

Abstract
Intron-22-inversion patients express the entire Factor VIII (FVIII)-amino-acid sequence intracellularly as 2 non-secreted polypeptides and have a positive “intracellular (I)-FVIII-CRM” status. Mutations conferring a positive I-FVIII-CRM status are associated with low inhibitor risk and are pharmacogenetically relevant because inhibitor risk may be affected by the nature of the therapeutic FVIII-protein (tFVIII), the affinity of any tFVIII-derived foreign peptide (tFVIII-fp) for any HLA class-II isomer (HLA-II) comprising individual major histocompatibility complex (MHC) repertoires, and the stability of any tFVIII-fp/HLA-II complex. We hypothesize that mutations conferring a completely or substantially negative I-FVIII-CRM status are pharmacogenetically irrelevant because inhibitor risk is high with any tFVIII and individual MHC repertoire.
Introduction
The development of neutralizing antibodies (“inhibitors”) to therapeutic Factor VIII (FVIII) proteins (tFVIIIs) is a serious complication of hemophilia A (HA) treatment, occurring in approximately 25% of patients with severe HA. Patient-, treatment-, and product-related variables contribute to inhibitor risk. The most salient patient-related determinant is the nature of the HA-causing FVIII gene (F8) mutation.1-3 The highest inhibitor incidence, 68% to 88%, occurs in patients with large F8 deletions, and the lowest (<10%) occurs in patients with missense mutations. This is consistent with the postulate that an endogenous FVIII polypeptide synthesized during immune system development can, through the induction of central tolerance, confer preexisting immunologic unresponsiveness to infused tFVIIIs. Patients with the intron-22-inversion (I22I) mutation2 have no circulating endogenously synthesized FVIII; thus, this mutation might be expected to be associated with a high rate of inhibitor development, but in large studies, only ∼20% of such subjects had developed inhibitors.4 We recently discovered a plausible mechanism for this conundrum5 and, in so doing, defined a new biomarker for the risk of immunogenicity of tFVIIIs, which we refer to as a patient’s “intracellular-FVIII-cross-reactive material” (I-FVIII-CRM) status. Other mutation types, obviously including missense mutations, but possibly other mutations, may be associated with a positive I-FVIII-CRM status. This biomarker identifies patients who may have a preexisting tolerance to tFVIIIs, a group of patients for whom pharmacogenomics may be most helpful.
Does the effect of F8 mutation type on endogenous FVIII synthesis underlie inhibitor risk?
As we alluded to before, the ability or inability of patients to induce and maintain tolerance toward their own (self-) FVIII protein underlies the well-established relationship between risk of developing inhibitors to tFVIIIs and underlying F8 mutation type, where risk increases roughly with the degree to which the gene is disrupted structurally and/or functionally. The physiologic mechanisms of central and peripheral immune tolerance require endogenous protein synthesis. Individuals first become tolerized to the FVIII protein they express during lymphocyte development.6 Patients with missense mutations are tolerant to their own endogenously expressed missense mutation–containing full-length FVIII (mm-FVIIIFL) proteins. These mm-FVIIIFL proteins are variably secreted and/or dysfunctional because of the nature and location of their mutant residues, but all may differ from the tFVIIIs by only one amino acid.7-10 Despite the existence of >980 distinct missense mutations, only ∼80 of these mutations have been identified in patients with inhibitors (HADB, http://hadb.org.uk). Overall, inhibitors develop in <10% of all HA patients with missense mutations, although the risk of developing inhibitors can be very high with a specific subset comprising <10 mutations, in so called “hot spots.”11
Although nonsynonymous single-nucleotide polymorphisms (ns-SNPs) allow the expression of fully functional endogenous FVIII proteins, in contrast to missense mutations, from an immunologic perspective they are akin in that they also encode proteins with amino acid sequences that differ from the sequences of the tFVIIIs at 1 to 3 residues.12 Mismatches between the infused and endogenous sequences in an individual expressing one or more permissive HLA class-II (HLA-II) molecules can elicit an alloimmune response.13 In our initial small study of African-American HA patients, those with the underlying black African-restricted haplotypes H3 and H4 were 3.6 times as likely to have developed inhibitors as were those with the underlying nonracially-restricted haplotypes H1 and H2, which were the same as those of white Europeans and those representing the recombinant-protein drugs currently approved for replacement therapy.14 Given that 2 subsequent studies,15,16 each with fewer African-descent subjects than in our initial study, did not confirm our observation, and because a third study is not yet complete, further investigation with a larger sample size is warranted. The third ongoing study has a larger sample of HA patients of African descent and it is hoped will be more definitive.5
Theoretically, exposures through blood transfusions and/or pregnancies to wild-type FVIII proteins that are allogeneically mismatched at residues encoded by nonsynonymous polymorphisms can elicit alloimmune responses that may crossreact with an individual’s endogenously expressed FVIII and cause the autoimmune bleeding disorder–acquired HA.17 Fortunately, this occurs very infrequently, presumably because any at-risk recipient or mother must have both a permissive HLA-II repertoire and a substantial exposure to the foreign FVIII molecule(s) in the presence of danger signals and other predisposing determinants.
Null-type F8 defects that result in the absence of circulating FVIII activity and antigen include multiexonic deletions, which preclude endogenous expression of most (or all) of a FVIII protein, and nonsense mutations, which, depending on their location, can result in decreased cytoplasmic levels of the F8 mRNA and/or can prevent translation of variable amounts of downstream-coding sequence (DCS). Transcription and/or translation of a full-length FVIII mRNA are prevented when the mutation is a deletion of an F8 exon or a premature termination codon (PTC): in these instances, establishment of complete immunologic unresponsiveness to any tFVIII is prevented. There are, however, a few moderating considerations. Some large deletions and duplications occur in-frame18 and the resultant mutant F8 may direct synthesis of most of an endogenous FVIII protein. Similarly, with “exon skipping,” the exons immediately flanking the exon bearing a PTC may be spliced together to generate a shorter but in-frame transcript. Both the deletion of in-frame exons and the exon-skipping mechanism allow for the production of somewhat smaller proteins that, although they are not secreted into plasma and/or are functional in coagulation, are still likely to produce immunologic unresponsiveness to corresponding sequences of the infused tFVIIIs.19,20
The F8 intron-22 inversion
The I22I mutation, which causes ∼40% to 45% of all cases of severe HA,1-3 results in truncation of the wild-type F8 transcription unit (Figure 1A) and inversion of exons 1 to 22 toward the telomere of the X-chromosome long arm (Figure 1B).21 Transcription of the I22-inverted F8 locus followed by primary mRNA processing yields a polyadenylated fusion transcript, F8I22I, containing FVIII exons 1 to 22 spliced to 2 cryptic (C) exons,21 referred to as Exons 23C and 24C (Figure 1B).5 Exon-23C contains 16 in-frame codons followed by an in-frame stop codon and 38 bp of untranslated sequence. Because this stop codon is situated <50 to 55 nucleotides upstream of the 3′-most exon-exon junction,21 the fusion transcript is not predicted to trigger nonsense-mediated mRNA decay.20 We recently demonstrated that F8I22I is translated, and it generates a protein with the 19-residue wild-type FVIII leader-peptide, the first 2124 amino acids of the mature FVIII protein, and 16 C-terminal non-FVIII amino acids encoded by exon-23C (ie, a fusion protein [FVIIII22I] containing 2159 amino acids).5
Figure 1.
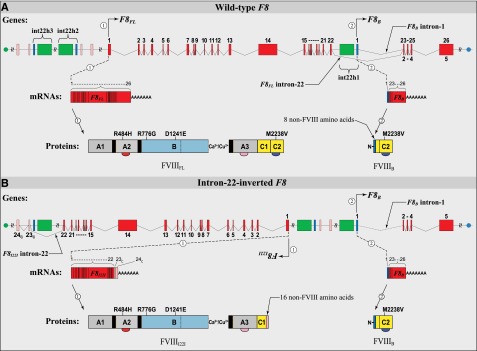
Structure and function of the wild-type and I22-inverted F8 loci: DNA, mRNA, and protein. (A) The wild-type F8 locus is comprised of 2 separately regulated transcription units that direct synthesis of 2 F8-exonic-sequence–containing polyadenylated transcript variants, 1 and 2, which are designated here as F8FL and F8B, respectively. F8FL contains all 9030 bases found in F8 exons 1 to 26 and encodes the full-length FVIII protein, FVIIIFL, whose mature circulating form, containing 2332 amino acid residues, is essential for normal blood coagulation. F8B contains 2598 bases —including 169 bases from its first exon, which is not found in F8FL, and 2429 bases from its last 4 exons (exons 2-5), which are identical to exons 23 to 26 of F8FL—and encodes wild-type FVIIIB, a recently identified intracellular protein comprised of 216 amino acids whose function, if any, remains unknown. Homologous recombination between int22h1 and int22h3 (depicted as green and dark blue rectangles, respectively) incompletely inverts F8FL. (B) The I22-inverted F8 locus is also comprised of 2 separately regulated transcription units that direct synthesis of 2 F8-exonic-sequence–containing polyadenylated transcript variants, 1 and 2, which are designated here as F8I22I and F8B, respectively. Because homologous recombination restores the genomic DNA sequence within which it occurs, it has long been suspected,2 but only recently proven experimentally,5 that the F8B gene and transcript, and hence the FVIIIB protein, are identical to that in healthy persons without HA. F8I22I contains the 6756 bases found in F8 exons 1 to 22 and, together with the 48 additional DCS bases in exon 23C, encodes the FVIIII22I protein, which contains 2159 translated amino acid residues and is trapped intracellularly.
Because the I22I arises through a homologous recombination event that restores completely the sequence within which it occurs—which includes the promoter and first exon of the nested F8B gene—transcription and translation of F8B is expected to be unaffected (ie, wild type).5,22 The F8B locus encodes a widely expressed, moderately abundant 2.6-kb transcript with exons 23 to 26 of F8 spliced in-frame to an unrelated first exon that has a Kozak’s consensus initiation codon.22 The F8B mRNA is predicted to encode a 216-amino-acid protein, FVIIIB, corresponding to exons 23 to 26 in F8. We have now identified the FVIIII22I and FVIIIB proteins in peripheral blood mononuclear cells (PBMCs) and liver sections from patients with I22I.5 Between them, these polypeptides contain the entire coding sequence for a full-length FVIII protein.
The last base of exon 22 corresponds to the third nucleotide of translated codon 2143, which encodes residue 2124 of the mature protein. Moreover, the first base of exon 23 corresponds to the first nucleotide of adjacent codon 2144, which encodes residue 2125 of the mature protein (Figure 2). Thus, with the I22I, truncation of F8 after exon 22 does not split a codon and every FVIII amino acid residue should be expressed.5
Figure 2.

Peptides from the Exon 22/Exon 23 junction and their affinity for common MHC-II variants. (A) Ribbon diagram of the structure of the C1 domain of FVIII highlighted to emphasize the I22I break point encoded by the 3′- and 5′-ends of exons 22 and 23, respectively. Y2105 and R2150 (red) represent the positions of recurrent mild HA-causing missense mutations that are strongly associated with inhibitor development. Residues 2106 to 2123 and 2126 to 2149 (blue) are 2 segments of C1 on either side of the inversion break point. M2124 and V2125 (orange and white, respectively) are the residues flanking the break point. (B) Forty-six amino acids comprising most of the C-terminal one-third of the C1 domain are shown together with the location of known HA-causing missense mutations; the presence (red asterisk) or absence (black minus sign) of inhibitor development in patients found to have one of these abnormalities with these is also indicated. Note that Y2105C and R2150H have been found in many unrelated alloimmunized HA patients and represent the N- and C-terminal missense mutations, respectively, closest to the exon 22/exon 23 junction, which have been identified in inhibitor patients. Although 18 additional missense mutations (green) have been identified more proximal than Y2105C and R2150H to the I22I break point, none of these patients has yet been reported to have developed inhibitors to date. (C) The immunogenicity potential of wild-type tFVIII-derived peptides encoded by the mRNA sequence spanning the exon 22/exon 23 junction is depicted as promiscuity scores.5,27 The binding affinities of commonly-occurring HLA-II proteins for peptides derived from the C1 domain region corresponding to the exon 22/exon 23 junction were predicted using the NetMHCIIpan-3.0 method (http://www.cbs.dtu.dk/services/NetMHCIIpan/28). The method predicts the binding affinity (in nM) for each 15-mer peptide–HLA-II complex. The population-level promiscuity scores for each 15-mer FVIII peptide is then defined as the (normalized) cumulative prevalence of common HLA-II proteins that bind it with high affinity (≤50 nM) in 3 populations: white European (blue), black African (green), and Global (red).
Results from quantitative real-time reverse transcriptase polymerase chain reaction–based assays showed that the cytoplasmic levels of the F8I22I mRNA are similar to those of the full-length wild-type F8 mRNA.5 The F8B transcript in I22I patients was also found to be present at levels similar to those of the F8B mRNA in persons without hemophilia.5 Because the FVIIII22I and FVIIIB proteins are found to be expressed in liver samples and PBMCs from HA patients with the I22I at similar levels to the FVIIIFL and FVIIIB proteins in normal persons,5 it is reasonable to postulate that these polypeptides provide central tolerance even though expression of these antigens in thymic tissue has not yet been demonstrated. There is a promiscuous expression of tissue-restricted self-antigens by medullary thymic epithelial cells. Thus, antigens expressed in the thymus mirror those of all tissues in the body.23,24 Recent transgenic mouse studies indicate that endothelial cells may be the only cell type capable of secreting their intracellularly-expressed FVIII protein. However, secretion of proteins may not be necessary to begin the process of tolerance induction. There are at least 2 documented mechanisms by which endogenously expressed intracellular polypeptides can be presented by the major histocompatibility complex (MHC)-II system, the type of antigen presentation necessary for inducing tolerance in CD4+ T cells. In 1 mechanism, peptides from endogenously expressed proteins can be displayed on antigen-presenting cell (APC) surfaces, bound to MHC-II molecules, through the cross-presentation process.25 In the other mechanism, the intracellularly expressed polypeptides are delivered to the MHC-II loading compartments of APCs in the form of apoptotic bodies that arise as a function of cellular senescence or targeted killing by cytolytic cells.26
However, peptides that are encoded by the nucleotide sequence spanning the exon 22/exon 23 junction (Met2124-Val2125) cannot be generated by patients with the I22I mutation (Figure 2). Fourteen 15-mer overlapping peptides could potentially be recognized as “foreign” if derived from the infused wild-type FVIII protein and presented by a patient’s repertoire of HLA-II receptors. However, not all foreign peptides elicit an immune response. A critical determinant for T cell–dependent immunization is the affinity with which any foreign peptide(s) derived from the infused protein binds to the distinct HLA-II repertoire on the surface of the patient’s APCs.29 Figure 2C illustrates the population-level “promiscuity scores,” a synthesis of (1) the frequencies of common (prevalence ≥0.5%) HLA-II variants in the specified populations, and (2) the predicted binding affinities of all overlapping FVIII peptides between positions 2090 and 2165 to those HLA-II variants. The set of overlapping peptides encoded by the nucleotide sequence spanning the exon 22/exon 23 junction (15 amino acids upstream and downstream of the green dotted line, Figure 2C) do not bind with high affinity to most of these HLA-II alleles. This analysis is supported by the clinical observation that although 15 individual missense mutations have been described in the region of the exon 22/exon 23 junction, none of these HA patients developed inhibitors (HADB, http://hadb.org.uk). The promiscuity scores also show that, farther upstream and downstream of the junction, there are regions where peptides bind with high affinity to many of these common HLA-II variants (Figure 2C). Note that these regions include amino acid positions 2105 and 2150, which correspond to sites of the highly recurrent mild HA-causing missense mutations Y2105C and R2150H, associated with a high inhibitor prevalence of 50% and 16%, respectively.11,27
We and others have wondered why the inhibitor rate in patients with the I22I is as high as ∼20% if almost all of the entire normal FVIII protein is present intracellularly. Although at the population level, the peptides encoded by the exon 22/exon 23 junction do not bind strongly (Kd <50 nM) to the most common HLA-II variants, the individual HLA-II repertoire of some patients binds with intermediate affinity (Kd <500 nM). Because all I22I patients had severe HA and were probably transfused heavily, in contrast to patients with missense mutations, the vast majority of whom have mild HA, the intermediate affinity of the junction peptides could be responsible for the immune response observed in some patients.
Intracellular FVIII cross-reacting material
I-FVIII-CRM appears to be a major determinant of immunologic tolerance to tFVIIIs in HA patients. We propose grouping F8 mutations into either “pharmacogenetically relevant” or “pharmacogenetically irrelevant” categories to guide inhibitor prediction and mitigation. Pharmacogenetics is the study of differences in individual responses to the same drug as a result of genetic variations in the patient population. Patients with pharmacogenetically relevant F8 mutations would thus be presumed to have immune tolerance to most FVIII proteins, and their risk of developing inhibitors may be related to such variables as the degree to which infused FVIII matches their endogenous FVIII, immune system differences that are highly patient-specific, and patterns of exposures to tFVIII.
Patients with pharmacogenetically irrelevant mutations would be presumed to have little or no tolerance to FVIII such that all FVIII replacement proteins would be risky in the setting of a permissive immune system and the necessary exposure level.
Currently available recombinant FVIII products represent only 2 of the 8 or more naturally occurring haplotypes of the human FVIII protein that result from combinations of several F8 ns-SNPs.12,14 We hypothesize that for HA patients with pharmacogenetically relevant mutations, a FVIII replacement product whose amino acid sequence is identical to that of the patient’s premutation haplotype may have lower immunogenicity potential than a mismatched FVIII product.
Patients with pharmacogenetically relevant mutations express most or all of the FVIII protein, whether secreted into the plasma or trapped in intracellular compartments. Because HLA-II variants vary greatly in their affinity for a given peptide, the probability of an alloimmune response developing in a patient with a specific pharmacogenetically relevant mutation will depend in part on whether any of the mismatched peptides derived from the infused FVIII protein can bind to the HLA-II isomers presented by the patient.27
Future directions
There has been significant improvement in the quality of FVIII replacement products over the last 2 decades, and numerous bioengineered coagulation factors are in the drug development pipeline.30,31 Currently, FVIII replacement products are dispensed interchangeably with the premise that each is equally suitable for all recipients. Until recently, there has been little deliberation on the effects of engineered differences in the amino acid sequence of the FVIII product on inhibitor risk. We wonder whether a B domain–deleted tFVIII would be safer for a patient with an in-frame multiexonic deletion involving exon 14. Results from ongoing studies may foster the development of recombinant FVIII products with varied primary sequences that have an improved safety profile for specific populations.
A personalized strategy is not only important for the choice of a FVIII treatment protein but also for the choice of a specific F8 for gene therapy.
Promiscuity scores help us understand, on a population level, the predicted inhibitor burden of a specific tFVIII product. For the care of the individual patient, however, specific combination of F8 mutation, FVIII haplotype, HLA-II repertoire, and possibly other immune system variants will have to be weighed.
Acknowledgments
The authors thank J. Lusher, V. La Terza, N. McCurley, S. Cole, G.S. Pandey, and K. Viel for help with the manuscript and/or for helpful comments.
This study was supported by the Modernization of Science Program of the Center for Biologics Evaluation and Research, United States Food and Drug Administration (Z.E.S.); and grants from the National Heart, Lung and Blood Institute, National Institutes of Health (1RC2-HL101851, HL-71130, HL-72533), the Bayer Healthcare Corporation, Bayer Hemophilia Awards Program, Baxter Healthcare Corporation, and the Clinical Translational Science Institute at the University of Southern California (T.E.H.).
Authorship
Contribution: Z.E.S., C.Y., and T.E.H. conducted the research leading to these concepts; Z.E.S., C.K.K., and T.E.H. wrote the manuscript; and J.N.L., C.Y., and T.N. edited the manuscript and contributed critical commentary.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The findings and conclusions in this article have not been formally disseminated by the US Food and Drug Administration and should not be construed to represent any agency determination or policy.
Correspondence: Tom E. Howard, Department of Pathology and Laboratory Medicine, Veterans Affairs Greater Los Angeles Healthcare System, Building 500, Room 1258, Los Angeles, CA 90073; e-mail: Tom.Howard@va.gov; and Zuben E. Sauna, Laboratory of Hemostasis, Division of Hematology Research and Review, Center for Biologics Evaluation and Research, Food and Drug Administration, Building 52, Room 4120, 10903 New Hampshire Ave, Silver Spring, MD 20993; e-mail: zuben.sauna@fda.hhs.gov.
References
- 1.Gouw SC, van den Berg HM, Oldenburg J, et al. F8 gene mutation type and inhibitor development in patients with severe hemophilia A: systematic review and meta-analysis. Blood. 2012;119(12):2922–2934. doi: 10.1182/blood-2011-09-379453. [DOI] [PubMed] [Google Scholar]
- 2.Oldenburg J, Pavlova A. Genetic risk factors for inhibitors to FVIII and FIX. Haemophilia. 2006;12(Suppl 6):15–22. doi: 10.1111/j.1365-2516.2006.01361.x. [DOI] [PubMed] [Google Scholar]
- 3.Oldenburg J, El-Maarri O, Schwaab R. Inhibitor development in correlation to factor VIII genotypes. Haemophilia. 2002;8(Suppl 2):23–29. doi: 10.1046/j.1351-8216.2001.00134.x. [DOI] [PubMed] [Google Scholar]
- 4.Antonarakis SE, Rossiter JP, Young M, et al. Factor VIII gene inversions in severe hemophilia A: results of an international consortium study. Blood. 1995;86(6):2206–2212. [PubMed] [Google Scholar]
- 5.Pandey GS, Yanover C, Miller-Jenkins LM, et al. PATH (Personalized Alternative Therapies for Hemophilia) Study Investigators. Endogenous factor VIII synthesis from the intron 22-inverted F8 locus may modulate the immunogenicity of replacement therapy for hemophilia A. Nat Med. 2013;19(10):1318–1324. doi: 10.1038/nm.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Starr TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Annu Rev Immunol. 2003;21:139–176. doi: 10.1146/annurev.immunol.21.120601.141107. [DOI] [PubMed] [Google Scholar]
- 7.Peerlinck K, Jacquemin MG, Arnout J, et al. Antifactor VIII antibody inhibiting allogeneic but not autologous factor VIII in patients with mild hemophilia A. Blood. 1999;93(7):2267–2273. [PubMed] [Google Scholar]
- 8.Fijnvandraat K, Turenhout EA, van den Brink EN, et al. The missense mutation Arg593 —> Cys is related to antibody formation in a patient with mild hemophilia A. Blood. 1997;89(12):4371–4377. [PubMed] [Google Scholar]
- 9.Thompson AR, Murphy ME, Liu M, et al. Loss of tolerance to exogenous and endogenous factor VIII in a mild hemophilia A patient with an Arg593 to Cys mutation. Blood. 1997;90(5):1902–1910. [PubMed] [Google Scholar]
- 10.Bogdanova N, Markoff A, Eisert R, et al. Spectrum of molecular defects and mutation detection rate in patients with mild and moderate hemophilia A. Hum Mutat. 2007;28(1):54–60. doi: 10.1002/humu.20403. [DOI] [PubMed] [Google Scholar]
- 11.Eckhardt CL, van Velzen AS, Peters M, et al. INSIGHT Study Group. Factor VIII gene (F8) mutation and risk of inhibitor development in nonsevere hemophilia A. Blood. 2013;122(11):1954–1962. doi: 10.1182/blood-2013-02-483263. [DOI] [PubMed] [Google Scholar]
- 12.Viel KR, Machiah DK, Warren DM, et al. A sequence variation scan of the coagulation factor VIII (FVIII) structural gene and associations with plasma FVIII activity levels. Blood. 2007;109(9):3713–3724. doi: 10.1182/blood-2006-06-026104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pandey GS, Yanover C, Howard TE, Sauna ZE. Polymorphisms in the F8 gene and MHC-II variants as risk factors for the development of inhibitory anti-factor VIII antibodies during the treatment of hemophilia a: a computational assessment. PLOS Comput Biol. 2013;9(5):e1003066. doi: 10.1371/journal.pcbi.1003066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Viel KR, Ameri A, Abshire TC, et al. Inhibitors of factor VIII in black patients with hemophilia. N Engl J Med. 2009;360(16):1618–1627. doi: 10.1056/NEJMoa075760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller CH, Benson J, Ellingsen D, et al. Hemophilia Inhibitor Research Study Investigators. F8 and F9 mutations in US haemophilia patients: correlation with history of inhibitor and race/ethnicity. Haemophilia. 2012;18(3):375–382. doi: 10.1111/j.1365-2516.2011.02700.x. [DOI] [PubMed] [Google Scholar]
- 16.Schwarz J, Astermark J, Menius ED, et al. Hemophilia Inhibitor Genetics Study Combined Cohort. F8 haplotype and inhibitor risk: results from the Hemophilia Inhibitor Genetics Study (HIGS) Combined Cohort. Haemophilia. 2013;19(1):113–118. doi: 10.1111/hae.12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tiede A, Eisert R, Czwalinna A, Miesbach W, Scharrer I, Ganser A. Acquired haemophilia caused by non-haemophilic factor VIII gene variants. Ann Hematol. 2010;89(6):607–612. doi: 10.1007/s00277-009-0887-3. [DOI] [PubMed] [Google Scholar]
- 18.Rost S, Löffler S, Pavlova A, Müller CR, Oldenburg J. Detection of large duplications within the factor VIII gene by MLPA. J Thromb Haemost. 2008;6(11):1996–1999. doi: 10.1111/j.1538-7836.2008.03125.x. [DOI] [PubMed] [Google Scholar]
- 19.White GC, II, Kempton CL, Grimsley A, Nielsen B, Roberts HR. Cellular immune responses in hemophilia: why do inhibitors develop in some, but not all hemophiliacs? J Thromb Haemost. 2005;3(8):1676–1681. doi: 10.1111/j.1538-7836.2005.01375.x. [DOI] [PubMed] [Google Scholar]
- 20.Isken O, Maquat LE. Quality control of eukaryotic mRNA: safeguarding cells from abnormal mRNA function. Genes Dev. 2007;21(15):1833–1856. doi: 10.1101/gad.1566807. [DOI] [PubMed] [Google Scholar]
- 21.Lakich D, Kazazian HH, Jr, Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nat Genet. 1993;5(3):236–241. doi: 10.1038/ng1193-236. [DOI] [PubMed] [Google Scholar]
- 22.Levinson B, Kenwrick S, Gamel P, Fisher K, Gitschier J. Evidence for a third transcript from the human factor VIII gene. Genomics. 1992;14(3):585–589. doi: 10.1016/s0888-7543(05)80155-7. [DOI] [PubMed] [Google Scholar]
- 23.Kyewski B, Klein L. A central role for central tolerance. Annu Rev Immunol. 2006;24:571–606. doi: 10.1146/annurev.immunol.23.021704.115601. [DOI] [PubMed] [Google Scholar]
- 24.Klein L, Hinterberger M, Wirnsberger G, Kyewski B. Antigen presentation in the thymus for positive selection and central tolerance induction. Nat Rev Immunol. 2009;9(12):833–844. doi: 10.1038/nri2669. [DOI] [PubMed] [Google Scholar]
- 25.Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12(8):557–569. doi: 10.1038/nri3254. [DOI] [PubMed] [Google Scholar]
- 26.Poon IKH, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. 2014;14(3):166–180. doi: 10.1038/nri3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yanover C, Jain N, Pierce G, Howard TE, Sauna ZE. Pharmacogenetics and the immunogenicity of protein therapeutics. Nat Biotechnol. 2011;29(10):870–873. doi: 10.1038/nbt.2002. [DOI] [PubMed] [Google Scholar]
- 28.Karosiene E, Rasmussen M, Blicher T, Lund O, Buus S, Nielsen M. NetMHCIIpan-3.0, a common pan-specific MHC class II prediction method including all three human MHC class II isotypes, HLA-DR, HLA-DP and HLA-DQ. Immunogenetics. 2013;65(10):711–724. doi: 10.1007/s00251-013-0720-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lazarski CA, Chaves FA, Jenks SA, et al. The kinetic stability of MHC class II:peptide complexes is a key parameter that dictates immunodominance. Immunity. 2005;23(1):29–40. doi: 10.1016/j.immuni.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 30.Sauna ZE, Pandey GS, Jain N, Mahmood I, Kimchi-Sarfaty C, Golding B. Plasma derivatives: new products and new approaches. Biologicals. 2012;40(3):191–195. doi: 10.1016/j.biologicals.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 31.Kimchi-Sarfaty C, Schiller T, Hamasaki-Katagiri N, Khan MA, Yanover C, Sauna ZE. Building better drugs: developing and regulating engineered therapeutic proteins. Trends Pharmacol Sci. 2013;34(10):534–548. doi: 10.1016/j.tips.2013.08.005. [DOI] [PubMed] [Google Scholar]