as a satiety signal in the central nervous system and has peripheral effects that are widespread, including angiogenesis, hematopoiesis, and regulation of bone formation.12,13 The human leptin receptor gene, ob-R, gives rise to different isoforms of the leptin receptor, generated by alternative messenger RNA (mRNA) splicing.14 One of these is a soluble form, which binds to circulating leptin and enhances signaling by reducing leptin clearance and degradation.15 There are also 4 transmembrane receptor isoforms, 1 long form and 3 short forms. The long-form receptor signals through the Janus kinase pathway, while the short forms signal through the mitogen-activated protein kinase pathway.16,17 Mutations in the leptin and/or leptin receptor genes have been shown to cause severe early-onset obesity, hyperphagia, and endocrine abnormalities.18,19
Previous studies have demonstrated higher circulating levels of leptin and lower levels of soluble leptin receptor in obese individuals compared to normal weight individuals.20–22 This was interpreted as leptin “resistance,” where a decrease in signaling capability of leptin at the level of the satiety center caused an increase in secretion of leptin.23 An increased level of circulating leptin was also shown in obese pregnant women compared to normal weight pregnant women, with the overall levels higher in pregnancy compared to the nonpregnant state.24
In addition to its systemic role in obesity, the placenta is a major site of leptin synthesis in the human,25,26 where it functions as an autocrine/paracrine hormone. The human placental leptin molecule is identical to that produced by adipose tissue.25 Its functions in the placenta and in pregnancy are still under debate, but it is thought to play an important role in fetal growth and development.27,28 However, there are limited data on placental leptin in pregnancies affected by obesity. Farley and colleagues showed no difference in placental leptin levels in obese women compared to lean women; however, the expression of the long form of the leptin receptor in the syncytiotrophoblast was significantly lower in the obese women.29 By analogy to the description of leptin resistance at the satiety center of the brain in nonpregnant obese women, this decrease was suggested to show placental leptin resistance and the reason for decreased sodium-dependent neutral amino acid transporter activity.29
In any event, it is agreed that the levels of leptin reaching the fetus are likely to be important as a regulator of fetal weight and subsequent adiposity in childhood.30 Indeed, higher umbilical cord blood levels of leptin correlate with placental weight and increased birth weight.31–33 However, the placental leptin from the syncytiotrophoblast contributes little to the fetus via the cord blood since most is secreted into the maternal circulation.34,35 This is confirmed by the lack of correlation between leptin concentrations in maternal serum and cord blood.36 Therefore, the correlation between higher cord leptin levels and increased birth weight cannot be explained by placental (syncytiotrophoblast) leptin production. As we seek to understand this complexity of how maternal obesity affects pregnancy and its outcomes, it appears to be important to examine concomitantly the systemic and placental autocrine/paracrine systems, using samples from a single set of patients. Thus, the aims of this study were to confirm some key published data and to gain new insights into both the systemic and intrauterine leptin systems with reference to the maternal body mass index (BMI) in such a single set of patients.
Materials and Methods
Patient Selection and Tissue Collection
Approval was obtained from the Western Institutional Review Board, and pregnant women at term (>37 weeks gestation) who were undergoing an elective prelabor cesarean section at Kapiolani Medical Center for Women and Children (Honolulu, Hawaii) were recruited between August and December 2012. Exclusion criteria included labor, premature rupture of membranes, multiple gestation, pregestational diabetes, acute infection at the time of delivery, maternal inflammatory/connective tissue/autoimmune disease, and maternal use of steroids. After meeting inclusion and exclusion criteria, a detailed consent form was signed by the patient prior to the performance of any study procedure or data collection.
The recruited patients were divided according to their prepregnancy BMI: lean (BMI 16.0-24.9), overweight (BMI 25.0-29.9), and obese (BMI ≥ 30). Information regarding the demographics of the mother (age, pregravid weight, height, substance use history, gravity, parity, gestational age at delivery, temperature at admission, medical history, and medication use), pregnancy complications (gestational diabetes, preeclampsia, intrauterine growth restriction, shoulder dystocia, and macrosomia), and neonatal information (gender, birth weight, Apgar scores, presence of respiratory distress syndrome, admission to the neonatal intensive care unit, hypoglycemia, hyperbilirubinemia, and neonatal malformation) were collected.
Fasting samples of maternal blood were collected into EDTA tubes prior to delivery, centrifuged and the supernatant plasma immediately stored at −80°C. Venous cord blood samples were similarly collected within 15 minutes of delivery and immediately centrifuged. All plasma samples were stored at −80°C until assayed. Two full-thickness samples of the placenta, excluding basal plate, were taken at the site of cord insertion within 15 minutes of delivery. One sample was fixed in neutral-buffered formaldehyde for 72 hours and stored in 70% ethanol until embedded in paraffin, the other sample was immediately stored at −80°C until used.
Maternal and Cord Plasma Analyses
Maternal and cord plasma leptin and the soluble leptin receptor were assayed using the Quantikine enzyme-linked immunosorbent assay kits (R&D Systems, Minneapolis, Minnesota) with intra-assay coefficient of variations of 3% and 4.9%, respectively. All samples were run in duplicate in a single assay.
Immunohistochemistry and Quantitation
Paraffin-embedded tissue blocks were serially sectioned (5 μm) onto slides, deparaffinized, hydrated in a graded series of xylenes and ethanols, and rinsed in distilled water. The slides were heated in sodium citrate buffer (10 mmol/L, pH 6.0) for 30 minutes for antigen retrieval. They were then treated for 20 minutes with 0.3% hydrogen peroxide to remove endogenous peroxidase activity. Nonspecific binding was blocked with 2.5% normal serum for 20 minutes, and the slides incubated for 60 minutes with predetermined dilutions of one of the following antibodies: polyclonal rabbit immunoglobulin G (IgG) for leptin (Santa Cruz Biotechnology, Santa Cruz, California; catalog number: sc-842, 0.25 μg/mL), polyclonal rabbit IgG that detects only the long form of the leptin receptor (Santa Cruz Biotechnology; catalog number: sc-1832, 0.5 μg/mL), or mouse monoclonal antibody that detects the cytoplasmic consensus region of all leptin receptor isoforms (Santa Cruz Biotechnology; catalog number: sc-8391, 4 μg/mL). Negative controls for each antibody were the nonimmune IgG (Vectastain ABC kit; Vector Labs, Burlingame, California) from the respective species at the same concentrations as the primary antibodies. After rinsing, the slides were incubated with biotinylated secondary antibody followed by the avidin-biotin complex (ABC) reagent (Vector Labs). The 3,3-diaminobenzidine substrate solution was used to visualize the antigens. Slides were rinsed and counterstained with Gill’s hematoxylin, then cleared and mounted in Pro-Texx mounting media (Lerner Laboratories, Pittsburgh, Pennsylvania).
A multispectral imaging system comprised of an Olympus BX51 microscope (Olympus America Inc, Mellville, New York) and a CRI Nuance spectral analyzer (Caliper Life Sciences, Hopkinton, Massachusetts) was used to obtain bright-field image cubes between 420 and 700 nm wavelength at 20-nm intervals. Inform software (version 1.4.0; PerkinElmer, Waltham, Massachusetts) was used to quantitate the unmixed image data. The average signal intensity per pixel was obtained from 5 different fields of villous tissue from each patient. The signal intensity for leptin in both syncytiotrophoblast and villous vascular endothelial cells was quantified at ×400. The signal intensity for leptin receptors in the syncytiotrophoblast was measured at ×400. The villous stroma cells staining for leptin receptor was quantified by object counting at ×100.
Isolation of RNA and Quantitative Real-Time Polymerase Chain Reaction
Placental tissues (2 mg) were pulverized with a BioPulverizer (BioSpec Products Inc, Bartletville, Oklahoma) and total RNA extraction performed using the RNeasy Fibrous MiniKit (QIAGEN Inc, Valencia, California) according to the product protocol. RNA quality was assessed by Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, California). Complementary DNA (cDNA) was prepared by reverse transcription in a total volume of 20 μL containing polymerase chain reaction (PCR) buffer II, 5.5 mmol/L magnesium chloride, 500 μmol/L each deoxynucleotide triphosphatase, 2.5 μmol/L random hexamers, 0.4 U/μL RNase inhibitor, 2.5 U/μL Multiscribe reverse transcriptase (Applied Biosystems Inc, Foster City, California), and 400 ng total RNA. This solution was incubated for 10 minutes at 25°C, 30 minutes at 48°C, and 5 minutes at 95°C. The cDNA was then amplified by PCR using specific primers for leptin, leptin receptor, and the housekeeping gene18S (Applied Biosystems, TaqMan Assays on Demand). StepOne Real-Time PCR System (Applied Biosystems) was used for real-time PCR according to the Applied Biosystems Inc. (ABI) protocol, where it was set at 1 cycle at 50°C for 2 minutes, 1 cycle at 95°C for 10 minutes, followed by 40 cycles at 95°C for 15 seconds, and 60°C for 1 minute. Each reaction was performed in triplicate, and the data were analyzed as described in the ABI User’s Bulletin no. 2 (http://www3.appliedbiosystems.com/cms/groups/mcb_support/documents/generaldocuments/cms_040980.pdf). The results were normalized to the expression of 18S in each sample and reported as mean ± standard deviation (SD) of relative gene expression.
Statistical Analysis
Data were analyzed using SAS statistical software (version 9.3; SAS Institute Inc, Cary, North Carolina). Continuous data were summarized as means ± SDs. The relationship of BMI with the intensity of immunostaining of placental leptin and leptin receptors was estimated using linear regression models. Because the levels of normalized RNA/18S for leptin and leptin receptor were not normally distributed, the Wilcoxon test was used to compare the median levels of lean and obese women. Statistical significant was set at P ≤ .05.
Results
Demographic Data
Patient demographics for 84 patients by BMI groups are shown in Table 1. There were no statistical differences in the mean age, average gestational age at delivery, gestational weight gain, and birth weight in lean, overweight, and obese women. The average gestational age at delivery was 39 weeks, and the average birth weight ranged from 3295 to 3590 g. The mean placental weight was statistically different by BMI (P = .002). Obese women had a higher mean placental weight (562 g) compared to lean (472 g) and overweight women (523 g).
Table 1.
Patient Demographics.
| Lean (N = 29) | Overweight (N = 24) | Obese (N = 31) | P Value | |
|---|---|---|---|---|
| Mean age, y | 33.40 ± 5.64 | 30.75 ± 5.33 | 30.74 ± 4.77 | .098 |
| BMI, kg/m2 | 21.14 ± 2.16 | 27.65 ± 1.31 | 37.28 ± 6.96 | <.001 |
| Mean gestational age at delivery, weeks | 38.99 ± 0.64 | 39.04 ± 0.53 | 39.23 ± 0.44 | .196 |
| Mean gestational weight gain, kg | 13.97 ± 5.32 | 15.45 ± 6.60 | 13.46 ± 6.77 | .493 |
| Mean birth weight, g | 3294.90 ± 408.80 | 3459.54 ± 538.30 | 3590.61 ± 448.05 | .052 |
| Mean placental weight, g | 472.00 ± 87.96 | 523.00 ± 98.77 | 562.23 ± 97.92 | .002 |
Maternal and Cord Plasma Leptin and Soluble Leptin Receptor
Maternal and cord plasma levels of leptin and its soluble receptor in relation to maternal BMI are shown in Figure 1. Maternal plasma leptin levels increased linearly as maternal BMI increased (P < .001). However, the levels of maternal plasma soluble receptor decreased as maternal BMI increased (P < .001; Figure 1A). Cord plasma leptin significantly increased with increasing maternal BMI (P = .02; Figure 1B) and birth weight (P = .006; Figure 1C). Cord plasma leptin was also significantly higher in female infants compared to male infants (P < .001; Figure 1D). However, there was no correlation between cord plasma leptin levels and those of maternal plasma (data not shown).
Figure 1.

Maternal and cord plasma leptin and soluble leptin receptor. A, Maternal plasma leptin levels increased linearly as maternal body mass index (BMI) increased (P < .001; open circle, solid line), plasma soluble receptor levels decreased as maternal BMI increased (P < .001; closed triangle, dash line). B, Cord plasma leptin significantly increased with increasing maternal BMI (P = .02) and (C) birth weight (P = .006). D, Cord plasma leptin was significantly higher in female infants compared to male infants (P < .001).
Placental Leptin: Protein and Gene Expression
Placental leptin was immunolocalized to the syncytiotrophoblast25,26 and the villous vascular endothelial cells as previously reported.27,37 Images of placental villous tissue immunostained with leptin in examples of a lean (Figure 2Aa) and an obese woman (Figure 2Ab) are shown together with the negative control (Figure 2Ac). When quantitatively compared, there was a trend for increased levels of leptin in the syncytiotrophoblast as maternal BMI increased, but this was not statistically significant (P = .156; Figure 2B). However, leptin levels in the villous vascular endothelial cells were significantly increased (P = .046) as a function of maternal BMI and this relationship was linear as shown in Figure 2C. There was no significant correlation either in syncytiotrophoblast or in vascular endothelial leptin expression by infant gender. In addition, there were no correlations between leptin levels and birth weight or placental weight in either the syncytiotrophoblast or villous vascular endothelial cells. The expression of leptin mRNA in the 2 extreme groups (lean and obese women) in the placental villous trophoblast showed no significant differences (Figure 2D).
Figure 2.

Immunostaining of leptin in placental tissue. A, Examples of placental sections immunostained for leptin from a lean (a) and obese woman (b), and negative control (c). Leptin immunostaining was localized to the syncytiotrophoblast (SN) and the villous vascular endothelial cells (EC). B, The quantitation of immunostaining in the syncytiotrophoblast showed a nonsignificant trend for increasing leptin levels with increasing maternal body mass index (BMI; P = .156). C, The quantitation in the villous vascular endothelial cells was significantly and linearly increased with increasing maternal BMI (P = .046). D, The expression of leptin messenger RNA (mRNA) in the 2 extreme groups (lean and obese women) in the placental villous trophoblast showed no significant differences (P = .5). Original magnification 400× for all sections.
Placental Leptin Receptors: Protein and Gene Expression
We first immunolocalized the long form of the leptin receptor in the placenta using the same antibody as previously used by Farley et al.29 This confirmed localization to the syncytiotrophoblast and cells in the villous stroma, previously identified as Hofbauer cells.38 Images of the placental villous tissue immunostained for the long-form leptin receptor in examples of a lean (Figure 3Aa) and an obese (Figure 3Ab) woman are shown together with the negative control (Figure 3Ac). Quantitation of this staining in the syncytiotrophoblast (Figure 3B) and the cells immunostained in the villous stroma (Figure 3C) showed no relationships to BMI. In addition, its levels failed to show any differences in relation to birth weight or infant gender (data not shown). The expression of mRNA in the placenta for the leptin receptor in the 2 extreme groups (lean and obese women) showed no significant difference (Figure 3D).
Figure 3.

Immunostaining of the long-form transmembrane leptin receptor in placental tissue. A, Examples of placental sections of a lean (a) and obese woman (b), and negative control (c). The long-form leptin receptor was localized to the syncytiotrophoblast (SN) and cells in the villous stroma (SC), previously identified as Hofbauer cells. B, Quantitation showed no statistical difference in the intensity of immunostaining in the syncytiotrophoblast (P = .5) and (C) villous stromal cells (0.69) by maternal body mass index (BMI). D, The expression of messenger RNA (mRNA) for the leptin receptor in the 2 extreme groups (lean and obese women) in the placental villous trophoblast showed no significant differences (P = .139). Original magnification 400× for all sections.
These results were markedly different from those reported by Farley et al,29 although the same antibody was utilized here. Therefore, we reimmunolocalized the leptin receptors using sections from the same patients with another antibody (sc-8391) that detects the long form and all the short isoforms of the leptin receptor. This showed similar staining in the syncytiotrophoblast and villous stroma as the antibody, which detected only the long form of the receptor (sc-1832). When this was quantitated, again there was no relationship between the level of immunostaining and the maternal BMI (data not shown).
Discussion
In this study, we first set out to confirm data previously reported by several different laboratories using a single set of patient samples. This also served to validate our sample set. We then sought to gain some new insights into placental leptin and its receptors and to show the effects of maternal obesity in gestation. Therefore, the results presented here first confirmed that maternal plasma leptin increases with increasing prepregnancy BMI. At the same time, the soluble leptin receptor in maternal plasma decreases. These relationships have been previously reported in both nonpregnant20 and pregnant women.39,40 Increased cord blood leptin levels with increasing maternal BMI and birth weight have also been confirmed. Previous studies have shown that large for gestational age infants had higher levels of cord blood leptin compared to normal weight infants.32,41 In addition, a meta-analysis of 44 studies showed a clear correlation between cord blood leptin levels and birth weight.33 The female infants in our study also had higher cord blood leptin levels than the male infants, which was also consistent with previous studies.31,42
For the first time, this study shows that in the placenta, there was a linear increase in the levels of leptin in the villous vascular endothelial cells as maternal BMI increased. However, there was only a trend, which was not significant, between leptin levels in the syncytiotrophoblast and maternal BMI, confirming previous work.29 We were able to distinguish and quantitate the levels of leptin from the immunostained villous vascular endothelium separately from that of the syncytiotrophoblast in the same tissue sections, by training the Inform Software program (PerkinElmer) to use pattern and density recognition. This was not a simple task, because both are single layers of cells along the edges of placental structures. The result is important because it is recognized that proteins produced by the villous vascular endothelium contribute to the fetal circulation, while the proteins from the syncytiotrophoblast tend to enter the maternal blood as suggested by Lea et al.27 Fetal leptin probably has an important role in fetal/neonatal growth regulation and may even be critical in the future development of metabolic dysfunction and childhood obesity. Higher cord leptin levels are associated with slower infant weight gain and less adiposity at 3 years of age, while higher levels at this age were associated with subsequent faster weight gain between 3 and 7 years of age.30 Another study using a retrospective cohort showed that maternal obesity doubles the risk of childhood obesity between the ages of 2 and 4 years,8 whereas those children exposed in utero to maternal obesity are at a higher risk of metabolic syndrome.9 Together these results suggest that perinatal leptin plays an important role in neonatal growth and the development of leptin resistance later in life. Although the origin of fetal leptin is still under debate, fetal adipose tissue undoubtedly contributes leptin to the fetal circulation.35 Our study suggests an additional significant placental contribution not from the syncytiotrophoblast but from the fetal vascular endothelium. The use of in vitro placental perfusion in 2 studies has suggested that the placental contribution of leptin to the fetal compared to the maternal circulation was only between 2% and 5% and that most placental (syncytotrophoblast) leptin entered the maternal circulation.34,35 Other studies have taken another approach and compared the leptin levels in venous and arterial umbilical cord blood, but the results have been contradictory. Two studies showed significantly higher leptin levels in umbilical venous cord blood compared to arterial cord blood,43,44 supporting the concept of a placental contribution to fetal leptin levels. However, another study showed no difference in the venous and arterial cord blood leptin levels.36 We have shown these possible relationships between maternal, placental, and fetal leptin to the fetal circulation in Figure 4.
Figure 4.
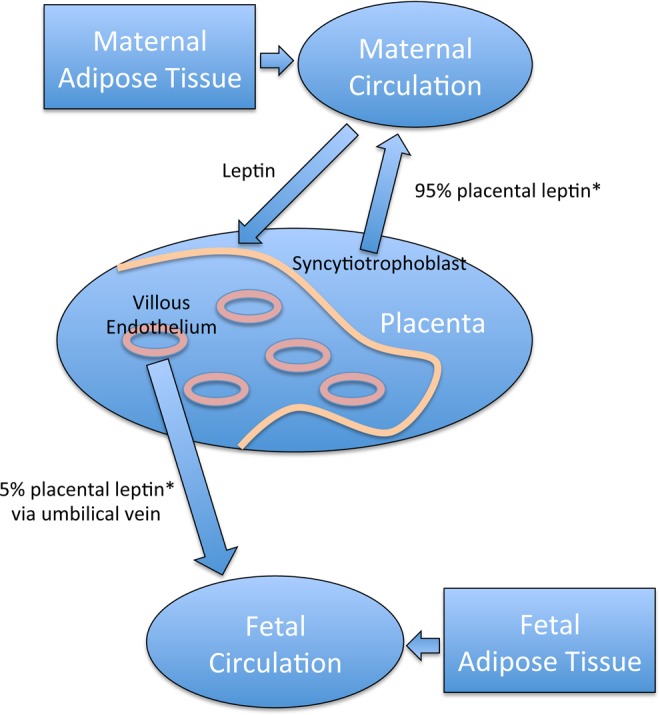
Possible relationships between maternal, placental, and fetal leptin. Placental leptin from the syncytiotrophoblast contributes 95% to the maternal circulation, while leptin from the villous endothelial cells contributes 5% to the fetal circulation. Maternal adipose tissue also secretes leptin that contributes to the maternal circulation and can target the placental trophoblast. Fetal adipose tissue contributes leptin to the fetal circulation. Placental leptin from the villous vascular endothelium shown in our study to be increased in obese women is secreted into the umbilical vein, where leptin levels were also higher. This would add to the fetal leptin levels and may directly affect fetal growth and development. * indicates reference 25.
Previous studies have localized the long form of the leptin receptor to the syncytiotrophoblast, which we confirmed here.27,29,37 However, in our study, we found no difference in the levels of its expression by maternal BMI, whereas decreased expression was reported in both syncytiotrophoblast and villous stroma in obese women compared to lean women.29 It was concluded in this study that similar to the leptin resistance at the satiety center of the brain in nonpregnant obese women, there was leptin resistance at the level of the placenta in obese pregnant women. We were unable to reproduce this using the same antibody for the long form of the leptin receptor with a larger cohort of patients. We further attempted to show this by using another antibody, which in addition to the long-form leptin receptor also detects the different short forms in the placenta.38 However, with this antibody and quantitation, there was similarly no downregulation or leptin-resistance evident in the syncytiotrophoblast. Thus, the significance of leptin function in the syncytiotrophoblast in the context of maternal BMI requires further work.
In addition to its localization to the syncytiotrophoblast using the antibody for the long form of the leptin receptor, we also detected receptors in the villous stroma, possibly Hofbauer cells, consistent with the report by Farley and colleagues.29 Indeed, it has been shown that the immunostaining of placental macrophages increased 2- to 3-fold in obese compared to lean women.45 However, our quantitation of the immunostaining of these cells failed to show any increase in expression with increasing obesity. These inconsistencies in receptor immunostaining may be due to different antigen retrieval procedures used in different studies and/or to the rather poor antibodies currently available for the detection of the different leptin receptor isoforms. There is certainly the need for further work to clarify leptin receptor expression in the placenta and the effects of maternal obesity, but future work should be carried out with antibodies of higher avidity, which would be more suitable for immunohistochemistry than those currently available.
In the present study, we also quantitated leptin and leptin receptor gene expression in the placental villous trophoblast. Unfortunately, because this method used pieces of tissue, which included all cell types, the dilution effect resulted in negative data. However, the major site of leptin production and action in the placenta is undoubtedly the syncytiotrophoblast, and the negative mRNA findings confirmed that there was indeed no increase in syncytiotrophoblast leptin mRNA in tissue from obese women or no reduction in the mRNA of its receptor. However, it is possible that by using a larger number of patients, significant differences in placental leptin/leptin receptor gene expression may be detected.
In conclusion, we have confirmed the relationships between maternal prepregnancy BMI and maternal leptin and the soluble leptin receptor and cord plasma leptin using a single set of patients. We have also confirmed the relationships between birth weight and cord plasma leptin and shown higher cord leptin levels in female versus male infants. Thus, validating our patient cohort. On the other hand, we were unable to confirm a decrease in expression of the leptin receptor in the syncytiotrophoblast with increased maternal BMI, making “placental leptin resistance” in obesity questionable. However, for the first time, we have identified the placental villous endothelial cells as the origin of increased placental leptin in obese women. This leptin would enter the fetal circulation, as shown here by higher cord blood levels in obesity. Together with the leptin from fetal adipose tissue, this is likely to be important in fetal growth and future childhood development.
Acknowledgments
The authors acknowledge Ms Sandra Yamamoto for her assistance with the laboratory aspect of this study. We also acknowledge Ms Ann Marie Savage and the nurses of the Family Birth Center at Kapiolani Medical Center for Women and Children for their valuable support. Finally, we acknowledge all the patients who donated their time and samples for this study.
Footnotes
Authors’ Note: This study was presented at the Society for Maternal Fetal Medicine Annual Meeting, New Orleans, LA, USA, February 6-8, 2012.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The funding for the study was provided by the Department of Obstetrics, Gynecology, and Women’s Health at University of Hawaii, John A. Burns School of Medicine. Statistical analyses were supported in part by grants from the National Institute of Minority Health and Health Disparities (U54MD007584 and G12MD007601) from the National Institutes of Health.
References
- 1. Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999-2010. JAMA. 2012;307 (5):491–497. [DOI] [PubMed] [Google Scholar]
- 2. Kristensen J, Vestergaard M, Wisborg K, Kesmodel U, Secher NJ. Pre-pregnancy weight and the risk of stillbirth and neonatal death. BJOG. 2005;112 (4):403–408. [DOI] [PubMed] [Google Scholar]
- 3. Weiss JL, Malone FD, Emig D, et al. Obesity, obstetric complications and cesarean delivery rate—a population-based screening study. Am J Obstet Gynecol. 2004;190 (4):1091–1097. [DOI] [PubMed] [Google Scholar]
- 4. Ovesen P, Rasmussen S, Kesmodel U. Effect of prepregnancy maternal overweight and obesity on pregnancy outcome. Obstet Gynecol. 2011;118 (2 pt 1):305–312. [DOI] [PubMed] [Google Scholar]
- 5. Watkins ML, Rasmussen SA, Honein MA, Botto LD, Moore CA. Maternal obesity and risk for birth defects. Pediatrics. 2003;111 (5 part 2):1152–1158. [PubMed] [Google Scholar]
- 6. Honein MA, Moore CA, Watkins ML. Subfertility and prepregnancy overweight/obesity: possible interaction between these risk factors in the etiology of congenital renal anomalies. Birth Defects Res A Clin Mol Teratol. 2003;67 (8):572–577. [DOI] [PubMed] [Google Scholar]
- 7. Stothard KJ, Tennant PW, Bell R, Rankin J. Maternal overweight and obesity and the risk of congenital anomalies: a systematic review and meta-analysis. JAMA. 2009;301 (6):636–650. [DOI] [PubMed] [Google Scholar]
- 8. Whitaker RC. Predicting preschooler obesity at birth: the role of maternal obesity in early pregnancy. Pediatrics. 2004;114 (1):e29–e36. [DOI] [PubMed] [Google Scholar]
- 9. Boney CM, Verma A, Tucker R, Vohr BR. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics. 2005;115 (3):e290–e296. [DOI] [PubMed] [Google Scholar]
- 10. Mohamed-Ali V, Pinkney JH, Coppack SW. Adipose tissue as an endocrine and paracrine organ. Int J Obes Relat Metab Disord. 1998;22 (12):1145–1158. [DOI] [PubMed] [Google Scholar]
- 11. Briana DD, Malamitsi-Puchner A. Reviews: adipocytokines in normal and complicated pregnancies. Reprod Sci. 2009;16 (10):921–937. [DOI] [PubMed] [Google Scholar]
- 12. Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395 (6704):763–770. [DOI] [PubMed] [Google Scholar]
- 13. Harris RB. Leptin—much more than a satiety signal. Annu Rev Nutr. 2000;20:45–75. [DOI] [PubMed] [Google Scholar]
- 14. Tartaglia LA. The leptin receptor. J Biol Chem. 1997;272 (10):6093–6096. [DOI] [PubMed] [Google Scholar]
- 15. Zastrow O, Seidel B, Kiess W, et al. The soluble leptin receptor is crucial for leptin action: evidence from clinical and experimental data. Int J Obes Relat Metab Disord. 2003;27 (12):1472–1478. [DOI] [PubMed] [Google Scholar]
- 16. Bifulco G, Trencia A, Caruso M, et al. Leptin induces mitogenic effect on human choriocarcinoma cell line (JAr) via MAP kinase activation in a glucose-dependent fashion. Placenta. 2003;24 (4):385–391. [DOI] [PubMed] [Google Scholar]
- 17. Perez-Perez A, Maymo J, Duenas JL, et al. Leptin prevents apoptosis of trophoblastic cells by activation of MAPK pathway. Arch Biochem Biophys. 2008;477 (2):390–395. [DOI] [PubMed] [Google Scholar]
- 18. Dubern B, Clement K. Leptin and leptin receptor-related monogenic obesity. Biochimie. 2012;94 (10):2111–2115. [DOI] [PubMed] [Google Scholar]
- 19. Farooqi IS, Wangensteen T, Collins S, et al. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N Engl J Med. 2007;356 (3):237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ogier V, Ziegler O, Mejean L, Nicolas JP, Stricker-Krongrad A. Obesity is associated with decreasing levels of the circulating soluble leptin receptor in humans. Int J Obes Relat Metab Disord. 2002;26 (4):496–503. [DOI] [PubMed] [Google Scholar]
- 21. van Dielen FM, van’t Veer C, Buurman WA, Greve JW. Leptin and soluble leptin receptor levels in obese and weight-losing individuals. J Clin Endocrinol Metab. 2002;87 (4):1708–1716. [DOI] [PubMed] [Google Scholar]
- 22. Hamed EA, Zakary MM, Ahmed NS, Gamal RM. Circulating leptin and insulin in obese patients with and without type 2 diabetes mellitus: relation to ghrelin and oxidative stress. Diabetes Res Clin Pract. 2011;94 (3):434–441. [DOI] [PubMed] [Google Scholar]
- 23. Caro JF, Sinha MK, Kolaczynski JW, Zhang PL, Considine RV. Leptin: the tale of an obesity gene. Diabetes. 1996;45 (11):1455–1462. [DOI] [PubMed] [Google Scholar]
- 24. Hauguel-de Mouzon S, Lepercq J, Catalano P. The known and unknown of leptin in pregnancy. Am J Obstet Gynecol. 2006;194 (6):1537–1545. [DOI] [PubMed] [Google Scholar]
- 25. Senaris R, Garcia-Caballero T, Casabiell X, et al. Synthesis of leptin in human placenta. Endocrinology. 1997;138 (10):4501–4504. [DOI] [PubMed] [Google Scholar]
- 26. Masuzaki H, Ogawa Y, Sagawa N, et al. Nonadipose tissue production of leptin: leptin as a novel placenta-derived hormone in humans. Nat Med. 1997;3 (9):1029–1033. [DOI] [PubMed] [Google Scholar]
- 27. Lea RG, Howe D, Hannah LT, Bonneau O, Hunter L, Hoggard N. Placental leptin in normal, diabetic and fetal growth-retarded pregnancies. Mol Hum Reprod. 2000;6 (8):763–769. [DOI] [PubMed] [Google Scholar]
- 28. Linnemann K, Malek A, Schneider H, Fusch C. Physiological and pathological regulation of feto/placento/maternal leptin expression. Biochem Soc Trans. 2001;29 (pt 2):86–90. [DOI] [PubMed] [Google Scholar]
- 29. Farley DM, Choi J, Dudley DJ, et al. Placental amino acid transport and placental leptin resistance in pregnancies complicated by maternal obesity. Placenta. 2010;31 (8):718–724. [DOI] [PubMed] [Google Scholar]
- 30. Boeke CE, Mantzoros CS, Hughes MD, et al. Differential associations of leptin with adiposity across early childhood. Obesity (Silver Spring). 2013;21 (7):1430–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Matsuda J, Yokota I, Iida M, et al. Serum leptin concentration in cord blood: relationship to birth weight and gender. J Clin Endocrinol Metab. 1997;82 (5):1642–1644. [DOI] [PubMed] [Google Scholar]
- 32. Vela-Huerta MM, San Vicente-Santoscoy EU, Guizar-Mendoza JM, Amador-Licona N, Aldana-Valenzuela C, Hernnandez J. Leptin, insulin, and glucose serum levels in large-for-gestational-age infants of diabetic and non-diabetic mothers. J Pediatr Endocrinol Metab. 2008;21 (1):17–22. [DOI] [PubMed] [Google Scholar]
- 33. Karakosta P, Chatzi L, Plana E, Margioris A, Castanas E, Kogevinas M. Leptin levels in cord blood and anthropometric measures at birth: a systematic review and meta-analysis. Paediatr Perinat Epidemiol. 2011;25 (2):150–163. [DOI] [PubMed] [Google Scholar]
- 34. Linnemann K, Malek A, Sager R, Blum WF, Schneider H, Fusch C. Leptin production and release in the dually in vitro perfused human placenta. J Clin Endocrinol Metab. 2000;85 (11):4298–4301. [DOI] [PubMed] [Google Scholar]
- 35. Lepercq J, Challier JC, Guerre-Millo M, Cauzac M, Vidal H, Hauguel-de Mouzon S. Prenatal leptin production: evidence that fetal adipose tissue produces leptin. J Clin Endocrinol Metab. 2001;86 (6):2409–2413. [DOI] [PubMed] [Google Scholar]
- 36. Schubring C, Englaro P, Siebler T, et al. Longitudinal analysis of maternal serum leptin levels during pregnancy, at birth and up to six weeks after birth: relation to body mass index, skinfolds, sex steroids and umbilical cord blood leptin levels. Horm Res. 1998;50 (5):276–283. [DOI] [PubMed] [Google Scholar]
- 37. Challier J, Galtier M, Bintein T, Cortez A, Lepercq J, Hauguel-de Mouzon S. Placental leptin receptor isoforms in normal and pathological pregnancies. Placenta. 2003;24 (1):92–99. [DOI] [PubMed] [Google Scholar]
- 38. Li RH, Poon SC, Yu MY, Wong YF. Expression of placental leptin and leptin receptors in preeclampsia. Int J Gynecol Pathol. 2004;23 (4):378–385. [DOI] [PubMed] [Google Scholar]
- 39. Lewandowski K, Horn R, O’Callaghan CJ, et al. Free leptin, bound leptin, and soluble leptin receptor in normal and diabetic pregnancies. J Clin Endocrinol Metab. 1999;84 (1):300–306. [DOI] [PubMed] [Google Scholar]
- 40. Nuamah MA, Sagawa N, Yura S, et al. Free-to-total leptin ratio in maternal plasma is constant throughout human pregnancy. Endocr J. 2003;50 (4):421–428. [DOI] [PubMed] [Google Scholar]
- 41. Schulz S, Hackel C, Weise W. Hormonal regulation of neonatal weight: placental leptin and leptin receptors. BJOG. 2000;107 (12):1486–1491. [DOI] [PubMed] [Google Scholar]
- 42. Tome MA, Lage M, Camina JP, Garcia-Mayor RV, Dieguez C, Casanueva FF. Sex-based differences in serum leptin concentrations from umbilical cord blood at delivery. Eur J Endocrinol. 1997;137 (6):655–658. [DOI] [PubMed] [Google Scholar]
- 43. Yura S, Sagawa N, Mise H, et al. A positive umbilical venous-arterial difference of leptin level and its rapid decline after birth. Am J Obstet Gynecol. 1998;178 (5):926–930. [DOI] [PubMed] [Google Scholar]
- 44. Lin KC, Hsu SC, Kuo CH, Zhou JY. Difference of plasma leptin levels in venous and arterial cord blood: relation to neonatal and placental weight. Kaohsiung J Med Sci. 1999;15 (12):679–685. [PubMed] [Google Scholar]
- 45. Challier JC, Basu S, Bintein T, et al. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta. 2008;29(3):274–281. [DOI] [PMC free article] [PubMed] [Google Scholar]