

Abstract
Hypercapnic acidosis, common in mechanically ventilated patients, has been reported to exert both beneficial and harmful effects in models of lung injury. Understanding its effects at the molecular level may provide insight into mechanisms of injury and protection. The aim of this study was to establish the effects of hypercapnic acidosis on mitogen-activated protein kinase (MAPK) activation, and determine the relevant signalling pathways. p44/42 MAPK activation in a murine model of ventilator-induced lung injury (VILI) correlated with injury and was reduced in hypercapnia. When cultured rat alveolar epithelial cells were subjected to cyclic stretch, activation of p44/42 MAPK was dependent on epidermal growth factor receptor (EGFR) activity and on shedding of EGFR ligands; exposure to 12% CO2 without additional buffering blocked ligand shedding, as well as EGFR and p44/42 MAPK activation. The EGFR ligands are known substrates of the matrix metalloprotease ADAM17, suggesting stretch activates and hypercapnic acidosis blocks stretch-mediated activation of ADAM17. This was corroborated in the isolated perfused mouse lung, where elevated CO2 also inhibited stretch-activated shedding of the ADAM17 substrate TNFR1 from airway epithelial cells. Finally, in vivo confirmation was obtained in a two-hit murine model of VILI where pharmacological inhibition of ADAM17 reduced both injury and p44/42 MAPK activation. Thus, ADAM17 is an important proximal mediator of VILI; its inhibition is one mechanism of hypercapnic protection and may be a target for clinical therapy.
Key points
Hypercapnia is common in mechanically ventilated patients with lung injury; while CO2 can ameliorate experimental lung injury, it can also cause harm.
Because hypercapnia can protect against ventilator-induced lung injury (VILI), understanding its impact on key signalling pathways may provide insight into the mechanisms of VILI.
We show that hypercapnia blocks stretch-mediated activation of p44/42 mitogen-activated protein kinase (MAPK) signalling in alveolar epithelial cells; this occurs through inhibition of sheddase (i.e. the metalloprotease, ADAM17), which releases ligands that bind to the epidermal growth factor receptor.
In vivo pharmacological blockade of ADAM17 reduces downstream MAPK activation and attenuates VILI in a two-hit mouse model.
Thus, hypercapnia uncovered a novel ADAM17-dependent mechanism of VILI, and this represents a potential therapeutic target.
Introduction
Mechanical ventilation is necessary in most patients with acute respiratory distress syndrome, but can contribute to lung damage, causing morbidity and mortality (ARDS Network, 2000; Gattinoni & Pesenti, 2005). Such ventilator-associated lung injury, termed ventilator-induced lung injury (VILI) when studied in laboratory models, has a variety of contributory mechanisms (Gattinoni et al. 2010). While reduced tidal volume is clearly associated with improved survival (ARDS Network, 2000), it commonly results in elevation of 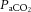 (Hickling et al. 1994), which may have independent positive or negative impact (Laffey & Kavanagh, 1999).
(Hickling et al. 1994), which may have independent positive or negative impact (Laffey & Kavanagh, 1999).
The mechanisms of VILI (Fan et al. 2013) include neutrophil infiltration, oxidative stress, eicosanoid production and intracellular signalling pathways such as nuclear factor-κB (NF-κB) (Liu et al. 2009) and mitogen-activated protein kinases (MAPKs) (Uhlig et al. 2002). Upregulation of the early growth response-1 (Egr1) gene by injurious ventilation contributes to VILI (Ngiam et al. 2010) and in cultured lung epithelial cells has been shown to be dependent on stretch-induced p44/42 MAPK activation (Copland & Post, 2007). MAPKs, comprising p44/42 MAPK, p38 MAPK and JNK, have been implicated in a variety of lung injury models initiated by high tidal volume (Uhlig et al. 2002; Dolinay et al. 2008; Ngiam et al. 2010), and MAPK phosphatases attenuate inflammatory responses in rodent lung injury (Park et al. 2012). In addition, cell culture models have linked MAPK activation to increased lung epithelial permeability in response to stretch and sepsis (Cohen et al. 2010). Pharmacological inhibition of p44/42 MAPK using PD98059 or U0126 has been shown to protect against VILI in mouse models (Li et al. 2007, 2013).
Activation of p44/42 MAPK in lung epithelial cells in response to mechanotransduction is dependent on activation of epidermal growth factor receptor (EGFR) (Correa-Meyer et al. 2002). EGF-like ligands, synthesized as transmembrane precursors, undergo shedding by metalloproteases (‘sheddases’), such that the ligands [e.g. heparin binding EGF-like growth factor (HB-EGF), transforming growth factor (TGF)α, amphiregulin (AREG), epiregulin (EREG)] are shed into the extracellular space where they bind and activate EGFR. A disintegrin and metalloprotease-17 [ADAM17, also known as tumour necrosis factor (TNF)α converting enzyme or TACE], has recently been identified as the major sheddase involved in release of these ligands (Shiomi et al. 2011). Accumulating knowledge of ADAM17 has demonstrated a role for this sheddase in lung injury in response to inflammatory stimuli (Arndt et al. 2011; Finigan et al. 2011; Dreymueller et al. 2012).
Deliberate elevation of CO2 can have varying effects in lung injury (Beitler et al. 2013; Curley et al. 2013), and while CO2 inhibits NF-κB activation (Takeshita et al. 2003) and Na+,K+-ATPase (Vadasz et al. 2008), understanding of its effects on cell signalling is incomplete. CO2, a highly soluble molecule, may affect multiple signalling systems.
While activation of sheddase activity followed by EGFR-mediated MAPK phosphorylation may contribute to VILI, the effects of hypercapnia on this pathway are unknown. We hypothesized that injurious cell stretch during mechanical ventilation would activate the sequential pathway components ADAM17, EGFR and p44/42 MAPK. Because these elements have been causally associated with lung injury, sheddase inhibition may be a therapeutic target and we further hypothesized that lung protection from hypercapnia would be associated with attenuation of this pathway.
Methods
Ethical approval
All animal procedures were reviewed and approved by the animal care committee of the Hospital for Sick Children (Toronto, ON, Canada) in accordance with the Guidelines of the Canadian Council on Animal Care.
Reagents
Primary antibodies were as follows: phospho-EGFR (Y1068), EGFR, phospho-p44/42 MAPK (T202/Y204) from Cell Signaling Technologies (New England Biolabs, Mississauga, ON, Canada); phospho-ADAM17 (T735) from AssaybioTech (Sunnyvale, CA, USA); ADAM17 from Thermo Scientific (Ottawa, ON, Canada); AREG, EREG and HB-EGF from Santa Cruz Biotechnology (Santa Cruz, CA, USA); and actin from Sigma-Aldrich (Oakville, ON, Canada). Cell culture media, HiPure plasmid isolation and Neon transfection kits were from Invitrogen (Burlington, ON, Canada). Inhibitor AG1478 was from Cell Signaling and TAPI from Calbiochem (Billerica, MA, USA).
Murine model of ventilator-induced lung injury
C57/BL6 male mice (20–25 g, Charles River, St Constant, QC, Canada) were anaesthetized (ketamine/xylazine 150/15 mg kg−1, i.p.), tracheostomized and ventilated using a computer-controlled small animal ventilator (SCIREQ; Flexivent, Montreal, Canada) as previously described (Peltekova et al. 2010). Baseline (protective) ventilation was VT 10 ml kg−1, positive end-expiratory pressure (PEEP) 2.0 cmH2O, frequency 135/min, on room air. Lung compliance was measured at baseline and hourly thereafter. Animals were randomized to four groups: to continue protective or receive injurious ventilation (peak inspiratory pressure of 27 cmH2O, PEEP 0 cmH2O, frequency 30–35/min) in the setting of normocapnia (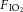 0.75,
0.75, 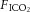 0, balance N2) or hypercapnia (
0, balance N2) or hypercapnia (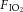 0.75,
0.75, 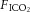 0.12, balance N2) for a period of 3 h (n = 5 per group in protective ventilation groups, n = 10 per group in injurious ventilation groups). After completion of the experiment, mice were killed by exsanguination under anaesthesia, bronchoalveolar lavage (BAL) performed for protein analysis, and lungs removed and snap frozen. Non-ventilated control mice (n = 5) were included for Western blot analyses.
0.12, balance N2) for a period of 3 h (n = 5 per group in protective ventilation groups, n = 10 per group in injurious ventilation groups). After completion of the experiment, mice were killed by exsanguination under anaesthesia, bronchoalveolar lavage (BAL) performed for protein analysis, and lungs removed and snap frozen. Non-ventilated control mice (n = 5) were included for Western blot analyses.
ADAM17 inhibition in vivo
C57/BL6 male mice were anaesthetized as above. Two doses (75 μg in 30 μl sterile saline) of TAPI-2 (Santa Cruz Biotechnology, Dallas, TX, USA) or vehicle (saline 30 μl) were given intratracheally and intravenously 1 h before the mouse received lipopolysaccharide (LPS; Ecoli 0111:54 Sigma Aldrich, 5 μg in 50 μl) intratracheally. The mouse was left to breathe spontaneously for 2 h while under general anaesthesia. An additional dose of TAPI-2 (75 μg) was then given intravenously. A tracheostomy was performed and baseline mechanical ventilation initiated as above. Initial compliance measurements were taken before starting injurious ventilation (VT 30 ml kg−1, PEEP 0 cmH2O and frequency 40/min) under normocapnia (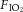 0.75, balance N2) for 3 h (n = 6 per group). Compliance measurements were taken hourly.
0.75, balance N2) for 3 h (n = 6 per group). Compliance measurements were taken hourly.
Rat primary alveolar epithelial cell isolation and stretch
Rat alveolar epithelial cells (AEC) were isolated as previously described (Gandhi et al. 2007). Briefly, 150–250 g male Sprague–Dawley rats were anaesthetized using ketamine/xylazine (80/8 mg kg−1). Rats were ventilated through a tracheal tube (tidal volume = 12 ml kg−1, PEEP = 1.0 cmH2O, frequency 38/min). A thoracotomy was performed, and heparin was injected into the right ventricle. After exsanguination, the lungs were perfused through the pulmonary artery and BAL was performed. ATII cells were loosened with elastase incubation (3 U ml−1; Worthington, Lakewood, NJ, USA) and then treated with trypsin inhibitor and DNase I (Worthington) before filtration through 100, 40 and 10 μm nylon mesh. Cells were purified based on differential adherence of cells to dishes coated with rat IgG (Sigma-Aldrich). Cells were seeded at 7.5 × 105 cells cm−2 on to laminin-coated Bioflex six-well plates (Flexcell International, Hillsborough, NC, USA) for stretch experiments. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 100 U ml−1 penicillin G sodium, and 100 μg ml−1 streptomycin sulphate. The culture medium was replaced 48 h after seeding to remove unattached cells. Twenty-four hours later, the medium was replaced by serum-free DMEM. The following morning, media was changed again using serum-free DMEM, which had been pre-equilibrated overnight in CO2 incubators under either 5% or 12% CO2 atmospheres. Inhibitors (AG1478, 150 nm; TAPI-1, 20 μm) or vehicle were added as indicated. Cells were incubated 60 min under either 5% or 12% CO2 and then remained under static condition or were subjected to cyclic stretch using a Flexcell FX-4000 Strain Unit (Flexcell International) for 10 min at a setting of 17% ΔSA equibiaxial strain at 0.5 Hz.
Western blots
Protein was isolated from lung tissue or AEC by extraction in RIPA buffer with protease and phosphatase inhibitors (20 mm Tris-HCl pH 8.0; 150 mm NaCl; 0.1% SDS; 1% Triton X-100; 0.5% sodium deoxycholate; 20 mm β-glycerophosphate; 10 mm NaF; 0.25 mm sodium orthovanadate, 1× Roche complete protease inhibitor (Roche Diagnostics, Laval, QC, Canada)). Protein concentrations were determined using the BioRad DC protein assay kit (BioRad Laboratories, Mississauga, ON, Canada). Proteins were size fractionated on SDS-PAGE gels and transferred to PVDF membrane. After blocking using 5% non-fat dry milk in TBST buffer [20 mm Tris pH 7.5; 140 mm NaCl; 0.1% (wt vol−1) Tween20], membranes were incubated overnight at 4°C with primary antibody in 5% bovine serum albumin in TBST buffer. Secondary antibody (horseradish peroxidase-conjugated) was diluted 1:10,000 in blocking buffer and incubated for 1 h at room temperature. All washes were in TBST. Enhanced chemiluminescence detection reagents were used according to the manufacturer's recommendations (Perkin Elmer, Waltham, MA, USA). Blots were stripped in 25 mm glycine-HCl pH 2; 1% SDS before being blocked and re-probed for normalization.
Quantitation of epidermal growth factor receptor ligand mRNAs
RNA was isolated from AEC cells following 10 or 30 min stretch using Trizol (Invitrogen) and further purified by LiCl precipitation. Relative quantitative real-time PCR was performed in duplicate on reverse transcribed cDNAs using an AB 7900HT Detection System (Applied Biosystems, Foster City, CA, USA) and Power SYBR Green (Applied Biosystems) reaction mix. Gene expression was calculated relative to 18S rRNA control and normalized to control cells held in static conditions under 5% CO2 using the comparative cycle threshold (ΔΔCt) method. Primers used were: HB-EGF: forward – 5′-GACCATGAAGCTGCTGCCGTCG-3′; reverse – 5′-ACCGGTCACCAACGCGGACA-3′; AREG: forward – 5′-GAGGCTGCGGCAAGAAAACGG-3′; reverse –5′ GTGTGGGTGTGGCTTGGCAGT-3′; EREG: forward – 5′-GTGTGGGTGTGGCTTGGCAGT-3′; reverse – 5′-CACTTGCGCCACACGGGGAT-3′.
Transfections/shedding assay
Plasmid DNAs encoding alkaline phosphatase (AP)-tagged expression vectors for the EGFR ligands HB-EGF, AREG, TGFα and EREG (Tokumaru et al. 2000) consist of a CMV-driven fusion protein consisting of the NH2-terminal HB-EGF signal sequence fused to full-length AP followed by the juxtamembrane site for ectodomain cleavage, transmembrane and cytoplasmic domains of the specified EGFR ligand. Transfection results in expression of AP in the ectodomain of the fusion protein, with release of AP activity into the culture medium dependent on ectodomain shedding. AEC (90–95% confluent) were trypsinized on day 2 after isolation, and subjected to electroporation (Neon transfection system; Invitrogen). Electroporated cells were seeded into Bioflex six-well plates at 9 × 105 cells well–1 in antibiotic-free DMEM/fetal bovine serum. Twenty-four hours after transfection, cell medium was replaced with serum-free medium; cells were entered into the stretch protocol as described above after 24 h serum starvation. Transfection efficiency was monitored by direct staining of fixed cells plated in 24-well plates with nitroblue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate after heat inactivation of endogenous APs.
Assessment of EGFR ligand shedding was performed as described (Wang et al. 2009) by pulldown of AP from 2 ml conditioned media using ConA-Sepharose (GE Healthcare Life Sciences, Baie d’Urfe, QC, Canada). Media was adjusted to contain 20 mm Tris pH 7.4; 1 mm MgCl2; 1 mm CaCl2; 1 mm MnCl2 to promote binding before addition of ConA beads and binding allowed to proceed overnight at 4°C in the presence of 1× Roche complete protease inhibitors with gentle inversion. ConA beads were centrifuged 5 min at 500 g, washed twice with ConA binding buffer and eluted in 20 μl 0.5 m α-methyl mannoside in 50 mm Tris pH 8.0. Supernatants were loaded on 7.5% SDS-PAGE in non-reducing SDS–sample buffer without previous boiling. SDS was removed from the gels by 30 min incubation in 2.5% (w/v) Triton X-100 and proteins allowed to renature for 30 min in AP buffer (100 mm Tris pH 9.5; 100 mm NaCl; 20 mm MgCl2). AP activity was visualized by staining with 0.2 mg ml−1 nitroblue tetrazolium/0.18 mg ml−1 5-bromo-4-chloro-3-indolyl phosphate at 37°C. Activity in matching cell lysates (direct lysis of monolayers in PBS containing 1% Triton X-100 and 1× Roche complete protease inhibitors) was measured by loading cell lysate on 7.5% SDS-PAGE and staining for AP activity as above. Intensity of staining was quantitated using ImageJ (NIH, Bethesda, MD, USA) and shedding expressed as AP activity in conditioned media divided by total AP (media + lysate) to control for transfection efficiency. The shedding assay was validated by demonstrating increased AP activity in supernatants of transfected (but not non-transfected) cells upon stimulation with phorbol 12-myristate 13-acetate, which was inhibitable by TAPI-1. In addition, no AP activity was detected in media from stretched, non-transfected cells, and TAPI-1 inhibited shedding of AP in response to stretch (data not shown).
Imaging of ADAM17 activity in isolated perfused lung
We applied real-time confocal microscopy in the isolated, perfused mouse lung to determine ADAM17-mediated TNFR1 shedding in intact alveoli as previously described (Rowlands et al. 2011).
Isolated perfused lung
We established the isolated perfused lung as previously described (Lindert et al. 2007; Islam et al. 2012, 2014). Briefly, we anaesthetized Swiss Webster mice (25 g) with 4% isoflurane followed by 80 mg kg−1 ketamine and 3 mg kg−1 xylazine intraperitoneally. We cannulated the pulmonary artery, left atrium and trachea and lungs were excised en bloc and then pump-perfused in Hepes (150 mmol l−1 Na+, 5 mmol l−1 K+, 1.0 mmol l−1 Ca2+, 1 mmol l−1 Mg2+ and 20 mmol l−1 Hepes at pH 7.4) containing 4% dextran (70 kDa) and 1% fetal bovine serum at pH 7.4 and osmolarity of 300 mosmol l–1 at a rate of 0.5 ml min−1 at 37°C. Pulmonary artery and left atrial pressures were held at 10 and 3 cmH2O, respectively. Lungs were constantly inflated through an airway cannula at airway pressure (Palv) of 5 cmH2O. For alveolar stretch we hyperinflate the lung by increased Palv from 5 to 15 cmH2O for 15 s (inflation to total lung capacity) (Perlman & Bhattacharya, 2007). The effects of CO2 were determined by continuously equilibrating the perfusion buffer to  of 40 mmHg (normocapnia) or 80 mmHg (hypercapnia).
of 40 mmHg (normocapnia) or 80 mmHg (hypercapnia).
Alveolar microinfusion
To label epithelial TNFR1surface expression, Alexa Fluor 633-conjugated TNFR1 antibodies against extracellular epitope (monoclonal antibody MCA2350; AbD Serotec, Raleigh, NC, USA) were infused by micropuncture (Kuebler et al. 2000; Lindert et al. 2007; Islam et al. 2014) of lung alveoli for 5 min followed by buffer wash to remove unbound antibody from the alveolar space. We carried out studies in alveoli that were not directly micropunctured.
Microscopy
Aveoli were viewed by laser scanning confocal microscopy (LSM 510; Carl Zeiss Microscopy, Heidelberg, Germany) using a ×40 water immersion objective (numerical aperture 0.80, Achroplan; Carl Zeiss Microscopy). We used light excitation at 488 nm for calcein green and at 633 nm for TNFR1. We viewed single alveoli in a 2 μm thick optical section at a 20 μm deep from the pleural surface. Alveolar fluorescence was quantified using MetaMorph software.
Statistical analysis
Data are presented as means ± s.d. Statistical differences were calculated with Sigmaplot v12.3 (Systat software Inc., San Jose, CA, USA) using one-way ANOVA followed by Holms–Sidak post-test unless otherwise indicated. P < 0.05 was considered significant.
Results
Lung injury (in vivo) and protection by inspired CO2
Baseline compliance was similar among all groups (data not shown). Average tidal volume at the start of injurious ventilation was 36.9 ml kg−1 in the hypercapnia group vs. 37.3 ml kg−1 in the normocapnia group (P = 0.58). Injurious ventilation induced a greater decrement in static lung compliance than protective ventilation (Fig. 1A); however, the decrement in lung compliance was less in the presence of hypercapnia (Fig. 1A). Similarly, lung microvascular leakage (BAL protein; Fig. 1B) was greater following injurious vs. protective ventilation, but reduced by hypercapnia.
Figure 1. Addition of 12% CO2 to inspired gas abrogates injury in mouse model of VILI.
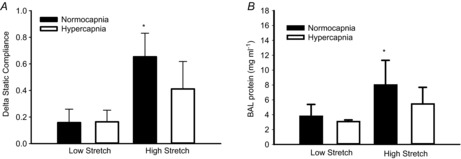
A, high VT mechanical ventilation under normocapnia induced a decrease in static compliance (*P < 0.05 vs. all other groups). B, high VT ventilation induced an increase in BAL protein only under normocapnia. *P < 0.05 vs. low stretch groups. n = 5 low stretch groups, n = 10 in high stretch groups. BAL, bronchoalveolar lavage.
Ventilator-induced lung injury: in vivo mitogen-activated protein kinases and inspired CO2
Because several MAPKs have been implicated in lung injury, we examined JNK, p38 MAPK and p44/42 MAPK in the early time-course of murine injurious ventilation (Fig. 2A). We found that only p44/42 MAPK showed both early and late activation (although this activation may be biphasic). Activation of JNK was transient (peaked at 5 min but returned to baseline levels within 30 min), while no significant activation of p38 MAPK was noted (Fig. 2A). Although transient activation of JNK may still be relevant via downstream cascades in VILI, we focused on p44/42 MAPK due to its longer-term activation, which may provide more opportunity for therapeutic targeting.
Figure 2. MAPK activation in mouse model of VILI.
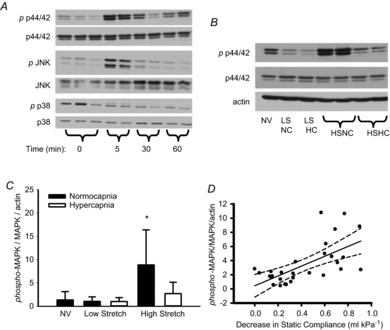
A, time course of MAPK activation during high stretch ventilation of mouse lung. B, representative Western blot demonstrates phosphorylation of p44/42 MAPK at 3 h in mechanically ventilated mouse lungs. C, p44/42 MAPK is activated by high VT only under normocapnia. *P < 0.05 vs. all other groups, n = 5 NV and LS groups, n = 10 HS groups. D, linear regression analysis indicating correlation between decrease in compliance and activation of p44/42 MAPK in ventilated mice. r2 = 0.406, P < 0.001, n = 30. HC, hypercapnia; HS high stretch; LS, low stretch; MAPK, mitogen-activated protein kinase; NC, normocapnia; NV, non-ventilated.
Following 3 h of injurious ventilation, p44/42 MAPK phosphorylation remained increased compared to protective ventilation (Fig. 2B). Hypercapnia resulted in less p44/42 MAPK activation (Fig. 2C). The degree of lung injury (i.e. decrement in lung compliance) positively correlated with the degree of activation of p44/42 MAPK (Fig. 2D).
Stretch-induced activation of p44/42 mitogen-activated protein kinase
To elucidate the roles of stretch and hypercapnic acidosis in the p44/42 MAPK activation pathway, we used primary AEC cultured at 5% (normocapnia) or 12% CO2 (hypercapnia) and subjected to cyclic stretch for 10 min. Western blot analysis of p44/42 MAPK phosphorylation (Fig. 3A) demonstrated activation by cyclic stretch under normocapnia, which was significantly reduced when cells were stretched under hypercapnia.
Figure 3. Cyclic stretch-induced activation of p44/42 MAPK in alveolar epithelial cells is dependent on EGFR activity; both are abrogated by hypercapnia.
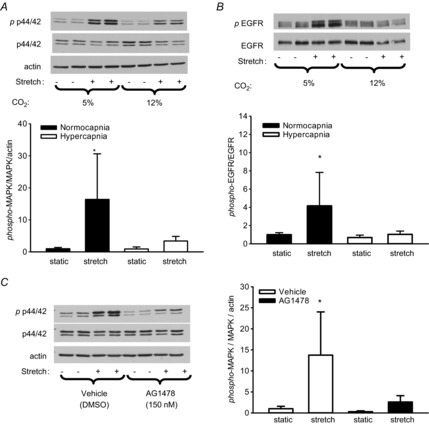
Western blots demonstrate phosphorylation of p44/42 MAPK (A) and of EGFR (B) induced by cyclic stretch under normocapnia (5% CO2 atmosphere) but not under hypercapnia (12% CO2). C, cyclic stretch-induced activation of p44/42 MAPK under normocapnia is blocked by 150 nm AG1478 *P < 0.05 vs. all other groups; n = 8. EGFR, epidermal growth factor receptor; MAPK, mitogen-activated protein kinase.
Because stretch can activate p44/42 MAPK via EGFR (Correa-Meyer et al. 2002), we examined EGFR phosphorylation in AEC subjected to cyclic stretch. Similar to MAPK phosphorylation, EGFR was activated by stretch under normocapnia, but not hypercapnia (Fig. 3B). The selective EGFR kinase inhibitor AG1478 (Fig. 3C) blocked stretch-induced activation of p44/42 MAPK, supporting EGFR dependence of stretch-induced p44/42 MAPK activation. In addition, EGFR activation in mouse lung in vivo exhibited a similar pattern of response to stretch and hypercapnia (Fig. 4), parallel to activation of p44/42 MAPK in vivo (Fig. 2) and consistent with EGFR and p44/42 MAPK in cultured AEC (Fig. 3).
Figure 4. EGFR activation in mouse model of VILI.
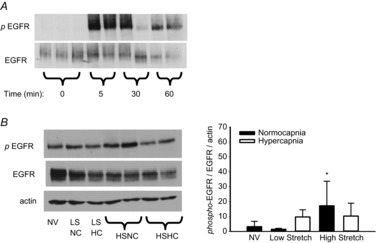
A, time course of EGFR activation during high stretch ventilation of mouse lung. B, following 3 h ventilation, EGFR remains significantly activated only in HSNC. †P < 0.05 vs. NV and LSNC, no other differences; n = 5 NV and LS groups, n = 10 HS groups. EGFR, epidermal growth factor receptor; HC, hypercapnia; HS high stretch; LS, low stretch; NC, normocapnia; NV, non-ventilated.
Impact of stretch and CO2 atmosphere on expression of epidermal growth factor receptor ligands
EGFR/MAPK signalling has been shown to include a positive autocrine feedback loop whereby mechanotransduction leads to activation of EGFR via ligand shedding, followed by a transcriptional increase in the gene expression of EGFR ligands (HB-EGF, AREG, EREG) (Kojic et al. 2010). Hence, we examined the effects of cyclic stretch and CO2 atmosphere on ligand mRNA expression in AEC. Cyclic stretch under normocapnia induced expression of all three mRNAs within 30 min (Fig. 5). Only EREG mRNA was induced by stretch under hypercapnia; however, this induction was significantly abrogated by hypercapnia.
Figure 5. Cyclic stretch-induced expression of EGFR ligand mRNAs is abrogated by hypercapnia.
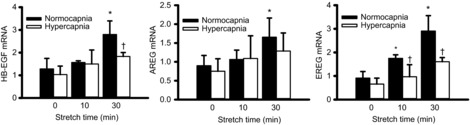
Relative quantitation by real-time polymerase chain reaction of mRNAs for HB-EGF, AREG and EREG, demonstrates induction within 30 min by cyclic stretch under normocapnia (5% CO2) (two-way ANOVA; *P < 0.05 compared to previous time point in same CO2 condition). This induction is reduced when cells are subjected to stretch under hypercapnia (12% CO2) (two-way ANOVA; †P < 0.05 vs. normocapnia at same time point; n = 4). AREG, amphiregulin; EREG, epiregulin; HB-EGF, heparin-binding epidermal growth factor-like growth factor.
Stretch-induced activation of sheddase
To confirm dependence on ligand shedding, we demonstrated that metalloprotease inhibition using the ADAM17 inhibitor TAPI-1 blocked stretch-induced EGFR and p44/42 MAPK activation (Fig. 6A and B). ADAM17 is synthesized as an inactive pro-enzyme, which undergoes post-translational modifications (including phosphorylation, glycosylation and maturation by cleavage of the propeptide) resulting in detection of multiple bands (80–130 kDa) on Western blots. Immunodetection of ADAM17 from AEC stretched under 5% or 12% CO2 suggested that neither stretch nor hypercapnic acidosis altered the expression, phosphorylation or maturation of ADAM17 protein (Fig. 7A).
Figure 6. Cyclic stretch activation of EGFR and p44/42 MAPK is dependent on ADAM17 activity.
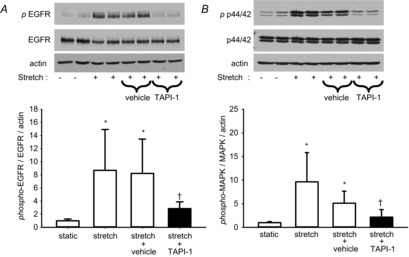
Western blots demonstrate activation of EGFR (A) and p44/42 MAPK (B) is blocked by TAPI-1. *P < 0.001 vs. static; †P < 0.05 vs. stretch+vehicle (dimethyl sulfoxide); n = 8. EGFR, epidermal growth factor receptor; MAPK, mitogen-activated protein kinase.
Figure 7. Hypercapnia reduces shedding of EGFR ligands in response to stretch via inhibition of ADAM17 activity.
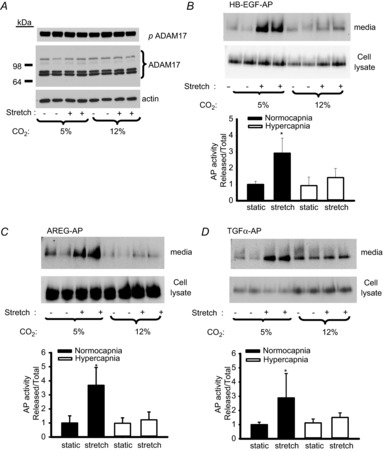
A, Western blot demonstrates that neither stretch nor CO2 affect ADAM17 protein phosphorylation, expression or maturation. B–D, alveolar epithelial cells were transfected with reporter plasmids encoding AP-tagged HB-EGF, AREG or TGFα. Shedding was quantitated as AP activity shed into media divided by total AP expression (media + cell lysate) for each well. *P < 0.05 vs. all other groups; n = 6. ADAM17, a disintegrin and metalloprotease-17; AP, alkaline phosphatase; AREG, amphiregulin; HB-EGF, heparin-binding epidermal growth factor-like growth factor; TGF, transforming growth factor.
Stretch-induced ligand release: impact of hypercapnia
We utilized AP-tagged EGFR ligands transiently transfected into AEC to assess shedding in response to cyclic stretch (Sahin et al. 2004). In AEC transfected with AP-tagged HB-EGF, cyclic stretch under normocapnia increased AP activity in the media approximately three-fold within 10 min; however, under hypercapnia, the stretch-induced shedding was blocked (Fig. 7B). Similar observations were made in cells transfected with AP-tagged AREG (Fig. 7C) and AP-tagged TGFα (Fig. 7D).
Effect of hypercapnia on alveolar stretch-induced ADAM17 activity in situ
To confirm effects of stretch and hypercapnic acidosis on ADAM17 activity we determined TNFR1 expression in situ by real-time confocal microscopy of live alveoli in mouse lung. To detect TNFR1 expression in alveolar epithelium, we utilized Alexa-633-conjugated anti-TNFR1 monoclonal antibody that ligates an extracellular domain of mouse TNFR1. Immunofluorescence at ≥50 grey levels at baseline conditions was accepted as TNFR1 surface expression and was stable for at least 1 h (Fig. 8A and C). The membrane-impermeable fluorescence quencher trypan blue (TB) abolished the TNFR1 fluorescence, which affirmed that the immunofluorescence was on the epithelial surface (data not shown). Alveolar stretch (a single 15 s hyperinflation) progressively decreased TNFR1 fluorescence (Fig. 8B–D), indicating that the stimulus induced ADAM17 activation. Stretch-induced TNFR1 shedding was inhibited by equilibration of supplemental CO2 (10%, hypercapnia) into the perfusate as compared with perfusate equilibrated with 5% CO2 (i.e. normocapnia) (Fig. 8B–D).
Figure 8. High CO2 inhibits INF-induced TNFR1 shedding in alveoli.
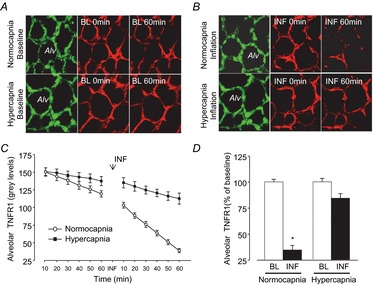
Confocal images (A and B) show alveolar epithelium stained with fluorescence-tagged TNFR1 monoclonal antibody (red) and cytosolic dye, calcein (green), and fluorescence intensity data (C and D) show real-time response to alveolar stretch (inflation). Tracings (C) and group data (D) show epithelial TNFR1 expression at BL and TNFR1 shedding response quantified by loss of TNFR1 fluorescence at 60 min. Hypercapnia abrogates inflation-induced TNFR1 shedding. Data are means ± s.e.m., n = 4 each bar (*P < 0.01 vs. corresponding BL). Alv, alveoli; BL, baseline; INF, inflation; TNFR1, tumour necrosis factor receptor 1.
Inhibition of ADAM17 protects in in vivo two-hit lung injury
To confirm the role of ADAM17 in an integrated model of VILI, we moved to a clinically relevant ‘two-hit’ murine model consisting of intratracheal LPS instillation before injurious ventilation. Mice treated with the ADAM17 inhibitor TAPI-2 developed less lung injury compared to vehicle-treated mice, as evidenced by less impairment of static compliance (Fig. 9A) and lower levels of BAL fluid protein (Fig. 9B). Effective blockade of ADAM17 was confirmed in the TAPI-2 group by greater than 10-fold suppression of soluble TNFα in BAL fluid (Fig. 9C). TAPI-2 treatment also significantly reduced the level of MCP-1 in BAL (405 ± 172.1 vs. 725 ± 139.0 pg ml−1, P = 0.008) although interleukin (IL)-6 (45 ± 12.1 vs. 59 ± 25 ng ml−1) and keratinocyte chemoattractant (KC) (36 ±6.3 vs. 36 ± 14.2 ng ml−1) were not significantly affected (n = 5 or 6 per group). Analysis of lung tissue from these mice showed that TAPI-2 successfully abrogated the increase in p44/42 MAPK activation, consistent with ADAM17-mediated activation of the EGFR/MAPK pathway in VILI (Fig. 9D).
Figure 9. Inhibition of ADAM17 with TAPI-2 protects mouse lung from LPS/VILI induced injury and decreases p44/42 MAPK activation.
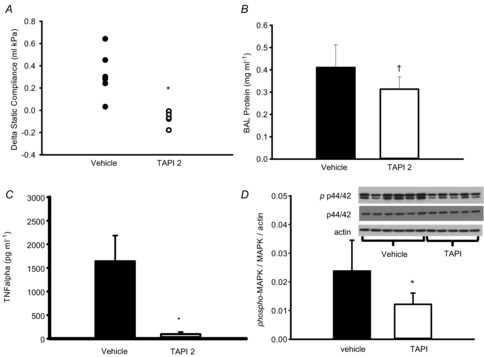
A, decrease in static compliance is prevented by TAPI-2 vs. vehicle. B, bronchoaveolar fluid protein is significantly lower in TAPI 2 vs. vehicle. C, BAL TNFα levels are suppressed by TAPI-2. D, Western blot demonstrating phosphorylation of p44/42 MAPK is significantly lower in TAPI 2 vs. vehicle. *P < 0.05, two-tailed t test, †P = 0.045 one-tailed t test; n = 5 or 6 per group. BAL, bronchoalveolar lavage; MAPK, mitogen-activated protein kinase; TNF, tumour necrosis factor.
Discussion
We show here for the first time that hypercapnia attenuates stretch-induced activation of p44/42 MAPK in lung epithelial cells, which occurs upstream at the level of ADAM17-mediated shedding of EGFR ligands. Important in vivo support for this derives from dose–response data whereby the degree of MAPK activation is proportional to the extent of lung injury, and because proximal pathway blockade (inhibition of ADAM17) is protective. These data provide new insights into the molecular pathogenesis of ventilator-associated lung injury, and may be important for patients with respiratory failure – the primary rationale for using mechanical ventilation – who frequently develop hypercapnia. The current data indicate that key effects of hypercapnia are through inhibiting stretch activation of ADAM17.
Hypercapnic acidosis inhibited the stretch-induced ADAM17 activity in cultured AEC because shedding of multiple reporters of ADAM17 activity (i.e. AP-tagged HB-EGF, AREG, TGFα) was reduced without any changes in ADAM17 protein expression or maturation. Similarly, stretch activated (and hypercapnic acidosis inhibited) the ADAM17-mediated shedding of TNFR1 in AEC in situ in isolated perfused lung. Finally, an integrated in vivo model of two-hit (i.e. LPS plus high stretch) murine lung injury was used to show that the pharmacological block of ADAM17 reduced both tissue p44/42 MAPK activation and overall lung injury. The current study demonstrates that the complete ADAM17 → EGFR→ p44/42 MAPK pathway is an important contributor to VILI, and that blockade of this pathway by proximal inhibition of ADAM17 is a mechanism by which hypercapnia may influence lung injury.
The role of ADAM17 in pulmonary inflammation and injury has been previously studied in non-epithelial cells. Using pharmacological inhibition and cell-specific knockouts in LPS-induced lung injury, endothelial ADAM17 has been shown to promote lung injury (Dreymueller et al. 2012) while knockout of myeloid ADAM17 decreased levels of alveolar neutrophils and inflammatory mediators (Arndt et al. 2011). Our data indicate a broader role for ADAM17 in lung injury: it mediates stretch-induced epithelial injury as well as a clinically relevant two-hit injury resulting from LPS and mechanical ventilation. This is important because lung injury in patients is usually initiated by a primary insult (usually sepsis), and thereafter worsened by mechanical ventilation that although necessary for oxygenation, contributes further to injury.
While exploring a signalling pathway upstream of our observation of p44/42 MAPK activation in VILI led us to ADAM17 as a target of inhibition in hypercapnic acidosis, ADAM17 acts on many other target proteins, including growth factors, cytokines, receptors and adhesion molecules (Pruessmeyer & Ludwig, 2009). Among these, there are several for which inhibition of ADAM17-dependent shedding might be expected to influence VILI, but the resulting net impact (i.e. overall benefit vs. harm) is complex and difficult to predict. For example, inhibition of shedding of L-selectin increases early steps in neutrophil influx (i.e. rolling) but ultimately decreases transmigration into tissue (Tang et al. 2011). Other adhesion molecule targets include E-selectin, Mac1, JAM-A and VE-cadherin, suggesting a complex net impact on endothelial permeability and neutrophil influx. Similarly, ADAM17-mediated shedding of TNFα and IL-6R would promote inflammatory responses while shedding of TNFR1 reduces signalling and provides a soluble antagonist for TNFα itself. Indeed, hypercapnia has been described as a ‘double-edged sword’ in lung injury (Curley et al. 2010a), and given the variety of substrates, ADAM17 inhibition could provide insight into hypercapnia's diverse effects.
The mechanisms by which ADAM17 activity is physiologically regulated (Gooz, 2010) are multifactorial and incompletely understood. ADAM17 possesses an inhibitory prodomain, which is removed by furin cleavage during maturation; it is also controlled via membrane localization of the enzyme and its substrates, by tissue inhibitor of metalloproteinase-3, and by reactive oxygen species. There is uncertainty regarding the significance of phosphorylation of ADAM17 in the maturation, trafficking or activity of the enzyme (Zhang et al. 2006; Schwarz et al. 2013). Our data indicate no effect of stretch or hypercapnia on levels, phosphorylation or maturation of ADAM17 protein, suggesting these interventions regulate enzymatic activity rather than expression or subcellular distribution. Similarly, ADAM17 has been shown to mediate EGFR ligand shedding in response to compressive stress in murine tracheal epithelial cells; however, the mechanotransduction pathway leading to ADAM17 activation is unknown (Shiomi et al. 2011). One possible hypothesis is that cyclic stretch, which generates reactive oxygen species in pulmonary epithelial cells (Chapman et al. 2005), activates redox regulation mechanisms influencing ADAM17 activity (Willems et al. 2010; Sham et al. 2013). The molecular mechanism by which hypercapnic acidosis impacts ADAM17 activity remains to be determined, and may be dependent on the hypercapnia itself or require the associated acidosis. Efforts to differentiate the contributions of CO2 versus acidosis in models of therapeutic hypercapnia are complicated in vivo due to innate physiological buffering responses. Although control of each factor independently is possible in in vitro experiments, the interpretation and relevance of such findings to the clinical context is limited and thus we have limited our cell culture experiments to hypercapnic acidosis as reflecting the clinical context that drove our hypothesis.
Available pharmacological inhibitors have limited selectivity for ADAM17 over other metalloproteases; indeed, such broad-spectrum metalloprotease inhibitors have thus far failed in clinical trials because of a high incidence of adverse side effects (Saftig & Reiss, 2011). More recently, studies have begun to focus on regulation of substrate selectivity, which could provide an opportunity for more selective blockade. For example, modulation at the substrate level has been shown in colonic epithelial cells, in which annexin A2 inhibits shedding of TNFα (but not of AREG or HB-EGF) following stimulation with phorbol 12-myristate 13-acetate or IL-1β (Tsukamoto et al. 2013). In addition, different protein kinase C isoforms specifically activate the cleavage of selected ADAM17 substrates without affecting overall protease activity; this provides additional insight into potential mechanisms whereby different triggers (e.g. reperfusion, stretch, sepsis) might select distinct substrates for cleavage (Dang et al. 2013). Our data showing similar effects on EGFR ligand and TNFR1 shedding suggest stretch and hypercapnic acidosis exert broad effects on multiple ADAM17 targets, but it will be important to extend analysis to additional cell types and substrates. In this context, understanding the precise roles of ADAM17 targets, both before and after shedding, will be key. Substrate selective inhibitors could allow fine-tuning of ADAM17 blockade, preferable to the apparently broad effect of hypercapnic acidosis.
To realize the benefits of low tidal volume ventilation, ‘permissive’ hypercapnia has become commonly accepted in mechanically ventilated patients. Hypercapnia exerts wide-ranging effects, which have been reviewed at the physiological, cellular and molecular levels (Curley et al. 2010b, 2011; Vadász et al. 2012). Key mechanistic findings include alterations in NF-κB activation and the resultant modulation of immune response, with contradictory reports of hypercapnia repressing (Takeshita et al. 2003; Contreras et al. 2012) or activating (Abolhassani et al. 2009; Oliver et al. 2012) NF-κB. There is also important evidence that hypercapnia impairs lung fluid reabsorption via inhibition of Na+,K+-ATPase (Vadasz et al. 2008) and that this inhibition is dependent on hypercapnia-induced p44/42 MAPK activity (Welch et al. 2010). This is in direct contrast to our findings of decreased p44/42 MAPK activation in hypercapnia, and probably results from differences in the model systems. For example, we measured effects of hypercapnia on stretch-induced p44/42 MAPK activation, rather than cells cultured under static condition; there are also differences in the time frames examined. Overall, the case for net harm vs. benefit in the critically ill remains the subject of debate (Beitler et al. 2013; Curley et al. 2013). Despite hypercapnia's definite disadvantages in the critically ill, it can provide an important tool in laboratory studies such as this to elucidate relevant pathophysiological pathways, as this study shows.
The inhibitory effect of hypercapnia on epithelial ADAM17 activity is clearly shown in our data. Our study employing stretch in multiple contexts (cultured primary cells, isolated perfused lung, intact animal in vivo) in both the absence and presence of additional inflammatory stimulus (LPS) suggests broad applicability of the findings. Although the relationship among these models has limitations (e.g. cyclic stretch in cell culture and in vivo models vs. static stretch in isolated perfused lung), the current data provide strong evidence that epithelial ADAM17 is activated, and injurious, in VILI. Understanding of the role of epithelial, endothelial vs. myeloid ADAM17 will require detailed studies using the relevant cell-specific knockouts (Peschon et al. 1998; Wang et al. 2013). None the less, such insights will be important due to the limited specificity of currently available pharmacological inhibitors of ADAM17, including the TAPI compounds used in our study, which can inhibit other ADAMs and matrix metalloproteases (Saftig & Reiss, 2011).
Acknowledgments
Plasmid DNAs encoding AP-tagged expression vectors for the EGFR ligands were created by Dr Shigeki Higashiyama (Ehime University, Japan) and generously provided with permission by Dr Carl Blobel (Cornell University, NY, USA).
Glossary
- ADAM17
a disintegrin and metalloprotease-17
- AEC
alveolar epithelial cells
- AREG
amphiregulin
- BAL
bronchoalveolar lavage
- EGFR
epidermal growth factor receptor
- EREG
epiregulin
- NF-κB
nuclear factor-κB
- HB-EGF
heparin binding EGF-like growth factor
- PEEP
positive end-expiratory pressure
- TGFα
transforming growth factor-α
- VILI
ventilator-induced lung injury
- VT
tidal volume
Additional information
Competing interests
None of the authors have any competing interests.
Author contributions
Experiments were performed in the laboratories of B.P.K. and J.B. G.O. conceived the study, conducted the study and analysed the data. D.E. and G.G. participated in study design, conducted experiments and analysed the data. J.B., M.P. and B.P.K. participated in study design, data analysis and interpretation. All authors participating in drafting or critically revising the manuscript and approved the final version.
Funding
This study was supported by Canadian Institutes of Health Research Operating Grant to B.P.K. (FRN 69006). B.P.K. holds the Dr Geoffrey Barker Chair in Critical Care Research.
References
- Abolhassani M, Guais A, Chaumet-Riffaud P, Sasco AJ, Schwartz L. Carbon dioxide inhalation causes pulmonary inflammation. Am J Physiol Lung Cell Mol Physiol. 2009;296:L657–L665. doi: 10.1152/ajplung.90460.2008. [DOI] [PubMed] [Google Scholar]
- The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000;342:1301–1308. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- Arndt PG, Strahan B, Wang Y, Long C, Horiuchi K, Walcheck B. Leukocyte ADAM17 regulates acute pulmonary inflammation. PLoS ONE. 2011;6:e19938. doi: 10.1371/journal.pone.0019938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beitler JR, Hubmayr RD, Malhotra A. CrossTalk opposing view: there is not added benefit to providing permissive hypercapnia in the treatment of ARDS. J Physiol. 2013;591:2767–2769. doi: 10.1113/jphysiol.2013.252619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman KE, Sinclair SE, Zhuang D, Hassid A, Desai LP, Waters CM. Cyclic mechanical strain increases reactive oxygen species production in pulmonary epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2005;289:L834–L841. doi: 10.1152/ajplung.00069.2005. [DOI] [PubMed] [Google Scholar]
- Cohen TS, Gray Lawrence G, Khasgiwala A, Margulies SS. MAPK activation modulates permeability of isolated rat alveolar epithelial cell monolayers following cyclic stretch. PLoS ONE. 2010;5:e10385. doi: 10.1371/journal.pone.0010385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras M, Ansari B, Curley G, Higgins BD, Hassett P, O'Toole D, Laffey JG. Hypercapnic acidosis attenuates ventilation-induced lung injury by a nuclear factor-kappaB-dependent mechanism. Crit Care Med. 2012;40:2622–2630. doi: 10.1097/CCM.0b013e318258f8b4. [DOI] [PubMed] [Google Scholar]
- Copland IB, Post M. Stretch-activated signaling pathways responsible for early response gene expression in fetal lung epithelial cells. J Cell Physiol. 2007;210:133–143. doi: 10.1002/jcp.20840. [DOI] [PubMed] [Google Scholar]
- Correa-Meyer E, Pesce L, Guerrero C, Sznajder JI. Cyclic stretch activates ERK1/2 via G proteins and EGFR in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2002;282:L883–L891. doi: 10.1152/ajplung.00203.2001. [DOI] [PubMed] [Google Scholar]
- Curley G, Contreras MM, Nichol AD, Higgins BD, Laffey JG. Hypercapnia and acidosis in sepsis: a double-edged sword? Anesthesiology. 2010a;112:462–472. doi: 10.1097/ALN.0b013e3181ca361f. [DOI] [PubMed] [Google Scholar]
- Curley G, Hayes M, Laffey JG. Can ‘permissive’ hypercapnia modulate the severity of sepsis-induced ALI/ARDS? Crit Care. 2011;15:212. doi: 10.1186/cc9994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curley G, Laffey JG, Kavanagh BP. Bench-to-bedside review: carbon dioxide. Crit Care. 2010b;14:220. doi: 10.1186/cc8926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curley GF, Laffey JG, Kavanagh BP. CrossTalk proposal: there is added benefit to providing permissive hypercapnia in the treatment of ARDS. J Physiol. 2013;591:2763–2765. doi: 10.1113/jphysiol.2013.252601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang M, Armbruster N, Miller MA, Cermeno E, Hartmann M, Bell GW, Root DE, Lauffenburger DA, Lodish HF, Herrlich A. Regulated ADAM17-dependent EGF family ligand release by substrate-selecting signaling pathways. Proc Natl Acad Sci U S A. 2013;110:9776–9781. doi: 10.1073/pnas.1307478110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinay T, Wu W, Kaminski N, Ifedigbo E, Kaynar AM, Szilasi M, Watkins SC, Ryter SW, Hoetzel A, Choi AM. Mitogen-activated protein kinases regulate susceptibility to ventilator-induced lung injury. PLoS ONE. 2008;3:e1601. doi: 10.1371/journal.pone.0001601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreymueller D, Martin C, Kogel T, Pruessmeyer J, Hess FM, Horiuchi K, Uhlig S, Ludwig A. Lung endothelial ADAM17 regulates the acute inflammatory response to lipopolysaccharide. EMBO Mol Med. 2012;4:412–423. doi: 10.1002/emmm.201200217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan E, Villar J, Slutsky AS. Novel approaches to minimize ventilator-induced lung injury. BMC Med. 2013;11:85. doi: 10.1186/1741-7015-11-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finigan JH, Faress JA, Wilkinson E, Mishra RS, Nethery DE, Wyler D, Shatat M, Ware LB, Matthay MA, Mason R, Silver RF, Kern JA. Neuregulin-1-human epidermal receptor-2 signaling is a central regulator of pulmonary epithelial permeability and acute lung injury. J Biol Chem. 2011;286:10660–10670. doi: 10.1074/jbc.M110.208041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi SG, Rafii B, Harris MS, Garces A, Mahuran D, Chen XJ, Bao HF, Jain L, Eaton DC, Otulakowski G, O'Brodovich H. Effects of cardiogenic edema fluid on ion and fluid transport in the adult lung. Am J Physiol Lung Cell Mol Physiol. 2007;293:L651–L659. doi: 10.1152/ajplung.00464.2006. [DOI] [PubMed] [Google Scholar]
- Gattinoni L, Pesenti A. The concept of “baby lung”. Intensive Care Med. 2005;31:776–784. doi: 10.1007/s00134-005-2627-z. [DOI] [PubMed] [Google Scholar]
- Gattinoni L, Protti A, Caironi P, Carlesso E. Ventilator-induced lung injury: the anatomical and physiological framework. Crit Care Med. 2010;38:S539–548. doi: 10.1097/CCM.0b013e3181f1fcf7. [DOI] [PubMed] [Google Scholar]
- Gooz M. ADAM-17: the enzyme that does it all. Crit Rev Biochem Mol Biol. 2010;45:146–169. doi: 10.3109/10409231003628015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickling KG, Walsh J, Henderson S, Jackson R. Low mortality rate in adult respiratory distress syndrome using low-volume, pressure-limited ventilation with permissive hypercapnia: a prospective study. Crit Care Med. 1994;22:1568–1578. doi: 10.1097/00003246-199422100-00011. [DOI] [PubMed] [Google Scholar]
- Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, Rowlands DJ, Quadri SK, Bhattacharya S, Bhattacharya J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med. 2012;18:759–765. doi: 10.1038/nm.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MN, Gusarova GA, Monma E, Das SR, Bhattacharya J. F-actin scaffold stabilizes lamellar bodies during surfactant secretion. Am J Physiol Lung Cell Mol Physiol. 2014;306:L50–L57. doi: 10.1152/ajplung.00252.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojic N, Chung E, Kho AT, Park JA, Huang A, So PT, Tschumperlin DJ. An EGFR autocrine loop encodes a slow-reacting but dominant mode of mechanotransduction in a polarized epithelium. FASEB J. 2010;24:1604–1615. doi: 10.1096/fj.09-145367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuebler WM, Parthasarathi K, Wang PM, Bhattacharya J. A novel signaling mechanism between gas and blood compartments of the lung. J Clin Invest. 2000;105:905–913. doi: 10.1172/JCI8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laffey JG, Kavanagh BP. Carbon dioxide and the critically ill – too little of a good thing? Lancet. 1999;354:1283–1286. doi: 10.1016/S0140-6736(99)02388-0. [DOI] [PubMed] [Google Scholar]
- Li LF, Chu PH, Hung CY, Kao WW, Lin MC, Liu YY, Yang CT. Lumican regulates ventilation-induced epithelial-mesenchymal transition through extracellular signal-regulated kinase pathway. Chest. 2013;143:1252–1260. doi: 10.1378/chest.12-2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LF, Liao SK, Ko YS, Lee CH, Quinn DA. Hyperoxia increases ventilator-induced lung injury via mitogen-activated protein kinases: a prospective, controlled animal experiment. Crit Care. 2007;11:R25. doi: 10.1186/cc5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindert J, Perlman CE, Parthasarathi K, Bhattacharya J. Chloride-dependent secretion of alveolar wall liquid determined by optical-sectioning microscopy. Am J RespirCell Mol Biol. 2007;36:688–696. doi: 10.1165/rcmb.2006-0347OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YY, Liao SK, Huang CC, Tsai YH, Quinn DA, Li LF. Role for nuclear factor-kappaB in augmented lung injury because of interaction between hyperoxia and high stretch ventilation. Transl Res. 2009;154:228–240. doi: 10.1016/j.trsl.2009.06.006. [DOI] [PubMed] [Google Scholar]
- Ngiam N, Peltekova V, Engelberts D, Otulakowski G, Post M, Kavanagh BP. Early growth response-1 worsens ventilator-induced lung injury by up-regulating prostanoid synthesis. Am J Respir Crit Care Med. 2010;181:947–956. doi: 10.1164/rccm.200908-1297OC. [DOI] [PubMed] [Google Scholar]
- Oliver KM, Lenihan CR, Bruning U, Cheong A, Laffey JG, McLoughlin P, Taylor CT, Cummins EP. Hypercapnia induces cleavage and nuclear localization of RelB protein, giving insight into CO2 sensing and signaling. J Biol Chem. 2012;287:14004–14011. doi: 10.1074/jbc.M112.347971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MS, He Q, Edwards MG, Sergew A, Riches DW, Albert RK, Douglas IS. Mitogen-activated protein kinase phosphatase-1 modulates regional effects of injurious mechanical ventilation in rodent lungs. Am J Respir Crit Care Med. 2012;186:72–81. doi: 10.1164/rccm.201109-1593OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltekova V, Engelberts D, Otulakowski G, Uematsu S, Post M, Kavanagh BP. Hypercapnic acidosis in ventilator-induced lung injury. Intensive Care Med. 2010;36:869–878. doi: 10.1007/s00134-010-1787-7. [DOI] [PubMed] [Google Scholar]
- Perlman CE, Bhattacharya J. Alveolar expansion imaged by optical sectioning microscopy. J Appl Physiol. 2007;103:1037–1044. doi: 10.1152/japplphysiol.00160.2007. [DOI] [PubMed] [Google Scholar]
- Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, Boyce RW, Nelson N, Kozlosky CJ, Wolfson MF, Rauch CT, Cerretti DP, Paxton RJ, March CJ, Black RA. An essential role for ectodomain shedding in mammalian development. Science. 1998;282:1281–1284. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- Pruessmeyer J, Ludwig A. The good, the bad and the ugly substrates for ADAM10 and ADAM17 in brain pathology, inflammation and cancer. Semin Cell Dev Biol. 2009;20:164–174. doi: 10.1016/j.semcdb.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Rowlands DJ, Islam MN, Das SR, Huertas A, Quadri SK, Horiuchi K, Inamdar N, Emin MT, Lindert J, Ten VS, Bhattacharya S, Bhattacharya J. Activation of TNFR1 ectodomain shedding by mitochondrial Ca2+ determines the severity of inflammation in mouse lung microvessels. J Clin Invest. 2011;121:1986–1999. doi: 10.1172/JCI43839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saftig P, Reiss K. The “A Disintegrin And Metalloproteases” ADAM10 and ADAM17: novel drug targets with therapeutic potential? Eur J Cell Biol. 2011;90:527–535. doi: 10.1016/j.ejcb.2010.11.005. [DOI] [PubMed] [Google Scholar]
- Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, Hartmann D, Saftig P, Blobel CP. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol. 2004;164:769–779. doi: 10.1083/jcb.200307137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz J, Broder C, Helmstetter A, Schmidt S, Yan I, Muller M, Schmidt-Arras D, Becker-Pauly C, Koch-Nolte F, Mittrucker HW, Rabe B, Rose-John S, Chalaris A. Short-term TNFalpha shedding is independent of cytoplasmic phosphorylation or furin cleavage of ADAM17. Biochim Biophys Acta. 2013;1833:3355–3367. doi: 10.1016/j.bbamcr.2013.10.005. [DOI] [PubMed] [Google Scholar]
- Sham D, Wesley UV, Hristova M, van der Vliet A. ATP-mediated transactivation of the epidermal growth factor receptor in airway epithelial cells involves DUOX1-dependent oxidation of Src and ADAM17. PLoS ONE. 2013;8:e54391. doi: 10.1371/journal.pone.0054391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiomi T, Tschumperlin DJ, Park J-A, Sunnarborg SW, Horiuchi K, Blobel CP, Drazen JM. TACE/ADAM17 mediates mechanotransduction in murine tracheal epithelial cells. Am J Respir Cell Mol Biol. 2011;45:376–385. doi: 10.1165/rcmb.2010-0234OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita K, Suzuki Y, Nishio K, Takeuchi O, Toda K, Kudo H, Miyao N, Ishii M, Sato N, Naoki K, Aoki T, Suzuki K, Hiraoka R, Yamaguchi K. Hypercapnic acidosis attenuates endotoxin-induced nuclear factor-kB activation. Am J Respir Cell Mol Biol. 2003;29:124–132. doi: 10.1165/rcmb.2002-0126OC. [DOI] [PubMed] [Google Scholar]
- Tang J, Zarbock A, Gomez I, Wilson CL, Lefort CT, Stadtmann A, Bell B, Huang LC, Ley K, Raines EW. Adam17-dependent shedding limits early neutrophil influx but does not alter early monocyte recruitment to inflammatory sites. Blood. 2011;118:786–794. doi: 10.1182/blood-2010-11-321406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokumaru S, Higashiyama S, Endo T, Nakagawa T, Miyagawa JI, Yamamori K, Hanakawa Y, Ohmoto H, Yoshino K, Shirakata Y, Matsuzawa Y, Hashimoto K, Taniguchi N. Ectodomain shedding of epidermal growth factor receptor ligands is required for keratinocyte migration in cutaneous wound healing. J Cell Biol. 2000;151:209–220. doi: 10.1083/jcb.151.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto H, Tanida S, Ozeki K, Ebi M, Mizoshita T, Shimura T, Mori Y, Kataoka H, Kamiya T, Fukuda S, Higashiyama S, Joh T. Annexin A2 regulates A disintegrin and metalloproteinase 17-mediated ectodomain shedding of pro-tumor necrosis factor-alpha in monocytes and colon epithelial cells. Inflamm Bowel Dis. 2013;19:1365–1373. doi: 10.1097/MIB.0b013e318281f43a. [DOI] [PubMed] [Google Scholar]
- Uhlig U, Haitsma JJ, Goldmann T, Poelma DL, Lachmann B, Uhlig S. Ventilation-induced activation of the mitogen-activated protein kinase pathway. Eur Respir J. 2002;20:946–956. doi: 10.1183/09031936.02.01612001. [DOI] [PubMed] [Google Scholar]
- Vadasz I, Dada LA, Briva A, Trejo HE, Welch LC, Chen J, Toth PT, Lecuona E, Witters LA, Schumacker PT, Chandel NS, Seeger W, Sznajder JI. AMP-activated protein kinase regulates CO2-induced alveolar epithelial dysfunction in rats and human cells by promoting Na,K-ATPase endocytosis. J Clin Invest. 2008;118:752–762. doi: 10.1172/JCI29723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadász I, Hubmayr RD, Nin N, Sporn PHS, Sznajder JI. Hypercapnia: A nonpermissive environment for the lung. Am J Respir Cell Mol Biol. 2012;46:417–421. doi: 10.1165/rcmb.2011-0395PS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Huang Z, Nayak PS, Matthews BD, Warburton D, Shi W, Sanchez-Esteban J. Strain-induced differentiation of fetal type II epithelial cells is mediated via the integrin alpha6beta1-ADAM17/tumor necrosis factor-alpha-converting enzyme (TACE) signaling pathway. J Biol Chem. 2013;288:25646–25657. doi: 10.1074/jbc.M113.473777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Maciejewski BS, Soto-Reyes D, Lee HS, Warburton D, Sanchez-Esteban J. Mechanical stretch promotes fetal type II epithelial cell differentiation via shedding of HB-EGF and TGF-alpha. J Physiol. 2009;587:1739–1753. doi: 10.1113/jphysiol.2008.163899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch LC, Lecuona E, Briva A, Trejo HE, Dada LA, Sznajder JI. Extracellular signal-regulated kinase (ERK) participates in the hypercapnia-induced Na,K-ATPase downregulation. FEBS Lett. 2010;584:3985–3989. doi: 10.1016/j.febslet.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems SH, Tape CJ, Stanley PL, Taylor NA, Mills IG, Neal DE, McCafferty J, Murphy G. Thiol isomerases negatively regulate the cellular shedding activity of ADAM17. Biochem J. 2010;428:439–450. doi: 10.1042/BJ20100179. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Thomas SM, Lui VW, Xi S, Siegfried JM, Fan H, Smithgall TE, Mills GB, Grandis JR. Phosphorylation of TNF-alpha converting enzyme by gastrin-releasing peptide induces amphiregulin release and EGF receptor activation. Proc Natl Acad Sci U S A. 2006;103:6901–6906. doi: 10.1073/pnas.0509719103. [DOI] [PMC free article] [PubMed] [Google Scholar]