

Abstract
At depolarized membrane potentials, the conductance of some voltage-gated K+ channels is reduced by C-type inactivation. This gating process is voltage independent in Kv1 and involves a conformational change in the selectivity filter that is mediated by cooperative subunit interactions. C-type inactivation in hERG1 K+ channels is voltage-dependent, much faster in onset and greatly attenuates currents at positive potentials. Here we investigate the potential role of subunit interactions in C-type inactivation of hERG1 channels. Point mutations in hERG1 known to eliminate (G628C/S631C), inhibit (S620T or S631A) or enhance (T618A or M645C) C-type inactivation were introduced into subunits that were combined with wild-type subunits to form concatenated tetrameric channels with defined subunit composition and stoichiometry. Channels were heterologously expressed in Xenopus oocytes and the two-microelectrode voltage clamp was used to measure the kinetics and steady-state properties of inactivation of whole cell currents. The effect of S631A or T618A mutations on inactivation was a graded function of the number of mutant subunits within a concatenated tetramer as predicted by a sequential model of cooperative subunit interactions, whereas M645C subunits increased the rate of inactivation of concatemers, as predicted for subunits that act independently of one another. For mutations located within the inactivation gate proper (S620T or G628C/S631C), the presence of a single subunit in a concatenated hERG1 tetramer disrupted gating to the same extent as that observed for mutant homotetramers. Together, our findings indicate that the final step of C-type inactivation of hERG1 channels involves a concerted, all-or-none cooperative interaction between all four subunits, and that probing the mechanisms of channel gating with concatenated heterotypic channels should be interpreted with care, as conclusions regarding the nature of subunit interactions may depend on the specific mutation used to probe the gating process.
Key points
C-type inactivation of voltage-gated K+ channels is caused by a conformational change in the selectivity filter that prevents ion conductance.
The role of subunit interaction during C-type inactivation of hERG1 K+ channels was characterized by using concatenated tetrameric channels containing a defined composition and stoichiometry of wild-type subunits and subunits with a mutation known to attenuate or accentuate inactivation gating.
Analysis of the kinetics and voltage dependence of steady-state inactivation for the concatenated channels indicated a variable extent of subunit interaction, dependent on the location of the mutation used to probe the gating process.
Mutations located in the selectivity filter or pore helix disrupted inactivation in a dominant-negative manner, suggesting that the final step of C-type inactivation of hERG1 K+ channels is mediated by a concerted, highly cooperative interaction between all four subunits.
Introduction
Inactivation of voltage-gated K+ (Kv) channels reduces outward K+ permeation during transient membrane depolarization to modulate action potential duration and the firing pattern of excitable cells. Two mechanisms of Kv channel inactivation, known as N-type and C-type are widely recognized. N-type inactivation is mediated by block of the central cavity of an open channel by a ‘ball’ domain located at the N-terminus of a Kv α-subunit (Hoshi et al. 1990, 1991) or an auxillary β-subunit (Rettig et al. 1994). N-terminal domains act fully independently of one another and only a single N-terminal domain is required to occlude the pore and prevent ion conductance (Zhou et al. 2001). C-type inactivation of Kv1 channels (Hoshi et al. 1991) has been attributed to a constriction of the narrow pore formed by the selectivity filter (Liu et al. 1996; Yellen et al. 1994), and is mediated by highly cooperative subunit interactions (Ogielska et al. 1995; Panyi et al. 1995; Yang et al. 1997). Recently it was proposed that C-type inactivation can occur in the absence of the pore constriction (Devaraneni et al. 2013) and might instead result from a subtle dilation of the pore at the outermost K+ binding site within the selectivity filter (Hoshi & Armstrong, 2013). This mechanism is compatible with the observation that raising the concentration of external K+ slows the rate of C-type inactivation (Pardo et al. 1992; Lopez-Barneo et al. 1993). In Kv1 channels such as Shaker, C-type inactivation is voltage independent and very slow with a time constant (τinact) of ∼1.5 s at 20°C (Hoshi et al. 1991). In contrast, C-type inactivation in hERG1 channels is fast and voltage-dependent with τinact that ranges from 2 to16 ms (Spector et al. 1996). The potential role of subunit interactions in fast C-type inactivation of hERG1 channels has not been investigated.
In the present study, we constructed concatenated hERG1 tetramers containing subunits that harbour a mutation that either disrupts or accentuates C-type inactivation. Several mutations in or near the pore helix/selectivity filter were selected for their known ability to inhibit inactivation gating, including the point mutations S620T (Ficker et al. 1998) and S631A (Schonherr & Heinemann, 1996), and the double mutation G628C/S631C (Smith et al. 1996). The single mutations T618A in the pore helix (Ferrer et al. 2011) or M645C in the S6 segment (Garg et al. 2011) accentuate inactivation by shifting its voltage dependence by −80 to −90 mV. Concatenated homotetrameric and heterotetrameric channels were constructed to contain a variable number of wild-type (WT) and mutant subunits with known stoichiometry. These concatemers were heterologously expressed in Xenopus laevis oocytes and two-microelectrode voltage clamp (TEVC) was used to determine the voltage dependence and kinetics of C-type inactivation gating.
Methods
Construction of hERG1 concatemers
WT and mutant forms of KCNH2 (HERG1, isoform 1a, NCBI Reference Sequence: NM_000238) cDNAs were cloned into the pSP64 poly(A) oocyte expression vector (Promega Corp., Madison, WI). Concatenated tetramers were engineered to contain a variable combination of WT and/or mutant KCNH2 cDNAs with defined positioning as shown in Fig. 1A. Four point mutations and one double mutation were chosen for study based on their known ability to either disrupt (S620T, S631A, G628C/S631C) or potentiate (T618A, M645C) inactivation gating. In the text and figures, concatenated tetramers formed by four WT or four mutant subunits are designated as WT4 and NM4, respectively, where N represents a native residue and M is a mutation (e.g. ST4 for a tetramer containing four subunits each harbouring a S620T mutation). Abbreviations for heterotypic tetramers indicate the relative positioning of the single WT and mutant subunits. For example, the ST1/WT1/ST1/WT1 channel was engineered to contain WT subunits in the second and fourth position together with subunits harbouring an S620T mutation in the first and third position of the tetramer (i.e. diagonal orientation of like subunits). Mutations were introduced into KCNH2 using the QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). Construction of dimers and fully concatenated tetramers was the same as previously described (Wu et al. 2014). All constructs were verified by DNA sequence analysis. Tetrameric KCNH2 plasmids were linearized with EcoR1 before in vitro transcription using the mMessage mMachine SP6 kit (Ambion Life Technologies, Grand Island, NY).
Figure 1. hERG1 concatenated tetramers and location of mutated residues characterized in this study.
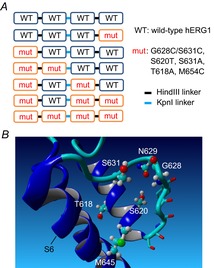
A, positioning of WT and mutant hERG1 subunits in seven different concatenated tetramers. The restriction enzymes used to link KCNH2 cDNAs are also indicated. B, location of residues in a hERG1 subunit that were mutated in this study, including Thr618 and Ser620 in the pore helix, Gly628 in the selectivity filter, Ser631 outside the selectivity filter, and Met645 in the S6 segment. Also shown is Asn629 that can interact with Ser620. Homology model of a single S5-S6 region of hERG1 was based on the KcsA channel structure (PDB ID: 1BL8) and prepared using YASARA (Krieger et al. 2009).
Isolation and voltage clamp of Xenopus oocytes
Procedures used for the surgical removal of ovarian lobes from Xenopus laevis and isolation of oocytes were approved by the University of Utah Institutional Animal Care and Use Committee and performed as described previously (Abbruzzese et al. 2010). Single stage 4 or 5 oocytes were injected with 10 ng cRNA encoding single hERG1 subunits and studied 1–3 days later. Some hERG1 tandem dimers and concatenated tetramers expressed poorly and therefore, oocytes were injected with 50 ng cRNA and studied 4–8 days later. The extracellular solution (‘98Na2K’) for most voltage clamp experiments contained (in mm): 98 NaCl, 2 KCl, 1 CaCl2, 1 MgCl2, 5 Hepes; pH was adjusted to 7.6 with NaOH. Ionic currents, filtered at 1 kHz and digitized at 2 kHz, were recorded using standard TEVC techniques (Goldin, 1991; Stühmer, 1992). Agarose cushion microelectrodes were prepared as described (Schreibmayer et al. 1994) and had a resistance of 0.3 MΩ when filled with 3 m KCl. For TEVC experiments, a GeneClamp 500 amplifier, Digidata 1322A data acquisition system and pCLAMP 8.2 software (Molecular Devices, Sunnyvale, CA) were used to produce command voltages and to record current and voltage signals.
Fully activated current–voltage (I–V) relationships were determined to characterize the voltage dependence of C-type inactivation. The pulse protocol consisted of an initial 1 s pulse (to +40 or +80 mV) followed by a 1 s pulse to a variable return potential (Vret) that ranged from +60 to −160 mV, depending on channel type. The peak value of tail current was determined by fitting the decay of the current trace to a one or two exponential function and extrapolating to the beginning of the return voltage step. Linear leak currents were subtracted and the resulting value of the peak tail current amplitude (Itail) was plotted as a function of Vret. To account for differences in channel expression, Itail values for each individual cell were normalized to the mean gmax for WT4 channels. The maximal slope conductance (gmax, in μA mV–1) of WT4 channel currents was estimated by fitting Itail measured at Vret ranging from −100 mV to −140 mV to a linear function. For oocytes expressing concatemers containing S620T or S631A mutant subunits, gmax was evaluated from Itail measured at Vret ranging from −40 mV to −120 mV.
Large outward K+ currents are associated with reduced hERG1 channel rectification, presumably because transient accumulation of extracellular K+ near the outer vestibule of the channel can inhibit C-type inactivation. For example, we previously reported that rectification of the I–V relationship measured for S620T and S631A hERG1 channels was reduced for large, compared to small outward currents (Casis et al. 2006). To minimize the effects of extracellular K+ accumulation, we optimized the amount of cRNA injection and time of expression to ensure that outward currents at +40 mV were <15 μA for those channel types that exhibited reduced inactivation (e.g. channels containing S620T, S631A or G628C/S631C subunits).
Channels containing subunits with either a T618A or M645C mutation exhibited enhanced inactivation and very small outward currents. Therefore, smaller stage 3 oocytes with reduced capacitance currents were used and cells were bathed in a high K+ solution to facilitate measurement of the rate of fast inactivating currents. The high K+ solution (‘96K4Na’) was prepared by modification of the standard extracellular solution: [K+] was increased to 96 mm and [Na+] was lowered to 4 mm.
Data analysis
Digitized data were analysed off-line with pCLAMP 8.2 and ORIGIN 8.5 (OriginLab, Northampton, MA) software. All data are presented as means ± s.e.m. (n = number of individual oocytes). Currents elicited with the fully activated I–V pulse protocol were used to estimate the voltage dependence of C-type inactivation. For each individual oocyte, Itail measured at a variable Vret was divided by the electrical driving force (Vret − Erev) then fitted with a Boltzmann function to determine the maximum value of Itail (Itail-max) by extrapolation to more negative potentials. Erev is the reversal potential for tail currents. Itail was normalized to the estimated value of Itail-max and the resulting relative conductance (g/gmax) was again plotted as a function of Vret and fitted with a standard Boltzmann function:
| (1) |
where F is the Faraday constant, R is the gas constant, T is the absolute temperature, V0.5 is the half-point (in mV) of the inactivation relationship, z is the equivalent charge for inactivation and c is the minimum value of g/gmax achieved at positive potentials.
We considered several models to examine whether hERG1 subunits act independently or cooperatively to induce C-type inactivation. For the independent model, where conformational changes in each of the individual subunits are assumed to contribute independently to inactivation gating, the predicted normalized inactivation curve for each concatenated tetramer is given by the product of two Boltzmann functions as follows:
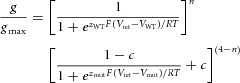 |
(2) |
where n is the number of WT subunits contained within a concatenated tetramer, zWT and zmut are the equivalent charge for inactivation, c is the minimum value of g/gmax and VWT and Vmut are the half inactivation voltages for concatenated homotypic WT and mutant channels, respectively. To define the voltage-dependent contribution of each subunit to the process of inactivation, the normalized inactivation curves for WT and mutant homotypic tetramers were each fit with a fourth power Boltzmann function. This approach is the same as that used previously to assess subunit interaction for activation of Kv1 channels (Hurst et al. 1992; Smith-Maxwell et al. 1998). Finally, the inactivation curves for heterotypic channels predicted by the independent model were fit with a standard Boltzmann function (eqn (1) and the calculated V0.5 and z values were used to estimate the free energy change associated with inactivation (ΔGinact):
| (3) |
As defined here, ΔGinact assumes that conformational changes in the channel that accompany inactivation are dominated by a single open to inactivated state transition. This is undoubtedly an oversimplification, as multiple open and inactivated states are necessary to fully describe single channel gating (Zou et al. 1997). Nonetheless, this approach greatly simplifies analysis and has proven useful for distinguishing widely divergent models (e.g. independent versus cooperative) of channel gating.
As described in a previous study of the conformational changes that lead to activation gate opening in Shaker K+ channels (Zandany et al. 2008), we considered two mechanistic models of subunit cooperativity. The standard cooperative model, also called the sequential cooperative model (Karpen & Ruiz, 2002; Zandany et al. 2008) assumes an equal energetic contribution from each subunit to the tetrameric channel function and describes the steady-state voltage dependence of activation (Hurst et al. 1992; Smith-Maxwell et al. 1998) and slow C-type inactivation (Ogielska et al. 1995; Panyi et al. 1995) in Kv channels. For this model, the predicted ΔGinact of a heterotetramer is the sum of the individual ΔGinact values contributed by each of the WT and mutant subunits as follows:
| (4) |
where n equals the number of WT subunits in a tetramer and ΔGWT4 and ΔGmut4 are the free energy values calculated for WT and mutant homotypic tetramers, respectively. Thus, the predicted voltage dependence of inactivation of heterotypic channels is,
| (5) |
and ΔGinact is a linear function of the number of WT or mutant subunits in a concatenated tetramer.
The concerted, all-or-none cooperative model of subunit interaction is exemplified by the final concerted step of Kv channel activation, where a simultaneous conformational change in all four subunits is required for channel opening (Sigworth, 1994; Zandany et al. 2008). This model predicts that a single mutant subunit will alter inactivation to the same extent as channels containing two, three or four mutant subunits per tetramer. For this model, ΔGinact of all the heterotetramers would be predicted to be the same as the homotypic mutant tetramer for mutations that disrupt inactivation (i.e. increase ΔGinact). Conversely, ΔGinact of all the heterotetramers would be predicted to be the same as WT4 channels for mutations that potentiate inactivation (i.e. reduce ΔGinact).
Results
A single G628C/S631C subunit is sufficient to eliminate C-type inactivation
The combined mutations of Gly628 and Ser631 to Cys eliminates inactivation of hERG1 channels (Smith et al. 1996). Gly628 is the outer Gly in the SVGFG K+ signature sequence of the selectivity filter and Ser631 is positioned just outside the selectivity filter (Fig. 1B). To determine if one or more subunits harbouring G628C/S631C (GCSC) mutations was required to attenuate inactivation, currents conducted by concatenated tetramers containing one, two, three or four mutant subunits were compared to WT4 homotypic channels. Currents were elicited in response to 4 s pulses applied in 10 mV increments to a Vt that ranged from −70 to +50 mV, and tail currents were measured at −70 mV. We previously reported that the biophysical properties of hERG1 whole cell currents conducted by concatenated WT4 channels was nearly identical to currents recorded from oocytes expressing single WT hERG1 subunits (Wu et al. 2014). Representative current traces at Vt of −20 and +20 mV for WT4, GCSC1/WT3 and GCSC4 channels are shown in Fig. 2A. For WT4 channels, the outward currents measured at the end of the test pulse (Itest) to −20 mV were approximately three times larger than Itest at +20 mV, a consequence of channel inactivation at the more depolarized transmembrane voltage. In contrast, Itest for GCSC1/WT3 and GCSC4 channels were about two times larger at +20 mV compared to −20 mV, reflecting reduced inactivation induced by the mutation. The Erev for WT4 channel currents was −95 mV and thus, tail currents were outward at the Vret of −70 mV. Erev for channels containing GCSC mutant subunits was −60 mV and tail currents were inward at −70 mV, indicating reduced K+ selectivity as previously reported (Smith et al. 1996). Normalized I–V (Itest–Vt) relationships for WT4, GCSC4 and four different heterotypic concatenated channels are plotted in Fig. 2B. Peak outward currents in response to a 4 s test pulse were normalized to the current measured at −20 mV for WT4 channels, or to 0 mV for GCSC4 and the heterotypic concatenated channels. The Itest–Vt relationship for WT4 channels was bell-shaped with a peak near −20 mV, and exhibited a negative slope conductance at more positive potentials. In contrast, the Itest–Vt relationship for channels containing one or more GCSC subunits was outwardly rectifying and linear for Vt > 0 mV (Fig. 2C). Thus, a single GCSC subunit eliminated inactivation, suggesting that concerted, all-or-none interactions between WT subunits are required for C-type inactivation of hERG1 channels.
Figure 2. A single G628C/S631C hERG1 subunit eliminates C-type inactivation.
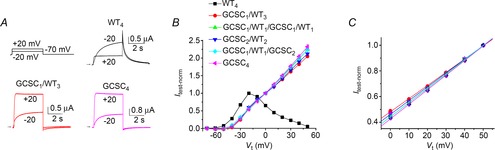
A, representative current traces for WT4, GCSC1/WT3 and GCSC4 concatenated tetramers recorded during 4 s pulses to −20 mV and +20 mV. Oocytes were bathed in 98Na2K extracellular solution. B, normalized current (Itest-norm) measured at the end of each 4 s test pulse plotted as a function of Vt for the indicated concatenated tetramers (n = 4–6). WT4 channel currents were normalized to Itest at −20 mV. GCSC4 and heterotypic tetramers were normalized to Itest at 0 mV. C, Itest-norm–Vt relationships for GCSC4 and heterotypic channels. Currents were normalized to Itest at +50 mV. All relationships were linear (continuous lines). Symbols refer to constructs as indicated in (B).
A single S620T subunit strongly attenuates C-type inactivation
Mutation of the pore helix residue Ser620 to Thr in hERG1 was reported to nearly abolish inactivation in hERG1 (Ficker et al. 1998). We find that this mutation greatly disrupts, but does not eliminate rectification when the level of heterologous channel expression is not excessive (Casis et al. 2006). Concatenated tetramers containing one or more S620T subunits were used to further probe the nature of subunit interaction during C-type inactivation of hERG1. Currents recorded using the fully activated Itail–Vret pulse protocol for WT4, ST1/WT3 and ST4 concatenated tetrameric channels are compared in Fig. 3A. Outward currents for both of the mutant channels were much larger compared to WT4 channels, indicating strong inhibition of inactivation. In Fig. 3B the fully activated Itail–Vret relationships for homotypic (WT4, ST4) and five different heterotypic channels are compared. To account for differences in level of channel expression between different constructs and individual oocytes, Itail values were normalized to the mean maximum slope conductance of WT4 channel currents. It is apparent from these plots that the presence of one or more S620T subunits within a concatenated tetramer was sufficient to attenuate inactivation greatly and caused a progressive positive shift in Erev, up to a maximum of 21 mV for ST4 channel currents, indicating reduced K+ ion selectivity. The fully activated Itail–Vret relationships for all the mutant tetramers were linear over the voltage range of −120 to 0 mV. At potentials >0 mV, the slope conductance for all the mutant channels was reduced. Plots of g/gmax versus Vret for all the constructs is summarized in Fig. 3C. When compared to WT4 channels, the half-point of this relationship was shifted by ≥ +83 mV for channels containing at least one S620T (Table 1). Inclusion of a single S620T subunit, either in the first or last position in a concatenated tetramer was equally capable of reducing inward rectification of the fully activated Itail–Vret relationships (Fig. 3B) and shifting the voltage dependence of rectification (Fig. 3C) to nearly the same extent achieved with homotypic ST4 channels. Variability in slope conductance of the Itail–Vret relationship observed at the most positive potentials could be explained in part by the contribution of endogenous outward currents relative to the amplitude of heterologously expressed hERG1 currents. To obtain a more quantitative measure of hERG1 channel rectification, we recorded ST1/WT3 and ST4 concatenated channel currents at potentials ranging from −50 to +140 mV, before and after block of hERG1 by 100 μm dofetilide, a highly specific hERG channel blocker. The Itail–Vret relationships for dofetilide-sensitive current are plotted in Fig. 3D and the calculated g/gmax–Vret relationships are plotted in Fig. 3E. The analysis indicates that g/gmax does not reach zero at very positive potentials for either channel type. The rectification of current of S620T mutant channels could result from highly attenuated inactivation, reduced open channel conductance at high potentials as previously reported for WT (Wang et al. 1996) and S631A (Zou et al. 1998) hERG1 channels expressed in oocytes, or a combination of both factors. Regardless of the underlying mechanism, it is clear that a single S620T subunit reduced channel rectification to the same extent as that obtained in the homotetrameric S620T channel. These findings indicate that C-type inactivation of hERG1 channels involves concerted, all-or-none interactions between WT subunits.
Figure 3. Single S620T hERG1 attenuates C-type inactivation.
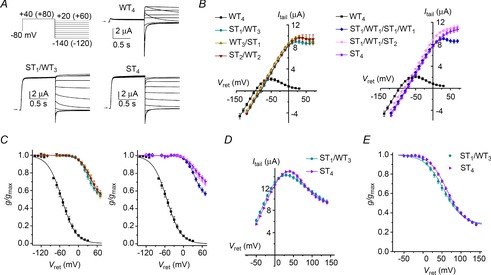
A, representative current traces for WT4, ST1/WT3 and ST4 concatenated hERG1 tetramers recorded during pulses to indicated voltages. The first pulse was to +40 mV for WT4 channels and +80 mV for channels containing mutant subunits. Vret was varied from +20 to −140 for WT4 channels and 60 to −120 mV for mutant channels. Oocytes were bathed in 98Na2K extracellular solution. B, fully activated Itail–Vret relationships for indicated concatenated tetramers (n = 5–12). Y-axis refers to mean tail currents (Itail, in μA) for WT4 channels. To adjust for variable expression, the maximum slope conductance (gmax) for each mutant channel type was scaled to match the gmax for WT4 channels. C, voltage dependence of rectification for concatenated tetramers as indicated by symbols in (B). Values of g/gmax near Erev were highly variable and omitted from these plots. Values of V0.5 and z determined from fitting data to Boltzmann function are summarized in Table 1. D, fully activated drug-sensitive Itail–Vret relationships of ST4 and ST1/WT3 channels (n = 4 for both). Itail represents current blocked by 100 μm dofetilide. E, voltage dependence of rectification for ST4 channels (V0.5 = +61.8 ± 2.2 mV; z = 1.08 ± 0.01) and ST1/WT3 channels (V0.5 = +52.4 ± 3.0 mV; z = 0.96 ± 0.03).
Table 1.
Summary of the voltage dependence of activation and inactivation for hERG1 concatenated tetramers
| Extracellular solution | |||||
|---|---|---|---|---|---|
| Channel type | V0.5 (mV) | z (e0) | 98Na2K | 96K4Na | n |
| WT4 | −52.9 ± 3.6 | 1.11 ± 0.02 | • | 5 | |
| ST1/WT3 | +29.8 ± 2.7 | 1.64 ± 0.07 | • | 12 | |
| WT3/ST1 | +39.9 ± 2.8 | 1.43 ± 0.04 | • | 6 | |
| ST2/WT2 | +41.8 ± 3.3 | 1.41 ± 0.10 | • | 12 | |
| ST1/WT1/ST1/WT1 | +36.1 ± 1.7 | 1.73 ± 0.09 | • | 11 | |
| ST1/WT1/ST2 | +37.5 ± 2.2 | 1.60 ± 0.10 | • | 9 | |
| ST4 | +39.7 ± 3.8 | 1.52 ± 0.15 | • | 7 | |
| SA1/WT3 | −0.1 ± 4.2 | 0.95 ± 0.04 | • | 7 | |
| SA2/WT2 | +3.7 ± 4.1 | 1.18 ± 0.02 | • | 5 | |
| SA1/WT1/SA1/WT1 | +2.9 ± 2.5 | 1.21 ± 0.06 | • | 7 | |
| SA1/WT1/SA2 | +15.2 ± 3.2 | 1.37 ± 0.08 | • | 7 | |
| SA4 | +24.3 ± 5.3 | 1.29 ± 0.04 | • | 9 | |
| WT4 | −50.5 ± 1.9 | 0.71 ± 0.03 | • | 5 | |
| TA1/WT3 | −96.7 ± 4.4 | 0.66 ± 0.01 | • | 4 | |
| TA2/WT2 | −109.2 ± 3.2 | 0.65 ± 0.01 | • | 5 | |
| TA1/WT1/TA1/WT1 | −112.2 ± 5.4 | 0.66 ± 0.03 | • | 5 | |
| TA1/WT1/TA2 | −124.8 ± 3.5 | 0.77 ± 0.02 | • | 5 | |
| TA4 | −134.1 ± 1.6 | 0.79 ± 0.02 | • | 5 | |
| MC1/WT3 | −101.4 ± 3.0 | 0.59 ± 0.02 | • | 5 | |
| MC2/WT2 | −106.5 ± 2.2 | 0.59 ± 0.02 | • | 5 | |
| MC1/WT1/MC1/WT1 | −104.8 ± 2.9 | 0.67 ± 0.03 | • | 4 | |
| MC1/WT1/MC2 | −122.8 ± 4.5 | 0.61 ± 0.03 | • | 6 | |
| MC4 | −141.4 ± 3.0 | 0.62 ± 0.03 | • | 6 | |
Data are expressed as means ± s.e.m. (n = number of oocytes).
Disruption of inactivation by S631A mutation indicates cooperative, but not fully concerted subunit interactions
The point mutation S631A was reported to abolish inactivation of N-truncated hERG1 channels (Schonherr & Heinemann, 1996), or shift the voltage dependence of C-type inactivation to far more positive potentials in full-length hERG1 channels (Zou et al. 1998). Ser631 is located just outside the selectivity filter and thus, mutation to Ala may attenuate inactivation gating by an indirect, allosteric mechanism. Concatenated tetramers containing one or more S631A subunits were used to probe for interaction with WT subunits. Currents recorded using the fully activated Itail–Vret pulse protocol for SA1/WT3 and SA4 concatenated tetrameric channels are compared in Fig. 4A. Mutant channels had reduced inactivation compared to WT4 channels, but unlike what was observed for channels harbouring S620T mutations, inactivation was suppressed much more in homotypic mutant (SA4) channels than in heterotypic channels containing only a single mutant subunit (SA1/WT3). In Fig. 4B, the fully activated Itail–Vret relationships for homotypic (WT4, SA4) and heterotypic channels are compared. The suppression of inactivation was a graded function of the number of S631A subunits present in a concatenated tetramer. Slope conductance was negative at potentials positive to −50 mV for WT4 channels versus −10 mV or higher voltages for the heterotypic channels. The voltage dependence of steady-state inactivation for all tetramers studies is summarized in Fig. 4C. The V0.5 was shifted to more positive potentials as the number of S631A subunits/tetramer was increased without a significant change in z (Table 1). Compared to WT4 channels, V0.5 was shifted by +53, +56, +68 and +77 mV when concatenated channels contained one, two, three or four S631A subunits, respectively. Over the voltage range investigated, the reduced inward rectification of S631A mutant channels is caused predominantly by attenuation of C-type inactivation. The contribution of nonlinear open channel conductance to rectification of the g/gmax is only significant for Vret > +40 mV (Zou et al. 1998) and thus has only a minor effect on the g/gmax–Vret relationships. The voltage dependence of C-type inactivation of the heterotypic channels predicted by the independent and standard cooperative models are shown in Figs 4D and 4E, respectively. Neither of these models matches well with the measured inactivation curves for the heterotypic channels shown in Fig. 4C. ΔGinact calculated from measured data and ΔGinact predicted for heterotypic channels by the independent and two cooperative models were plotted together as a function of the number of WT subunits per tetramer (Fig. 4F). The ΔGinact values for measured data lie between the values predicted by the two cooperative models. The same was true after correcting g/gmax–Vret relationships for the contribution of nonlinear open channel conductance or assuming that inactivation reduced g/gmax to zero at high voltage for all the heteromeric tetramers (not shown). Thus, when S631A mutations were used to probe channel gating, we would conclude that highly cooperative, but not fully concerted (i.e. all-or-none) subunit interactions mediate C-type inactivation of hERG1 channels.
Figure 4. Graded inhibition of hERG1 inactivation by S631A subunits.
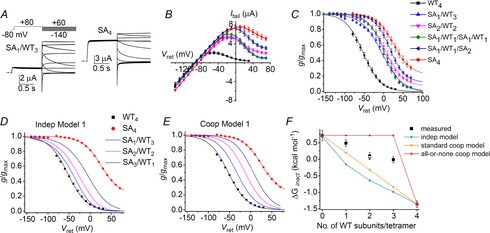
A, representative current traces for SA1/WT3 and SA4 concatenated tetramers recorded during pulses to indicated voltages. Oocytes were bathed in 98Na2K extracellular solution. B, fully activated Itail–Vret relationships for concatenated tetramers as indicated in the key in (C) (n = 5–9). Y-axis refers to mean WT4 channel currents (in μA). To adjust for variable expression, the gmax for each mutant channel type was scaled to match the gmax for WT4 channels. C, voltage dependence of inactivation for concatenated tetramers as indicated. Values of g/gmax at −90 mV (near Erev) were omitted. Values of V0.5 and z determined from fitting data to a Boltzmann function are summarized in Table 1. D and E, inactivation curves (g/gmax–Vret relationships) of WT4 and SA4 channels (same as mean data plotted in C) together with curves predicted for three heterotypic channels by the independent model (D) and the standard cooperative model (E). F, ΔGinact calculated from data (squares, n = 5–9) plotted as a function of the number of WT subunits contained in a concatenated tetramer is compared to ΔGinact predicted by the indicated independent and cooperative models. Open square represents SA1/WT1/SA1/WT1 channels.
Subunit interactions probed with point mutations that increase the rate of inactivation
C-type inactivation of homomeric hERG1 channels is potentiated by mutation of the pore helix residue Thr618 to Ala (Ferrer et al. 2011) or by mutation of the S6 segment residue Met645 to Cys (Garg et al. 2011). A consequence of this effect is that outward currents conducted by these mutant channels are barely detectable in oocytes bathed in an extracellular solution containing low levels of K+. To facilitate measurement of currents conducted by channels containing T618A or M645C mutant subunits, oocytes were bathed in 96K4Na extracellular solution. Channel inactivation was assessed with a fully activated Itail–Vret pulse protocol as before, but the magnitude of inward currents was greatly enhanced by the increased electrochemical driving force. Exemplary current traces using this protocol to measure currents conducted by WT4, TA1/WT3 and TA4 channels and their corresponding Itail–Vret relationships are shown in Fig. 5A and B, respectively. Note that outward tail currents are virtually absent. The voltage dependence of steady-state inactivation determined from the fully activated Itail–Vret relationships is summarized in Fig. 5C and Table 1 for WT4, TA4 and four different heteromeric concatenated channels. A single T618A subunit resulted in about half of the total shift in V0.5 observed for the TA4 channels. There was no difference in V0.5 when two T618A mutant subunits were positioned adjacent or diagonal to one another. The ΔGinact calculated from measured data and ΔGinact predicted for heterotypic channels by the independent and two cooperative models are plotted as a function of the number of T618A subunits per tetramer in Fig. 5D. The ΔGinact values predicted by the independent model and the standard cooperative model are very similar to each other because there is considerable overlap of the voltage range for inactivation for WT4 and TA4 channels and the slope of the inactivation curves are shallow (z values are small). The measured ΔGinact values for the heteromeric channels containing either one or three T618A subunits match best with the independent model, but channels with two T618A subunits match best with the standard cooperative model. In addition to the difficulty in distinguishing between the two model predictions, assuming that inactivation of these mutant channels can be reduced to a single O to I transition may be overly simplified (e.g. inactivation is a multistep process and the final step is not rate-limiting in the mutant channels), nullifying the ΔG analysis. Moreover, the calculated ΔG values may be confounded by our inability to measure directly the maximum conductance of these highly inactivated mutant channels. Therefore, we employed a different method, analysis of the kinetics of inactivation, to probe the nature of subunit interactions.
Figure 5. Effects of T618A subunits on the voltage dependence of hERG1 inactivation.
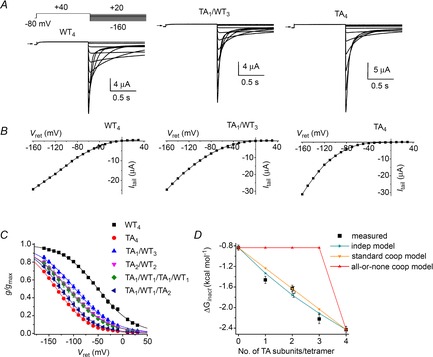
A, representative current traces for WT4, TA1/WT3 and TA4 concatenated tetramers recorded during pulses to indicated voltages. Oocytes were bathed in 96K4Na extracellular solution. B, fully activated Itail–Vret relationships for currents shown in (A). C, voltage dependence of inactivation for indicated concatenated tetramers. Values of V0.5 and z determined from fitting data to a Boltzmann function are summarized in Table 1. D, ΔGinact calculated from data (squares, n = 4–5) plotted as a function of the number of mutant T618A subunits contained in a concatenated tetramer is compared to ΔGinact predicted by the indicated independent and cooperative models. Open square represents TA1/WT1/TA1/WT1 channels.
A triple voltage pulse protocol (Spector et al. 1996) was used to measure the onset rate of hERG1 current inactivation. From a holding potential of −80 mV, channels were first activated/inactivated by a pulse to +40 mV, then allowed to recover from inactivation during a brief second pulse to −200 mV. After recovery from inactivation and before significant deactivation at −200 mV (3–6 ms), a third pulse was applied to a positive potential (20–100 mV) during which time open channels rapidly inactivate. For these experiments, currents were filtered at 5 kHz and digitized at 10 kHz. The rate of hERG1 inactivation for outward currents elicited by the third pulse of a three-pulse protocol was determined by fitting the rate of ionic current decay to a single exponential function:
| (6) |
Where I is the inactivating current, A1 is the steady-state current, A2 is the amplitude of inactivating current and τinact is the time constant for current decay. At the depolarized potentials examined, A1 was negligible after subtraction of leak currents. To reduce interference from the decay of capacitance currents that merge with inactivating ionic currents in TEVC recordings, the current fitting region was initiated 2 ms after the onset of the depolarizing third pulse. Assuming a two-state model for hERG1 channel inactivation 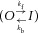 , then:
, then:
| (7) |
At sufficiently positive voltages (≥ +20 mV), kb is assumed to be negligible. Thus, the inactivation rate constant k can be estimated by 1/τinact. If an independent conformational change in any one of the subunits is sufficient to cause inactivation, then k would be the sum of the individual rate constants contributed by each of the subunits within a tetramer (Ogielska et al. 1995):
| (8) |
where n is the number of WT subunits contained in a tetramer and kWT and kmut are the inactivation rate constants for the WT and mutant concatenated tetramers, respectively. If C-type inactivation is a cooperative process, then the inactivation rate of a heterotypic channel would be exponentially related to the sum of the energetic contributions of each of the WT and mutant subunits (Ogielska et al. 1995) and log k would be a linear function of the number of mutant subunits contained with a tetramer.
The fitting of inactivating hERG1 currents with a single exponential function is illustrated in Fig. 6A. The time constant of inactivation (τinact) was determined for a range of Vt (+20 to +100 mV) for WT4, TA4 and four heterotypic channels. The value 1/τinact for inactivation increased as a function of Vt and the number of T618A subunits per tetramer (Fig. 6B). The calculated k at +60 mV and +80 mV for the heteromeric channels matched the values for k predicted by the standard cooperative model (linear relationship between log k and number of T618A subunits per tetramer), but not for k predicted by the independent model (Fig. 6C and D).
Figure 6. Cooperative T618A subunit interactions mediate accelerated rate of hERG1 inactivation.
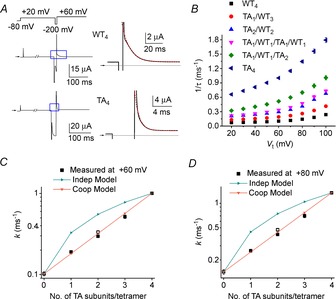
A, triple pulse voltage clamp protocol and corresponding currents recorded from oocytes expressing WT4 or TA4 hERG1 channels. Left panels show currents recorded in response to the indicated voltage steps. The first pulse was applied for 1 s and the second was 5 ms in duration for WT4 and 4.5 ms for TA4 channels. Expanded view of area designated by blue box is shown in right-hand panels. Inactivating current elicited during third pulse to +60 mV was fitted to a single exponential function (dotted red curves) to estimate τinact. B, plot of 1/τinact as a function of voltage for indicated concatenated tetramers (n = 4–7). C and D, estimated forward rate constants for inactivation (k) and k predicted by two models are plotted as a function of the number of T618A subunits in a concatenated tetramer for Vt of +60 mV (C) and +80 mV (D). Open square represents TA1/WT1/TA1/WT1 channels.
Channel inactivation was next probed using M645C, a mutation located in the S6 segment. Tail currents recorded in response to a variable Vret for WT4, MC1/WT3 and MC4 channels are shown in Fig. 7A and corresponding Itail–Vret relationships are shown in Fig. 7B. The voltage dependence of steady-state inactivation is summarized in Fig. 7C and Table 1 for WT4, MC4 and four heteromeric concatenated channels. Similar to T618A, a single M645C subunit resulted in about half of the total shift in V0.5 observed for the MC4 channels. There was a small difference in V0.5 when two M645C mutant subunits were positioned adjacent or diagonal to one another. The ΔGinact calculated from measured data and that predicted for heterotypic channels by the independent and cooperative models are plotted as a function of the number of M645C subunits per tetramer in Fig. 7D. ΔGinact values predicted by the independent and standard cooperative models are almost indistinguishable, again because of the considerable overlap in the voltage range of inactivation for WT and mutant homomeric channels. Therefore, we analysed inactivation kinetics to probe for potential subunit interactions. Currents used to characterize the onset of hERG1 inactivation are illustrated in Fig. 8A for WT4 and MC4 channels. The mean values for 1/τinact as a function of Vt for WT4, MC4 and four heterotypic channels are plotted in Fig. 8B. The calculated and predicted values of k as a function of the number of M645C subunits per tetramer at +60 mV and +80 mV are plotted in Fig. 8C and D. With only one exception (MC1/WT1/MC1/WT1 channels), the measured values of k match those predicted by the independent model.
Figure 7. Effects of M645C subunits on the voltage dependence of hERG1 inactivation.
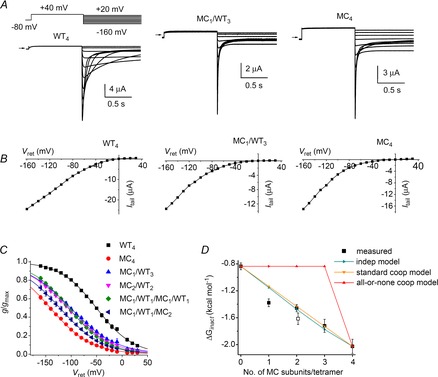
A, representative current traces for WT4, MC1/WT3 and MC4 concatenated tetramers recorded during pulses to indicated voltages. Oocytes were bathed in 96K4Na extracellular solution. B, fully activated Itail–Vret relationships for currents shown in (A). C, voltage dependence of inactivation for indicated concatenated tetramers. Values of V0.5 and z determined from fitting data to Boltzmann function are summarized in Table 1. D, ΔGinact calculated from data (squares, n = 4–6) plotted as a function of the number of mutant M645C subunits contained in a concatenated tetramer together with ΔGinact predicted by the indicated independent and cooperative models. Open square represents MC1/WT1/MC1/WT1 channels.
Figure 8. Independent M645C subunit interactions mediate accelerated rate of hERG1 inactivation.
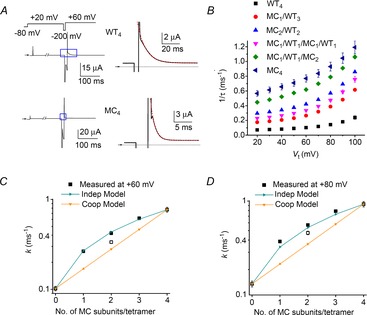
A, triple pulse voltage clamp protocol and corresponding currents recorded from oocytes expressing WT4 or MC4 hERG1 channels. Left panels show currents recorded in response to the indicated voltage steps. The first pulse was 1 s in duration and the second was 5 ms in duration for WT4 and 3.5 ms for MC4 channels. Expanded view of area designated by blue box is shown in right-hand panels. Inactivating current elicited during third pulse to +60 mV was fitted with a single exponential function (dotted red curves) to estimate τinact. B, plot of 1/τinact as a function of voltage for indicated concatenated tetramers (n = 4–10). C and D, estimated forward rate constants for inactivation (k) and k predicted by indicated models are plotted as a function of the number of M645C subunits in a concatenated tetramer at +60 mV (C) and +80 mV (D). Open square represents MC1/WT1/MC1/WT1 channels.
Discussion
Despite intense scrutiny, the molecular mechanisms and structural basis by which C-type inactivation prevents ion permeation in Kv channels remain uncertain. We studied the effects of several different mutations incorporated into one or more subunits of concatenated tetramers to probe the nature of subunit interactions during C-type inactivation of hERG1 channels. The study of concatenated tetramers can be complicated by unanticipated co-assembly of multiple units (McCormack et al. 1992; Hurst et al. 1995; Sack et al. 2008). However, proper assembly can be achieved if subunits that are nearly identical to one another (e.g. WT subunits plus subunits with a single point mutation) are linked together (Hurst et al. 1992; Sack et al. 2008). As discussed recently (Wu et al. 2014), several of our findings with hERG1 tetrameric channels indicate that formation of multimerized concatamers is unlikely and that formation of multimerized concatemers probably did not unduly complicate analysis of our experimental findings. The biophysical properties of currents recorded from cells expressing WT4 channels or single WT hERG1 subunits are very similar (Wu et al. 2014). Concatenated hERG1 tetramers expressed with a similar efficiency regardless of the number of mutant subunits per tetramer. With one exception, the positioning of two mutant subunits (S620T, S631A or T618A) in a diagonal versus an adjacent orientation did not affect biophysical properties of the channel. MC1/WT1/MC1/WT1 channels inactivated slower than MC2/WT2 channels, suggesting that when positioned adjacent to one another mutant subunits may weakly cooperate to accelerate inactivation.
The presence of a single S620T or G628C/S631C mutant subunit in a tetramer was sufficient to suppress or eliminate inactivation to the same extent as their respective homotypic mutant channels, indicative of fully concerted, all-or-none subunit interactions. This finding is similar to the subunit interactions that mediate channel opening of Shaker Kv1 channels, where a mutation (G466P) in the glycine hinge of a single subunit is sufficient to induce channels to open at more negative potentials to the same extent achieved by mutation of Gly466 in all four subunits (Zandany et al. 2008). S631A and T618A subunits altered gating in a manner consistent with subunit cooperativity, whereas M645C subunits accelerated the rate of inactivation as expected if subunits acted independently. A simple explanation for this apparent discrepancy would be that these mutations interfere with different steps of the inactivation pathway. Rate equilibrium free energy relationship analysis suggests that C-type inactivation of hERG1 involves sequential conformational changes in channel segments in the following order: S5, S5P, S4, S45 linker, S6, pore helix, selectivity filter (Wang et al. 2011; Perry et al. 2013a,b). Below we relate the location of the residues mutated in our study to their functional effects on subunit interactions associated with inactivation gating of hERG1.
Based on their location in the pore helix and selectivity filter, mutation of Ser620 (S620T) and Gly628 (G628C/S631C) would be predicted to disrupt the final step of C-type inactivation gating. Molecular dynamics simulations provide plausible explanations for how these mutations attenuate or prevent C-type inactivation of hERG1 channels. Interaction between Ser620 and Asn629 appear to be the critical determinant of whether the selectivity filter gate is in an open (conducting) or closed (inactivated) state. Modelling by Stansfeld et al. (2008) suggests that formation of an intrasubunit disulphide bond between G628C and S631C prevents the structural change in the selectivity filter required for C-type inactivation to occur. Ser620 and Asn629 form an H-bond with a water molecule behind the selectivity filter and this interaction favours the conducting state of the selectivity filter gate. Mutation of Ser620 to Thr stabilizes this interaction and promotes the conducting state of the selectivity filter (Stansfeld et al. 2008). In a more recent model, the collapsed (non-conductive) state of the selectivity filter was most likely when the distance between the side chains of Ser620 and Asn629 was reduced to a distance that favoured formation of a bidentate H-bond (Kopfer et al. 2012). In the S620T mutant, the additional methyl group adds steric bulk, which may alter a potential hydrogen bond network, whereas steric hindrance introduced by the disulphide bond formed between Cys628 and Cys631 prevents H-bonding between Ser620 and Asn629. Our finding that a single S620T subunit within a hERG1 concatemer suppressed inactivation as effectively as four mutant subunits suggests that preventing a single intra-subunit Ser620-Asn629 interaction is sufficient to disrupt gating fully. These results also indicate that inactivation is all-or-nothing with respect to the requirement for Ser620–Asn629 bonding; all four bonds must be formed for rapid C-type inactivation to occur over the normal voltage range.
Ser631 is located just outside the selectivity filter. Ser631 in hERG1 is homologous to Thr449 in Shaker. Mutation of Ser631 to Ala in hERG1 slows the rate of inactivation and shifts the voltage dependence of inactivation to more positive potentials, whereas mutation of Thr449 to Ala in Shaker accelerates the rate of voltage independent inactivation (Lopez-Barneo et al. 1993). Nonetheless, characterization of concatemers incorporating these mutations indicates that subunits interact cooperatively during C-type inactivation for both hERG1 and Shaker (Ogielska et al. 1995) channels. The side chain of Thr618 faces towards S5 and away from the inactivation gate (Fig. 1B). Thus, S631A and T618A mutations may affect a rate-limiting step in the inactivation sequence that is mediated by cooperative conformational changes in individual subunits and that occurs earlier than the fully concerted final step. We recently reported that inhibition of hERG1 inactivation by ICA-105574 is also mediated by cooperative subunit interactions (Wu et al. 2014). This hERG1 agonist binds to a hydrophobic pocket located between two adjacent subunits and formed by specific residues in S5, S6 and the backside of the pore helix (Garg et al. 2011).
Our finding that the location of the mutation used to probe C-type inactivation provides insights into subunit cooperativity during sequential steps in the inactivation process echoes previous studies on activation gating in other Kv channels. Activation of Kv channels is initiated by a two-stepped outward movement of the voltage sensors in each of the four subunits. The first step occurs independently in each subunit while the second step involves cooperative subunit interactions (Mannuzzu & Isacoff, 2000). In most studies where mutant subunits were used to characterize channel activation, findings have been consistent with a standard (i.e. sequential) model (Zandany et al. 2008) of cooperative subunit interactions (Hurst et al. 1992; Tytgat & Hess, 1992; Zagotta et al. 1994a,b). However, a point mutation of the gating hinge (G466P) in a single subunit of concatenated tetrameric Shaker channels causes the same negative shift in the voltage dependence of activation as that measured for channels containing only mutant subunits, consistent with predictions of the concerted, all-or-none model of cooperative subunit interactions (Zandany et al. 2008).
Two mutations (T618A or M645C) that accelerated the rate and shifted the voltage dependence of inactivation of hERG1 channels were compared. To facilitate study of highly inactivated mutant channels, currents were recorded from oocytes bathed in a high K+ solution. The slope of the inactivation curves are very shallow (z = 0.6–0.7) when currents are measured under these ionic conditions and, therefore, ΔGinact for heterotypic concatemers predicted by the independent model were very similar to those predicted by the standard cooperative model. The pattern of ΔGinact as a function of the number of mutant subunits per tetramer did not match one model better. Therefore, we characterized the effect of the mutations on the rate of inactivation (k) as the two models predicted greater differences in k for the heteromeric channels. We could not measure outward currents from these channels when cells were bathed in a low external K+ bath solution. Thus, a limitation of our analysis is the untested assumption that the kinetics of inactivation of WT and mutant channels were slowed to a similar extent by the high external K+ solution. When probed with T618A or M645C mutations, analysis of the kinetics of inactivation indicated cooperative or independent subunit interactions, respectively. It is unclear how the T618A mutation alters inactivation gating, but molecular modelling indicates that the side chain of Thr618 interacts with Trp568 in the S5 segment and mutation of this residue (W568L) eliminates inactivation, suggesting that S5 movement during channel activation may be coupled to the inactivation gate via interaction between these two residues (Ferrer et al. 2011). Met645 is located in the equivalent position in the S6 segment as Ala463 in Shaker. Mutation of the Shaker residue (A463V) increases the rate of C-type inactivation by two orders of magnitude (Hoshi et al. 1991). The inactivation rate of Shaker channels formed from WT/A463V concatenated dimers was best described by transition state energy additivity (standard cooperative model). Similar effects were observed for the equivalent mutation (A413V) in Kv1.3 channels (Panyi et al. 1995). In contrast, we found that mutation of Met645 to Cys in hERG1 altered the rate of inactivation in a manner predicted for independent subunit interactions. Thus, probing hERG1 channel gating with two different mutations (T618A or M645C) that both accelerate inactivation rate suggested different types of subunit interactions, as did the mutation of residues located in the equivalent positions in the S6 segment of different K+ channels (M645C in hERG1, A463V in Shaker and A413V in Kv1.3). The M645C mutation may accelerate the rates of both an independent step in the inactivation pathway of hERG1 and the cooperative step involving the gate structure (i.e. pore helix/selectivity filter).
In summary, incorporation of mutant subunits that disrupt or enhance inactivation into concatenated tetramers with defined stoichiometry has provided insights into the molecular mechanisms of C-type inactivation gating in hERG1 channels. Mutation of single residues just outside the selectivity filter (S631A) or in the pore helix (T618A) suggests that highly cooperative subunit interactions mediate fast inactivation, consistent with earlier studies of C-type inactivation with Kv1 channels. Disruption of the Ser620–Asn629 interaction by the S620T mutation or formation of a disulphide bridge between Gly628 and Ser631 presumably interferes with a final, concerted conformational change in all four subunits that prevents ion permeation. Our findings also suggest that probing the mechanisms of channel gating with concatenated heterotypic channels should be interpreted with care as conclusions regarding the nature of subunit interactions may depend on the specific mutation used to probe the gating process.
Acknowledgments
None declared.
Glossary
- ΔGinact
free energy change associated with inactivation
- Erev
reversal potential
- GCSC
Gly628Cys/Ser631Cys
- gmax
maximal slope conductance
- hERG1
human ether-a-go-go-related gene 1
- Itail
peak value of tail current
- Itest
current at the end of a test pulse
- MC
Met645Cys
- SA
Ser631Ala
- ST
Ser620Thr
- TA
Thr618Ala
- TEVC
two-microelectrode voltage clamp
- τinact
time constant of inactivation
- Vret
return potential
- V0.5
half-point of inactivation curve
- WT
wild-type
Additional information
Competing interests
None declared.
Author contributions
W.W. was involved in the design of the experiments, collection, analysis and interpretation of data, and writing of the manuscript. A.G. participated in plasmid construction, analysis and interpretation of data. M.C.S. was involved in the conception and design of the experiments, analysis and interpretation of data, and writing of the manuscript. All authors have read and approved of the final version of the manuscript.
Funding
This work was supported by National Institutes of Health/National Heart, Lung, and Blood Institute (R01 HL055236 to M.C.S.).
References
- Abbruzzese J, Sachse FB, Tristani-Firouzi M, Sanguinetti MC. Modification of hERG1 channel gating by Cd2+ J Gen Physiol. 2010;136:203–224. doi: 10.1085/jgp.201010450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casis O, Olesen SP, Sanguinetti MC. Mechanism of action of a novel human ether-a-go-go-related gene channel activator. Mol Pharmacol. 2006;69:658–665. doi: 10.1124/mol.105.019943. [DOI] [PubMed] [Google Scholar]
- Devaraneni PK, Komarov AG, Costantino CA, Devereaux JJ, Matulef K, Valiyaveetil FI. Semisynthetic K+ channels show that the constricted conformation of the selectivity filter is not the C-type inactivated state. Proc Natl Acad Sci U S A. 2013;110:15698–15703. doi: 10.1073/pnas.1308699110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer T, Cordero-Morales JF, Arias M, Ficker E, Medovoy D, Perozo E, Tristani-Firouzi M. Molecular coupling in the human ether-a-go-go-related gene-1 (hERG1) K+ channel inactivation pathway. J Biol Chem. 2011;286:39091–39099. doi: 10.1074/jbc.M111.292060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficker E, Jarolimek W, Kiehn J, Baumann A, Brown AM. Molecular determinants of dofetilide block of HERG K+ channels. Circ Res. 1998;82:386–395. doi: 10.1161/01.res.82.3.386. [DOI] [PubMed] [Google Scholar]
- Garg V, Stary-Weinzinger A, Sachse F, Sanguinetti MC. Molecular determinants for activation of human ether-a-go-go-related gene 1 potassium channels by 3-nitro-N-(4-phenoxyphenyl) benzamide. Mol Pharmacol. 2011;80:630–637. doi: 10.1124/mol.111.073809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin AL. Expression of ion channels by injection of mRNA into Xenopus oocytes. Methods Cell Biol. 1991;36:487–509. doi: 10.1016/s0091-679x(08)60293-9. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Armstrong CM. C-type inactivation of voltage-gated K+ channels: Pore constriction or dilation? J Gen Physiol. 2013;141:151–160. doi: 10.1085/jgp.201210888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- Hurst RS, Kavanaugh MP, Yakel J, Adelman JP, North RA. Cooperative interactions among subunits of a voltage-dependent potassium channel. Evidence from expression of concatenated cDNAs. J Biol Chem. 1992;267:23742–23745. [PubMed] [Google Scholar]
- Hurst RS, North RA, Adelman JP. Potassium channel assembly from concatenated subunits: effects of proline substitutions in S4 segments. Receptors Channels. 1995;3:263–272. [PubMed] [Google Scholar]
- Karpen JW, Ruiz M. Ion channels: does each subunit do something on its own. Trends Biochem Sci. 2002;27:402–409. doi: 10.1016/s0968-0004(02)02124-2. [DOI] [PubMed] [Google Scholar]
- Kopfer DA, Hahn U, Ohmert I, Vriend G, Pongs O, de Groot BL, Zachariae U. A molecular switch driving inactivation in the cardiac K+ channel HERG. PLoS One. 2012;7:e41023. doi: 10.1371/journal.pone.0041023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger E, Joo K, Lee J, Raman S, Thompson J, Tyka M, Baker D, Karplus K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins. 2009;77(Suppl 9):114–122. doi: 10.1002/prot.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Jurman ME, Yellen G. Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron. 1996;16:859–867. doi: 10.1016/s0896-6273(00)80106-3. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, Hoshi T, Heinemann SH, Aldrich RW. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- Mannuzzu LM, Isacoff EY. Independence and cooperativity in rearrangements of a potassium channel voltage sensor revealed by single subunit fluorescence. J Gen Physiol. 2000;115:257–268. doi: 10.1085/jgp.115.3.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack K, Lin L, Iverson LE, Tanouye MA, Sigworth FJ. Tandem linkage of Shaker K+ channel subunits does not ensure the stoichiometry of expressed channels. Biophys J. 1992;63:1406–1411. doi: 10.1016/S0006-3495(92)81703-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogielska EM, Zagotta WN, Hoshi T, Heinemann SH, Haab J, Aldrich RW. Cooperative subunit interactions in C-type inactivation of K channels. Biophys J. 1995;69:2449–2457. doi: 10.1016/S0006-3495(95)80114-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panyi G, Sheng Z, Tu L, Deutsch C. C-type inactivation of a voltage-gated K+ channel occurs by a cooperative mechanism. Biophys J. 1995;69:896–903. doi: 10.1016/S0006-3495(95)79963-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo LA, Heinemann SH, Terlau H, Ludewig U, Lorra C, Pongs O, Stühmer W. Extracellular K+ specifically modulates a rat brain K+ channel. Proc Natl Acad Sci U S A. 1992;89:2466–2470. doi: 10.1073/pnas.89.6.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry MD, Ng CA, Vandenberg JI. Pore helices play a dynamic role as integrators of domain motion during Kv11.1 channel inactivation gating. J Biol Chem. 2013a;288:11482–11491. doi: 10.1074/jbc.M113.461442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry MD, Wong S, Ng CA, Vandenberg JI. Hydrophobic interactions between the voltage sensor and pore mediate inactivation in Kv11.1 channels. J Gen Physiol. 2013b;142:275–288. doi: 10.1085/jgp.201310975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettig J, Heinemann SH, Wunder F, Lorra C, Parcej DN, Dolly JO, Pongs O. Inactivation properties of voltage-gated K+ channels altered by presence of β-subunit. Nature. 1994;369:289–294. doi: 10.1038/369289a0. [DOI] [PubMed] [Google Scholar]
- Sack JT, Shamotienko O, Dolly JO. How to validate a heteromeric ion channel drug target: assessing proper expression of concatenated subunits. J Gen Physiol. 2008;131:415–420. doi: 10.1085/jgp.200709939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonherr R, Heinemann SH. Molecular determinants for activation and inactivation of HERG, a human inward rectifier potassium channel. J Physiol. 1996;493.3:635–642. doi: 10.1113/jphysiol.1996.sp021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreibmayer W, Lester HA, Dascal N. Voltage clamping of Xenopus laevis oocytes utilizing agarose-cushion electrodes. Pflugers Arch. 1994;426:453–458. doi: 10.1007/BF00388310. [DOI] [PubMed] [Google Scholar]
- Sigworth FJ. Voltage gating of ion channels. Q Rev Biophys. 1994;27:1–40. doi: 10.1017/s0033583500002894. [DOI] [PubMed] [Google Scholar]
- Smith-Maxwell CJ, Ledwell JL, Aldrich RW. Role of the S4 in cooperativity of voltage-dependent potassium channel activation. J Gen Physiol. 1998;111:399–420. doi: 10.1085/jgp.111.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PL, Baukrowitz T, Yellen G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature. 1996;379:833–836. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- Spector PS, Curran ME, Zou A, Keating MT, Sanguinetti MC. Fast inactivation causes rectification of the IKr channel. J Gen Physiol. 1996;107:611–619. doi: 10.1085/jgp.107.5.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfeld PJ, Grottesi A, Sands ZA, Sansom MS, Gedeck P, Gosling M, Cox B, Stanfield PR, Mitcheson JS, Sutcliffe MJ. Insight into the mechanism of inactivation and pH sensitivity in potassium channels from molecular dynamics simulations. Biochemistry. 2008;47:7414–7422. doi: 10.1021/bi800475j. [DOI] [PubMed] [Google Scholar]
- Stühmer W. Electrophysiological recording from Xenopus oocytes. Methods Enzymol. 1992;207:319–339. doi: 10.1016/0076-6879(92)07021-f. [DOI] [PubMed] [Google Scholar]
- Tytgat J, Hess P. Evidence for cooperative interactions in potassium channel gating. Nature. 1992;359:420–423. doi: 10.1038/359420a0. [DOI] [PubMed] [Google Scholar]
- Wang DT, Hill AP, Mann SA, Tan PS, Vandenberg JI. Mapping the sequence of conformational changes underlying selectivity filter gating in the Kv11.1 potassium channel. Nat Struct Mol Biol. 2011;18:35–41. doi: 10.1038/nsmb.1966. [DOI] [PubMed] [Google Scholar]
- Wang S, Morales MJ, Liu S, Strauss HC, Rasmusson RL. Time, voltage and ionic concentration dependence of rectification of h-erg expressed in Xenopus oocytes. FEBS Lett. 1996;389:167–173. doi: 10.1016/0014-5793(96)00570-4. [DOI] [PubMed] [Google Scholar]
- Wu W, Sachse FB, Gardner A, Sanguinetti MC. Stoichiometry of altered hERG1 channel gating by small molecule activators. J Gen Physiol. 2014;143:499–512. doi: 10.1085/jgp.201311038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Yan Y, Sigworth FJ. How does the W434F mutation block current in Shaker potassium channels? J Gen Physiol. 1997;109:779–789. doi: 10.1085/jgp.109.6.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G, Sodickson D, Chen T-Y, Jurman ME. An engineered cysteine in the external mouth of a K+ channel allows inactivation to be modulated by metal binding. Biophys J. 1994;66:1068–1075. doi: 10.1016/S0006-3495(94)80888-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Shaker potassium channel gating. III: Evaluation of kinetic models for activation. J Gen Physiol. 1994a;103:321–362. doi: 10.1085/jgp.103.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Dittman J, Aldrich RW. Shaker potassium channel gating. II: Transitions in the activation pathway. J Gen Physiol. 1994b;103:279–319. doi: 10.1085/jgp.103.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandany N, Ovadia M, Orr I, Yifrach O. Direct analysis of cooperativity in multisubunit allosteric proteins. Proc Natl Acad Sci U S A. 2008;105:11697–11702. doi: 10.1073/pnas.0804104105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Morais-Cabral JH, Mann S, MacKinnon R. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 2001;411:657–661. doi: 10.1038/35079500. [DOI] [PubMed] [Google Scholar]
- Zou A, Curran ME, Keating MT, Sanguinetti MC. Single HERG delayed rectifier K+ channels in Xenopus oocytes. Am J Physiol Heart Circ Physiol. 1997;272:H1309–H1314. doi: 10.1152/ajpheart.1997.272.3.H1309. [DOI] [PubMed] [Google Scholar]
- Zou A, Xu QP, Sanguinetti MC. A mutation in the pore region of HERG K+ channels reduces rectification by shifting the voltage dependence of inactivation. J Physiol. 1998;509:129–138. doi: 10.1111/j.1469-7793.1998.129bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]